

Abstract
Adoptive T-cell therapy holds great promise for the treatment of metastatic melanoma. However, prohibitive costs associated with current technology required for culture and expansion of tumor-reactive T-cells, the need for intense preconditioning regimens to induce lymphopenia, and the unpredictable anti-tumor effect of adoptively transferred T-cells remain significant impediments for its clinical implementation. Here we report a simplified combinatorial approach that involves short activation of CD8+ T cells in the presence of IL-12 followed by adoptive transfer into tumor bearing animals after a single injection of cyclophosphamide. This approach resulted in complete eradication of B16 melanoma, and the establishment of long term immunological memory capable of fully protecting mice after a second B16 melanoma challenge. The activated donor cells were unique because they simultaneously exhibited traits for cytotoxic effector function, central memory-like, homing, and senescence. After tumor eradication and within three months after transfer, CD8+ cells exhibited a conventional memory CTL phenotype. Moreover, these memory CTLs acquired functional attributes characteristic of memory stem cells, including the ability to resist chemotherapy-induced toxicity. Our results suggest that short-term T-cell receptor signaling in the presence of IL-12 promotes promiscuous qualities in naïve CTL which - upon transfer into lymphopenic hosts- are sufficient to eradicate tumors and generate life-long tumor-specific memory.
Keywords: Pmel, melanoma, IL-12, ACT, T cell therapy, memory, CD8
Introduction
While the prognosis of localized melanoma is favorable with surgical resection alone, stage 4 metastatic melanoma has a very poor prognosis with a median overall survival of approximately 11 months with chemotherapy and 5-year survival of only 5% [1]. The recently FDA approved CTLA-4 monoclonal antibody, ipilimumab, when combined as first line therapy in patients with metastatic melanoma with chemotherapy was able to prolong overall survival by only 2 months (11.2 vs 9.1 months; hazard ratio [HR] for death, 0.72; P < .001),and 3 year survival by only 8% (20.8% vs 12.2%) [2]. Melanoma tumors often overexpress tissue-specific developmental antigens that can be recognized by the immune system [3]. Therefore, immunologists have devised and begun to refine three major experimental therapies. They include using therapeutic vaccinations that use modified tumor cells and dendritic cells, lowering the threshold of immune activation by reducing negative controls exercised by CTLA-4 and regulatory T-cells (TReg) [3], and employing adoptive cell therapy (ACT) directed against the tumor. The major conceptual hurdles and goals of ACT employing CD8+ CTL against melanoma and other tumors are two-fold. First, tumor-specific CTL should be primed and expanded in vitro in such a manner that they will act as effector CTL in vivo by seeking out tumor cells and killing them upon recognition. Second, tumor-specific memory CTL that can self renew and, if needed, reacquire effector function should develop in vivo for life-long protection of the host against reccurrence of the tumor.
Rather elegant in vivo immunological studies have shown that the division of a single naïve CTL is sufficient to defeat a viral infection by give rise to effector CTL and memory CTL [4, 5]. In some circumstances, the acquisition of the effector CTL stage by naïve CTL may be required prior to their further development into memory CTL [6, 7]. Because ACT with naïve CTL usually fails to eradicate tumors, we begin this report with the investigation of the tumoricidal activity of the progeny of CTL primed in vitro under distinct culture conditions as well as in the context of prior conditioning of the host which is known to improve ACT for several reasons [8-15]. Our results indicate that CTL priming and expansion protocols for ACT must take into consideration that the activation of CTL results in developmentally different fates [16, 17] which will determine their response to lymhopenic environments, their ability to lyse the tumor, and the development of anti-tumor immunity. These outcomes were preferentially endorsed by the quality and quantity or length of the exogenous signals [18-21]. Our report specifically explores the activation and control of naïve CTL by T-cell receptor signals and exogenous cytokine at the time of the initial in vitro activation. We found that a brief antigen-dependent activation of naïve CD8+ T cells in the presence of IL-12 followed by exposure to a lymphopenic environment results in a novel differentiation program that clears B16 melanoma tumors and generates cells that will protect against a tumor re-challenge.
Materials and methods
Mice
C57BL/6 (Thy1.1-) and Pmel-1 transgenic [22] (Thy1.1+Vβ13+) mice were purchased from Jackson Laboratory (Bar Harbor, ME). All animals were housed under specific pathogen-free conditions in accordance with Institutional and Federal guidelines at the University of Miami and the experiments were approved by the local IACUC.
Antibodies and flow cytometry
All antibodies were purchased from BD Biosciences. They were used at concentrations recommended by their manufacturers and the stained cell samples were examined on an LSR-II (BD Biosciences). The analysis was performed with FlowJo (Treestar, Ashland, OR). To assess the efflux capability of the cells, they were incubated with 2.5 μM daunorubicin (Sigma) for 20 min at 37°C prior to staining with APC anti-mouse CD8 PE anti-mouse Thy1.1 mAbs. Daunorubicin fluorescence was detected at 610/15 nm.
Melanoma culture and tumor growth
B16-F10 cells, derived from a gp100+ spontaneous murine melanoma cell line, were obtained from American Type Culture Collection (ATCC) (Manassas, VA). The cells were cultured in RPMI 1640 containing 10% FBS, 0.1% penicillin/streptomycin, 0.2% L-glutamine, 0.05% 2-mercaptoethanol, 0.01% sodium pyruvate, 0.1% HEPES and 0.1% nonessential amino acids. Melanoma tumors were established by subcutaneous (s.c.) injection of 2.5 × 105 B16-F10 cells in the right flank. Tumors were measured using calipers every other day. Mice with tumors larger than 400mm2 were euthanized.
Ex vivo CD8+T cell activation and adoptive cell transfer
Cell suspensions from spleen and lymph nodes of Pmel-1 mice were adjusted to 1×106 cells/ml in complete RPMI and activated with 1 μg/ml of cognate peptide (KVPRNQDWL) (American Peptide; CA). Where indicated, IL-12 (R&D Systems) was added at the time of priming to provide 10 ng/ml. Three days after activation, so-called “early” Pmel cells were harvested. To generate so-called “late” Pmel cells, early Pmel cells were recultured for four more days in the presence of 10ng/ml of IL-2 (R&D Systems). One day prior to ACT, wild type C57BL6 mice bearing 7-day-old B16-F10 tumors were conditioned by a single intraperitoneal injection of 4 mg CTX (Sigma). 24 hours later, adoptive immunotherapy (ACT) was performed by intravenous injection of 5 × 106 Pmel cells that had been primed under various conditions. To determine their persistence, blood, bone marrow, spleen, and lymph node suspensions were stained with mAb against Thy1.1, CD8 and and donor Pmel cells were identified as CD8+ Thy1.1+ cells by flow cytometry and compared to the endogenous CD8+Thy1.1-CTL.
Gene expression analysis
Early Pmelsham and early Pmel12 were harvested from tissue culture flasks. Memory Pmel cells were sorted (CD8+Thy1.1+) from spleen suspensions from recipients of early Pmel12 cells 90 days after adoptive transfer. Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany), and cDNA was synthesized from 1 mg of RNA using the Omniscript RT Kit (Qiagen), as per manufacturer's instructions. Gene expression was measured using quantitative real-time PCR and TaqMan probes (Applied Biosystems, Foster City, CA, USA) in a final reaction volume of 20 ml. Ribosomal 18s RNA was used as the internal standard. RT-PCR was performed on a StepOne real-time PCR system (Applied Biosystems). The relative quantification of the target transcripts normalized to the endogenous control was determined by the comparative Ct method. Relative changes in gene expression between samples were analyzed using the 2-ddCt method.
Statistical analyses
p values were calculated using student t test and a significant difference among experimental groups was defined as a p value of <0.05. Cumulative survival was calculated using a Kaplan-Meier curve. The relationship between donor Pmel cells and tumor size was examined by scatter plot analysis and descriptive statistics as well as by fitting a regression model.
Results
Anti-melanoma activity of early Pmel12 cells synergizes with preconditioning of the host
The most frequently used syngeneic mouse melanoma model is the B16 tumor. This tumor expresses several melanoma associated antigens including gp100 that is recognized by the transgenic Pmel-1 T-cell receptor in C57BL/6 mice [22]. The striking biological and molecular similarities of this model to human melanoma have been recently discussed [23]. Because this model is also known to be resistant to consistent cures by various immunotherapeutical strategies it was chosen in the following studies. Initially, we noticed that ACT with naïve Pmel cells activated for three days in the presence of IL-12 (early Pmel12) significantly delayed the tumor growth of established B16 melanoma tumors (see Figure 1A) as compared to other culture conditions (Figure 1B). However, eventually all tumors progressed and the animals succumbed to the disease.
Figure 1.

Antitumor activity of CTL primed in the presence of IL-12 synergizes with host preconditioning. (A) Naïve Pmel cells were activated in in vitro as indicated. Tumor progression was monitored in mice receiving primed cells as indicated (B) and in mice pre-treated with a single dose of CTX (C). Experiments shown in B and C represent a minimum of three individual observations with 5 animals per group. (D) Cumulative survival of mice from indicated groups.
Next, we decided to augment the efficacy of ACT by preconditioning the host [11]. Tumor bearing animals received a single intraperitoneal injection of 4 mg of cyclophosphamide (CTX). This treatment has been shown to induce a transient lymphopenia that lasts for three days and is completely resolved after 14 days [24]. 24 hours after CTX injection, early Pmelsham, early Pmel12 , late Pmelsham or late Pmel12 cells were adoptively transferred into the mice (Figure 1A) and tumor progression was monitored (Figure 1C). CTX treatment on its own delayed tumor growth and it was further delayed by the transfer of late Pmelsham and, more impressively, by late Pmel12 or early Pmelsham. In sharp contrast, when early Pmel12 were transferred into CTX treated mice the tumor was quickly eradicated and the animals survived (>70 days after ACT; p=<0.001 as compared to CTX and p=0.003 as compared to both PBS and CTX) (Figure 1D).
Because of the long-term survival of the experimental group receiving CTX and early Pmel12 (individual mice were kept alive for more than 120 days) we assessed whether the animals had developed immunity against the tumor. Indeed, these animals resisted a second challenge with the B16 melanoma 50 days after adoptive therapy (Figure 1D). These results suggested that only naïve tumor-specific CD8+ T cells primed for three days in the presence of IL-12 were optimally differentiated to respond to the host environment 24 hours post conditioning with CTX.
Circulation of transferred cells after ACT correlates with tumor regression
In patients receiving ACT for melanoma the persistence of adoptively transferred CTL has been suggested to correlate with their anti-tumor activity [25]. To this end, we collected blood samples from mice with distinctly different tumor burdens, ranging from palpable - but not measurable - to >400 mm2. When we compared the tumor size to the number of circulating Pmel cells, lower percentages of donor cells seemed to coincide with larger tumors (Figure 2A). To expand this observation to a scatter plot, blood and tumors from 15 independent mice were assessed on various days post ACT (Figure 2B). Despite considerable variation in the tumor sizes (mean 160.04, std. dev. 190.51; median 75.33, IQR: 0 to 310.00) their inverse correlation to the percentages of circulating Pmel cells (mean 2.68, std. dev. 2.51; median 1.74, IQR: 0.30 to 4.80) seemed significant. Using a linear regression without intercept to the reciprocal of the donor Pmel percentage (using the 15 independent observations from days 39 to 45), the resulting model explained approximately 81% of the variation in tumor size (R2=0.809, p<0.001 model fit) and supported the idea that the persistence of donor cells is linked to tumor regression.
Figure 2.
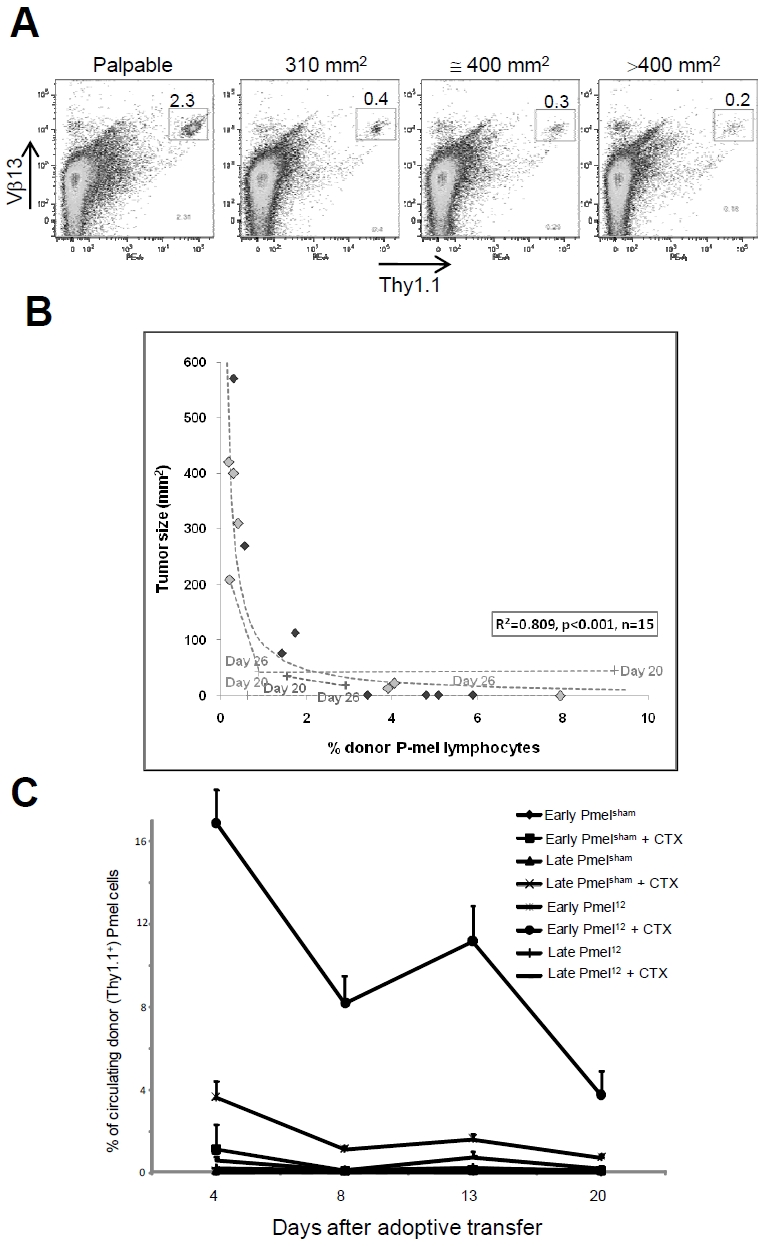
Tumor regression and eradication correlate with persistence of donor cells in circulation after ACT. Lymphodepletion enhances the survival of early Pmel12 which inversely correlates with tumor burden. (A). Representative scatter plot of frequency of donor Pmel cells (Vβ13+Thy1.1+) in mice with the indicated tumor burden. (B) Scatter plot analysis of the relationship between tumor burden and frequency of donor Pmel cells in 15 mice. (C) Frequency of donor cells in mice treated as indicated. Results shown are the average of 5 independent measurements ± SDEV, per group. Adoptively transferred mice were bled at the indicated time points and the percentage of circulating donor cells (Thy1.1+) was determined by flow cytometry.
Next, we examined the impact of the preconditioning of the host on the persistence of Pmel primed under different conditions. The number of circulating early Pmel12 was significantly elevated in lymphopenic hosts (Figure 2C) and it paralleled the superior anti-tumor activity shown in Figure 1C. The donor cells were detectable in circulation as early as day 4 and remained readily detectable beyond 3 weeks post ACT (approximately 4% of all circulating lymphocytes). When early Pmel12 were transferred into lymphocomplete hosts, however, their persistence was significantly reduced and they reached the detection limit three weeks post ACT. The comparison of these conditions suggests a synergistic interplay between in vitro priming conditions and factor(s) available in preconditioned hosts.
Progeny of early Pmel12 are dynamically established in preconditioned hosts
Long-term persistence of the donor cells and immunological anti-tumor memory (Figure 3 and Figure 1D) suggested that some donor cells had acquired characteristics associated with self-renewing memory CTL, such as a CD44lo/Sca-1hi phenotype [26, 27]. To that end, we determined the number of donor-derived CTL and their anatomical location after ACT of preconditioned hosts (Figure 3). Progeny of the transferred early Pmel12 were present in the examined lymphoid compartments for more than 31 days and CD44lo/Sca-1hi cells comprised the majority of the donor cells in contrast to the host cells (Figure 4). Interestingly, their relative organ distribution changed with time. Initially, they equivalently accumulated in spleen, blood and lymph nodes but after four weeks they had preferentially seeded the lymph nodes and bone marrow, organs known to support self-renewing memory CTL [28]. These findings suggested that the offspring of early Pmel12 undergoes extensive changes for at least one month in lymphopenic hosts.
Figure 3.

Donor cells dynamically engraft the host after ACT. Wild type mice adoptively transferred with early Pmel12 cells were sacrificed at the indicated time points, and cell suspensions from spleen, lymph nodes, bone marrow, and peripheral blood were prepared. Cell suspensions were stained with anti-thy1.1, CD8, CD44 and Sca-1 mAbs. Expression of CD44 and Sca-1 was determined on both host (Thy1.1-) and donor (Thy1.1+) CD8+ populations. Frequency of donor Pmel cells is shown as the average donorhost ratio±SDEV (n=3). P values were calculated using a two sided Student t test. The experiment shown is representative of three repeats.
Figure 4.

Distribution of CD44l0Sca-1hi donor Pmel cells. Wild type mice adoptively transferred with early Pmel12 cells were sacrificed, and cell suspensions from spleen, lymph nodes, bone marrow, and peripheral blood were prepared. Cell suspensions were stained with anti-thy1.1, CD8, CD44 and Sca-1 mAbs. Expression of CD44 and Sca-1 was determined on both host (Thy1.1) and donor (Thy1.1+) CD8+ populations. Frequency of donor Pmel cells is shown as the average donorhost ratio±SDEV (n=3) 5 and 31 days after adoptive transfer (A). Gating strategy to determine the distribution of CD44l0Sca-1hi host and donor cells (B).
Early Pmel12 exhibit unusual traits
Having identified a hereto unreported immunotherapy approach that cures 100% of established B16 melanomas, we were interested in characterizing underlying reasons for the effectiveness of the ACT. Based on the potent in vivo activity of early Pmel12 (in contrast to early Pmelsham, late Pmelsham, and late Pmel12; Figure 1 and data not shown) it became relevant to characterize the apparently unique activation of early Pmel12. Based on flow cytometry, the cell population seemed to represent central memory CTL, i.e., CD62L+, CD44+ CTL as we and others reported previously [29, 30]. On the other hand, the cells also expressed high levels of the IL-2Ra (CD25) as well as the cytokine IFN-g and the cytotoxic granule molecule granzyme B (Figure 5A) suggesting that they also had acquired traits of active effector CTL. To shed light on this apparent discrepancy, we used real time RT-PCR to compare the expression of several molecules in early Pmel112 with early Pmelsham representing conventional effectors (Figure 5B). We included probes for a) CTL effector molecules (perforin and granzyme B); b) transcription factors critical for the various fates of activated CTL (Runx3, Eomes, T-bet, prdm1, bcl-6 and TCF-1 [31-39]; c) members of the gc-cytokine receptor cytokine receptor family (CD25, CD127 and CD132); d) a master regulator of mitochondrial biogenesis (ppargc1a and ppargc1b) to interrogate energy metabolism [40]; e) the multidrug efflux molecule abcb1b to monitor the recently described chemotherapy resistance of memory CTL [41]; and f) p19 arf because of its negative control of self-renewing ability and promotion of senescence [42] as well as Klrg1 whose increased expression correlates with the progressive differentiation of CTL into short-lived effector cells [43]. We found that some genes were underexpressed while others were overexpressed (left versus right of Figure 5B). Here we focus only on statistically significant differences. It was interesting that the t-box transcription factor ratio between eomes and tbet was inverted because t-bet has been proposed to divert central-memory CTL into effector and effector-memory CTL consistent with their expression of IFN-g and granzyme B (Figure 5A). We also found that the CTL memory transcription factor tcf7 was strongly repressed while the expression of the effector function transcription factor prdm1 was elevated. Also elevated were the terminal differentiation marker Klrg1 and even much more so p19arf. Taken together, these markers pointed to the generation of a late effector stage CTL by IL-12 at the time of transfer, which seemed to contradict conventional wisdom that effector cells are less suitable for ACT[44]. Also, these cells expressed lymph node homing receptor CD62L used by naïve and memory CTL as we have previously shown [29]. Taken all together, Pmel12 seemed to be uniformly made up of promiscuous CTL traits.
Figure 5.

Promiscuous traits of early Pmel12 cells at the time of ACT. (A) Naïve, early Pmelsham and Pmel12 cells were incubated with mAbs against the indicated parameters. Cells were analysed by flow cytometry and representative histograms are shown. (B) Gene expression analysis by real time rtPCR was performed on mRNA extracted from early early Pmel-sham and Pmel12 cells. Expression is depicted as the ratio of Pmel12 to Pmelsham. Data shown is the average of three independent measurements ± SDEV. P values were calculated using a two sided Student t test.
Adoptively transferred early Pmel12 eventually convert into conventional memory CTL
Next, we wanted to know whether the unusually programmed donor cells and their progeny would continue to maintain unusual properties after ACT. If not, they should have differentiated into resting memory CTL by the time of the analysis (1-3 months after transfer). In support of the latter possibility, the eomes/t-bet ratio was reversed, and CD25, granzyme B as well as p19 arf expression were abolished. Moreover, the IL-7R (CD127) and tcf7 were upregulated. Yet, mRNA expression of the abcb1b transporter that contributes to the survival of long-lived memory CD8+ T cells [45] was not significantly upregulated (Figure 6A). Therefore, we also investigated its functional activity by evaluating the cells' ability to efflux a fluorescent anthracy-cline (daunorubicin; [41]). After allowing the cells to efflux for 30 min (Figure 6B), a significant percentage of the early Pmel12 progeny had removed daunorubicin in contrast to early Pmel12 prior to ACT (20% versus 1.7%). These differences were not intrinsic to the transgenic cells because the efflux ability of naïve Pmel cells and the host's own cells compared favorably to each other (7.7% versus 8.6%). This functional assessment together with the gene expression results and the dynamic homing of the transferred CTL suggested that critical memory characteristics were not acquired in vitro prior to ACT but later during their further differentiation in vivo.
Figure 6.

Long term Pmel12 survivors display late effector/memory-like phenotype and resist daunorubicin induced toxicity. (A) Gene expression analysis of memory associated markers was performed on early Pmel12 and sorted long term Pmel survivors. Expression is depicted as the ratio of long term Pmel12 survivors to early Pmel12. Data shown is the average of three independent experiments ± SDEV. P values were calculated using the Student t test. (B) Cell suspensions of naive, early Pmelsham, early Pmel12, and splenocytes from wt recipient of early Pmel12 cells 90 days after adoptive transfer, were incubated in the presence of daunorubicin and stained with mAbs against CD8+ and Thy1.1. The subset of CD8+ positive cells that effluxed daunorubicin is shown in the gate on each panel. Data represents three separate experiments.
Engrafted early Pmel12 are capable of immune reconstitution
Based on the accumulating evidence that early Pmel12 acquired conventional memory CTL traits in vivo, we hypothesized that the cells also may have acquired the ability to self renew because self renewal is an intrinsic characteristic of memory CTL providing life-long protection [41]. To that end we assessed their ability to reestablish themselves after a second CTX treatment of the animals. Seven and 10 days later (day 47 and day 50), we determined the percentage of donor CTL progeny (Thy1.1+) and host CTL progeny (Thy1.1-) by flow cytometry (Figure 7A, fourth row) as compared to their steady state levels without the second CTX treatment. While on both days the total CTL remained reduced to approximately 40% of their numbers prior the second CTX treatment (day 47: 1.76 + 6.15 / 5.45 +7.85; day 50: 2.40 + 5.07 15 / 5.45 +7.85) progeny of early Pmel12 began to preferentially recover from 22% to 32% of the total CTL. This fast recovery is consistent with their direct development from memory CTL rather than a thymus-dependent recovery that is expected to be responsible for the recovery of naïve host cells.
Figure 7.

Long term Pmel12 cells are capable of peripheral CTL reconstitution followed chemotherapy induced lymphodepletion. (A) Wild type recipients of early Pmel12 received 4 mg of CTX i.p. 40 days after adoptive transfer or were left untreated. Frequencies of donor and CD8+ T cells on peripheral blood were determined by flow cytometry at the indicated time points. (B) Unmanipulated (no CTX) or reconstituted (+ CTX) wt recipients were vaccinated with 100 μg of gp100 peptide i.p. Mice were bled 3 and 45 days after vaccination (d53 and d85 after adoptive transfer, respectively) and the frequency of donor Pmel cells was assayed by flow cytometry. Dot blots shown are representative of at least 5 independent observations.
We also compared the antigen-dependent replicative potentials of the progeny of donor CTL in CTX conditioned mice with mice that received a second CTX treatment (Figure 7B). To that end, Pmel12 recipient mice were challenged with the Pmel antigen (i.p. injection of 100 μg of gp100 peptide). Antigen-dependent expansions were approximately 5-fold irrespective of the second CTX treatment. Similarly, and exactly as expected for normal CTL responses [46], the cells contracted equally well to slightly elevated numbers. These results suggest that memory CTL derived from Pmel12 are capable of immune reconstitution of functional CTL.
The possibility that Pmel progeny recovered from contaminating transgenic stem cells present in the cultures used for ACT was excluded because the in vitro cultures required antigen receptor stimulation to detect any progeny in vivo (Figure 8A) and to effectively control de progression of tumors (Figure 8B).
Figure 8.

Long term Pmel12 cells are not the progeny of a stem cell progenitor present in the spleen. Splenocytes from wt C57BL/6 (Thy1.1-/Vb13-) and Pmel (Thy1.1+/Vb13+) were cultured in the presence of IL-12 for 3 days. Early Pmel12 were also generated as positive control. Wt recipient mice bearing 7 day-old B16 melanoma tumors were injected i.p with 4 mg of CTX and 24 hrs later received 5 × 106 of the cells described above. Frequency of donor cells (Thy1.1+/CD8+) was determined by flow cytometry, and data is shown from one representative mouse per group (A). Tumor progression was followed in the same mice. Each data point represents the average of 5 independent measurements ± SDEV (B).
Discussion
CD8+ T cells optimally suited for anti-tumor ACT should be able to travel to the site of the tumor, recognize the malignant cells, and erradicate them. In addition, some of the transferred CTL should persist as memory CTL lifelong to ensure anti-tumor immunity. Here, we described a simple strategy for in vitro CTL priming and host conditioning that fulfills these demands based on its reproducible tumor eradication and generation of immunity in a melanoma mouse model known to be fairly resistant to immunotherapy.
Our study investigated in detail the role of the specific stage of CTL activation/differentiation at the time of ACT and their transition into long-term anti-tumor immunity in vivo. It is worthwhile mentioning that our differentiation strategy employed naïve CTL, rather than tumor infiltrating lymphocytes (TIL). The former, but not the latter, are easily obtained and may provide more reproducible results because TIL of late stage tumors may be dysregulated by multiple mechanisms [47]. On the other hand, the choice of naïve T-cells requires redirecting their specificity to known tumor antigens. Experiments addressing the latter strategy are underway in several laboratories, including ours.
Our investigations demonstrated that an early stage of CTL activation (day 3) can be differentially programmed by cytokine to benefit from the environment of CTX treated hosts. Only cells primed in the presence of IL-12, but not IL-2 (Figure 1 and data not shown), achieved the therapy goal. Likewise, extending the in vitro culture to a total of seven days generated a different cell stage (MD-M, MP and MGL; unpublished observation) and failed to eradicate the tumor (Figure 1).
Over the past decade many elegant studies have shed molecular light on how activation of CTL proceeds in vivo upon viral infection. An early stage has been defined as memory precursors that are IL-7R high and KLRG1 low [43, 48]. During further activation and differentiation in vivo most of them may acquire cytotoxic activity and eventually become terminal effectors before they die or exhaust [46, 49, 50] while few will survive to give rise to long-term memory CTL. Unexpectedly, we found that the cells most effective in the adoptive transfer acquired an unusual gene expression profile if they represented an identical stage at the time of their analysis by real-time RT-PCR rather than a combination of distinct populations. The former, but not the latter, is supported by the results from the flow cytometry because the histograms primarily displayed single populations (Figure 5A and data not shown). Transcription factor expression, such as prdm1, and surface molecule expression such as CD25 (Figure 5), corresponded to strongly activated effector CTL while klrg1 and p19arf accumulation suggested they were terminally differentiating into senescent and/or exhausted cells [46, 49, 50]. Similarly perplexing, they expressed CD62L suggesting that they are homing to lymph nodes thought to be reserved for quiescent naïve and central memory CTL. Interestingly, the high expression of t-bet by these cells has been associated previously not only with the effector stages of T-cells but specifically also to their homing to tumors in situ [51]. Taken all together, our in vitro activation of CTL seems unlikely to proceed analogously to their physiological activation in vivo. Perhaps it should be expected that CTL capable of efficiently seeding CTX treated hosts are distinct from CTL stages defined according to anti-viral CTL responses. Additional correlative experiments in our laboratories point to a unique role of the tumor suppressor p19arf for the IL-12 dependent in vitro activation/ differentiation (M.D.-M., M.P. and M.G.L. unpublished observation).
The success of the ACT strategy was contingent on a synergy between the use of IL-12 to prime naïve CTL and the conditioning of the host with CTX. Notably, a lymphodepletion regimen of a single bolus of CTX without a second drug or total body irradiation was sufficient to convert the limited anti-tumor activity of transferred early Pmel12 into their capability of complete tumor rejection and, ultimately, anti-tumor immunity. This finding could have important clinical implications because it is often suggested that the efficacy of adoptively transferred T cells is proportional to the intensity of lymphodepletion [52, 53] while intensification comes at the expense of higher toxicities. We are currently addressing whether the IL-12 primed CD8+ T cells are “supraoptimally fit” and thus require a less rigorous conditioning regimen. Our current working hypothesis is that the synergistic action is due to the in vitro generation of a transient CTL stage that is optimally responsive in vivo to the cytokine milieu in the CTX treated host [54]. Based on the known pleiotropic effects of CTX, other explanations may include its induction of lymphopenia [55], its reduction of Tregs [56] and its systematic activation of DC or even its induction of a highly immunogenic form of tumor cell lysis [57]. These explanations are not exclusive of each other and individual mechanism may become critical at different time points after the adoptive transfer.
Our results show a strong correlation between the levels of circulating donor cells and tumor regression with ineffective populations disappearing by day 8 after the ACT (Figure 2). Similar findings have been described in patients receiving ACT for melanoma. In these patients, transferred cells with long-term persistence in circulation expressed high levels of CD127 (IL-7R), which suggested an IL-7-dependent survival mechanism [58]. The cells we used for ACT did not significantly upregulate the IL-7R (Figure 5B) and it was upregulated only among their long-term surviving progeny (Figure 6A). Similarly, the transcription factor TCF-7 that is known to promote long-term memory and longevity of memory CTL [38], was down regulated in the donor cells, but it became the most highly induced gene in their long-term surviving progeny Figure 6A. The high expression of TCF-7 and IL-7R by the Pmel cells remaining in the animals for at least nine months after cure (data not shown) led us to investigate whether long-term survival of the adoptively transferred CTL became associated with the acquisition of memory stem-cell like traits that were reported to reside within a CD44low/Sca-1high population [26]. Indeed, we found that a significantly larger population of CD44low/Sca-1high cells existed among the donor population compared to the endogenous CTL (Figure 4). These cells also preferentially homed to the lymph node and bone marrow (Figure 3), the two organs in which memory CTL are preferentially maintained [28]. Functionally these cells were even able to efflux daunorubicin and to propagate themselves upon lymphopenia-inducing cytotoxic chemotherapy, a trait they share with hematopoietic stem cells [59]. This cumulative evidence suggests that the donor derived CTL evolving after ACT had acquired stem-cell like features necessary to maintain long-term memory in vivo. Finally, these cells were readily responsive to a second antigen and tumor challenges (Figure 7 and Figure 1).
In conclusion, this report describes a simple and extremely potent strategy for ACT of CD8+ T -cells in a pre-clinical mouse melanoma model. The two key components entail the brief activation of naïve CTL in the presence of IL-12 in vitro and their transfer into tumor-bearing animals treated with a single dose of CTX. The key results include that an apparently novel differentiation stage of the transferred CTL promotes both tumor eradication and the generation of self-renewing memory CTL. Our results also suggest that transcription factors driving CTL activation and differentiation may become useful bio-markers to predict the efficacy of cells for ACT prior to their infusion and that a threshold of donor cells needs to be detectable in the blood circulation within the first week long term tumor eradication.
Acknowledgments
This work was supported by The Dodson Multidisciplinary Immunotherapy Institute at the Sylvester Cancer Center and by the National Institute of Health Grants R01CA083672 (to D.J.C) and K01CA134927 (to C.M.D-M). We thank Despina Kolonias for her technical assistance.
References
- 1.Schadendorf D, Ugurel S, Schuler-Thurner B, Nestle FO, Enk A, Brocker EB, Grabbe S, Rittgen W, Edler L, Sucker A, Zimpfer-Rechner C, Berger T, Kamarashev J, Burg G, Jonuleit H, Tuttenberg A, Becker JC, Keikavoussi P, Kampgen E, Schuler G. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol. 2006;17:563–570. doi: 10.1093/annonc/mdj138. [DOI] [PubMed] [Google Scholar]
- 2.Robert C, Thomas L, Bondarenko I, O'Day S, M DJ, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH, Jr, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 3.Sang M, Wang L, Ding C, Zhou X, Wang B, Lian Y, Shan B. Melanoma-associated antigen genes - an update. Cancer Lett. 302:85–90. doi: 10.1016/j.canlet.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 4.Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, Busch DH. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity. 2007;27:985–997. doi: 10.1016/j.immuni.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 5.Reiner SL, Sallusto F, Lanzavecchia A. Division of labor with a workforce of one: challenges in specifying effector and memory T cell fate. Science. 2007;317:622–625. doi: 10.1126/science.1143775. [DOI] [PubMed] [Google Scholar]
- 6.Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T-cell progenitors. Nature. 2008;452:356–360. doi: 10.1038/nature06672. [DOI] [PubMed] [Google Scholar]
- 7.Bannard O, Kraman M, Fearon DT. Secondary replicative function of CD8+ T cells that had developed an effector phenotype. Science. 2009;323:505–509. doi: 10.1126/science.1166831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111–117. doi: 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bracci L, Moschella F, Sestili P, La Sorsa V, Valentini M, Canini I, Baccarini S, Maccari S, Ramoni C, Belardelli F, Proietti E. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin Cancer Res. 2007;13:644–653. doi: 10.1158/1078-0432.CCR-06-1209. [DOI] [PubMed] [Google Scholar]
- 10.Schiavoni G, Mattei F, Di Pucchio T, Santini SM, Bracci L, Belardelli F, Proietti E. Cyclophos-phamide induces type I interferon and augments the number of CD44(hi) T lymphocytes in mice: implications for strategies of chemoimmunotherapy of cancer. Blood. 2000;95:2024–2030. [PubMed] [Google Scholar]
- 11.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, Surh CD, Rosenberg SA, Restifo NP. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, Palmer DC, Boni A, Muranski P, Yu Z, Gattinoni L, Antony PA, Rosenberg SA, Restifo NP. Microbial trans-location augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117:2197–2204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Awwad M, North RJ. Cyclophosphamide (Cy) -facilitated adoptive immunotherapy of a Cy-resistant tumour. Evidence that Cy permits the expression of adoptive T-cell mediated immunity by removing suppressor T cells rather than by reducing tumour burden. Immunology. 1988;65:87–92. [PMC free article] [PubMed] [Google Scholar]
- 14.Hoover SK, Barrett SK, Turk TM, Lee TC, Bear HD. Cyclophosphamide and abrogation of tumor-induced suppressor T cell activity. Cancer Immunol Immunother. 1990;31:121–127. doi: 10.1007/BF01742376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikezawa Y, Nakazawa M, Tamura C, Takahashi K, Minami M, Ikezawa Z. Cyclophosphamide decreases the number, percentage and the function of CD25+ CD4+ regulatory T cells, which suppress induction of contact hypersensitivity. J Dermatol Sci. 2005;39:105–112. doi: 10.1016/j.jdermsci.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 16.van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. 2001;2:423–429. doi: 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 17.Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. 2001;2:415–422. doi: 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bevan MJ. In a radiation chimaera, host H-2 antigens determine immune responsiveness of donor cytotoxic cells. Nature. 1977;269:417–418. doi: 10.4049/jimmunol.176.1.677. J Immunol 2006; 176: 5-6. [DOI] [PubMed] [Google Scholar]
- 19.Xiao Z, Casey KA, Jameson SC, Curtsinger JM, Mescher MF. Programming for CD8 T cell memory development requires IL-12 or type I IFN. J Immunol. 2009;182:2786–2794. doi: 10.4049/jimmunol.0803484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teixeiro E, Daniels MA, Hamilton SE, Schrum AG, Bragado R, Jameson SC, Palmer E. Different T cell receptor signals determine CD8+ memory versus effector development. Science. 2009;323:502–505. doi: 10.1126/science.1163612. [DOI] [PubMed] [Google Scholar]
- 21.Zehn D, Turner MJ, Lefrancois L, Bevan MJ. Lack of original antigenic sin in recall CD8(+) T cell responses. J Immunol. 184:6320–6326. doi: 10.4049/jimmunol.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becker JC, Houben R, Schrama D, Voigt H, Ugurel S, Reisfeld RA. Mouse models for melanoma: a personal perspective. Exp Dermatol. 19:157–164. doi: 10.1111/j.1600-0625.2009.00986.x. [DOI] [PubMed] [Google Scholar]
- 24.Salem ML, Kadima AN, El-Naggar SA, Rubinstein MP, Chen Y, Gillanders WE, Cole DJ. Defining the ability of cyclophosphamide preconditioning to enhance the antigen-specific CD8+ T-cell response to peptide vaccination: creation of a beneficial host microenvironment involving type I IFNs and myeloid cells. J Immunother. 2007;30:40–53. doi: 10.1097/01.cji.0000211311.28739.e3. [DOI] [PubMed] [Google Scholar]
- 25.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–675. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stemberger C, Neuenhahn M, Gebhardt FE, Schiemann M, Buchholz VR, Busch DH. Stem cell-like plasticity of naive and distinct memory CD8+ T cell subsets. Semin Immunol. 2009;21:62–68. doi: 10.1016/j.smim.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, Paulos CM, Muranski P, Restifo NP. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herndler-Brandstetter D, Landgraf K, Jenewein B, Tzankov A, Brunauer R, Brunner S, Parson W, Kloss F, Gassner R, Lepperdinger G, Grubeck-Loebenstein B. Human Bone Marrow Hosts Polyfunctional Memory CD4+ and CD8+ T Cells with Close Contact to IL-15-Producing Cells. J Immunol. doi: 10.4049/jimmunol.1100243. [DOI] [PubMed] [Google Scholar]
- 29.Diaz-Montero CM, EI Naggar S, AI Khami A, EI Naggar R, Montero AJ, Cole DJ, Salem ML. Priming of naive CD8+ T cells in the presence of IL-12 selectively enhances the survival of CD8(+)CD62L (hi) cells and results in superior anti-tumor activity in a tolerogenic murine model. Cancer Immunol Immunother. 2008;57:563–572. doi: 10.1007/s00262-007-0394-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lisiero DN, Soto H, Liau LM, Prins RM. Enhanced Sensitivity to IL-2 Signaling Regulates the Clinical Responsiveness of IL-12-Primed CD8+ T Cells in a Melanoma Model. J Immunol. doi: 10.4049/jimmunol.1003317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Egawa T, Tillman RE, Naoe Y, Taniuchi I, Littman DR. The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J Exp Med. 2007;204:1945–1957. doi: 10.1084/jem.20070133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Setoguchi R, Tachibana M, Naoe Y, Muroi S, Akiyama K, Tezuka C, Okuda T, Taniuchi I. Repression of the transcription factor Th-POK by Runx complexes in cytotoxic T cell development. Science. 2008;319:822–825. doi: 10.1126/science.1151844. [DOI] [PubMed] [Google Scholar]
- 33.Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, Groner Y, Rao A. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. 2009;206:51–59. doi: 10.1084/jem.20081242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Intlekofer AM, Takemoto N, Kao C, Banerjee A, Schambach F, Northrop JK, Shen H, Wherry EJ, Reiner SL. Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J Exp Med. 2007;204:2015–2021. doi: 10.1084/jem.20070841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Y, Ju S, Chen E, Dai S, Li C, Morel P, Liu L, Zhang X, Lu B. T-bet and eomesodermin are required for T cell-mediated antitumor immune responses. J Immunol. 185:3174–3183. doi: 10.4049/jimmunol.1000749. [DOI] [PubMed] [Google Scholar]
- 37.Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 11:114–120. doi: 10.1038/ni.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 33:229–240. doi: 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. Cutting Edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J Immunol. 2006;177:7515–7519. doi: 10.4049/jimmunol.177.11.7515. [DOI] [PubMed] [Google Scholar]
- 40.Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turtle CJ, Swanson HM, Fujii N, Estey EH, Riddell SR. A distinct subset of self-renewing human memory CD8+ T cells survives cytotoxic chemotherapy. Immunity. 2009;31:834–844. doi: 10.1016/j.immuni.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG, Thomas NE, Sharpless NE. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell. 2009;8:439–448. doi: 10.1111/j.1474-9726.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mizutani T, Masuda M, Nakai E, Furumiya K, Togawa H, Nakamura Y, Kawai Y, Nakahira K, Shinkai S, Takahashi K. Genuine functions of P-glycoprotein (ABCB1) Curr Drug Metab. 2008;9:167–174. doi: 10.2174/138920008783571756. [DOI] [PubMed] [Google Scholar]
- 46.Wirth TC, Xue HH, Rai D, Sabel JT, Bair T, Harty JT, Badovinac VP. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity. 33:128–140. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, Klebanoff CA, Johnson LA, Kerkar SP, Yang S, Muranski P, Palmer DC, Scott CD, Morgan RA, Robbins PF, Rosenberg SA, Restifo NP. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 117:808–814. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 49.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 50.Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73:975–983. doi: 10.1002/cyto.a.20643. [DOI] [PubMed] [Google Scholar]
- 51.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 52.Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, Yu Z, Rosenberg SA, Restifo NP. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother. 33:1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, Restifo NP. Increased intensity lymphodepletion and adoptive immunotherapy–how far can we go? Nat Clin Pract Oncol. 2006;3:668–681. doi: 10.1038/ncponc0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerlach C, van Heijst JW, Schumacher TN. The descent of memory T cells. Ann N Y Acad Sci. 1217:139–153. doi: 10.1111/j.1749-6632.2010.05830.x. [DOI] [PubMed] [Google Scholar]
- 55.Brode S, Cooke A. Immune-potentiating effects of the chemotherapeutic drug cyclo-phosphamide. Crit Rev Immunol. 2008;28:109–126. doi: 10.1615/critrevimmunol.v28.i2.20. [DOI] [PubMed] [Google Scholar]
- 56.Nakahara T, Uchi H, Lesokhin AM, Avogadri F, Rizzuto GA, Hirschhorn-Cymerman D, Panageas KS, Merghoub T, Wolchok JD, Houghton AN. Cyclophosphamide enhances immunity by modulating the balance of dendritic cell subsets in lymphoid organs. Blood. 115:4384–4392. doi: 10.1182/blood-2009-11-251231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, Sanchez M, Lorenzi S, D'Urso MT, Belardelli F, Gabriele L, Proietti E, Bracci L. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 71:768–778. doi: 10.1158/0008-5472.CAN-10-2788. [DOI] [PubMed] [Google Scholar]
- 58.Huang J, Khong HT, Dudley ME, El-Gamil M, Li YF, Rosenberg SA, Robbins PF. Survival, persistence, and progressive differentiation of adoptively transferred tumor-reactive T cells associated with tumor regression. J Immunother. 2005;28:258–267. doi: 10.1097/01.cji.0000158855.92792.7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med. 2005;11:1299–1305. doi: 10.1038/nm1326. [DOI] [PubMed] [Google Scholar]