

Abstract
Type 2 diabetes is a complex metabolic disorder characterized by high blood glucose in the context of insulin resistance and relative insulin deficiency by β-cell failure. Even if the mechanisms underlying the pathogenesis of β-cell failure are still under investigation, recent increasing genetic, experimental, and clinical evidence indicate that hyperactivation of the unfolded protein response (UPR) to counteract metabolic stresses is closely related to β-cell dysfunction and apoptosis. Signaling pathways of the UPR are “a double-edged sword” that can promote adaptation or apoptosis depending on the nature of the ER stress condition. In this paper, we summarized our current understanding of the mechanisms and components related to ER stress in the β-cell pathogenesis of type 2 diabetes.
1. Introduction
Modern lifestyle, with overconsumption of energy-rich foods and reduced physical activity, has increased the rate of type 2 diabetes (T2D). T2D is a major cause of morbidity and mortality, decreasing both life quality and expectancy of affected individuals. Obesity is linked to insulin resistance and T2D [1]. In order to adapt to an increased metabolic load in obesity and insulin resistance, the normal pancreatic islets usually increase beta-cell mass through an increase in β-cell proliferation and neogenesis, as well as beta cell hypertrophy [2, 3] and enhancing β-cell function [4, 5]. However, failure of adaptation to the increased metabolic load results in a progressive decline in β-cell functions and cell death. As a consequence, individuals progress from normal glucose tolerance to impaired glucose tolerance and finally to established T2D [6, 7]. Accumulating evidence indicates that β-cell loss in T2D results from intertwined stress responses of gluco-/lipotoxicity, oxidative stress, and ER stress [6–14]. However, detailed molecular mechanisms underlying β-cell dysfunction and death remain to be clarified.
2. The Unfolded Protein Response in β Cells
The endoplasmic reticulum (ER) is a major subcellular compartment involved in calcium storage, lipid production, and protein biosynthesis in which a variety of extracellular signaling molecules and protein receptors critical for cellular homeostasis are properly folded, assembled, matured, and finally transported to their destination to function. These processes rely on the protein folding activity of chaperones densely populated in the ER [13]. However, folding activity can be overwhelmed with the amount of proteins imported into the ER under the instance of “ER stress”, during which unfolded proteins accumulate in the ER and trigger downstream signaling pathways, which is called the unfolded protein response (UPR) [15]. The UPR is triggered by three ER stress signaling transducers—PKR-like ER kinase (PERK, EIF2AK3), inositol requiring 1α (IRE1α), and activating transcription factor 6α (ATF6α)—on the ER membrane, resulting in attenuation of protein translation and transcriptional activation of UPR genes [15]. In addition, cells activate a pathway to dispose of misfolded proteins from the ER, termed “ER-associated degradation (ERAD)” [16]. Regulation of these processes from biosynthesis to degradation is required for protein homeostasis, and disruption in these processes can lead to terminal misfolding and/or aggregation of proteins in the ER. Then, terminally misfolded proteins which cannot be dealt by ERAD machineries need to be cleared from the ER by an additional process such as autophagy [17]. Thus, the adaptive pathways maintain cellular function and avoid apoptosis during ER stress. However, if ER stress is severe and chronic, UPR-mediated efforts to correct the protein folding defect fail, and the apoptotic pathway is preferentially activated over time [18, 19].
Increasing evidence indicates that ER stress is associated with a variety of diseases including diabetes, neurodegenerative disease, cancer, bipolar disease, liver disease, cardiac disease, muscle degeneration, autoimmune disease, and others [20, 21]. Several scientists have found evidence that T2D may be an example of an important human disease caused by ER stress [22, 23]. T2D occurs in patients who fail to compensate for insulin resistance by increasing insulin secretion. Therefore, pancreatic β-cell dysfunction and apoptosis are central to T2D pathogenesis. In this paper, we will discuss the complex ER stress responses responsible for β-cell protection as well as dysfunction and death during T2D.
2.1. Three Stress Response Pathways in the UPR
Cells have evolved an intertwined three cellular pathway termed “the unfolded protein response” to prevent accumulation of misfolded proteins in the ER lumen. ER stress such as misfolded protein accumulation is sensed by the luminal domains of three ER transmembrane proteins: PERK, IRE1α and ATF6α. Then activated stress sensors initiate the complex signaling pathways (Figure 1) [15].
Figure 1.
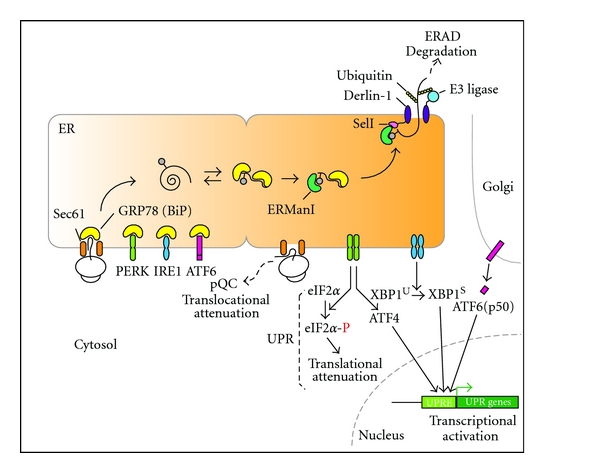
Activation of cellular responses during ER stress. In the resting state, newly synthesized polypeptides are cotranslationally translocated from the ribosome to the inside of the ER, in which GRP78 (BiP) plays two very important roles. First, GRP78 interacts and stabilizes polypeptides entering the ER and facilitates their proper folding, assembly, and maturation. Second, GRP78 interacts with PERK, IRE1α, and ATF6α, making them stay monomeric and functionally inactive on the membrane. However, these interactions are sensitive to protein folding status and can be easily disrupted by accumulation of misfolded proteins in the ER, resulting in activation of several pathways for protecting cells from accumulation of misfolded proteins: UPR, ERAD, and pQC. The pQC pathway is characterized by substrate-specific inhibition of protein translocation during ER stress, resulting in efficient degradation of mistranslocated proteins in the cytosol. This pathway is physiologically important in terms of controlling protein quantity in damaged ER. Following dissociation of GRP78 from ER stress sensors under ER stress, cells activate the UPR pathways to transfer signals to the nucleus and cytosol. PERK and IRE1α are autophosphorylated and modify their downstream signaling molecules, eIF2α phosphorylation, and Xbp1 mRNA splicing, respectively. Phosphorylated eIF2α attenuates general protein translation in short. In addition, accumulation of phosphorylated eIF2α induces ATF4 expression. Together with ATF4, spliced XBP1 and cytosolic fragments of ATF6α (p50) transcriptionally activate various UPR genes involved in either adaptation or apoptosis during ER stress.
2.1.1. PERK Pathway
During ER stress, PERK is dissociated from GRP78 (BiP), an abundant ER chaperone, then multimerizes and autophosphorylates [24]. Activation of PERK leads to phosphorylation of the alpha subunit of eukaryotic initiation factor 2 (eIF2α), which is an early response required for the attenuation of global protein translation in response to ER stress aimed to prevent further overload of the nascent polypeptides to be folded in the ER lumen [15, 25, 26]. PERK, on the other hand, induces efficient translation of several specific transcripts (such as cationic amino acid transporter 1 (cat-1), growth arrest, and DNA damage 34 (GADD34), ATF5, and ATF4) even under the condition of significant eIF2α phosphorylation [27–29]. Among them, translational increase of ATF4 induces expression of several genes involved in ER protein folding, ERAD, amino acid biosynthesis and transport function, and antioxidative stress response. Thus, translational inhibition to general mRNA transcripts but translational activation to specific mRNA transcripts by PERK is an important component of the UPR-mediated adaptation pathways to ER stress [29, 30]. Therefore, PERK activity and eIF2α phosphorylation are particularly important to maintain function of pancreatic β-cells, which continuously synthesize and secrete large amounts of insulin molecules according to physiological fluctuation of blood glucose [29, 31]. Several studies discussed below have shown that disruption of their activities increases susceptibility of β cells to death and induces β-cell failure associated with insulin resistance in T2D [23, 32, 33]. In contrast, PERK also induces expression of CHOP through transcriptional activation by ATF4 [34], an important proapoptotic gene of ER stress-mediated β-cell death [35, 36]. Therefore, it is now believed that the PERK-mediated signaling pathway may behave like a binary switch determining life and death of β cell depending on the nature of ER stress condition.
2.1.2. IRE1α Pathway
The luminal domain of PERK is functionally interchangeable in transmitting ER stress signal with IRE1α, another ER sensor, from which GRP78 is released upon exposure to ER stress [24]. IRE1α, like PERK, is autophosphorylated and dimerized by its cytoplasmic kinase domain as misfolded proteins in the ER accumulate, leading to activation of the C-terminal ribonuclease domain and specific cleavage of mRNA encoding a basic leucine zipper containing transcription factor X-box-binding protein (XBP1) [37–39]. The spliced Xbp1 mRNA encodes a strong transcription factor (XBP1s) for many UPR genes important in protein folding, trafficking, secretion, and ER-associated degradation [40–42]. Therefore, the transcriptional function of XBP1s is important for many professional secretory cells, particularly, β cells [42, 43]. Thus, the IRE1α-XBP1 pathway contributes to restoring ER homeostasis to meet protein folding demand and protein transport [44]. Beside homeostatic function, IRE1α, under chronic ER stress, also activates proapoptotic c-Jun N-terminal kinase (JNK) signaling pathway and interacts with members of the B-cell lymphoma 2 (BCL2) family, causing cellular dysfunction and apoptosis [15, 45]. Moreover, it has been shown more recently that endonuclease activity of IRE1α cleaves ER-localized mRNAs, including proinsulin mRNA, resulting in β-cell dysfunction and apoptosis [46, 47].
2.1.3. ATF6 Pathway
The stress sensors mediating the third UPR pathway activating transcription factor α (ATF6α) and ATF6β, a structural homologue of ATF6α [48], are also associated with GRP78 and retained in the ER membrane. During ER stress, both proteins released from GRP78 traffic to the Golgi apparatus [49, 50] from which their active cytosolic fragments (p50ATF6α and p60ATF6β) are generated by S1P and S2P protease and migrate into the nucleus [51]. Although the activation mode of ATF6β during ER stress seems the same to ATF6α and biochemical studies to ATF6β suggest it has similar biological functions to ATF6α, analysis of mouse embryonic fibroblasts (MEFs) deficient in ATF6α or ATF6β revealed that ATF6α but not ATF6β is responsible for transcriptional induction of ER chaperones including GRP78 and that p50ATF6α heterodimerized with XBP1s are capable of binding both ER stress response element (ERSE) and UPR elements (UPRE) conserved in the promoters of UPR genes, resulting in significant activation of genes to restore proper ER function, protein folding, and ERAD [52, 53]. However, double knockout of ATF6α and ATF6β caused embryonic lethality whereas ATF6α-or ATF6β-deficient mice are dispensable for embryonic and postnatal development, respectively, these results suggest that ATF6α and ATF6β possess at least an overlapping function which is essential for mouse development [52, 53]. Although ATF6α-null murine model did not show pancreatic β-cell demise from functional deficiency of ATF6α [52, 53], hyperactivation of ATF6α decreases insulin gene expression via upregulation of the orphan nuclear receptor small heterodimer partner (SHP; NR0B2) which has been shown to play a role in β-cell dysfunction [54].
3. ERAD and Non-ERAD Mechanisms
The UPR involves three distinct mechanisms, namely transcriptional induction of ER-resident chaperones to facilitate protein folding, translational attenuation to decrease the demand of protein folding, and ERAD to degrade the unfolded proteins accumulated in the ER lumen. Emerging data now indicate that the function of the UPR restoring ER homeostasis is facilitated by both ERAD and non-ERAD (such as autophagy and preemptive quality control) mechanisms to remove aggregated proteins from the ER and reduce new substrates during stress. Thus, efficient removal of misfolded proteins is essential to protect cells from ER stress. Here, we will review the protein degradation pathways associated with the ER.
3.1. ER-Associated Degradation (ERAD) Pathway
Eukaryotic cells have protein quality and quantity control systems aimed to dispose of misfolded proteins from the ER [55]. Consequently, chaperones in the ER distinguish physical differences between properly folded and unfolded proteins in the ER [56, 57]. Hsp70-type (such as GRP78) and glycan-dependent chaperones (such as calreticulin and calnexin) bind unfolded proteins and contribute to maintain solubility of substrates, leading to remodeling of proteins that have incorrect conformation [57–59]. Efficient removal of misfolded proteins by the ERAD pathway seems to be very specific because it directly deals with specific substrates and, apparently, is essential to protect cells from ER stress and restore proper ER function. Several distinct steps complete this pathway. First, although the mechanism selecting substrates is still under investigation, when cells recognize terminally misfolded proteins unable to acquire their native structures [60], ER-mannosidase I (ERManI) and mannosidase-like proteins (EDEMs) flag a target glycoprotein for degradation and activate the degradation pathway thereafter [61–63]. Second, substrates are retrotranslocated across the ER membrane via multicomplex channels, including Sec61, Derlin-1, or E3 ubiquitin ligase family members [64–66]. Third, target proteins are ubiquitylated by an E3 ligase. Finally, target proteins are then removed from the ER membrane and transported to the proteasome for degradation. Although the importance of the ERAD mechanism in pancreatic β-cells has not been studied extensively, recent studies suggest that defective protein degradation by reduction of ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1) activity can compromise viability in β cells in T2D. Downregulation of UCH-L1 expression and activity in β cells induces ER stress and apoptosis [67]. In addition, E3 ubiquitin ligase HRD1 may have a protective role as an ubiquitin ligase for ATF6α [68], which inhibits hyperactivation of ATF6α in the islets of WFS1-deficient mice.
3.2. Autophagy
While ERAD controls the degradation of smaller units of unfolded and misfolded proteins, larger aggregates and long-lived proteins are detoxified via degradation in the lysosome, a process called autophagy [69]. Autophagy was originally identified as a dynamic process for degradation of cytosolic organelles [70]. Now it has also been addressed as an additional degradation pathway for proteins strongly linked to the UPR pathway [69]. For example, the phosphorylation of eIF2α is also required for the induction of autophagy [71]. Therefore, ER stress stimulates autophagy as an adaptive response to clean up terminally misfolded proteins from the ER.
3.3. Preemptive Quality Control (pQC)
In addition to typical quality control pathway in mammals such as ERAD, a new degradation pathway for secretory proteins has recently been discovered. During acute ER stress, some secretory and membrane proteins are rerouted in a signal sequence-selective manner from its normal fate of being translocated into the ER to a pathway of proteasome-mediated degradation. Their cotranslational rerouting to the cytosol for degradation reduces the burden of misfolded substrates entering the ER, termed this process pre-emptive quality control (pQC) [72] For example, prion protein (PrP) is mistranslocated and rerouted to the cytosol for immediate degradation by the proteasome during ER stress. This process is largely regulated by the specific signal sequence of proteins [72, 73].
Efficient UPR pathway activated at the early stage of ER stress readily remodel misfolded proteins and restore proper ER function. As ER stress is excessive and prolonged, terminally misfolded proteins are disposed of from the ER by the ERAD pathway. At the same time, the pQC pathway reroutes misfolded proteins from the ER to the cytosol for degradation, leading to a reduction in the burden of misfolded substrates entering the ER. Therefore, when the UPR and/or ERAD pathways are compromised, the pQC pathway is apparently beneficial for cells under ER stress (Figure 1). Furthermore, to remove large aggregations, cells activate autophagy by which a large portion of aggregations can be transported directly to lysosomes for degradation without passing through the Golgi (Figure 2). However, terminally misfolded proteins often accumulate and aggregate in the ER. When the previously mentioned protein degradation mechanisms are not functionally efficient in dealing with the increasing amount of substrates, cells fail to be rescued from accumulation of misfolded proteins in the ER and cytosol. This may activate several ER stress-mediated pro-apoptotic pathways resulting in the death of stressed cells [74].
Figure 2.
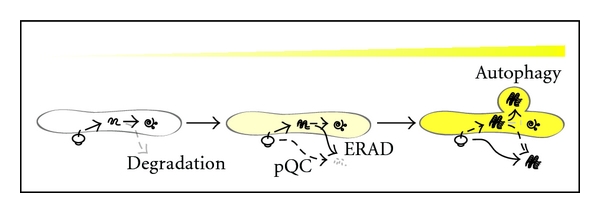
Potential activation mechanisms for disposing misfolded proteins from the ER. Misfolded proteins in the ER can be disposed of by serial activation of ERAD, pQC and autophagy according to the degree of protein misfolding and aggregation. In general, a small amount of protein is spontaneously misfolded and efficiently degraded in the ER even in the resting state. Under ER stress, accumulation of misfolded proteins in the ER activates pQC to reduce the burden of proteins in damaged ER as well as the ERAD pathway to dispose of misfolded proteins from the ER. However, under prolonged ER stress, ERAD does not efficiently dispose of and degrade protein aggregates from the ER, resulting in activation of an alternative way to clean them up from the ER, called “autophagy”. Activation of these pathways is aimed at increasing ER capacity for protecting cells from misfolded proteins.
4. Roles of UPR-Related Genes in β Cells
4.1. PERK/eIF2α Pathway
A rare human autosomal recessive genetic disorder, the Wolcott-Rallison syndrome, is characterized by early infancy diabetes, multiple epiphyseal dysplasia, and growth retardation [75, 76]. These patients have endocrine and exocrine insufficiency and pancreatic atrophy with reduced number of β-cells [77, 78]. This syndrome was found to be associated with mutations in the Perk gene. In addition to that, linkage between diabetes and Perk gene was reported in Scandinavian families [79] and South Indian populations [80].
In addition to human studies, investigations using PERK−/− mice and mice (Ser51Ala) with mutation in the phosphorylation site of eIF2α showed a potential relationship between ER stress and β-cell function [29, 30, 81]. Pancreatic β cells developed normally in whole body Perk-null mice but showed a diabetic phenotype soon after birth mainly due to β-cell death [30, 81, 82]. In these studies, ER distention was observed in pancreatic β cells. In addition, there was more proinsulin production in Perk−/− β cells challenged by high glucose, causing them to experience higher translational loads and higher levels of ER stress than wild type. These results suggested that ER overload and unresolved ER stress may cause β-cell death in those mice. Conditional deletion of Perk at different developmental stages showed that PERK expression in β-cells is not required postnatally in adult mice to maintain glucose homeostasis, whereas its expression is a prerequisite for fetal/neonatal β cell proliferation and differentiation [82].
Similar to Perk−/− mice, eIF2α homozygous mutant mice showed deficiency in pancreatic β cells in the embryonic stage, and they died within 18 hr after birth [29]. Moreover, the β cells of late embryonic stage in the mutant mice have reduced insulin contents and show severe distension of the ER [31, 33]. The results suggest that function of eIF2α phosphorylation may be required for β-cell differentiation and proliferation, along with embryonic β cells of Perk−/− mice. However, recent results showed that eIF2α phosphorylation but not PERK expression in β cells is required at the adult stage to maintain β-cell functions and glucose homeostasis. The absence of eIF2α phosphorylation in β cells caused defective intracellular trafficking of ER cargo proteins, increased oxidative damage, and reduced expression of stress response and β-cell-specific genes and apoptosis due to heightened and unregulated proinsulin translation [33]. Since Ser51Ala homozygous mutation in eIF2α (A/A) does not allow any compensatory phosphorylation by other eIF2α kinases (such as heme-regulated inhibitor kinase (HRI, EIF2AK1), general control of nitrogen metabolism kinase 2 (GCN2, EIF2AK4), and dsRNA-activated protein kinase (PKR, EIF2AK2)) [83] in adult β cells in response to physiological stimuli, whereas other eIF2α kinase in PERK-deficient adult β cells might compensate the requirements for eIF2α phosphorylation. This characteristic might contribute to phenotypic discrepancies between Perk−/− and A/A mice. The heterozygous mutant (S/A) mice did not spontaneously develop diabetes; however, on a 45% high fat diet, these mice showed glucose intolerance and insulin secretion defect in β cells [31]. These results suggested that translational regulation through eIF2α phosphorylation is required to maintain functional integrity of the ER. This hypothesis was further demonstrated by a study using mice with conditional A/A mutation in β cells [33]. The A/A mice were rescued by transgenic expression of wild-type eIF2α cDNA, which could be specifically deleted in β cells by tamoxifen-inducible Cre recombinase. As early as 3 weeks after deletion of wild-type eIF2α cDNA, β cells showed significantly distended ER and swollen mitochondria [33].
4.2. WFS1
Wolfram syndrome (WFS) is a rare autosomal recessive neurodegenerative disorder characterized by early onset diabetes, optic atrophy, and hearing impairment [84]. This syndrome is genetically linked with mutations in the Wfs1 gene that encodes the protein wolframin [85, 86]. As in human cases, mice deficient in the Wfs1 gene developed glucose intolerance and overt diabetes due to insufficient insulin secretion. Pancreatic β cells in mutant mice experienced ER stress shown by phosphorylation of eIF2α and spliced form of XBP1 [87–89]. Recent study suggests that WFS1 may affect maturation of plasma membrane proteins or stability of ER membrane proteins. Deficiency of WFS1 in β cells destabilizes Na+/K+ ATPase β1 subunit and E3 ubiquitin ligase HRD1 [68, 90]. Thus, wolframin may be involved in the ER folding and assembly of subunits of oligomeric proteins. In addition, Wolframin may suppress ATF6α hyperactivation in β cells by stabilizing HRD1, which brings ATF6α to the proteasome. Therefore, WFS may have a role as a negative regulator of chronic or unresolvable ER stress.
4.3. P58IPK
Although p58IPK (IPK, inhibitor of protein kinase) was first known to be an inhibitor of the PKR, its function has been shown to inhibit another eIF2α kinase, PERK. However, recent evidence suggests that p58IPK serves as a cochaperone in the ER lumen for the Hsp70 family member BiP [91]. Mice lacking the p58IPK gene showed gradual onset of glucosuria and hyperglycemia mainly due to apoptosis of pancreatic β cells [92]. In addition, p58IPK deletion in Akita mice (carrying a C96Y mutation in the Ins2 gene) exacerbate the diabetic phenotype [93]. These results indicate that chaperoning ability in the ER is important to preserve ER function in β cells.
4.4. ATF6α
ATF6α was also found to be associated with β-cell function in genetic studies. In a study of Pima Indians, a native American population with a high prevalence of type II diabetes, [94] they found an association of variants in ATF6 with T2D [95]. In another study conducted in a Dutch cohort, they also found that the majority of single nucleotide polymorphisms (SNPs) in Aft6 allele were significantly associated with impaired fasting glucose, impaired glucose tolerance, and T2D [96]. Furthermore, associated variants differed from those identified in the Pima Indians. Since ATF6α is important for protective cell response to accumulation of unfolded and misfolded proteins in the ER, disturbances of this process might contribute to β-cell apoptosis. However, there is no direct evidence of pancreatic β-cell demise in the ATF6α null murine model.
5. Causes of ER Stress in β Cell
Increasing evidence indicates that ER stress is one of the main causes of β-cell dysfunction and death [6–14]. However, it is not clear what is the main cause of ER stress in β cells, and which subpathway of UPR is responsible for this process. In this section, we will describe the potential sources and mechanisms for ER stress-mediated β-cell demise.
5.1. Lipotoxicity-Mediated ER Stress
Hyperlipidemia (elevated serum lipid levels) also results from sustained insulin resistance. It is thought that chronically elevated levels of circulating free fatty acids (FFAs) are putative mediators of progressive β-cell dysfunction and death in T2D [97]. When FFA levels are elevated twofold above the basal upon lipid infusion, obese nondiabetic individuals showed a significant reduction in glucose-stimulated insulin release [98]. It has recently been shown that a saturated long-chain FFA palmitate induces ER stress in both clonal and primary murine and human β cells, whereas unsaturated long-chain FFAs do or do not induce ER stress to a lesser content [10, 11, 99, 100]. Palmitate preferentially activates both PERK and IRE1α pathways [101–103]. However, it is uncertain whether palmitate can specifically activate the ATF6α branch because both palmitate and oleate induce total Xbp1 mRNA, and the expression of the known ATF6α target gene Hspa5 (Grp78) is controversial [102, 104]. How saturated FFAs activate the unfolded protein response has not been answered. Several recent studies indicate that palmitate triggers ER stress in β cells through perturbation in ER Ca2+ handling. Calcium-specific dye assays revealed palmitate depletes ER Ca2+ and slows ER Ca2+ uptake in β cells [104]. Although palmitate-mediated ER Ca2+ depletion increased misfolded protein accumulation as the mechanism of the sarco-/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitors thapsigargin and cyclopiazonic acid, two commonly known synthetic ER stressors, the detailed molecular mechanism of ER Ca2+ depletion by palmitate is not well known. Other reports indicated palmitate rapidly increases the saturated lipid content of the ER, leading to compromised ER morphology and integrity and thereby may directly or indirectly induce ER stress [100, 105]. For example, impairment of lipid content control of the ER by palmitate hampers ER-to-Golgi protein trafficking of vesicular stomatitis virus G protein (VSVG), contributing to the unfolded protein response through accumulation of misfolded proteins in the ER lumen [106]. Interestingly, increased fatty acid desaturation by stearoyl coenzyme A desaturase 1 (SCD1) reduced palmitate-induced cell death in MIN6 β cells and human embryonic kidney (HEK) cells [107]. In contrast, knockdown of SCD in INS-1 β cells decreased desaturation of palmitate to monounsaturated fatty acid (MUFA), lowered fatty acid partitioning into complex neutral lipids, and augmented palmitate-induced ER stress and apoptosis [108]. Furthermore, the importance of lipid content control of the ER by SCD in β cells was further manifested by studies of diabetic murine models. First, loss of SCD1 worsens diabetes in leptin-deficient obese mice [109]. Second, SCD1 and SCD2 mRNA expression were shown to be induced in islets from prediabetic hyperinsulinemic Zucker diabetic fatty (ZDF) rats, whereas several fatty acid desaturases including SCD1 mRNA levels were markedly reduced in diabetic ZDF rat islets [108].
5.2. Misfolded Protein-Mediated ER Stress
Since proinsulin represents up to 20% of the total mRNA and 30–50% of the total protein synthesis in the β cell [110–112], misfolded mutant insulin proteins might be a potent cause of ER stress. The Akita mutant in both mouse and human, which carries a cysteine 96 to tyrosine substitution in the Ins2 gene, showed hyperglycemia and a reduced β cell mass [35]. This missense mutation disrupts a disulfide bond formation of mature insulin causing incorrect folding of proinsulin in the ER. Accumulation of the mutant insulin induced cell death mainly through ER stress evidenced by upregulated expression of ER stress marker genes such as BiP, spliced XBP1s, activated ATF6α, and CHOP in the Akita mutant islets and β-cell lines [113–115]. In heterozygous, but not homozygous, Akita's mutant mice, the homozygous disruption of CHOP delayed diabetes development suggested that β-cell death is partially CHOP dependent [93].
Similar to Akita diabetes due to accumulation of misfolded proinsulin, ER accumulation of islet amyloid polypeptide (IAPP, amylin) oligomers may contribute to β-cell loss in T2D [12]. Human IAPP is an 89 amino acid protein that undergoes processing to a 37-amino acid amyloidogenic peptide coexpressed and cosecreted with insulin by β cells. A study showed that islet amyloid is present at autopsy in over than 90% of patients with T2D [8]. Moreover, increased expression of ER stress marker genes such as BiP and CHOP was observed in islets of human T2D patients [22]. The IAPP-driven ER stress theory was exemplified by animal studies overexpressing human IAPP in β cells. Human IAPP but not murine IAPP forms toxic oligomers and triggers ER stress-induced apoptosis in β cells of both rat and mouse murine models [12]. Therefore, intracellular deposit of human IAPP toxic oligomer could be a link between ER stress and β-cell death in human T2D.
5.3. High Glucose-Mediated ER Stress
Insulin resistance in T2D causes blood glucose levels to remain high [116]. In a high blood glucose state, called hyperglycemia, the β-cell increases its metabolic activity, which eventually leads to cellular stress. This in turn may further impair β-cell function and survival, a process called glucotoxicity [117]. It has been shown that glucotoxicity is mediated at least in part by excess generation of reactive oxygen species (ROS) [117, 118]. When excess glucose is available to the β cell, excessive ROS can be generated in β cells by several biochemical pathways including mitochondrial oxidative phosphorylation, ER oxidative folding pathway, and other alternative metabolism pathways; overflowed glucose is shunted (such as glucosamine and hexosamine metabolism and sorbitol metabolism) [117, 118]. Elevated ROS perturbs insulin synthesis and secretion by decreasing the expression and activity of key transcription factors such as PDX-1 and MafA, which regulate proinsulin genes and other multiple genes involved in β-cell differentiation, proliferation, and survival [119].
Accumulating evidence suggests that protein folding in the ER and production of ROS are closely linked events [120]. In several reports, prolonged UPR activation leads to the accumulation of ROS via two sources: the UPR-regulated oxidative protein folding machinery in the ER and oxidative phosphorylation in mitochondria [118, 121]. In the ER lumen, oxidative protein folding is conducted by protein disulfide isomerase (PDI) and a family of ER oxidoreductases 1 (ERO1) that catalyze disulfide bond formation in folding proteins. In this reaction, an oxidant flavin adenine dinucleotide (FAD)-bound ERO1 oxidizes PDI, which then subsequently oxidizes folding proteins directly. FAD-bound ERO1 then passes two electrons to molecular oxygen, perhaps resulting in the production of ROS such as hydrogen peroxide or peroxide [120]. Overexpression of a misfolded protein CPY (yeast vacuolar protein carboxypeptidase Y) activates the UPR, causes oxidative stress, and induces apoptosis. However, removal of all cysteine residues in CPY reduced oxidative stress and cell death [122]. Therefore, oxidative protein folding in the ER can be a source of ROS generation. It is known that ER stress increase leakage of Ca2+ from the ER lumen through mainly the inositol-1,4,5-trisphosphate receptor (IP3R) [21]. Recent studies suggest that ER Ca2+ leakage may occur by oxidation-induced activation of the Ca2+ release channel IP3R during ER stress and oxidative stress [123]. Increases in cytosolic Ca2+ can stimulate mitochondrial ROS production through multiple mechanisms [124]. Introduced by Ca2+ uniporter or mitochondrial ryanodine receptor, Ca2+ stimulates the TCA cycle and nitric oxide synthase (NOS), which subsequently generates nitric oxide. Nitric oxide and Ca2+ inhibits respiration complex I, III, or IV, which enhance ROS generation. High levels of ROS generation within the mitochondria then further increase Ca2+ release from the ER. In turn, Ca2+ also dissociates cytochrome c from the inner membrane cardiolipin, which triggers permeability transition pore (PTP) opening and cytochrome c release across the outer membrane. Now the vicious cycle of ER Ca2+ release and mitochondrial ROS production activates cytochrome-c-mediated apoptosis. The ER of diabetic pancreatic β cells synthesizing great quantities of proinsulin to maintain normoglycemia can be an important site of ROS production because correct folding of proinsulin absolutely depends on the formation of disulfide bond by oxidative protein folding. This theory is partially supported by recent reports that PERK and eIF2α phosphorylation-deficient β cells having reduced UPR responses showed increased proinsulin synthesis due to losing of translational control; thereby, large accumulation of proinsulin in the ER attributed to accumulated ROS in β cells possibly by both ER oxidative protein folding machinery and Ca2+ mediated-mitochondria activation [33, 82]. Thus, excess ROS and the mitochondrial cell death pathway may induce β-cell death. Therefore, the finding that increased proinsulin synthesis causes oxidative damage in β cells may reflect events in β-cell failure associated with insulin resistance in T2D [74].
Under high glucose conditions, it is possible that increased proinsulin biosynthesis may overwhelm the ER protein folding capacity leading to UPR activation. Chronic exposure (more than 24 hrs) of β cells to high-glucose caused hyperactivation of IRE1α showing Xbp1 mRNA splicing, whereas acute expose (1–3 hrs) to high glucose activated IRE1α without Xbp1 mRNA splicing [47, 118, 125]. In the chronic high-glucose state, activated IRE1α degrades proinsulin mRNA contributing to the reduction of proinsulin biosynthesis [47, 125]. Moreover, recent reports suggest that IRE1α's RNase causes endonucleolytic decay of many ER's localized mRNAs, including those encoding chaperones, thereby culminating in cellular dysfunction and death of several mammalian cells including β cells [46, 47, 125]. However, the studies also revealed that activated IRE1α at the physiological level may have a beneficial effect aiding in the enhancement of proinsulin biosynthesis in pancreatic β cells with the induction of a subset of downstream genes of IRE1α [47]. Thus, depending on the forms of ER stress, β cells may generate binary signaling of life and death through IRE1α.
It is well established that high blood sugar amplifies FFA-mediated toxicity in β cells [117, 118]. Although why glucose exacerbates β-cell lipotoxicity is not well known, a recent report suggests that chronic hyperglycemia may amplify fatty acid-induced ER stress in β cells [101]. The study showed that high glucose amplifies palmitate-mediated stimulation of the IRE1α and PERK pathways. Glucose stimulates the mammalian target of rapamycin complex 1 (mTORC1), an important nutrient sensor involved in the regulation of cellular stress, and the activated mTORC1 mediates amplification of fatty acid's lipotoxicity by increasing IRE1α protein levels and activating the JNK pathway, leading to increased β-cell apoptosis.
6. Concluding Remarks
Increasing evidence indicates that hyperactivation of the UPR indispensible for ER homeostasis has a role in β cell dysfunction and death during the progression of T2D and genetic forms of diabetes. Therefore, it is currently believed that ER stress-related diseases including T2D occur from adaptation to apoptosis of stressed cells. The complete understanding of this molecular mechanism responsible for life and death will shed light on future T2D prevention and treatment.
Acknowledgments
This work was supported by a Korea Research Foundation grant funded by the Korean Government (MOEHRD) (BRL-2010-0001199). The authors apologize for the exclusion of relevant peer-reviewed articles due to size limitations. The authors thank Dr. Zac Callaway for assistance with the manuscript preparation.
References
- 1.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 2.Steil GM, Trivedi N, Jonas J-C, et al. Adaptation of β-cell mass to substrate oversupply: enhanced function with normal gene expression. American Journal of Physiology. 2001;280(5):E788–E796. doi: 10.1152/ajpendo.2001.280.5.E788. [DOI] [PubMed] [Google Scholar]
- 3.Jetton TL, Lausier J, LaRock K, et al. Mechanisms of compensatory β-cell growth in insulin-resistant rats: roles of Akt kinase. Diabetes. 2005;54(8):2294–2304. doi: 10.2337/diabetes.54.8.2294. [DOI] [PubMed] [Google Scholar]
- 4.Liu YQ, Jetton TL, Leahy JL. β-cell adaptation to insulin resistance. Increased pyruvate carboxylase and malate-pyruvate shuttle activity in islets of nondiabetic zucker fatty rats. Journal of Biological Chemistry. 2002;277(42):39163–39168. doi: 10.1074/jbc.M207157200. [DOI] [PubMed] [Google Scholar]
- 5.Chen C, Hosokawa H, Bumbalo LM, Leahy JL. Mechanism of compensatory hyperinsulinemia in normoglycemic insulin- resistant spontaneously hypertensive rats. Augmented enzymatic activity of glucokinase in β-cells. Journal of Clinical Investigation. 1994;94(1):399–404. doi: 10.1172/JCI117335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 7.Prentki M, Nolan CJ. Islet β cell failure in type 2 diabetes. Journal of Clinical Investigation. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cnop M, Welsh N, Jonas J-C, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(supplement 2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 9.Poitout V, Robertson RP. Minireview: secondary β-cell failure in type 2 diabetes—a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143(2):339–342. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 10.Cnop M, Igoillo-Esteve M, Cunha DA, Ladrière L, Eizirik DL. An update on lipotoxic endoplasmic reticulum stress in pancreatic β-cells. Biochemical Society Transactions. 2008;36(5):909–915. doi: 10.1042/BST0360909. [DOI] [PubMed] [Google Scholar]
- 11.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine Reviews. 2008;29(1):42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 12.Haataja L, Gurlo T, Huang C-J, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocrine Reviews. 2008;29(3):303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with β-cell failure and diabetes. Endocrine Reviews. 2008;29(3):317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robertson AP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. Journal of Biological Chemistry. 2004;279(41):42351–42354. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]
- 15.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 16.Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nature Cell Biology. 2005;7(8):766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 17.Kruse KB, Brodsky JL, McCracken AA. Autophagy: an ER protein quality control process. Autophagy. 2006;2(2):135–137. doi: 10.4161/auto.2.2.2388. [DOI] [PubMed] [Google Scholar]
- 18.Rutkowski DT, Arnold SM, Miller CN, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biology. 2006;4(11):p. e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends in Biochemical Sciences. 2007;32(10):469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66(2):S102–S109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 21.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nature Reviews Drug Discovery. 2008;7(12):1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 22.Laybutt DR, Preston AM, Åkerfeldt MC, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50(4):752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- 23.Marchetti P, Bugliani M, Lupi R, et al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia. 2007;50(12):2486–2494. doi: 10.1007/s00125-007-0816-8. [DOI] [PubMed] [Google Scholar]
- 24.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature Cell Biology. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 25.Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annual Review of Cell and Developmental Biology. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- 26.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiological Reviews. 2006;86(4):1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 27.Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular Cell. 2000;6(5):1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 28.Jiang HY, Wek SA, McGrath BC, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Molecular and Cellular Biology. 2004;24(3):1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scheuner D, Song B, McEwen E, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Molecular Cell. 2001;7(6):1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 30.Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in Perk-/- mice reveals a role for translational control in secretory cell survival. Molecular Cell. 2001;7(6):1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 31.Scheuner D, Mierde DV, Song B, et al. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nature Medicine. 2005;11(7):757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- 32.Herbert TP. PERK in the life and death of the pancreatic β-cell. Biochemical Society Transactions. 2007;35(5):1205–1207. doi: 10.1042/BST0351205. [DOI] [PubMed] [Google Scholar]
- 33.Back SH, Scheuner D, Han J, et al. Translation attenuation through eIF2α phosphorylation prevents oxidative stress and maintains the differentiated state in β cells. Cell Metabolism. 2009;10(1):13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic- reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 35.Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. Journal of Clinical Investigation. 2002;109(4):525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. Journal of Clinical Investigation. 2008;118(10):3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calfon M, Zeng H, Urano F, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415(6867):92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 38.Shen X, Ellis RE, Lee K, et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107(7):893–903. doi: 10.1016/s0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 40.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21(1):81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and Cellular Biology. 2003;23(21):7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Acosta-Alvear D, Zhou Y, Blais A, et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Molecular Cell. 2007;27(1):53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 43.Lipson KL, Fonseca SG, Ishigaki S, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metabolism. 2006;4(3):245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 44.Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Current Opinion in Cell Biology. 2011;23(2):143–149. doi: 10.1016/j.ceb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 46.Han D, Lerner AG, Vande Walle L, et al. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138(3):562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lipson KL, Fonseca SG, Ishigaki S, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metabolism. 2006;4(3):245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 48.Li MQ, Baumeister P, Roy B, et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Molecular and Cellular Biology. 2000;20(14):5096–5106. doi: 10.1128/mcb.20.14.5096-5106.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the er to the Golgi. Journal of Biological Chemistry. 2002;277(15):13045–13052. doi: 10.1074/jbc.M110636200. [DOI] [PubMed] [Google Scholar]
- 50.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Developmental Cell. 2002;3(1):99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 51.Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Molecular Cell. 2000;6(6):1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 52.Wu J, Rutkowski DT, Dubois M, et al. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Developmental Cell. 2007;13(3):351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 53.Yamamoto K, Sato T, Matsui T, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Developmental Cell. 2007;13(3):365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 54.Seo HY, Yong DK, Lee KM, et al. Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression via up-regulation of orphan nuclear receptor small heterodimer partner. Endocrinology. 2008;149(8):3832–3841. doi: 10.1210/en.2008-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hegde RS, Ploegh HL. Quality and quantity control at the endoplasmic reticulum. Current Opinion in Cell Biology. 2010;22(4):437–446. doi: 10.1016/j.ceb.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nature Reviews Molecular Cell Biology. 2003;4(3):181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 57.Fewell SW, Travers KJ, Weissman JS, Brodsky JL. The action of molecular chaperones in the early secretory pathway. Annual Review of Genetics. 2001;35:149–191. doi: 10.1146/annurev.genet.35.102401.090313. [DOI] [PubMed] [Google Scholar]
- 58.Trombetta ES, Parodi AJ. Quality control and protein folding in the secretory pathway. Annual Review of Cell and Developmental Biology. 2003;19:649–676. doi: 10.1146/annurev.cellbio.19.110701.153949. [DOI] [PubMed] [Google Scholar]
- 59.Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92(3):351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 60.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annual Review of Biochemistry. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 61.Hosokawa N, Wada I, Hasegawa K, et al. A novel ER α-mannosidase-like protein accelerates ER-associated degradation. EMBO Reports. 2001;2(5):415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakatsukasa K, Nishikawa SI, Hosokawa N, Nagata K, Endo T. Mnl1p, an α-mannosidase-like protein in yeast Saccharomyces cerevisiae, is required for endoplasmic reticulum-associated degradation of glycoproteins. Journal of Biological Chemistry. 2001;276(12):8635–8638. doi: 10.1074/jbc.C100023200. [DOI] [PubMed] [Google Scholar]
- 63.Jakob CA, Bodmer D, Spirig U, et al. Htm1p, a mannosidase-like protein, is involved in glycoprotein degradation in yeast. EMBO Reports. 2001;2(5):423–430. doi: 10.1093/embo-reports/kve089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hebert DN, Bernasconi R, Molinari M. ERAD substrates: which way out? Seminars in Cell and Developmental Biology. 2010;21(5):526–532. doi: 10.1016/j.semcdb.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 65.Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase hrd1p. Cell. 2010;143(4):579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ye Y, Shibata Y, Kikkert M, Van Voorden S, Wiertz E, Rapoport TA. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(40):14132–14138. doi: 10.1073/pnas.0505006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Costes S, Huang C-J, Gurlo T, et al. β-cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes. 2011;60(1):227–238. doi: 10.2337/db10-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fonseca SG, Ishigaki S, Oslowski CM, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. Journal of Clinical Investigation. 2010;120(3):744–755. doi: 10.1172/JCI39678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hetz C, Thielen P, Matus S, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes and Development. 2009;23(19):2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stromhaug PE, Klionsky DJ. Approaching the molecular mechanism of autophagy. Traffic. 2001;2(8):524–531. doi: 10.1034/j.1600-0854.2001.20802.x. [DOI] [PubMed] [Google Scholar]
- 71.Kouroku Y, Fujita E, Tanida I, et al. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death and Differentiation. 2007;14(2):230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 72.Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell. 2006;127(5):999–1013. doi: 10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hegde RS, Kang SW. The concept of translocational regulation. Journal of Cell Biology. 2008;182(2):225–232. doi: 10.1083/jcb.200804157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaufman RJ, Back SH, Song B, Han J, Hassler J. The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation in β-cells. Diabetes, Obesity and Metabolism. 2010;12(supplement 2):99–107. doi: 10.1111/j.1463-1326.2010.01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wolcott CD, Rallison ML. Infancy-onset diabetes mellitus and multiple epiphyseal dysplasia. The Journal of Pediatrics. 1972;80(2):292–297. doi: 10.1016/s0022-3476(72)80596-1. [DOI] [PubMed] [Google Scholar]
- 76.Stoss H, Pesch HJ, Pontz B. Wolcott-Rallison syndrome: diabetes mellitus and spondyloepiphyseal dysplasia. European Journal of Pediatrics. 1982;138(2):120–129. doi: 10.1007/BF00441137. [DOI] [PubMed] [Google Scholar]
- 77.Thornton CM. Autopsy findings in the Wolcott-Rallison syndrome. Pediatric Pathology and Laboratory Medicine. 1997;17(3):487–496. [PubMed] [Google Scholar]
- 78.Castelnau P, Le Merrer M, Diatloff-Zito C, Marquis E, Tête MJ, Robert JJ. Wolcott-Rallison syndrome: a case with endocrine and exocrine pancreatic deficiency and pancreatic hypotrophy. European Journal of Pediatrics. 2000;159(8):631–633. doi: 10.1007/pl00008394. [DOI] [PubMed] [Google Scholar]
- 79.Holm P, Julier C, Kockum I, et al. A genomewide scan for type 1-diabetes susceptibility in scandinavian families: identification of new loci with evidence of interactions. American Journal of Human Genetics. 2001;69(6):1301–1313. doi: 10.1086/324341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Allotey RA, Mohan V, McDermott MF, et al. The EIF2AK3 gene region and type I diabetes in subjects from South India. Genes and Immunity. 2004;5(8):648–652. doi: 10.1038/sj.gene.6364139. [DOI] [PubMed] [Google Scholar]
- 81.Zhang P, McGrath B, Li S, et al. The PERK eukaryotic initiation factor 2α kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Molecular and Cellular Biology. 2002;22(11):3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic β cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metabolism. 2006;4(6):491–497. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 83.Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxidants and Redox Signaling. 2007;9(12):2357–2371. doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- 84.Barrett TG, Bundey SE. Wolfram (DIDMOAD) syndrome. Journal of Medical Genetics. 1997;34(10):838–841. doi: 10.1136/jmg.34.10.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khanim F, Kirk J, Latif F, Barrett TG. WFS1/wolframin mutations, wolfram syndrome, and associated diseases. Human Mutation. 2001;17(5):357–367. doi: 10.1002/humu.1110. [DOI] [PubMed] [Google Scholar]
- 86.Cryns K, Sivakumaran TA, Van den Ouweland JMW, et al. Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Human Mutation. 2003;22(4):275–287. doi: 10.1002/humu.10258. [DOI] [PubMed] [Google Scholar]
- 87.Yamada T, Ishihara H, Tamura A, et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic β-cells. Human Molecular Genetics. 2006;15(10):1600–1609. doi: 10.1093/hmg/ddl081. [DOI] [PubMed] [Google Scholar]
- 88.Ishihara H, Takeda S, Tamura A, et al. Disruption of the WFS1 gene in mice causes progressive β-cell loss and impaired stimulus—secretion coupling in insulin secretion. Human Molecular Genetics. 2004;13(11):1159–1170. doi: 10.1093/hmg/ddh125. [DOI] [PubMed] [Google Scholar]
- 89.Riggs AC, Bernal-Mizrachi E, Ohsugi M, et al. Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia. 2005;48(11):2313–2321. doi: 10.1007/s00125-005-1947-4. [DOI] [PubMed] [Google Scholar]
- 90.Zatyka M, Ricketts C, da Silva Xavier G, et al. Sodium-potassium ATPase β1 subunit is a molecular partner of Wolframin, an endoplasmic reticulum protein involved in ER stress. Human Molecular Genetics. 2008;17(2):190–200. doi: 10.1093/hmg/ddm296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rutkowski DT, Kang SW, Goodman AG, et al. The role of p58IPK in protecting the stressed endoplasmic reticulum. Molecular Biology of the Cell. 2007;18(9):3681–3691. doi: 10.1091/mbc.E07-03-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ladiges WC, Knoblaugh SE, Morton JF, et al. Pancreatic β-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes. 2005;54(4):1074–1081. doi: 10.2337/diabetes.54.4.1074. [DOI] [PubMed] [Google Scholar]
- 93.Oyadomari S, Yun C, Fisher EA, et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell. 2006;126(4):727–739. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 94.Hanson RL, Ehm MG, Pettitt DJ, et al. An autosomal genomic scan for loci linked to type II diabetes mellitus and body-mass index in Pima Indians. American Journal of Human Genetics. 1998;63(4):1130–1138. doi: 10.1086/302061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thameem F, Farook VS, Bogardus C, Prochazka M. Association of amino acid variants in the activating transcription factor 6 gene (ATF6) on 1q21-q23 with type 2 diabetes in Pima Indians. Diabetes. 2006;55(3):839–842. doi: 10.2337/diabetes.55.03.06.db05-1002. [DOI] [PubMed] [Google Scholar]
- 96.Meex SJR, Van Greevenbroek MMJ, Ayoubi TA, et al. Activating transcription factor 6 polymorphisms and haplotypes are associated with impaired glucose homeostasis and type 2 diabetes in Dutch Caucasians. Journal of Clinical Endocrinology and Metabolism. 2007;92(7):2720–2725. doi: 10.1210/jc.2006-2280. [DOI] [PubMed] [Google Scholar]
- 97.Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM: genetic and clinical implications. Diabetes. 1995;44(8):863–870. doi: 10.2337/diab.44.8.863. [DOI] [PubMed] [Google Scholar]
- 98.Carpentier A, Mittelman SD, Bergman RN, Giacca A, Lewis GF. Prolonged elevation of plasma free fatty acids impairs pancreatic β- cell function in obese nondiabetic humans but not in individuals with type 2. Diabetes. 2000;49(3):399–408. doi: 10.2337/diabetes.49.3.399. [DOI] [PubMed] [Google Scholar]
- 99.Cnop M, Ladrière L, Igoillo-Esteve M, Moura RF, Cunha DA. Causes and cures for endoplasmic reticulum stress in lipotoxic β-cell dysfunction. Diabetes, Obesity and Metabolism. 2010;12(supplement 2):76–82. doi: 10.1111/j.1463-1326.2010.01279.x. [DOI] [PubMed] [Google Scholar]
- 100.Cnop M. Fatty acids and glucolipotoxicity in the pathogenesis of Type 2 diabetes. Biochemical Society Transactions. 2008;36(3):348–352. doi: 10.1042/BST0360348. [DOI] [PubMed] [Google Scholar]
- 101.Bachar E, Ariav Y, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G. Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic β-cells via activation of mTORC1. PLoS ONE. 2009;4(3, article e4954) doi: 10.1371/journal.pone.0004954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic β-cell apoptosis. Endocrinology. 2006;147(7):3398–3407. doi: 10.1210/en.2005-1494. [DOI] [PubMed] [Google Scholar]
- 103.Kharroubi I, Ladrière L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic β-cell apoptosis by different mechanisms: role of nuclear factor-κB and endoplasmic reticulum stress. Endocrinology. 2004;145(11):5087–5096. doi: 10.1210/en.2004-0478. [DOI] [PubMed] [Google Scholar]
- 104.Cunha DA, Hekerman P, Ladrière L, et al. Initiation and execution of lipotoxic ER stress in pancreatic β-cells. Journal of Cell Science. 2008;121(14):2308–2318. doi: 10.1242/jcs.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. Journal of Lipid Research. 2006;47(12):2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 106.Preston AM, Gurisik E, Bartley C, Laybutt DR, Biden TJ. Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse beta cells by promoting protein overload. Diabetologia. 2009;52(11):2369–2373. doi: 10.1007/s00125-009-1506-5. [DOI] [PubMed] [Google Scholar]
- 107.Busch AK, Gurisik E, Cordery DV, et al. Increased fatty acid desaturation and enhanced expression of stearoyl coenzyme A desaturase protects pancreatic β-cells from lipoapoptosis. Diabetes. 2005;54(10):2917–2924. doi: 10.2337/diabetes.54.10.2917. [DOI] [PubMed] [Google Scholar]
- 108.Green CD, Olson LK. Modulation of palmitate-induced endoplasmic reticulum stress and apoptosis in pancreatic β-cells by stearoyl-CoA desaturase and Elovl6. American Journal of Physiology. 2011;300(4):640–649. doi: 10.1152/ajpendo.00544.2010. [DOI] [PubMed] [Google Scholar]
- 109.Flowers JB, Rabaglia ME, Schueler KL, et al. Loss of stearoyl-CoA desaturase-1 improves insulin sensitivity in lean mice but worsens diabetes in leptin-deficient obese mice. Diabetes. 2007;56(5):1228–1239. doi: 10.2337/db06-1142. [DOI] [PubMed] [Google Scholar]
- 110.Schuit FC, In ’t Veld PA, Pipeleers DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(11):3865–3869. doi: 10.1073/pnas.85.11.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schuit FC, Kiekens R, Pipeleers DG. Measuring the balance between insulin synthesis and insulin release. Biochemical and Biophysical Research Communications. 1991;178(3):1182–1187. doi: 10.1016/0006-291x(91)91017-7. [DOI] [PubMed] [Google Scholar]
- 112.Vander Mierde D, Scheuner D, Quintens R, et al. Glucose activates a protein phosphatase-1-mediated signaling pathway to enhance overall translation in pancreatic β-cells. Endocrinology. 2007;148(2):609–617. doi: 10.1210/en.2006-1012. [DOI] [PubMed] [Google Scholar]
- 113.Wang J, Takeuchi T, Tanaka S, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic β-cell dysfunction in the Mody mouse. Journal of Clinical Investigation. 1999;103(1):27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Allen JR, Nguyen LX, Sargent KEG, Lipson KL, Hackett A, Urano F. High ER stress in β-cells stimulates intracellular degradation of misfolded insulin. Biochemical and Biophysical Research Communications. 2004;324(1):166–170. doi: 10.1016/j.bbrc.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 115.Nozaki JI, Kubota H, Yoshida H, et al. The endoplasmic reticulum stress response is stimulated through the continuous activation of transcription factors ATF6 and XBP1 in Ins2+/Akita pancreatic β cells. Genes to Cells. 2004;9(3):261–270. doi: 10.1111/j.1356-9597.2004.00721.x. [DOI] [PubMed] [Google Scholar]
- 116.Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YDI. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37(8):1020–1024. doi: 10.2337/diab.37.8.1020. [DOI] [PubMed] [Google Scholar]
- 117.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and β-cell dysfunction. Endocrine Reviews. 2008;29(3):351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Leibowitz G, Bachar E, Shaked M, et al. Glucose regulation of β-cell stress in type 2 diabetes. Diabetes, Obesity and Metabolism. 2010;12(supplement 2):66–75. doi: 10.1111/j.1463-1326.2010.01280.x. [DOI] [PubMed] [Google Scholar]
- 119.Robertson RP. Oxidative stress and impaired insulin secretion in type 2 diabetes. Current Opinion in Pharmacology. 2006;6(6):615–619. doi: 10.1016/j.coph.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 120.Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. Journal of Cell Biology. 2004;164(3):341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants and Redox Signaling. 2007;9(12):2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 122.Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Molecular Cell. 2004;15(5):767–776. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 123.Li G, Mongillo M, Chin KT, et al. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. Journal of Cell Biology. 2009;186(6):783–792. doi: 10.1083/jcb.200904060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American Journal of Physiology. 2004;287(4):C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 125.Lipson KL, Ghosh R, Urano F. The role of IRE1α in the degradation of insulin mRNA in pancreatic β-cells. PLoS ONE. 2008;3(2, article e1648) doi: 10.1371/journal.pone.0001648. [DOI] [PMC free article] [PubMed] [Google Scholar]