

Abstract
Although the molecular mechanisms of TNF signaling have been largely elucidated, the principle that regulates the balance of life and death is still unknown. We report here that the death domain kinase RIP, a key component of the TNF signaling complex, was cleaved by Caspase-8 in TNF-induced apoptosis. The cleavage site was mapped to the aspartic acid at position 324 of RIP. We demonstrated that the cleavage of RIP resulted in the blockage of TNF-induced NF-κB activation. RIPc, one of the cleavage products, enhanced interaction between TRADD and FADD/MORT1 and increased cells' sensitivity to TNF. Most importantly, the Caspase-8 resistant RIP mutants protected cells against TNF-induced apopotosis. These results suggest that cleavage of RIP is an important process in TNF-induced apoptosis. Further more, RIP cleavage was also detected in other death receptor-mediated apoptosis. Therefore, our study provides a potential mechanism to convert cells from life to death in death receptor-mediated apoptosis.
Keywords: TNF, apoptosis, NF-κB, RIP, caspase
The proinflammatory cytokine, tumor necrosis factor (TNF), has an important role in diverse cellular events such as septic shock, induction of other cytokines, cell proliferation, differentiation, and apoptosis (Tracey and Cerami 1993; Vandenabeele et al. 1995). TNF is a member of the TNF family that includes FasL, lymphotoxin (LT), CD27L, OX40, CD30L, and CD40L (Smith et al. 1994; Nagata and Golstein 1995). Although many of the TNF-mediated processes can be regulated by either one of its receptors, TNF-R1 and TNF-R2, apoptosis is mainly induced through TNF-R1 (Tartaglia and Goeddel 1992). Both TNF-R1 and TNF-R2 belong to the TNF/NGF receptor superfamily and are characterized by multiple cysteine-rich domains in their extracellular regions (Tartaglia and Goeddel 1992; Nagata and Golstein 1995). Along with several other members of this superfamily such as Fas, DR3, DR4, and DR5, TNF-R1 is also known as a death receptor (Itoh and Nagata 1993; Tartaglia et al. 1993). These death receptors share a conserved cytoplasmic domain known as the death domain, as this region is critical for transduction of their ligand-induced death signals (Nagata 1997; Ashkenazi and Dixit 1998).
Recently, the distinct signal transduction pathways for TNF-R1-mediated responses have been uncovered, largely due to identifying the proteins that are recruited into the TNF receptor complex on TNF binding to TNF-R1 (Hsu et al. 1995, 1996a,b). The binding of TNF to TNF-R1 leads to the trimerization of TNF-R1 and the recruitment of TNF-R1-associated death domain protein (TRADD) into the receptor complex (Hsu et al. 1995). Then TRADD serves as a platform to recruit other proteins into the complex. Three of these adapter proteins, FADD/MORT1 (FAS-associated death domain protein), TRAF2 (TNFR-associated factor 2), and RIP (the death domain kinase) have been shown to interact with TRADD directly (Rothe et al. 1994, 1995; Boldin et al. 1995; Chinnaiyan et al. 1995; Hsu et al. 1995, 1996a,b; Stanger et al. 1995). Disruption of FADD/MORT1 protein expression completely blocks TNF-induced apoptosis (Yeh et al. 1998; Zhang et al. 1998). The essential role of FADD/MORT1 in TNF-induced apoptosis is thought to be the recruitment of Caspase-8 and subsequent its activation (Boldin et al. 1996; Varfolomeev et al. 1998). The active Caspase-8 then initiates a caspase cascade, which results in apoptosis (Faleiro et al. 1997; Cryns and Yuan 1998). Recently BID(BH3-interacting domain death agonist), a member of the Bcl2 family, has been identified as a target of Caspase-8 (Li et al. 1998; Luo et al. 1998). The cleavage of BID leads to cytochrome c release and activation of Caspase-9 (Li et al. 1998; Luo et al. 1998). Although FADD/MORT1 is essential for TNF-induced apoptosis, the recruitment of RIP and TRAF2 is responsible for activation of NF-κB and AP-1 respectively (Liu et al. 1996; Ting et al. 1996; Lee et al. 1997; Natoli et al. 1997; Reinhard et al. 1997; Yeh et al. 1997; Kelliher et al. 1998). The indispensable role of RIP in TNF-induced NF-κB activation was suggested by generating RIP-deficient Jurkat cells and RIP−/− mice (Ting et al. 1996; Kelliher et al. 1998). Consistent with the previous finding that NF-κB activation protects cells from TNF-induced apoptosis, RIP−/− MEF cells are hypersensitive to TNF treatment (Kelliher et al. 1998).
When cells are exposed to TNF treatment, two transcription factors, NF-κB and AP-1 are activated (Brenner et al. 1989; Osborn et al. 1989). Activation of these two transcriptional factors leads to induction of many other cytokines and immunoregulatory proteins and is pivotal for many inflammatory responses (Siebenlist et al. 1994; Karin et al. 1997; Baeuerle 1998). Whereas NF-κB is activated through NIK and IKK (DiDonato et al. 1997; Malinin et al. 1997; Mercurio et al. 1997; Regnier et al. 1997), AP-1 activity is regulated by MAP kinases such as JNK and p38 (Karin et al. 1997). Inactive NF-κB is located in the cytoplasm because its interaction with the inhibitory proteins, IκBs, masks its nuclear translocation signal (Siebenlist et al. 1994; Baeuerle and Baltimore 1996). On stimuli, IκBs are phosphorylated by IKK at their regulatory region and are rapidly degraded after polyubiquitination. The degradation of IκBs leads to the release of NF-κB and allows NF-κB to translocate into the nucleus, where it activates its target genes (Baeuerle and Baltimore 1996). In response to TNF treatment, NF-κB activation protects cells against TNF-induced apoptosis, whereas AP-1 has little effect (Beg and Baltimore 1996; Liu et al. 1996; Van Antwerp et al. 1996; Wang et al. 1996). It has been shown that several of NF-κB's target genes, including cIAP-1, cIAP-2, and IEX-1L have such anti-apoptotic properties (Wang et al. 1998; Wu et al. 1998).
Despite the rapid progress in elucidation of the molecular mechanisms of TNF signaling, the principle that regulates the balance of life and death in response to TNF is still unclear. In this study we found that receptor-interacting protein (RIP) is cleaved by Caspase-8 when cells undergo TNF-induced apoptosis. The cleavage of RIP abolished its NF-κB inducing ability. In addition, one of the cleavage products, RIPc, enhanced TRADD and FADD interaction, whereas wild-type RIP interfered with this association. Overexpression of RIPc increased TNF-induced apoptosis. Moreover, the cleavage-resistant RIP mutants protected cells against TNF-induced apoptosis. We also observed RIP cleavage in Fas and TRAIL-mediated apoptosis. These results demonstrated that RIP cleavage has an important role in shifting cells from life to death in response to TNF treatment and provided a potential mechanism for the regulation of death receptor-mediated apoptosis.
Results
Cleavage of RIP is specific for TNF-induced apoptosis
To investigate the possible changes in TNF signaling effectors during TNF-induced apoptosis, cell extracts from HeLa cells after different treatments were subjected to Western blot analysis with anti-TRADD, anti-TRAF2, anti-RIP, and anti-FADD antibodies. Normally, HeLa cells are not susceptible to TNF-induced death; however, addition of cycloheximide (CHX) renders HeLa cells TNF sensitive. Whereas no obvious changes of TRADD, TRAF2, and FADD protein levels were detected, the RIP protein is decreased when cells undergo apoptosis (Fig. 1A; data not shown). Surprisingly, a 42-kD band was also observed with the same anti-RIP antibody (Fig. 1A). This new 42-kD fragment appeared to be the death domain-containing (carboxy-terminal) portion of RIP as the anti-RIP antibody only recognizes the death domain of RIP when different segments of RIP were tested (data not shown). The new RIP fragment was solely generated in apoptotic cells as it was neither detected in control cells nor cells treated with TNF or CHX (Fig. 1A). Because CHX blocks de novo protein synthesis, this result suggested that the 42-kD fragment is the product of RIP cleavage (RIPc; see Fig. 2). A similar observation was also made in HEK293 cells and Jurkat cells treated with TNF in the presence of CHX (data not shown). In the case of human breast carcinoma MCF7 cells that are TNF sensitive, RIP cleavage occurred following TNF treatment in the absence of CHX (Fig. 1B). These results indicated that RIP was selectively cleaved during TNF-induced apoptosis. Importantly, RIP cleavage was detected as early as 1 hr after treatment, and the amount of RIP cleavage correlated with the percentage of apoptotic cells (Fig. 1C).
Figure 1.

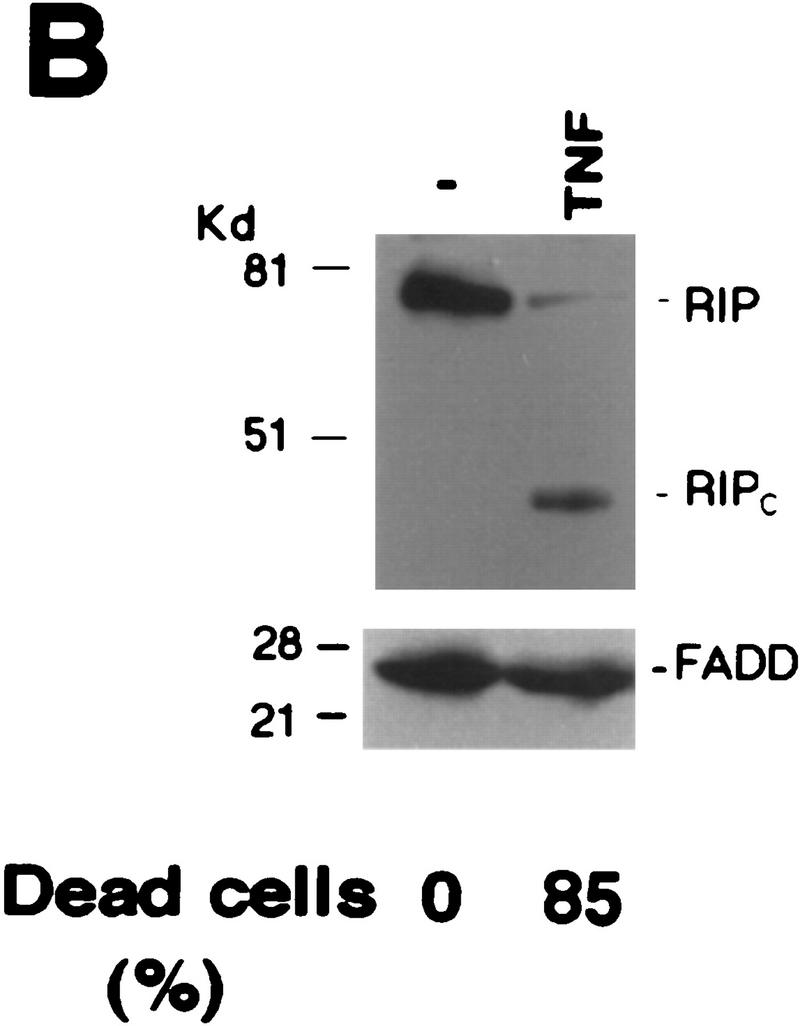


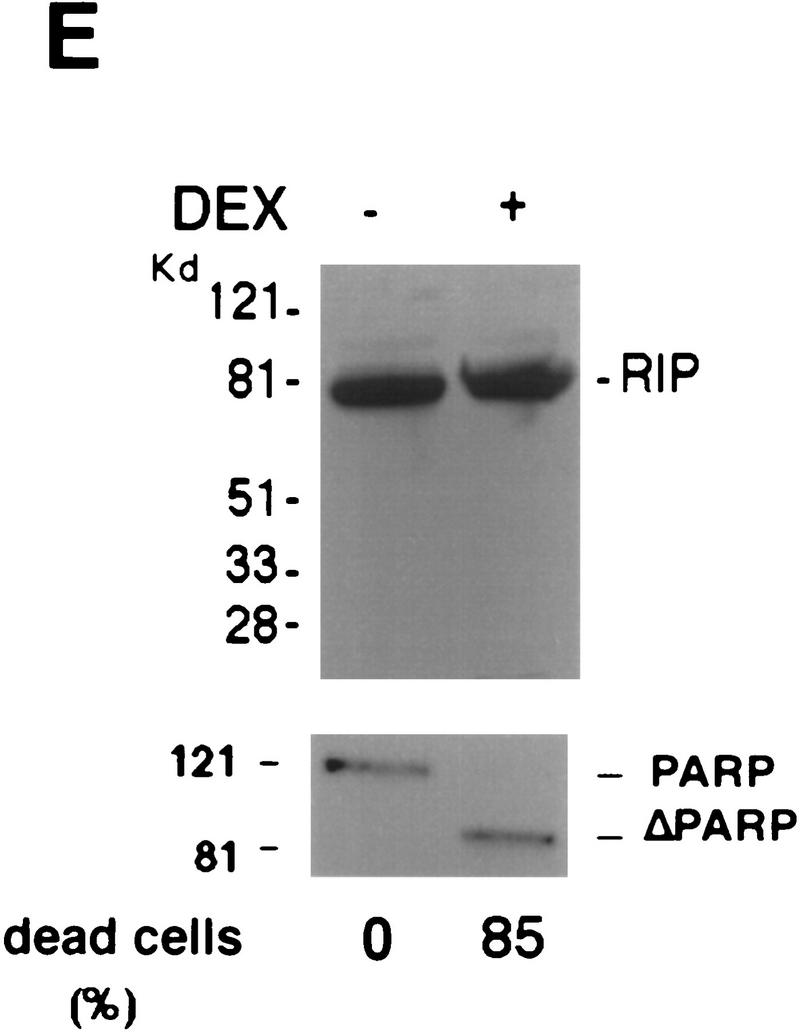
Cleavage of RIP in TNF-induced apoptosis. (A) HeLa cells were treated with TNF (15 ng/ml) (lane 2); CHX (10 μg/ml) (lane 3); or TNF (15 ng/ml) + CHX (10 μg/ml) (lane 4) for 10 hr. Cell extracts were resolved on SDS-PAGE and then Western blotted for RIP (top) and FADD (bottom). Untreated cell extract was loaded as a control (lane 1). (B) MCF7 cells were treated with TNF (15 ng/ml) for 18 hr. RIP and FADD were detected by Western blot. (C) Time course of RIP cleavage in TNF induced apoptosis in HeLa cells. Cells were treated with TNF (15 ng/ml) + CHX (10 μg/ml) and incubated for the indicated time periods. RIP and FADD were detected by Western blot analysis. (D) HeLa cells were treated with TNF (15 ng/ml) + CHX (10 μg/ml) for 6 hr (lane 2); UVC (20 J/m2, followed by overnight culture) (lane 3); and staurosporine (1 μm) overnight (lane 4). Cell extracts were resolved on SDS-PAGE and Western blotted for RIP (top), PARP (middle), and FADD (bottom). Untreated cell extract was loaded as a control (lane 1). (E) 2B4 cells were treated with DEX (1 μm) for 10 hr, and RIP and PARP were detected by Western blot analysis. Cell death was determined by trypan blue exclusion staining. The percentages of dead cells are at the bottom of the panels. Data are normalized to the rate of spontaneous cell death occurring in untreated cells (<5%). The positions of the molecular mass markers are indicated in kD at left of panels.
Figure 2.




RIP is cleaved by a caspase. (A) HeLa cells were pre-treated with 10 μm of DEVD–CHO (lane 4) or Z–VAD–FMK (lane 3) for 1 hr, or left untreated (lanes 1,2). This was followed by treatment with TNF (15 ng/ml) + CHX (10 μg/ml) (lanes 2–4) for 6 hr. Cell extracts were resolved on SDS-PAGE and Western blotted for RIP (top), PARP (middle), and FADD (bottom). Untreated cells extract was loaded as a control (lane 1). The percentages of dead cells are at the bottom of the panel. (B) 35S-Labeled RIP protein was incubated with apoptotic (lane 3) or normal (lane 2) HeLa cell extracts at 30°C for 3 hr, resolved on SDS-PAGE, and visualized by autoradiography. Nontreated RIP protein was loaded as a control (lane 1). The resultant cleavage products are indicated with arrows. (C) 35S-Labeled Myc-tagged RIP protein was incubated with RIP-depleted apoptotic (lane 6) or normal (lane 5) HeLa cell extracts at 30°C for 3 hr, transferred onto a nitrocellular filter, visualized by autoradiograph, and probed with anti-RIP and anti-Myc to detect the carboxy- and amino-terminal portion of RIP, respectively. Nontreated RIP protein was loaded as a control (lane 4). The resultant cleavage products are indicated with arrows. The normal and apoptotic (with or without RIP depletion) HeLa cell extracts were loaded as controls (lanes 1–3). (D) Apoptotic cell extracts were preincubated with 10 μm of DEVD–CHO (lane 4), Z–VAD–FMK (lane 3), or Z–FA–FMK (lane 5) for 15 min, mixed with 35S-labeled RIP protein, and incubated at 30°C for 3 hr. The result was visualized by autoradiograph. The resultant cleavage products are indicated with arrows. The positions of the molecular mass markers are indicated in kD at left of panels.
To test whether this cleavage of RIP is specific for TNF-induced death, we treated HeLa cells with other apoptosis-inducing agents such as staurosporine, which is a protein kinase C inhibitor, and UV, which is a genotoxic agent. As shown in Figure 1D (lanes 3,4) staurosporine and UV did not induce RIP cleavage, although all of the treated cells underwent apoptosis and poly (ADP ribose) polymerase (PARP), a substrate of Caspase-3, was cleaved completely (Lazebnik et al. 1994). To further examine the specificity of RIP cleavage, Western blotting was performed with cell extracts from murine T-cell hybridoma 2B4 cells with or without dexamethasome (DEX) treatment. Again, no RIP cleavage was detected, despite the fact that most cells were killed (Fig. 1E). Taken together, these results suggest that RIP cleavage is unique to TNF-induced apoptosis.
RIP is cleaved at D324 by Caspase-8
Because caspases have key roles in the initiation and execution of cellular death machinery (Cohen 1997; Nagata 1997; Cryns and Yuan 1998), we examined whether RIP cleavage is caspase-dependent by using specific caspase inhibitors. Before treating HeLa cells with TNF plus CHX, either the caspase inhibitor Z–VAD–FMK or DEVD–CHO, both of which have an inhibitory effect on TNF-induced apoptosis (Cohen 1997), was added into culture medium. Z–VAD–FMK specifically represses Caspase-1, Caspase-8, and other caspases, whereas DEVD–CHO inhibits Caspase-3 and related caspases (Cohen 1997; Li et al. 1998; Luo et al. 1998). As shown in Figure 2A, RIP cleavage was blocked completely by addition of Z–VAD–FMK and partially inhibited by DEVD–CHO. Interestingly, DEVD–CHO also failed to entirely block TNF-induced death (Fig. 2A). As a control, Z–FA–FMK, a cathepsin B inhibitor, showed no effect at all on RIP cleavage (data not shown). To confirm that RIP is a direct target of caspases, we performed in vitro cleavage assays with 35S-labeled RIP as the substrate. In this assay, RIP was cleaved in apoptotic cell extract only (Fig. 2B). To verify that the two fragments of cleaved RIP represent the two portions of RIP, we performed Western blot experiments following in vitro cleavage assay. Because the 35S-labeled RIP is amino-terminal Myc-tagged (Hsu et al. 1996b), anti-Myc antibody was used to detect the amino-terminal portion of RIP. To assure that the detected carboxy-terminal portion of RIP by anti-RIP antibody is from in vitro-cleaved RIP, we depleted the endogenous RIP and the carboxy-terminal portion of RIP from the apoptotic extract (Fig. 2C, middle, lane 3). As shown in Figure 2C, the anti-RIP antibody recognized the upper fragment (Fig. 2C, left, lane 6; middle, lane 2,6) whereas the anti-Myc antibody detected the smaller fragment (Fig. 2C, left, lane 6; right, lane 6). Therefore, the two cleavage products represented the carboxy- and amino-terminal portions of RIP, which were referred as RIPc and RIPn, respectively (Fig. 2C). Furthermore, the addition of Z–VAD–FMK in apoptotic cell extracts abolished RIP cleavage, whereas Z–FA–FMK did not (Fig. 2D). DEVD–CHO had a better inhibitory effect than it did in cell culture (Fig. 2A). These results indicated that RIP is cleaved directly by a Z–VAD–FMK sensitive caspase.
A hallmark of caspases is that these proteases selectively recognize an aspartic acid (D) as the P1 residue at the cleavage site (Cohen 1997; Cryns and Yuan 1998). To determine the cleavage site of RIP, we generated a series of RIP mutants in which certain aspartic acids were substituted with other amino acids. Given the sizes of the two cleavage fragments, we assumed that the cleavage site is located in the central portion of RIP. Therefore, we introduced point mutations to eliminate the aspartic acids at positions 248, 250, 251, 300, and 324 and three RIP mutants—RIP(D248A, D250A, D251P), RIP(D300V) and RIP(D324K)—were generated (Fig. 3A). These RIP mutants were then in vitro-translated in the presence of [35S]methionine and subjected to a cleavage assay in apoptotic cell extract as described in Figure 2. Similar to the wild-type RIP, RIP(D248A, D250A, D251P), and RIP(D300V) were cleaved into two fragments of 42 and 40 kD (Fig. 3B, lanes 7–12). There was no cleavage, however, detected in the RIP(D324K) mutant under the same conditions (Fig. 3B, lanes 4–6). No cleavage of RIP(D324K) was also confirmed by Western blotting as described in Figure 2C (data not shown). This mutant was also resistant to cleavage in vivo when it was introduced into HeLa cells (Fig. 3C). In this experiment we used RIP (1-558) and RIP (1-558, D324K) expression vectors. The death domain of RIP was deleted in these two constructs, as ectopic expression of full-length RIP protein usually causes cell death and results in low level of RIP expression. These results suggested that the aspartic acid at position 324 is the cleavage site of RIP. Interestingly, the proceeding sequence of this site, 321LQLD324, matches perfectly with LXXD, the preferred cleavage site of Caspase-8 (Cohen 1997; Li et al. 1998; Luo et al. 1998).
Figure 3.





Mapping of the caspase cleavage site in RIP. (A) Schematic illustration of substitution mutants of RIP constructs. The kinase and death domains of RIP are shown as shaded and solid bars, respectively. The amino acid substitutions of each mutant are indicated. (B) 35S-Labeled RIP proteins were incubated with apoptotic HeLa cell extract (lanes 3,6,9,12) or normal HeLa cell extract (lanes 2,5,8,11) at 30°C for 3 hr, resolved on SDS-PAGE and visualized by autoradiograph. Nontreated RIP proteins were loaded as controls (lanes 1,4,7,10). (Lanes 1–3) Wild-type RIP; (lanes 4–6) RIP(D324K); (lanes 7–9) RIP (D248A, D250A, D252P); (lanes 10–12) RIP(D300V). The resultant cleaving products are indicated with arrows. (C) HeLa cells were transfected with Myc-tagged RIP(1–558) (lanes 1,2) or RIP(1–558, D324K) (lanes 3,4). Cells were treated with TNF + CHX (lanes 2,4) or remained untreated (lanes 1,3). Myc-tagged RIP proteins were detected by Western blot analysis with anti-Myc antibody. The position of Myc-tagged RIP (1–558)/ RIP (1–558, D324K) is indicated. RIPn indicates the resultant Myc-tagged amino-terminal portion of RIP (1–324 amino acids). (D) 35S-Labeled RIP proteins were incubated with Caspase-3 (lanes 2,6), Caspase-8 (lanes 3,7), or apoptotic HeLa cell extract at 37°C for 2 hr, resolved on SDS-PAGE and visualized by autoradiograph. Nontreated RIP proteins were loaded as controls (lanes 1,5). The resultant cleavage products are indicated with arrowheads. (E) Fifty microliters of apoptotic cell extract was incubated with the indicated antibodies bound on protein A resin at 4°C for 12 hr. The resin was removed by centrifugation and the supernatants were applied in cleavage assays as described in B. The positions of the molecular mass markers are indicated in kD at left of panels.
Next we investigated whether Caspase-8 is responsible for RIP cleavage. Because both Caspase-8 and Caspase-3 have critical roles in TNF-induced apoptosis (Cohen 1997; Nagata 1997), recombinant Caspase-3 and Caspase-8 were tested in an in vitro cleavage experiment. As shown in Figure 3D, Caspase-8 cleaved wild-type RIP into two fragments, which were identical to those generated by the apoptotic cell extract. Although Caspase-3 also cleaved at the same site as Caspase-8 did, it produced a much different and more sophisticated cleavage of RIP. Importantly, Caspase-8 was unable to cleave RIP (D324K) (Fig. 3D). In contrast, the cleavage pattern of RIP(D324K) by Caspase-3 largely remained the same and only the Caspase-8-like cleavage disappeared (Fig. 3D). Considering that Caspase-3 is deficient in MCF7 cells (Srinivasan et al. 1998), yet RIP was cleaved as efficiently as in other cell lines (Fig. 1B), it is unlikely that Caspase-3 is involved in RIP cleavage in vivo. In addition, the inhibitor Z–VAD–FMK totally abrogated RIP cleavage by Caspase-8 (data not shown). We also performed immune-depletion experiments with anti-Caspase-8 or anti-Caspase-3 antibody. As shown in Figure 3E, the depletion of Caspase-8 from the apoptotic extract completely abolished its ability to cleave RIP whereas the removal of caspase-3 had no effect. This result further supported that Caspase-8 is the protease that cleaves RIP in TNF-induced apoptosis.
Cleavage of RIP results in the blockage of NF-κB activation and the enhancement of TRADD and FADD interaction
One of the essential functions of RIP is to mediate TNF-induced NF-κB activation, and overexpression of RIP activates NF-κB (Hsu et al. 1996b; Kelliher et al. 1998). To see whether cleavage of RIP affects its ability to mediate NF-κB activation, we constructed expression vectors of RIPn(1–324) and RIPc(325–671) and performed luciferase assays after they and a NF-κB reporter plasmid were introduced into HEK293 cells. To verify that a D324K mutation does not alter RIP's ability to activate NF-κB, expression vectors of RIP, RIP(D324K), RIP(1–558), and RIP(1–558, D324K) were also transfected into HEK293 cells, respectively, to measure their functions in terms of NF-κB activation. In the case of RIP, RIP(D324K) and RIPc, as overexpression of these proteins kills the majority of transfected cells (data not shown; see Fig. 5A, below), the expression vector of poxvirus protein CrmA was also included in cotransfection experiments (Ray et al. 1992). As shown in Figure 4A, substituting Asp-324 with a Lys in RIP did not show any consequence on NF-κB activation, as both RIP(D324K) and RIP(1–558, D324K) induced similar folds of NF-κB activation as their wild-type counterparts did. The cleavage of RIP, however, eradicated its ability to robustly activate NF-κB because RIPn exerted no activity and RIPc only showed marginal activity (Fig. 4A). Previously it has been shown that the death domain portion (559–671) of RIP functions as a dominant-negative mutant and its overexpression blocks TNF-induced NF-κB activation (Hsu et al. 1996b). To examine whether RIPc has the same effect, we performed similar experiments as described above with the expression vectors of RIPc, RIPn, and RIP(559–671). After transfection (24 hr), half transfection samples were treated with TNF. As shown in Figure 4B, whereas RIPn had no effect, RIPc inhibited TNF-induced NF-κB activation almost as sufficiently as the dominant-negative mutant RIP(559–671) did. Taken together, these results suggested that cleavage of RIP eliminates its competence to transduce TNF signaling and one of the cleavage products, RIPc, represses TNF-induced NF-κB activation.
Figure 5.

Effects of RIP cleavage on apoptosis. (A) HeLa cells were cotransfected with pRSV–LacZ and different RIP constructs with (open bars) or without CrmA (solid bars) as indicated. Twenty-four hours post-transfection, the cells were fixed and stained with X-gal. Viable blue cells, determined by their sizes and shapes, were counted from 20 randomly selected fields. Data shown represent three independent experiments. The difference of viable cells of wild-type RIP vs. RIP (D324K) transfection is statistically significant (P < 0.005). (B) Duplicates of MCF7 cells were cotransfected with pRSV–LacZ and different RIP constructs. Twenty-four hours post-transfection, half of the cells were treated with 15 ng/ml TNF for 18 hr and then fixed and stained with X-gal. Alive blue cells were counted from 20 randomly selected fields for each sample. Percentage of viable cells for each plasmid was calculated by dividing the number of viable transfected cells after treatment with the number of viable transfected cells before treatment. Data shown represent three independent experiments. The differences of viable cells of wild-type versus RIP (D324K) and RIPc versus vector transfections are statistically significant (both P < 0.05). (C) MCF7 cells were cotransfected with pRSV–LacZ and different RIP constructs as indicated. Cells were treated and counted as described in B. The difference of viable cells of wild-type vs. RIP (D324K) transfection is statistically significant (P < 0.005).
Figure 4.




Effects of RIP cleavage on NF-κB activation and TRADD/FADD interaction. (A) Cells were co-transfected with p2xNF-κB–Luc, pRSV–lacZ and different RIP constructs as indicated or an empty vector. In the experiments described in the right panel, the expression vector of CrmA was also included. Twenty-four hours post-transfection, the cells were collected. Luciferase assay was conducted and normalized as described (Liu et al. 1996). The results were the average of three duplicated experiments. (B) Cells were cotransfected with p2xNF-κB–Luc, CrmA and different RIP constructs. Twenty-four hours post-transfection, half of the cells were treated with TNF (15 ng/ml) for 10 hr (solid bars). (Open bars) Nontreated cells. Luciferase activities were detected and normalized. The results shown were the average of three duplicated experiments. (C) HEK293 cells were cotransfected with FLAG–TRADD (2.5 μg), FADD (2.5 μg), CrmA (1 μg), along with increasing amounts of Xpress-tagged RIP (1.5, 3, 4.5 μg in lanes 2, 3, and 4, respectively) or RIPc (1.5, 3, 4.5 μg in lanes 6, 7, and 8, respectively). The total amount of DNA in each transfection was adjusted to 10 μg with the empty vector. Cells were collected at 24 hr after transfection. Expressions of Flag–TRADD and FADD were determined by Western blot (bottom). Immunoprecipitation experiments were performed with anti-Flag (M2) antibody, and coprecipitated FADD and Xpress-tagged RIP proteins were detected by Western blotting with anti-FADD and anti-Xpress, respectively (top). (D). HeLa cells (2 × 107) were treated with TNF (15 ng/ml) + CHX (10 μg/ml) for the indicated time periods, immunoprecipitation experiments were performed with anti-TRADD antibody, and coprecipitated RIP, FADD, and TRADD proteins were detected by Western blot. (Lane 1) One percent of the cell extract from the 480-min sample as an input control. The blot of FADD was exposed for a longer time to visualize the precipitated FADD protein. Cell death was determined by trypan blue exclusion staining. The percentages of dead cells are shown at the bottom of the panel. The positions of the molecular mass markers are indicated in kD on the left of C and D. (*IgG heavy chain).
To initiate TNF-induced apoptosis, it is thought that FADD needs to be recruited into the TNF-R1 complex through the death domain of TRADD (Hsu et al. 1996a). Because the interaction between RIP and TRADD is also mediated by their death domains (Hsu et al. 1996b), it is possible that RIP may compete with FADD for binding to TRADD. To test this possibility, expression vectors of FADD and Flag–TRADD were cotransfected with increasing amounts of either wild-type RIP or RIPc. Then co-immune-precipitation experiments were performed with anti-Flag antibody. As shown in Figure 4C (top), whereas the presence of increasing amounts of wild-type RIP weakened TRADD and FADD interaction, RIPc appeared to strengthen this interaction. In each sample, comparable expression level of TRADD or FADD was detected as shown in the bottom panel of Figure 4C. These data implied that the cleavage of RIP might also accelerate apoptosis by enhancing TRADD–FADD interaction. To further test this notion, we investigated the endogenous TRADD and FADD interaction when RIP is cleaved. As seen in Figure 4D, whereas the intact RIP was recruited transiently into the TRADD complex, RIPc showed prolonged interaction with TRADD (top panel). This sustained RIPc and TRADD interaction correlated with increasing cell death in response to TNF treatment. Most importantly, increasing recruitment of FADD into the TRADD complex was detected when the intact RIP was released from the complex (Fig. 4D, middle).
Ectopic expression of D324K mutants of RIP protects cells against TNF-induced apoptosis
Because overexpression of RIP induced cell death and RIP was cleaved in transfected cells (data not shown), it is possible that cleavage-resistant RIP(D324K) may cause cell death to a lesser extent. To address this question, HeLa cells were cotransfected with different RIP expression vectors and a LacZ reporter plasmid. In the presence of CrmA, which blocks cell death induced by overexpression of RIP (Liu et al. 1996), expression of RIP, RIP(D324K), or RIPc displayed similar transfection efficiency (Fig. 5A). Without CrmA, however, whereas expression of RIP or RIPc killed >90% transfected cells (94.8% and 92.4%, respectively), ∼30% of RIP(D324K)-transfected cells were still viable (Fig. 5A). The differenc between wild-type RIP and RIP(D324K) is statistically significant (χ2 test, P < 0.005). This result suggested that substituting Asp-324 with a Lys in RIP impaired its capacity to induce cell death. This may be attributable to uncleavable RIP(D324K) constitutively activating NF-κB.
To investigate the effect of RIP cleavage on TNF-induced apoptosis further, similar experiments were performed in MCF7 cells without CrmA. MCF7 cells were chosen because they are TNF-sensitive without CHX. This will allow us to avoid using CHX, as CHX inhibits the expression of transfected proteins as well as the expression of their downstream genes. Although ectopic expression of death domain containing RIP proteins kills the majority of transfected cells, there are still a certain amount of transfected cells viable (Fig. 5A,B). In these experiments with MCF7 cells, the remaining transfected cells were challenged with TNF. As shown in Figure 5B, 18 hr after TNF treatment, ∼40% of RIP-transfected cells were viable whereas 65% of RIP(D324K)-transfected cells were still alive. Also, whereas the control vector showed 20% viable cells, only 8% of RIPc-transfected cells were alive in response to TNF treatment (Fig. 5B). Therefore, in remaining transfected cells, although expression of RIP provided a certain level of protection, RIP(D324K) had a much greater anti-apoptotic effect. More importantly, the presence of RIPc rendered cells more susceptible to TNF-induced apoptosis. To further confirm this observation and to rule out that those remaining transfected cells may have defects in their apoptotic machinery—although this is unlikely because the remaining RIPc transfected cells had greater response to TNF—we performed the same experiments but with RIP(1–558) and RIP(1–558, D324K) constructs. As shown in Figure 5C, compared with the 20% of transfected cells that survived in the vector control, expression of RIP(1-558) and RIP(1-558, D324K) rescued 43% and 65% transfected cells from TNF treatment, respectively. Expression of RIPn did not exhibit any effect on TNF-induced apoptosis at all. Although expression of RIP or RIP(1–558) activated NF-κB and provided some level of protection against TNF-induced apoptosis, cleavage of these two proteins disabled them to continue activating NF-κB and reduced their protective effect. In contrast, RIP(D324K) and RIP(1–558, D324K) were kept intact and had greater protection effect.
RIP is also cleaved in Fas and TRAIL induced apoptosis
To examine whether RIP is also cleaved during apoptosis mediated by other death receptors, we performed Western blotting with cell extracts from anti-Fas or TRAIL-treated cells. As shown in Figure 6A, RIP cleavage was observed in anti-Fas-treated Jurkat cells. When HeLa cells were induced to undergo apoptosis with TRAIL, which binds to DR4 and DR5 (Chaudhary et al. 1997; Pan et al. 1997; Walczak et al. 1997), similar cleavage of RIP was also detected (Fig. 6B). In both cases, there was no detectable change of FADD and TRAF2 (data not shown). These results implied that cleavage of RIP might be a common process during death receptor-mediated apoptosis. To further evaluate this notion, we investigated whether there is any difference of sensitivity between wild-type Jurkat and RIP-deficient Jurkat cells (Ting et al. 1996) in response to anti-Fas and TRAIL treatment. As shown in Figure 6C, similar to TNF/CHX-induced death, the extent of cell death triggered by anti-Fas or TRAIL in RIP-deficient Jurkat cells was notably higher than that in wild-type Jurkat cells. As a control, UV-induced death was also measured in these two lines and no difference was noticed (Fig. 6C). This is consistent with the early observation that UV did not induce RIP cleavage (Fig. 2A). Accordingly, RIP cleavage may represent a unique regulatory step in all death receptor-mediated apoptosis.
Figure 6.



Cleavage of RIP in Fas and TRAIL induced apoptosis. (A) Jurkat cells were treated with anti-Fas (IgM, 250 ng/ml) for 6 hr (lane 2) and RIP protein was detected by Western blot analysis. (Lane 1) Untreated cell extract. The percentages of dead cells were shown at the bottom of the panel. (B) HeLa cells were treated with TRAIL (0.5 μg/ml) for 2 hr (lane 2) and RIP protein was detected by Western blot analysis. (Lane 1) Untreated cell extract. The percentages of dead cells are shown at the bottom of the panel. The positions of the molecular weight markers are indicated in kD at left. (C) Wild-type or RIP-deficient Jurkat cells were treated with TNF (15 ng/ml) + CHX (10 μg/ml), TRAIL (0.5 μg/ml), anti-Fas (250 ng/ml), or UVC (20J/m2), and followed by 6 hr culture. Cell death was determined by trypan blue exclusion staining. The percentages of dead cells shown are the average of three independent experiments. The probabilities in these experiments between wild-type RIP (open bars) and RIP− (solid bars) cells are nontreated, >0.9; TNF/CHX, <0.025; TRAIL, <0.025; Fas, <0.005; and UV, >0.1.
Discussion
Cleavage of RIP is essential for transition from life to death in TNF-induced apoptosis
Despite the fact that TNF can induce apoptosis by recruiting FADD into TNF-R1 complex, TNF is not cytotoxic to most types of cells (Smith et al. 1994). Because activation of NF-κB protects cells against apoptosis (Beg and Baltimore 1996; Liu et al. 1996; Van Antwerp et al. 1996; Wang et al. 1996), it has been proposed that the balance between life and death is regulated by NF-κB (Van Antwerp et al. 1998). Inhibition of NF-κB activation enhances apoptosis in response to TNF and many other apoptosis inducing agents (Beg and Baltimore 1996; Liu et al. 1996; Van Antwerp et al. 1996; Wang et al. 1996). In spite of much progress in understanding of TNF signaling, for a given cell, the mechanism that controls the outcome of TNF treatment is still largely unknown. Our work described in this paper provides a potential mechanism through which the transition from life to death is achieved when cells are induced to undergo apoptosis by TNF. As show in Figure 7, the death domain kinase RIP, the key effector that transduces TNF signal to NF-κB activation, is specifically cleaved by Caspase-8. The cleavage of RIP disables it to deliver TNF signal and subsequently, abolish the induction of anti-apoptosis factors. In addition, RIPc, one of the cleavage products, promotes apoptosis directly by enhancing TRADD and FADD interaction. Therefore, besides BID and downstream caspases (Cohen 1997; Li et al. 1998; Luo et al. 1998), RIP is another critical substrate of Caspase-8. Whereas cleavage of BID and downstream caspases amplifies apoptotic signal, cleavage of RIP shuts off the protective pathway and enhances killing.
Figure 7.

Regulation of TNF-R1signaling pathways during apoptosis. When cells undergo TNF-induced apoptosis, RIP is cleaved by Caspase-8. Although cleavage of BID and other caspases activates death pathways, cleavage of RIP neutralizes the protective effect of NF-κB. RIP mediates TNF-induced NF-κB activation through IκB kinase (IKK) (Baeuerle and Baltimore 1996) and TRAF2 transduces TNF signaling by activating MEKK/JNKK/JNK pathway (Liu et al. 1996).
Our observation that the cleavage of RIP is specific for TNF and other death receptor-mediated apoptosis (Fig. 1, 2, and 6) suggests that the cleavage of RIP is an early active, rather than a late passive, event during apoptosis. Although all of the tested agents including Dexamethasone, staurosporine, and genotoxic agent (UV) induced massive apoptosis and activation of caspases, the cleavage of RIP is barely detected (Fig. 2). These results, plus the fact that Caspase-3 cleaved RIP at multiple sites in vitro but is unable to do so in vivo (Fig. 3), strongly indicate that Caspase-8 mediated cleavage of RIP is highly selective in vivo and is spatially and timely regulated. Therefore, the cleavage of RIP represents a key regulatory step in the triggering of cell death during TNF-induced apoptosis. Further support for this notion is provided by evidence that the uncleavable RIP mutants showed elevated cytoprotection against TNF (Fig. 5). Constitutive activation of NF-κB by RIP(D324K) mutant may have a large role in its efficiency against TNF-induced cell death. It is also possible that the presence of uncleaved full-length RIP(D324K) interfers the interaction between TRADD and FADD and leads to diminished levels of TNF-induced apoptosis.
The cleavage of RIP by Caspase-8 also generates another fragment, RIPn, which contains the entire kinase domain of RIP. The role of RIP kinase activity in TNF signaling is still unknown but it is clear that this activity is dispensable for NF-κB activation (Hsu et al. 1996b; Ting et al. 1996). Consistent with this finding, RIPn has no role in TNF-induced NF-κB activation (Fig. 4). The possible function of RIPn in apoptosis is under study. To explore other possible functions of RIPc, we also studied whether RAIDD (Duan and Dixit 1997), an adapter molecule for RIP, interacts with wild-type RIP and RIPc differently. We found that wild-type RIP and RIPc bound to RAIDD equally well (data not shown). Probably, cleavage of RIP has no effect on the interaction between RIP and RAIDD.
Recently, it has been reported that TRAF2 was degraded on CD30 ligation and the down regulation of TRAF2 sensitized cells to TNF cytotoxicity (Duckett and Thompson 1997). More evidence is necessary, however, to prove that TRAF2 degradation happens under physiological conditions, as this finding was made by ectopic expression of CD28–CD30 chimera. We did not detect TRAF2 degradation in TNF-induced apoptosis. It is possible that TRAF2 degradation is specific to CD30 signaling. Because RIP has a more essential role in NF-κB activation in response to TNF than TRAF2 does, TRAF2 may not be the key target needed to be eliminated for cells undergoing TNF-induced apoptosis.
Is RIP cleavage a general mechanism for death receptor-mediated apoptosis?
RIP was identified by its interaction with Fas; however, Fas-mediated apoptosis does not require RIP (Stanger et al. 1995; Kelliher et al. 1998). Subsequently, RIP was found to be a key effector in the TNF-R1 and DR3 signal complex (Ashkenazi and Dixit 1998). Whether RIP is a component of the TRAIL receptor complex remains as an open question. Regardless of the presence of RIP in these death receptor complexes, RIP cleavage also occurred in apoptosis induced by Fas and TRAIL (Fig. 6). Moreover, RIP-deficient cells are more sensitive to cell death mediated by these two death receptors (Fig. 6). Consistent with our observation, it is reported recently that activation of NF-κB protects cells from Fas-mediated apoptosis (Zheng and Lenardo 1999). Because RIP is not cleaved in UV-induced cell death and UV kills both wild-type Jurkat and RIP-deficient Jurkat cells with equal efficiency (Figs. 1D and 6C), it seems that the protective effect of RIP is specific to death receptor-mediated apoptosis. Interestingly, RIP is cleaved more efficiently by anti-Fas treatment than by TNF treatment (data not shown). Because FasL is a more effective death factor than TNF, it is intriguing to speculate that the efficiency of RIP cleavage may be a critical factor in determining the efficiency of apoptosis by different death receptors. Further study on the mechanism of death receptor signaling will be the key to fully understand the role of RIP cleavage in death receptor-mediated apoptosis.
Concluding remarks
Regulation of TNF signaling is complex and is achieved through multiple steps. For instance, silencer of death domain (SODD) prevents TNF-R1 self-aggregation by interacting with the death domain of TNF-R1 (Jiang et al. 1999). This regulation ensures that TNF-R1 will not aggregate in the absence of TNF. Then, on TNF binding to its receptor, the recruitment of different effectors into the TNF receptor complex in different cellular contexts represents another level of control in TNF signaling (Baker and Reddy 1998). In this study, we identified a new mechanism through which the balance between life and death in response to TNF is regulated. We showed that RIP, is a critical substrate of Caspase-8, as is the case with BID and downstream caspases (such as Caspase-3). The cleavage of RIP results in the blockage of NF-κB activation and the enhancement of TNF-induced apoptosis. Therefore, our study provided a new possible regulation of TNF signaling and that this regulation may be crucial in converting cells from life to death in response to TNF.
Materials and methods
Reagents
Anti-RIP antibody, anti-PARP antibody, anti-Myc antibody, active recombinant Caspase-3 and Caspase-8 were purchased from Pharmingen. Anti-Fas (IgM) and anti- FADD were from Medical & Biological Laboratories and Transduction Laboratories, respectively. Anti-TRADD, anti-Caspase 3, anti-HA and anti-Xpress antibodies were from Santa Cruz. Rabbit anti-Caspase 8 was kindly provided by Dr. G.M. Cohen (Sun et al. 1999). The caspase inhibitors Z–VAD–FMK and DEVD–CHO, Cathepsin B inhibitor I (Z–FA–FMK) and staurosporine were purchased from Calbiochem. Recombinant TRAIL was purchased from BIOMOL.
Cell culture and transfection
HeLa and HEK293 cells were cultured in Dulbecco's modified Eagle medium (DMEM). MCF7 and Jurkat cells were cultured in RPMI 1640 medium. The media were supplemented with 10% fetal calf serum, 2 mm glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were transfected with Lipofectamine (GIBCO) as described previously (Liu et al. 1996).
Induction of apoptosis and determination of cell viability
For induction of apoptosis, cells were treated with TNF (15 ng/ml) plus CHX (10 μg/ml), anti-Fas, TRAIL, staurosporine or UV as indicated in the figure legends. Cell viability was determined by trypan blue exclusion assay.
Plasmids
The mammalian expression plasmids RIP, TRADD, and FADD have been described previously (Hsu et al. 1996b; Liu et al. 1996). Substitution RIP mutants were constructed by site-directed mutagenesis using the QuikChange site directed mutagenesis kit (Stratagene). In RIP (D248A, D250A, D251P), the aspartic acids at positions 248, 250, and 251 were substituted by alaline, alaline, and proline, respectively. In RIP (D300V), the aspartic acid at position 300 was replaced by a valine. In RIP(D324K), the aspartic acid at positions 324 was replaced by a lysine. The RIP-N and RIP-C expression plasmids were constructed in pcDNA vector by PCR. All constructs were confirmed by DNA sequencing.
Western blot analysis and coimmunoprecipitation
After treatment with different reagents as described in the legends to Figures 1, 2, 4, and 6, cells were collected and lysed in M2 buffer (20 mm Tris at pH 7, 0.5% NP-40, 250 mm NaCl, 3 mm EDTA, 3 mm EGTA, 2 mm DTT, 0.5 mm PMSF, 20 mm β-glycerol phosphate, 1 mm sodium vanadate, 1 μg/ml leupeptin). Fifty micrograms of the cell lysates was fractionated by SDS-polyacrylamide gels and Western blotted. The proteins were visualized by enhanced chemiluminescence (ECL), according to the manufacturer's (Amersham) instructions.
For immunoprecipitation assays, 293 cells transfected transiently with each of the plasmids were lysed in lysis buffer (20 mm Tris-HCl at pH 7.5, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 30 mm NaF, 2 mm sodium pyrophosphate, 10 μg/ml aprotinin, 10 μg/ml leupeptin) and the amounts of expressed proteins were determined by Western blotting. The lysates were mixed and precipitated with anti-FLAG antibody (M2) and protein A–Sepharose beads by incubation at 4°C overnight. The beads were washed three times with lysis buffer and the bound proteins were resolved in 10% SDS-PAGE. Detection was accomplished by Western blot analysis. For immunoprecipitation assays of endogenous proteins, 5 × 107 of HeLa cells were treated with TNF (15 ng/ml) plus cycloheximide (10 μg/ml) as indicated in the legend of Figure 4, lysed in lysis buffer and precipitated with 2 μg of anti-TRADD antibody.
In vitro cleavage of RIP protein
HeLa cells (5 × 107) were treated with 15 ng/ml human TNF and 10 μm cycloheximide for 4 hr at 37°C, then washed twice with cold DMEM and resuspended in 400 μl extract buffer (10 mm HEPES at pH 7.0, 40 mm glycerol phosphate, 50 mm NaCl, 2 mm MgCl, 1 mm DTT, 5 mm EDTA, 1 mm PMSF and 1μg/ml leupeptin). After freezing and thawing four times, the sample was centrifuged at 12,000g for 15 min at 4°C. The supernatant was again centrifuged at 100,000g for 60 min. The aliquots of cell extract (supernatants) were stored at −80°C. The RIP depleted extract was prepared by incubating 50 μl of apoptotic cell extract with 2 μg of anti-RIP bound on protein A resin at 4°C for 2 hr. The resin was removed by centrifugation and the supernatants were applied in protein cleavage assay.
35S-Labeled, wild-type, and mutant RIP proteins were prepared by coupled transcription and translation using the TNT-coupled reticulocyte lysate system (Promega). One microliter of 35S-labeled protein was mixed with 5 μl (∼50 μg of total proteins) of apoptotic cell extract and incubated at 37°C for 2 hr. The reaction was terminated by the addition of SDS-PAGE loading buffer. The resultant proteins were resolved in 4%–20% SDS-PAGE and visualized by autoradiography. For the caspase inhibition assay, 10 μm of each inhibitor was incubated with cell extract at 37°C for 15 min before the addition of 35S-labeled RIP proteins. In the experiments with recombinant caspases, the reaction was carried out in 25 μl of extract buffer with 50 ng of each recombinant caspase and 1 μl of 35S-labeled RIP proteins. For capspase depletion assay, 10 μl of anti-Caspase-3, anti-Caspase 8, or anti-HA sera was bound to 20 μl of protein A resin. Fifty microliters of apoptotic cell extract was incubated with the antibodies at 4°C for 12 hr. The resin was moved by centrifugation and the supernatants were applied in protein cleavage assay.
Apoptosis assay
HeLa or MCF-7 cells were co-transfected with pRSV–LacZ plus different RIP constructs as indicated in the figure legends. Twenty-four hours post-transfection, the cells were treated with 15 ng/ml of human TNF for 18 hr. Cells were fixed and stained as described (Liu et al. 1996).
Luciferase assay
Cells were cotransfected with p2xNF-κB Luc and different RIP constructs as indicated in the figure legends. Cells were collected and luciferase assay was conducted as described (Liu et al. 1996).
Acknowledgments
We thank Drs. A. Ting and B. Seed for the RIP-deficient Jurkat cells; Dr. G.M. Cohen for anti-Caspase 8 antibody. We also thank J. Lewis for his assistance in manuscript preparation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL zgliu@helix.nih.gov; FAX (301) 402-1997.
References
- Ashkenazi A, Dixit VM. Death receptors: Signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA. IκB-NF-κB structures: At the interface of inflammation control. Cell. 1998;95:729–731. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D. NF-κB: Ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- Baker SJ, Reddy EP. Modulation of life and death by the TNF receptor superfamily. Oncogene. 1998;17:3261–3270. doi: 10.1038/sj.onc.1202568. [DOI] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-κB in preventing TNFα-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D. A novel protein that interacts with the death domain of Ras/APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1 and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Brenner DA, O'Hara M, Angel P, Chojkier M, Karin M. Prolonged activation of jun and collagenase genes by tumor necrosis factor-alpha. Nature. 1989;337:661–663. doi: 10.1038/337661a0. [DOI] [PubMed] [Google Scholar]
- Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-κB pathway. Immunity. 1997;7:821–830. doi: 10.1016/s1074-7613(00)80400-8. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Cohen GM. Caspases: The executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns V, Yuan J. Proteases to die for. Genes & Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Duan H, Dixit VM. RAIDD is a new ‘death’ adapter molecule. Nature. 1997;385:86–89. doi: 10.1038/385086a0. [DOI] [PubMed] [Google Scholar]
- Duckett CS, Thompson CB. CD30-dependent degradation of TRAF2: Implications for negative regulation of TRAF signaling and the control of cell survival. Genes & Dev. 1997;11:2810–2821. doi: 10.1101/gad.11.21.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faleiro L, Kobayashi R, Fearnhead H, Lazebnik Y. Multiple species of CPP32 and Mch2 are the major active caspases present in apoptotic cells. EMBO J. 1997;16:2271–2281. doi: 10.1093/emboj/16.9.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H, Shu H-B, Pan M-G, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996a;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Hsu H, Huang J, Shu H-B, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996b;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Itoh N, Nagata S. A novel protein domain required for apoptosis: Mutational analysis of human FAS antigen. J Biol Chem. 1993;268:10932–10937. [PubMed] [Google Scholar]
- Jiang Y, Woronicz JD, Liu W, Goeddel D. Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science. 1999;283:543–546. doi: 10.1126/science.283.5401.543. [DOI] [PubMed] [Google Scholar]
- Karin M, Liu ZG, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Lee SY, Reichlin A, Santana A, Sokol KA, Nussenzweig MC, Choi Y. TRAF2 is essential for JNK but not NF-κB activation and regulates lymphocyte proliferation and survival. Immunity. 1997;7:703–713. doi: 10.1016/s1074-7613(00)80390-8. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu C-J, Yuan J. Cleavage of BID by Caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: Cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Natoli G, Costanzo A, Ianni A, Templeton DJ, Woodgett JR, Balsano C, Levrero M. Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science. 1997;275:200–203. doi: 10.1126/science.275.5297.200. [DOI] [PubMed] [Google Scholar]
- Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor κB. Proc Natl Acad Sci. 1989;86:2336–2340. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, O'Rourke KAM, Chinnaiyan R, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111–113. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- Ray CA, Black RA, Kronheim SR, Greenstreet TS, Sleath PR, Salvessen GS, Pickup DJ. Viral inhibition of inflammation: Cowpox virus encodes an inhibitor of the interleukin-1β converting enzyme. Cell. 1992;69:587–604. doi: 10.1016/0092-8674(92)90223-y. [DOI] [PubMed] [Google Scholar]
- Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- Reinhard C, Shamoon B, Shyamala V, Williams LT. Tumor necrosis factor alpha-induced activation of c-jun amino-terminal kinase is mediated by TRAF2. EMBO J. 1997;16:1080–1092. doi: 10.1093/emboj/16.5.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- Rothe M, Pan M-G, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculovirus inhibitor or apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- Smith CA, Farrah T, Goodwin RG. The TNF receptor superfamily of cellular and viral proteins: Activation, costimulation, and death. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Li F, Wong A, Kodandapani L, Smidt R, Krebs JF, Fritz LC, Wu JC, Tomaselli KJ. Bcl-xl functions downstream of caspase-8 to inhibit Fas- and tumor necrosis factor receptor 1-induced apoptosis of MCF7 breast carcinoma cells. J Biol Chem. 1998;273:4523–4529. doi: 10.1074/jbc.273.8.4523. [DOI] [PubMed] [Google Scholar]
- Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: A novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81:513–523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- Sun XM, MacFarlane M, Zhuang J, Wolf BB, Green DR, Cohen GM. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J Biol Chem. 1999;274:5053–5060. doi: 10.1074/jbc.274.8.5053. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–153. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Ayres TM, Wong GHW, Goeddel DV. A novel domain within the 55 Kd TNF receptor signals cell death. Cell. 1993;74:845–853. doi: 10.1016/0092-8674(93)90464-2. [DOI] [PubMed] [Google Scholar]
- Ting AT, Pimentel-Muinos FX, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15:6189–6196. [PMC free article] [PubMed] [Google Scholar]
- Tracey KJ, Cerami A. Tumor necrosis factor, other cytokines and disease. Annu Rev Cell Biol. 1993;9:317–343. doi: 10.1146/annurev.cb.09.110193.001533. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Verma IM, Green DR. Inhibition of TNF-induced apoptosis by NF-κB. Trends Cell Biol. 1998;8:107–111. doi: 10.1016/s0962-8924(97)01215-4. [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Declercg W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: Structure and function. Trends Cell Biol. 1995;5:392–399. doi: 10.1016/s0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, Timour MS, Gerhart MJ, Schooley KA, Smith CA, Goodwin RG, Rauch CT. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386–5397. doi: 10.1093/emboj/16.17.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-κB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wu MX, Ao Z, Prasad KV, Wu R, Schlossman SF. IEX-1L, an apoptosis inhibitor involved in NF-κB-mediated cell survival. Science. 1998;281:998–1001. doi: 10.1126/science.281.5379.998. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, Ohashi P, Rothe M, Goeddel DV, Mak TW. Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV, Mak TW. FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–1958. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- Zhang J, Cado D, Chen A, Kabra NH, Winoto A. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 1998;392:296–300. doi: 10.1038/32681. [DOI] [PubMed] [Google Scholar]
- Zheng LX, Lenardo M. NF-κB regulates Fas/APO-1/CD95- and TCR-mediated apoptosis of T lymphocytes. Eur J Immunol. 1999;29:878–886. doi: 10.1002/(SICI)1521-4141(199903)29:03<878::AID-IMMU878>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]