

Abstract
A palladium-catalyzed hydroalkylation reaction of allylic amine derivatives by alkylzinc reagents is reported. This reductive cross-coupling yields anti-Markovnikov products using a variety of allylic amine protecting groups. Preliminary mechanistic studies suggest that a reversible β-hydride elimination/hydride insertion process furnishes the primary Pd-alkyl intermediate, which then undergoes transmetallation followed by reductive elimination to form a new sp3-sp3 carbon-carbon bond.
The functionalization of Pd-alkyl intermediates has classically been difficult due to their propensity to undergo β-hydride elimination. Advances in this field have focused primarily on three general approaches to functionalize the Pd-alkyl: 1) rapid oxidation of the intermediate to allow for either substitution or reductive elimination of the resultant high oxidation state Pd complex,1 2) ligand control to slow β-hydride elimination2 and/or 3) substrate control through the use of stabilizing interactions such as the formation of π-allyl3 or π-benzyl intermediates.1c,4 Using the latter approach, our lab has reported a series of cross-coupling reactions of π-allyl5 (A, Scheme 1) or π-benzyl (B) intermediates6 that are formed by in situ generation of a Pd-hydride (through either alcohol oxidation or decomposition of a Pd-alkyl) and subsequent insertion into a conjugated alkene.
Scheme 1. Pd-Alkyl Stabilization Using Conjugated Alkenes.

One advantage of such a tactic to prepare Pd-alkyl intermediates is the utilization of an alkene as a surrogate for an alkyl electrophile.2a Also, by using a conjugated alkene, a stabilized Pd-alkyl is ultimately formed as the thermodynamic product through reversible β-hydride elimination/hydride insertion processes,6a enabling cross-coupling as well as the formation of a single constitutional isomer. Unfortunately, this approach is limited to alkene coupling partners, which provide formal stabilization. Specifically, without stabilization, reversible β-hydride elimination/hydride insertion processes lead to complex mixtures of alkene isomers and coupled products.6 Therefore, to overcome this limitation, we envisioned applying alternative stabilizing motifs that could enable the selective reductive cross-coupling of a non-conjugated alkene and an organometallic, thereby significantly expanding the scope of alkene substrates.
To this effect, a functional group that can act as a versatile synthetic handle and can be easily incorporated into the alkene substrate would be most attractive. A recent report by Feringa and coworkers7 demonstrated the use of phthalimide-protected allylic amines to achieve aldehyde-selective anti-Markovnikov Wacker oxidations (Scheme 2). It should be noted that the phthalimide group is uniquely suited in directing the Wacker oxidation to the anti-Markovnikov position, as other allylic amine derivatives lead to mixtures of products.7 The authors suggest the high selectivity may be attributed to chelation of the phthalimide group to the palladium catalyst prior to oxypalladation. Encouraged by Feringa's results, the evaluation of allylic phthalimides was pursued in order to hypothetically direct the formation of and stabilize the resultant Pd-alkyl complex for subsequent cross-coupling. Herein, we report the development and preliminary mechanistic investigation of a highly selective anti-Markovnikov hydroalkylation reaction of allylic amine derivatives, wherein, a broad range of functional groups at the allylic position are permitted.
Scheme 2. Feringa and Coworkers' Wacker Oxidation and Our Proposed Hydroalkylation Approach PG = protecting group R′ = PG or H.

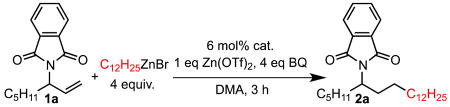
Preliminary investigations utilized the optimal conditions for the previously reported Pd(IiPr)(OTs)2-catalyzed hydroalkylation of styrene derivatives6c with organozinc reagents. Using allylic phthalimide 1a, an 80% GC yield of 2a was delivered in high selectivity for the linear product (entry 3, Table 1). Control studies of the reaction conditions involving the removal of the NHC ligand (entry 4) and the use of Pd(MeCN)2Cl2 as the catalyst (entry 5) resulted in similar yields and selectivity, prompting the use of these simplified conditions in further optimization. Lowering the temperature and the utilization of molecular sieves resulted in excellent yield and 17:1 selectivity favoring the linear over the branched product (entry 7).
Table 1. Reaction Optimization.
 | ||||||
|---|---|---|---|---|---|---|
| entry | catalyst | temp. | additive | % conv.a | % yielda | (I:b)b |
| 1 | no palladium | RT | -- | <2 | -- | -- |
| 2c | Pd(IiPr)(OTs)2 | RT | Cu(OTf)2, O2 | <2 | -- | -- |
| 3 | Pd(IiPr)(OTs)2 | RT | -- | 85 | 80 | 15:1 |
| 4 | Pd(MeCN)2(OTs)2 | RT | -- | 86 | 81 | 16:1 |
| 5 | Pd(MeCN)2Cl2 | RT | -- | 88 | 80 | 17:1 |
| 6 | Pd(MeCN)2Cl2 | 0 °C | -- | 95 | 86 | 17:1 |
| 7 | Pd(MeCN)2Cl2 | 0 °C | 3 Å MS | 99 | 91 | 17:1 |
Measured by GC using an internal standard.
Ratio of linear:branched product isomers determined by GC. Yields are a combination of both linear and branched products.
Zn(OTf)2 and BQ were not added.
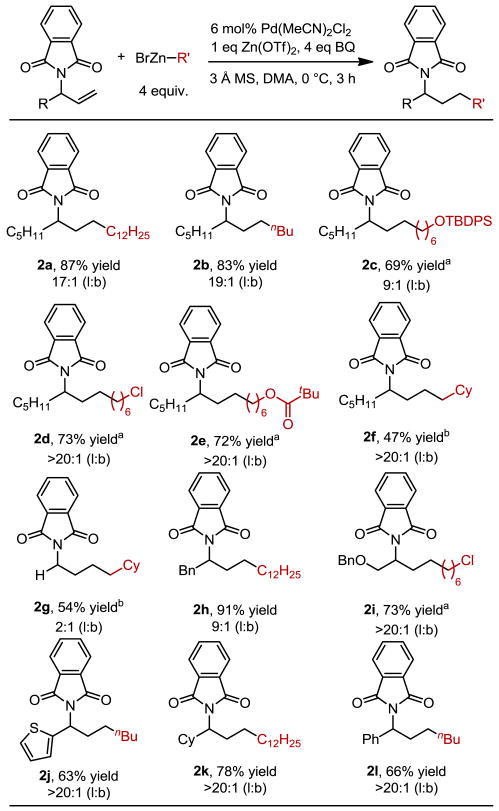
Evaluation of the scope initially focused on the use of various organozinc reagents with 1a. Simple alkyl organozinc reagents perform well, giving linear products 2a and 2b in high selectivity. Functionalized organozinc reagents were then examined, which led to products containing a TBDPS-protected alcohol (2c), an alkyl chloride (2d), and an ester (2e). Methylcyclohexyl zinc bromide, which contains substitution at the β-position, gave product 2f in 46% yield with excellent selectivity. We next examined the effect of varying groups on the allylic phthalimide substrate. Lower selectivity for the linear product (2:1) was observed when the substrate lacks additional substitution at the carbon bearing the phthalimide group (2g). Introduction of a benzyl group (2h) resulted in a 91% yield with modestly diminished selectivity for the linear product. Substrates containing a benzyl ether (2i) and a heteroaromatic (2g) both gave moderate yields with >20:1 selectivity. Although these substrates could potentially competitively coordinate to palladium, they produce the anti-Markovnikov product selectively. Substrates containing larger groups adjacent to the phthalimide (2k, 2l) also gave good yields with high selectivity for the linear product.
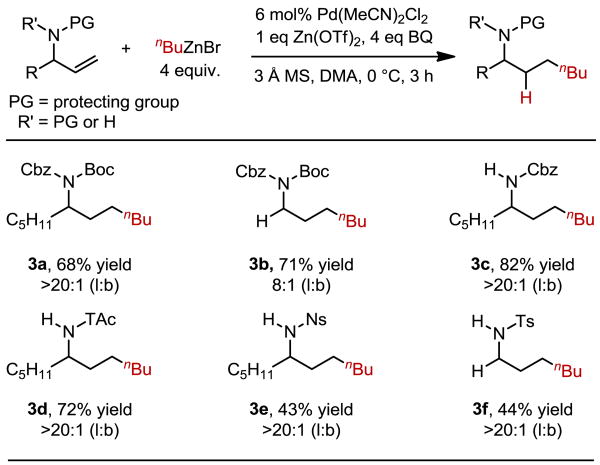
It was initially believed, based on Feringa's report, that allylic phthalimides were uniquely suited to stabilize the resultant Pd-alkyl intermediates, potentially through chelation,7,8 and selectively deliver the linear product. Therefore, it was surprising that under our optimized conditions, a variety of electronically disparate amine protecting groups also gave the linear hydroalkylation product (Table 3). Several attractive and commonly employed amine protecting groups were evaluated including doubly protected N-Cbz-N-Boc allylic amines (3a, 3b) suggesting that this strategy may be more general than anticipated. Additionally, a variety of singly protected amines were tolerated under the reaction conditions, including a Cbz protected amine, which gave the coupled product in 82% yield with >20:1 selectivity (3c) for the linear product. Electron poor protecting groups such as trichloroacetamide (TAc, 3d) and sulfonamide protected amines gave the cross-coupled products in excellent selectivity (3e, 3f). These cases are especially interesting in that the Lewis basicity of the carbonyl or sulfonyl groups should be significantly reduced, raising questions about our previous hypothesis involving chelation to the catalyst. Finally, the use of N-tosyl allyl amine, which also lacks additional substitution at the amine-bearing carbon, gave the anti-Markovnikov product in >20:1 selectivity (3f) in contrast to the related phthalimide protected product 2g.
Table 3. Scope of Protecting Groups.
|
Ratio of linear:branched product isomers determined by GC, 1H NMR integration, and/or yields of isolated products. All yields represent an average of two experiments on at least 0.5 mmol scale.
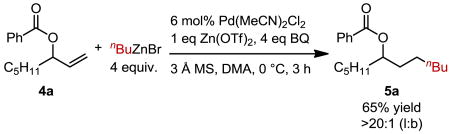
Encouraged by the successful coupling of various protected allylic amines, we subjected a protected allylic alcohol, a ubiquitous intermediate in organic synthesis, to our reductive cross-coupling reaction conditions. To our delight, a benzoyl-protected allylic alcohol (4a) gave the corresponding product 5a in 65% yield and >20:1 selectivity (eq. 1). Submission of dodecene to the reaction conditions resulted in alkene isomerization and trace amounts of product isomers, suggesting that allylic substitution is required to achieve selectivity for the anti- Markovnikov product.9
 |
(1) |
Considering the breadth of protecting groups allowed and the generally high selectivity for the anti-Markovnikov product, an important mechanistic consideration is whether insertion into the Pd-hydride is selectively directed by the allylic amine. To probe this, a perdeuterated alkylzinc reagent was prepared and submitted to the hydroalkylation reaction conditions (Scheme 3). Surprisingly, 91% deuterium incorporation results with a similar amount of deuterium incorporation observed at both alkene carbons. This suggests that, after formation of a Pd-hydride (deuteride, C) via transmetallation with the organozinc reagent and β-hydride elimination,6c coordination and insertion of the allylic amine (A) can result in either a 1,2-insertion (D′) or a 2,1-insertion (D). In the case of a 2,1-insertion, intermediate D rearranges to the primary Pd-alkyl complex (F) via a β-hydride elimination (E) followed by a migratory insertion. Primary Pd-alkyl intermediates F and D′ then undergo transmetallation followed by reductive elimination. It should be noted that no deuterium is incorporated into the allylic position, which is consistent with the observed stereochemical retention when using an enantioenriched substrate (Scheme 3). The combination of this information with the ability to successfully use electron poor allylic amine derivatives suggests that the success of this reaction arises from a slowing of β-hydride elimination at the allylic position in combination with a relatively fast cross-coupling of the primary Pd-alkyl intermediate.
Scheme 3. Deuterium Labeling Study and Evaluation of Retention of Enantiomeric Excess.

In summary, we have reported a Pd-catalyzed hydroalkylation reaction of various protected allylic amines, which we have also extended to an allylic alcohol derivative. This process features the ability to incorporate a remote stereocenter in regions devoid of other functionality. Initial mechanistic studies suggest a reversible β-hydride elimination/hydride insertion process to arrive at a primary Pd-alkyl intermediate, which predominantly undergoes cross-coupling. While the precise role of the protecting group on the amine remains unclear, it is required to achieve selective anti-Markovnikov addition products. Future work will be focused on exploring the synthetic utility of this method and elucidating the features required to control β-hydride elimination in related processes.
Supplementary Material
Table 2.
Palladium-Catalyzed Cross-Coupling of Pthalimide Protected Allylic Amines with Alkylzinc Reagents.
|
Ratio of linear:branched product isomers determined by GC, 1H NMR integration, and/or yields of isolated products.
8 equiv. of BrZn-R was used.
The reaction was performed at rt. All yields represent an average of two experiments on at least 0.5 mmol scale.
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS RO1 GM3540). We are grateful to Johnson Matthey for the gift of various Pd salts. We also would like to thank Susanne Podhajsky for preforming key preliminary experiments.
Footnotes
Supporting Information Available: Optimization data, experimental procedures, and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Xu LM, Li BJ, Yang Z, Shi ZJ. Chem Soc Rev. 2010;39:712–733. doi: 10.1039/b809912j. [DOI] [PubMed] [Google Scholar]; (b) Kalyani D, Sanford MS. J Am Chem Soc. 2008;130:2150–2151. doi: 10.1021/ja0782798. [DOI] [PubMed] [Google Scholar]; (c) Kalyani D, Satterfield AD, Sanford MS. J Am Chem Soc. 2010;132:8419–8427. doi: 10.1021/ja101851v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Satterfield AD, Kubota A, Sanford MS. Org Lett. 2011;13:1076–1079. doi: 10.1021/ol103121r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wang A, Jiang H, Chen H. J Am Chem Soc. 2009;131:3846–3847. doi: 10.1021/ja900213d. [DOI] [PubMed] [Google Scholar]; (f) Liu G, Stahl SS. J Am Chem Soc. 2006;128:7179–7181. doi: 10.1021/ja061706h. [DOI] [PubMed] [Google Scholar]; (g) Alexanian EJ, Lee C, Sorensen EJ. J Am Chem Soc. 2005;127:7690–7691. doi: 10.1021/ja051406k. [DOI] [PubMed] [Google Scholar]; (h) Desai LV, Sanford MS. Angew Chem, Int Ed. 2007;46:5737–5740. doi: 10.1002/anie.200701454. [DOI] [PubMed] [Google Scholar]; (i) Welbes LL, Lyons TW, Cychosz KA, Sanford MS. J Am Chem Soc. 2007;129:5836–5837. doi: 10.1021/ja071204j. [DOI] [PubMed] [Google Scholar]; (j) Streuff J, Hövelmann CH, Nieger M, Muñiz K. J Am Chem Soc. 2005;127:14586–14587. doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]; (k) Qiu S, Xu T, Zhou J, Guo Y, Liu G. J Am Chem Soc. 2010;132:2856–2857. doi: 10.1021/ja909716k. [DOI] [PubMed] [Google Scholar]; (l) Li Y, Song D, Dong VM. J Am Chem Soc. 2008;130:2962–2964. doi: 10.1021/ja711029u. [DOI] [PubMed] [Google Scholar]; (m) Park CP, Lee JH, Yoo KS, Jung KW. Org Lett. 2010;12:2450–2452. doi: 10.1021/ol1001686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Frisch AC, Beller M. Angew Chem, Int Ed. 2005;44:674–688. doi: 10.1002/anie.200461432. [DOI] [PubMed] [Google Scholar]; (c) Ishiyama T, Abe S, Miyaura N, Suzuki A. Chem Lett. 1992:691–694. [Google Scholar]; (d) Zhou J, Fu GC. J Am Chem Soc. 2003;125:12527–12530. doi: 10.1021/ja0363258. [DOI] [PubMed] [Google Scholar]; (e) Netherton MR, Dai C, Neuschütz K, Fu GC. J Am Chem Soc. 2001;123:10099–10100. doi: 10.1021/ja011306o. [DOI] [PubMed] [Google Scholar]; (f) Netherton MR, Fu GC. Angew Chem, Int Ed. 2002;41:3910–3912. doi: 10.1002/1521-3773(20021018)41:20<3910::AID-ANIE3910>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]; (g) Menzel K, Fu GC. J Am Chem Soc. 2003;125:3718–3719. doi: 10.1021/ja0344563. [DOI] [PubMed] [Google Scholar]
- 3.(a) Bäckvall JE, Bystroem SE, Nordberg RE. J Org Chem. 1984;49:4619. [Google Scholar]; (b) Bäckvall JE, Nystroem JE, Nordberg RE. J Am Chem Soc. 1985;107:3676. [Google Scholar]; (c) Bäckvall JE, Vaagberg JO. J Org Chem. 1988;53:5695. [Google Scholar]; (25) Bar GLJ, Lloyd-Jones GC, Booker-Milburn KI. J Am Chem Soc. 2005;127:7308. doi: 10.1021/ja051181d. [DOI] [PubMed] [Google Scholar]; (d) Du H, Yuan W, Zhao B, Shi Y. J Am Chem Soc. 2007;129:7496. doi: 10.1021/ja072080d. [DOI] [PubMed] [Google Scholar]; (e) Du H, Zhao B, Shi Y. J Am Chem Soc. 2008;130:8590. doi: 10.1021/ja8027394. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhao B, Du H, Cui S, Shi Y. J Am Chem Soc. 2010;132:3523. doi: 10.1021/ja909459h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Tamaru Y, Hojo M, Higashimura H, Yoshida Z-i. Angew Chem, Int Ed Engl. 1986;25:735–737. [Google Scholar]; (b) Tamaru Y, Hojo M, Kawamura S, Yoshida Z. J Org Chem. 1986;51:4089. [Google Scholar]; (c) Rodriguez A, Moran WJ. Eur J Org Chem. 2009:1313–1316. [Google Scholar]; (d) Parrish JP, Jung YC, Shin SI, Jung KW. J Org Chem. 2002;67:7127–7130. doi: 10.1021/jo020159p. [DOI] [PubMed] [Google Scholar]
- 5.Liao L, Sigman MS. J Am Chem Soc. 2010;132:10209–10211. doi: 10.1021/ja105010t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Gligorich KM, Cummings SA, Sigman MS. J Am Chem Soc. 2007;129:14193–14195. doi: 10.1021/ja076746f. [DOI] [PubMed] [Google Scholar]; (b) Urkalan KB, Sigman MS. Angew Chem, Int Ed. 2009;48:3146–3149. doi: 10.1002/anie.200900218. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Urkalan KB, Sigman MS. J Am Chem Soc. 2009;131:18042–18043. doi: 10.1021/ja908545b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Werner EW, Urkalan KB, Sigman MS. Org Lett. 2010;12:2848–2851. doi: 10.1021/ol1009575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiner B, Baeza A, Jerphagnon T, Feringa BL. J Am Chem Soc. 2009;131:9473–9474. doi: 10.1021/ja902591g. [DOI] [PubMed] [Google Scholar]
- 8.(a) Enzmann A, Eckert M, Ponikwar W, Polborn K, Schneiderbauer S, Beller M, Beck W. Eur J Inorg Chem. 2004:1330–1340. [Google Scholar]; (b) Fairlamb IJS, Kapdi AR, Lee AF, Sánchez G, López G, Serrano JL, García L, Pérez J, Pérez E. Dalton Trans. 2004:3970–3981. doi: 10.1039/b413886d. [DOI] [PubMed] [Google Scholar]
- 9.Internal alkenes were also submitted to the reaction conditions, which resulted in <10% yield of a mixture of product isomers.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.