

Abstract
Protein p6 is a nonspecific DNA-binding protein occurring in high abundance in phage φ29-infected cells. Here, we demonstrate a novel role for this versatile histone-like protein: its involvement in regulating the viral switch between early and late transcription. p6 performs this role by exhibiting a reciprocal functional interaction with the regulatory protein p4, also phage encoded, which is required for repression of the early A2b and A2c promoters and activation of the late A3 promoter. On the one hand, p6 promotes p4-mediated repression of the A2b promoter and activation of the A3 promoter by enhancing binding of p4 to its recognition site at PA3; on the other, p4 promotes p6-mediated repression of the A2c promoter by favoring the formation of a stable p6–nucleoprotein complex that interferes with RNA polymerase binding to PA2c. We propose that the observed interplay between proteins p6 and p4 is based on their DNA architectural properties.
Keywords: Bacteriophage φ29, transcriptional regulation, histone-like protein, architectural factors
The process of transcription, like other DNA transactions such as replication and recombination, occurs on an inherently stiff molecule (see Shore and Baldwin 1983; Wang and Giaever 1988). However, DNA must be at least locally flexible for the appropriate nucleoprotein complexes to assemble. This requirement is partially fulfilled by the participation of DNA-binding proteins that have the ability to remodel DNA (Nash 1990, 1996; Grosschedl et al. 1994; Werner and Burley 1997). Such architectural factors are found in both eukaryotes and prokaryotes and include specific as well as nonspecific DNA-binding proteins.
Nonspecific architectural factors presumably regulate transcription by introducing conformational changes that either hinder or assist DNA recognition by sequence-specific control proteins. In prokaryotes, there is substantial evidence that factors capable of altering general DNA flexibility such as the major histone-like proteins HU and H-NS can also function as transcriptional modulators. Thus, HU has been shown to stimulate lac repressor–operator binding and CAP binding to lac DNA (Flashner and Gralla 1988), to stabilize Mu repressor binding to the Mu early operator (Betermier et al. 1995), to aid in GalR-mediated repression of the gal operon (Aki and Adhya 1997), and to displace the LexA repressor from its DNA-binding sites (Preobrajenskaya et al. 1994). Likewise, H-NS has been reported to influence transcription of a number of genes involved in diverse biological processes (Falconi et al. 1993; Zuber et al. 1994; Atlung and Ingmer 1997; Williams and Rimsky 1997).
In eukaryotes, the abundant nonhistone chromosomal protein HMG-1 (and the highly homologous protein HMG-2) also functions as an architectural element in the assembly of several transcriptional nucleoprotein complexes (for review, see Grosschedl et al. 1994; Bustin and Reeves 1996). Thus, HMG-1 and HMG-2 have been shown to enhance the sequence-specific DNA binding of a variety of proteins, such as steroid receptors (Oñate et al. 1994; Verrier et al. 1997; Boonyaratanakornkit et al. 1998); the POU domain-containing proteins Oct1, Oct2, and Oct6 (Zwilling et al. 1995); HOX proteins (Zappavigna et al. 1996); and the tumor suppressor p53 (Jayaraman et al. 1998). The ability of HU, H-NS, and HMG-1 to bind DNA nonspecifically but in a conformation-dependent manner (Bianchi et al. 1989; Bracco et al. 1989; Yamada et al. 1991; Pontiggia et al. 1993) and to bend DNA on binding (Hodges-Garcia et al. 1989; Oñate et al. 1994; Spurio et al. 1997) have been related to their effects as transcriptional modulators (see Flashner and Gralla 1988; Oñate et al. 1994; Atlung and Ingmer 1997; Williams and Rimsky 1997; Jayaraman et al. 1998).
Protein p6 from Bacillus subtilis phage φ29 is an early phage protein that resembles in various aspects the type of general architectural factors described above. It is a small DNA-binding protein, abundant in infected cells, that binds DNA as a dimer and recognizes a structural feature rather than a specific sequence in DNA (for review, see Salas and Rojo 1993). Initial binding of p6 to DNA occurs preferentially at DNA regions showing a tendency to bend with a defined periodicity. Such a structural feature is found at the ends of the phage genome, where p6 imposes bends on the DNA every 12 bp forming a compact nucleoprotein complex that plays an essential role in the initiation of φ29 DNA replication. In this complex the DNA forms a right-handed solenoid wrapping around the multimeric p6 core (Serrano et al. 1993). Nevertheless, under conditions that favor binding of p6 to DNA (low salt and saturating protein concentrations), p6–nucleoprotein complexes are observed covering most of the φ29 genome, suggesting that p6 may also have a structural role in organizing the genome (Gutiérrez et al. 1994). In agreement with this hypothesis, p6 concentrations at late times of infection can be as high as 1 mm, an amount sufficient to saturate not only the φ29 replication origins but also the entire intracellular viral DNA (Abril et al. 1997).
If p6 covers most of the φ29 genome in vivo, it is likely that the complex it forms with DNA affects other processes besides the initiation of DNA replication. In fact, binding of p6 to the right genomic end represses transcription from the early C2 promoter (PC2) by blocking access to RNA polymerase (RNAP) (Whiteley et al. 1986; Barthelemy et al. 1989). Other φ29 promoters, however, appear to be less sensitive to repression by p6 (Barthelemy et al. 1989). The highly dynamic nature of the p6–DNA nucleoprotein complexes, with continuous assembly and disassembly due to the low binding affinity of p6, probably allows processes such as replication and transcription to occur efficiently in the nucleoprotein complex (see Gutiérrez et al. 1994). In fact, as with other general architectural factors like HU, p6-induced DNA distortions could even facilitate DNA recognition by the specific proteins performing these DNA transactions. With this in mind, we investigated whether p6 has any effect on the transcriptional activity of the φ29 regulatory protein p4.
Transcription of phage φ29 is subject to a gene expression program involving two stages (for review, see Salas and Rojo 1993). The phage-encoded sequence-specific DNA-binding protein p4 regulates the switch between the early and late stages of transcription by repressing the two main early promoters A2b (PA2b) and A2c (PA2c), and activating the late A3 promoter (PA3) (see Salas and Rojo 1993; Rojo et al. 1998). Protein p4 binds upstream of RNAP at PA2c and PA3, and by interacting with the carboxy-terminal domain of the RNAP α subunit, it represses and activates transcription, respectively (Mencía et al. 1996a,b; Monsalve et al. 1996a,b). These activities of p4, whose binding to DNA is accompanied by a sharp bend in the DNA, could conceivably be modulated by agents that promote changes in DNA topology such as protein p6. We find that not only does p6 affect specific recognition by p4 but, even more interestingly, p4 also modulates the DNA-binding specificity of p6. At PA3, p6 favors binding of p4, thus having a positive effect on PA3 activation and PA2b repression. Binding of p4 to its site at PA3, in turn, promotes formation of a stable p6–nucleoprotein complex at PA2c that prevents RNAP from binding to this promoter. Therefore, in the presence of p4 and high concentrations of p6, repression of PA2c is mediated directly by p6, and only indirectly by p4. Overall, our results reveal a previously unreported key role for the histone-like protein p6 in regulating the early–late transcriptional switch in bacteriophage φ29.
Results
Protein p6 forms a nucleoprotein complex of low stability at the DNA region containing PA2b, PA2c, and PA3
PA2b, PA2c, and PA3 are clustered in a small central 219-bp region of the φ29 genome that contains two binding sites for protein p4 (see Fig. 1A). We examined the binding pattern of protein p6 to this DNA region, prior to analyzing whether p4 binding to any of its recognition sites is influenced by p6. Figure 1B shows a DNase I protection assay using a 366-bp DNA fragment (labeled at the early strand) that includes PA2b, PA2c, and PA3 in the presence of increasing amounts of p6 and in the absence or presence of RNAP. Under similar incubation conditions, a multimeric p6–DNA complex is formed at the right end of the φ29 genome at a p6 concentration of 1.4 μm (Freire et al. 1994). Not surprisingly, given the moderately higher specificity that p6 shows for the genome ends, the p6 concentration required for it to bind to the DNA region containing PA2b, PA2c, and PA3 was about 7- to 10-fold higher than that required at the genome ends (Fig. 1B, lanes 4,5). It should be noted that the highest p6 concentration used (14.4 μm) is about seven times greater than that required to cover all the DNA molecules present in the reaction.
Figure 1.
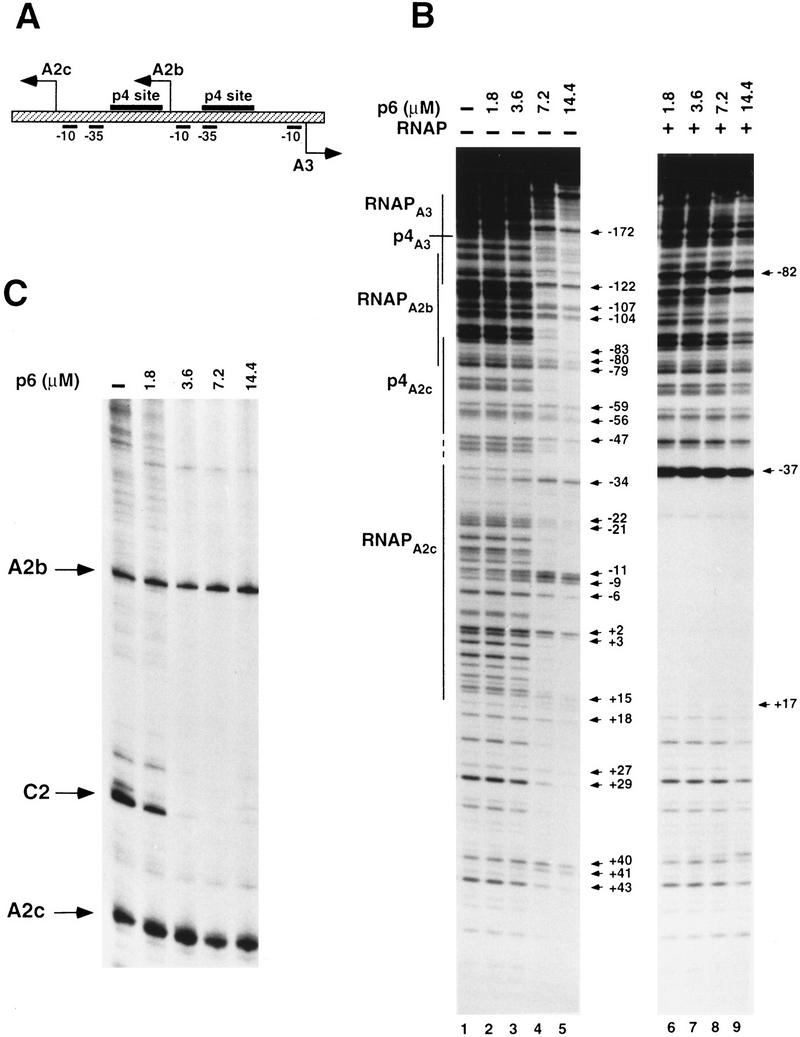
(A) Graphic illustrating the relative location of PA2c, PA2b, and PA3 and the positions of the two p4 binding sites in the 366-bp fragment used in B. The transcription initiation sites for these promoters are located 77 bp (PA2c), 174 bp (PA2b), and 298 bp (PA3) from the left end of the DNA fragment. The p4-binding site at PA3 is centered at position −82 relative to the transcription start site, at PA2c it is centered at position −71. The A3 promoter lacks a distinct −35 box, and the p4-binding site at PA3 overlaps with the −35 box of PA2b. (B) Binding pattern of protein p6 to the DNA region containing PA2c, PA2b, and PA3. DNase I footprinting was performed with a 366-bp DNA fragment including the three promoters labeled at the early strand (see Materials and Methods). The locations of the p4- and RNAP-binding sites are marked with lines. The broken vertical line spans a region where the footprint observed with RNAP alone and with RNAP plus p4 differ (Monsalve et al. 1996a). The DNA was incubated with the indicated amounts of p6 (lanes 1–5) or preincubated with those amounts of p6 prior to addition of RNAP (20 nm) (lanes 6–9). The arrows in the left panel indicate the positions that become either hypersensitive or unprotected in the presence of p6 (numbered relative to the transcription start site at PA2c). The positions marked by arrows in the right panel are indicative of RNAP binding to PA2b (hypersensitivity at position −82 relative to the transcription start site at PA3) and to PA2c (hypersensitivity at position −37 relative to the transcription start site at PA2c and protection from this position to +17). (C) Expression of PA2b and PA2c in the presence of p6. Transcripts were detected by primer extension using complete φ29 DNA as template for in vitro transcription. Transcription from the C2 promoter, whose expression is subject to repression by p6, was followed as control.
The nucleoprotein complex formed at the genome ends in the presence of p6 generates a characteristic pattern of DNase I hypersensitive bands regularly spaced every 24 bp that are flanked by protected regions; occasionally, unprotected or moderately hypersensitive sites are also observed at the center of the protected regions (Prieto et al. 1988). The DNase I footprint observed in Figure 1B (lanes 4,5) shows slightly hypersensitive or merely unprotected bands spaced about 24 bp apart, with some unprotected or moderately hypersensitive sites in the middle. This DNase I digestion pattern bears more resemblance to the footprint observed at the genome ends at high ionic strength (Prieto et al. 1988). These results suggest that the complex formed by p6 with the DNA fragment bearing PA2b, PA2c, and PA3 is similar in structure to that formed at the DNA ends, although less stable. In agreement with the lower stability of this p6–DNA complex, the 366-bp DNA fragment preincubated with increasing concentrations of p6 and then incubated with RNAP showed a footprint pattern (even at the highest p6 concentration) that is characteristic of RNAP bound to PA2b (hypersensitive band at −82 relative to the PA3 start site) and to PA2c (hypersensitive band at −37 and protection from about −55 to +17 relative to the PA2c start site) (Fig. 1B, lanes 6–9). Under these conditions, RNAP is not seen to be bound at PA3 as this requires the simultaneous presence of RNAP and p4 (Rojo and Salas 1995; Rojo et al. 1998). Thus, at the p6 concentrations tested, p6 was displaced from the DNA by RNAP molecules bound to PA2b and PA2c.
Figure 1C shows the differences in the p6-binding affinities between the DNA region containing PA2b, PA2c, and PA3 and the right φ29 genome end as reflected in transcriptional repression. Whereas transcription from the C2 promoter (located very close to the right end of the genome) was completely inhibited at p6 concentrations of 3.6 μm, transcription from PA2b and PA2c was not affected at any of the p6 concentrations shown, as would be expected from the results presented in Figure 1B (lanes 6–9). Protein p6 concentrations as high as 20 μm are required to observe a slight inhibition of PA2b and PA2c transcription (data not shown). We can therefore conclude that p6 by itself binds to the DNA region where protein p4 exerts its regulatory effect, although it shows a considerably lower affinity than for the genome ends. Moreover, the p6–nucleoprotein complex formed at the DNA region containing PA2b, PA2c, and PA3 is not sufficiently stable to interfere with RNAP binding.
Repression of PA2c and activation of PA3 is more efficient in the presence of protein p6
Protein p4 regulates expression from the early A2c and A2b promoters and the late A3 promoter in vitro by binding to two DNA recognition sites: the sequence 5′-CTTTTT-(N15)-AAAATG-3′ upstream of PA3, and the sequence 5′-CAAAGT-(N15)-TAAAAG-3′ upstream of PA2c (see Monsalve et al. 1998; Rojo et al. 1998). The target site at PA3 is a high-affinity binding site that overlaps with the −35 region of PA2b (see Fig. 1A). Binding of p4 to this site has a dual effect: On the one hand, it results in repression of PA2b due to the exclusion of RNAP from this promoter; on the other, it leads to the activation of PA3 due to interactions between p4 and RNAP and the further stabilization of the holoenzyme at this promoter lacking a consensus −35 box (Salas and Rojo 1993; Rojo and Salas 1995; Mencía et al. 1996a,b). Stable binding of p4 to the low-affinity target site at PA2c (centered at position −71 from the PA2c transcriptional start site) requires the interaction of p4 with RNAP. This interaction causes repression of PA2c by an overstabilization of RNAP at the promoter that prevents promoter clearance (Monsalve et al. 1996a,b). Figure 2A is a primer extension assay that shows the behavior of these promoters as a function of p4 concentration (lanes 3–8). For simplicity, only expression from PA2c and PA3 is shown. Expression from PA2b is omitted because its repression is regulated by the same p4-binding event as the activation of PA3, the two promoters showing precisely inverse expression patterns. Transcription from the C2 promoter, whose expression is affected by p6 but not by p4, was also followed as a control. Activation of PA3 was lost when p4 concentrations lower than 160 nm were used. Similarly, repression of the A2c promoter by at least 50% was observed only at p4 concentrations ≥160 nm.
Figure 2.

(A) Effect of p6 on activation of PA3 and repression of PA2c by p4. The complete φ29 genome was used for in vitro transcription reactions including RNAP (20 nm) and p4 (at the indicated concentrations) or p4 and p6 (14.4 μm). In reactions containing p6, the template DNA was preincubated with the protein for 10 min at 37°C. Transcripts corresponding to PA2c, PA3, and PC2 (as control for p6 activity) were detected by primer extension. (B) Quantitative representation of the data shown in A. PA2c repression is expressed as percent activity remaining in the presence of p4 (▴) or p4 and p6 (□). PA3 activation refers to the levels of transcription observed in the presence of p4 (▴) or p4 and p6 (□) relative to those obtained in their absence. Numbers for PA2c and PA3 transcription obtained from lanes 1-8 were corrected using PC2 transcription as a standard.
To investigate whether protein p6 has any effect in modulating the transcriptional activities of protein p4, we followed expression of PA2c and PA3 in the presence of p6 and the same range of p4 concentrations used above (Fig. 2A, lanes 9–14). The concentration of p6 added was 14.4 μm, a concentration at which it is able to form complexes with DNA without affecting expression of PA2c (Fig. 1B,C; Fig. 2A, lane 2). As expected, transcription from PC2 was repressed in all of the reactions containing p6. Differences in the expression from both PA2c and PA3 were observed in the reactions containing p6 relative to those lacking it (Fig. 2A, cf. lanes 3–8 and lanes 9–14; see also Fig. 2B). First, in the presence of p6, significant repression of PA2c was observed at concentrations of p4 that by themselves had negligible or no effect (Fig. 2A, cf. lanes 6,7 and lanes 12,13; Fig. 2B). Second, at concentrations of p4 that produce some degree of PA2c repression, the level of repression was much stronger when p6 was also present. Thus, whereas full repression of PA2c was never observed even at the highest p4 concentration used in reactions lacking p6, the activity of PA2c was completely blocked at p4 concentrations ranging from 160 to 660 nm when p6 was also included (Fig. 2A,B). Third, in the presence of p6, activation of PA3 occurred at p4 concentrations where no activation was observed when p6 was absent (Fig. 2A, cf. lanes 6–8 and lanes 12–14; Fig. 2B). These results suggest that the presence of protein p6 may indeed stabilize or facilitate DNA binding of protein p4 or its interaction with RNAP, because the effective concentrations of p4 required for its transcriptional activities are at least eightfold lower when p6 is also present.
Protein p6 enhances binding of protein p4 to PA3 whereas protein p4 favors the formation of a stable p6–nucleoprotein complex at PA2c
To investigate how protein p6 exerts its effect on the transcriptional regulation of PA2c and PA3 by protein p4, we performed DNase I footprinting over a range of p4 concentrations, and in the absence or in the presence of p6 (Fig. 3). The same DNA fragment as in Figure 1 was used (also labeled at the early strand). As expected, when RNAP was the only protein incubated with the DNA fragment, binding of RNAP to PA2b and PA2c was detected (Fig. 3, lane 2; Rojo and Salas 1995; Monsalve et al. 1996a). When p4 (330 nm) and RNAP were simultaneously present, the set of hypersensitive bands that characterize binding of p4 immediately upstream of RNAP at PA2c and PA3 was observed; in addition, the −82 hypersensitive band at PA2b was lost as a consequence of p4 binding at PA3 (Rojo and Salas 1995; Monsalve et al. 1996a). Because the presence of p4 is required for stabilization of RNAP at PA3, protection downstream of the p4-binding site at this promoter was now observed because of the binding of RNAP to PA3. These footprint features were gradually lost as the p4 concentration decreased and, at 80 nm p4, the DNase I digestion pattern was almost the same as that obtained with RNAP alone (Fig. 3, lanes 4–8; Table 1). These results are consistent with the transcription data shown in Figure 2, where no effect on transcription of PA2c and PA3 was detected at p4 concentrations <80 nm.
Figure 3.
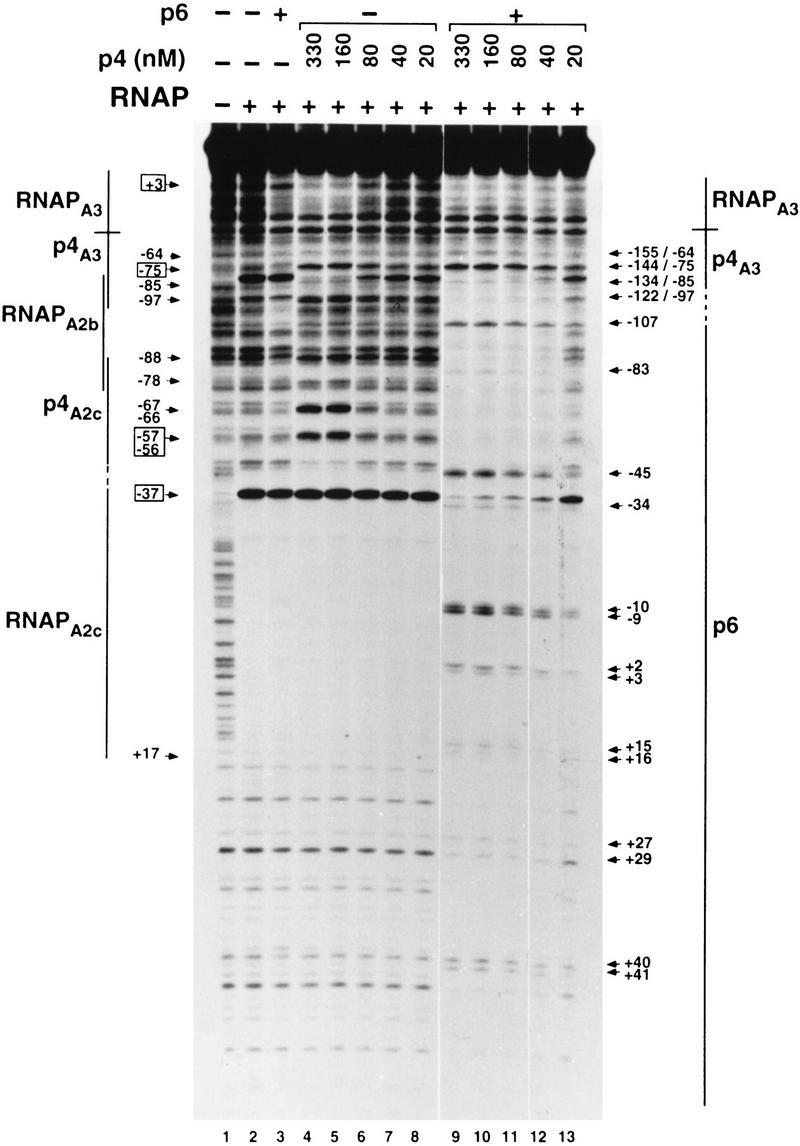
Effect of p6 on the binding of p4 and RNA polymerase to PA2c and PA3. In the reactions with p6, the 366-bp DNA fragment was preincubated with the protein (14.4 μm) for 10 min at 37°C. Protein p4 was present at the indicated concentrations and RNAP at 20 nm. The binding sites for p4, RNAP, and p6 are indicated by lines. Broken lines indicate regions where footprints overlap. Binding of RNAP to PA2c generates hypersensitivity at position −37 and protection from about −55 to +17 (numbered relative to the transcription start site at PA2c). Binding of p4 to PA2c generates the set of hypersensitivities marked above the RNAP-binding site at this promoter. For simplicity, the hypersensitivity at position −82 (relative to the transcription start site at PA3) indicative of RNAP binding to PA2b is not marked. The −64, −75, −85, and −97 hypersensitivities (relative to the transcription start site at PA3) originate from binding of p4 to its site at PA3. Binding of RNAP to PA3 generates protection of a DNA region immediately downstream of the p4-binding site. Arrows at right indicate DNase I hypersensitivities created on binding of protein p6 (numbered relative to the transcription start site at PA2c) and binding of protein p4 to PA3 (numbered relative to the transcription start sites at PA2c/PA3). Numbers in boxes are the specific bands that were densitometrically scanned to obtain data presented in Table 1.
Table 1.
Binding of protein p4 and RNA polymerase to PA3 and PA2c in the absence or presence of protein p6
| Protein p4 (nm)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 330
|
160
|
80
|
40
|
20
|
330
|
160
|
80
|
40
|
20
|
||
|
|
− protein p6
|
|
+ protein p6
|
||||||||
| RNAPA3 | 100 | 89 | 25 | 0 | 0 | 100 | 100 | 96 | 91 | 66 | |
| p4A3 | 100 | 85 | 17 | 0 | 0 | 100 | 100 | 100 | 89 | 43 | |
| RNAPA2c | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 5 | 10 | 35 | |
| p4A2c | 100 | 90 | 14 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
Data were obtained by densitometric scanning of the specific bands at positions marked in boxes in Fig. 3. The numbers represent relative percentages obtained after correcting for sample loading variations and background. A value of 100 was arbitrarily assigned to the maximum binding (maximum protection for the +3 band for RNAPA3; maximum hypersensitivities for the bands at −75 for p4A3, −56/−57 for p4A2c, and −37 for RNAPA2c).0 corresponds to no binding, i.e., values equal to background.
Unexpectedly, when the DNA fragment was preincubated with protein p6 prior to the addition of p4 and RNAP, we observed a footprint pattern substantially different from that obtained when the DNA fragment was incubated only with RNAP and p4. Most remarkably, the footprint features indicative of p4 and RNAP binding to PA2c, which were quite evident at 330 nm and 160 nm of p4 with p6 absent, disappeared (Fig. 3, cf. lanes 4–8 and lanes 9–13; see also Table 1). Instead, a pattern of DNase I protection with the characteristics of p6 binding was observed extending from a DNA region immediately upstream of the p4 recognition site at PA3. The positions of the hypersensitive and the unprotected sites basically coincide with those observed when this DNA fragment was incubated with p6 alone (see Fig. 1B, lanes 4,5), indicating that p6 binds with the same phase irrespective of the presence or absence of p4. Nevertheless, some of the bands become more protected whereas some others become more hypersensitive, pointing to slight changes in the nucleoprotein complex formed in the presence of p4. In fact, our data indicate that the p6–nucleoprotein complex formed is more stable in the presence of protein p4 than in its absence. Thus, whereas p6 alone was unable to block binding of RNAP to PA2c, p6 was able to do so in the presence of p4. Even when the DNA fragment was preincubated with p4 (660 nm) and RNAP prior to adding p6, the footprint pattern observed was that characteristic of p6 binding upstream of PA3 and extending through PA2c (not shown). Thus, the p6–DNA complex formed under these conditions is sufficiently stable to even displace a prebound p4–RNAP complex from PA2c. It should be noted that although the footprint assay presented here was performed in the absence of nucleotides (to visualize binding of RNAP to PA3, which would otherwise escape the promoter), the same results regarding PA2c were obtained in the presence of nucleotides. Basically, the footprint that characterizes protein p4 holding the RNAP at PA2c (Monsalve et al. 1996a) was replaced by that of protein p6 bound to this promoter (data not shown).
Very interestingly, our results reveal that the enhanced repression of PA2c observed in the presence of both p4 and p6 is not due to the stabilization of the p4–RNAP–DNA complex but, rather, to the direct repression of PA2c by p6. The higher levels of PA2c repression achieved when not only p4 but also p6 was present (see Fig. 2) would then indicate that the complex formed by p6 at PA2c is more effective in blocking RNAP binding than is p4 in retaining RNAP at the promoter. The finding that this nucleoprotein complex forms even at low p4 concentrations explains why repression of PA2c in the presence of both p4 and p6 was observed at p4 concentrations at which p4 alone did not produce any significant effect (i.e., 80 and 40 nm; see Fig. 2).
In contrast to the effect of protein p6 at PA2c, the DNase I digestion pattern at PA3 suggested an increased binding affinity of protein p4 for its site at PA3 in the presence of p6 (Fig. 3; Table 1). In the absence of p6, the features accompanying p4 binding at PA3 (DNase I hypersensitivities at positions −64, −75, −85, and −97, lack of hypersensitivity at position −82, and protection by RNAP of PA3) were lost at ∼80 nm (or less) of p4, in agreement with the drop in PA3 activity observed previously at this protein concentration (see Fig. 2). In the presence of p6, however, these features were maintained at lower p4 concentrations, explaining why activation of the A3 promoter persists under these conditions. The data presented in Table 1 would indicate that p6 enhances the binding affinity of p4 for its site at PA3 by at least fourfold. The hypersensitivities observed when p4 binds to PA3 are thought to be a consequence of the strong bend induced on the DNA on binding by p4 (see Rojo and Salas 1995). In the presence of p4 and p6, the −97 hypersensitive position, located at the side of the p4 recognition sequence from which binding of p6 is detected, became weaker. This suggests that a small change in the curvature induced by p4 probably occurs so as to adapt to the structural changes associated with binding of p6 to this DNA region.
Binding of protein p4 to its site at PA3 is required for p6 to form a stable nucleoprotein complex at PA2c
Although the experiments that revealed the effect of p4 on the binding of p6 to PA2c included RNAP, the simultaneous presence of RNAP is not necessary. Figure 4 shows the DNase I digestion pattern of reactions containing the 366-bp DNA fragment, increasing concentrations of p6, and a fixed amount of p4 (80 nm) but no RNAP. In the absence of p4, as much as 14.4 μm p6 was required for efficient binding of p6 to this DNA (see Fig. 1B). In contrast, under similar incubation conditions but when p4 was also present, binding of p6 was detected at p6 concentrations about eightfold lower (1.8 μm) (Fig. 4). This concentration is close to that required for p6 to form the characteristic nucleoprotein complex at the genome ends (1.4 μm) (Freire et al. 1994). Moreover, the nucleoprotein complex formed in the presence of both p4 and p6 produced the same DNase I digestion pattern as that observed when RNAP was also included. Thus, the presence of RNAP is not essential for p4 to exert its effect on the binding of p6 to this DNA region. Because p4 binds with very low affinity to its site at PA2c and with high affinity to its site at PA3 in the absence of RNAP (Rojo and Salas 1995; Monsalve et al. 1996a), these observations suggest that it is the binding of p4 to the latter site that is important in facilitating binding of p6.
Figure 4.
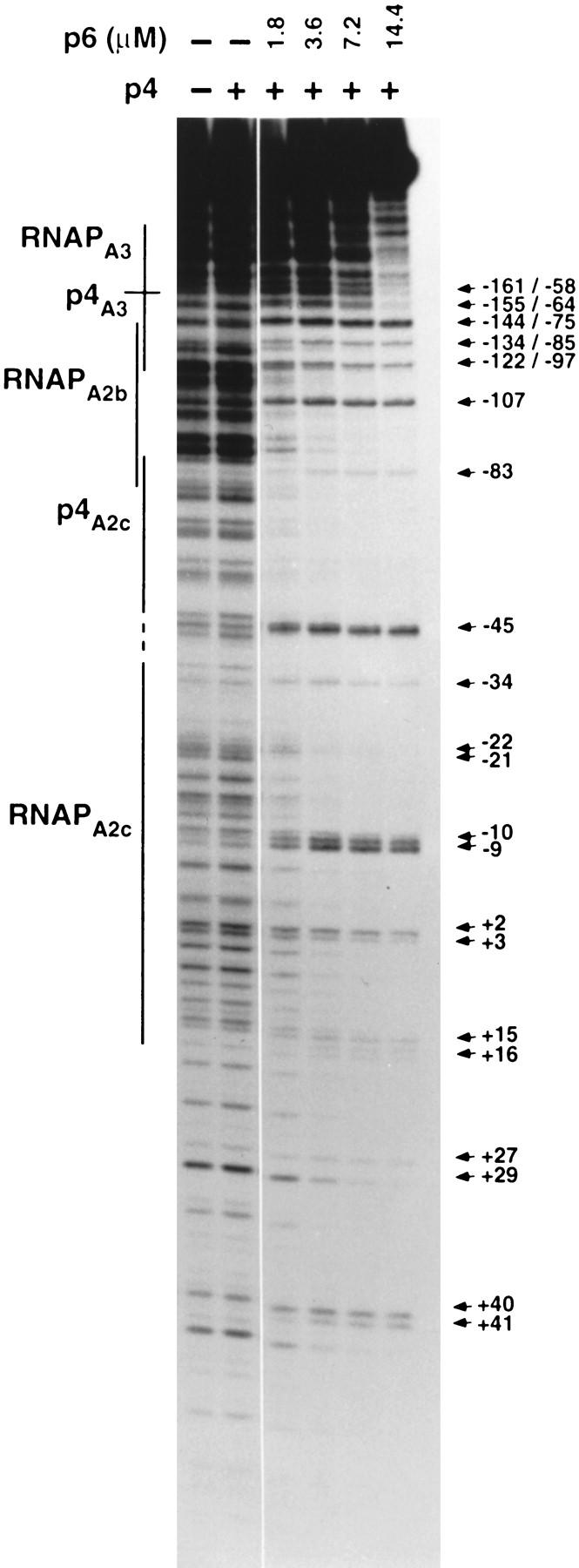
Complex formation by p6 in the presence of p4 but in the absence of RNA polymerase. DNase I footprinting was performed using the 366-bp DNA fragment. Protein p6 was present at the indicated concentrations and p4 was present at 60 nm. The locations of the p4- and RNAP-binding sites are indicated by lines. Arrows at right indicate DNase I hypersensitivities created on binding of p4 to PA3 numbered relative to the transcription start sites at PA2c/PA3 (the −58 hypersensitivity appears when p4 binds PA3 in the absence of RNAP) and upstream binding of p6 (positions −107 to +41, numbered relative to the transcription start site at PA2c).
To address this question more directly, we have compared the transcription and DNase I footprinting patterns of the 366-bp DNA fragment containing the PA3 and PA2c p4-binding sites with those observed for an analogous 198-bp DNA fragment lacking the PA3 p4-binding site (Figs. 5,6). As a control, a DNA fragment containing PC2 was also included in the transcription reactions. Figure 5 (lanes 1–4) shows that the DNA fragment containing PA2c and PA3 behaved like the full-length φ29 genome from a transcriptional point of view. Thus, at 80 nm p4, repression of PA2c and activation of PA3 was only noticeable when both p4 and p6 were present. In contrast, when the DNA fragment lacking the p4 recognition site at PA3 was used (lanes 5–8), no change in the activity of PA2c was observed when both p4 and p6 were present (Fig. 5, cf. lanes 4 and 8). In agreement with the transcriptional results, p6 lost the ability to displace the p4–RNAP complex from PA2c when the p4-binding site at PA3 was lacking (Fig. 6, cf. lanes 5 and 10). It should be noted that, in the absence of p4, p6 will bind with the same affinity and phasing to both DNA fragments (data not shown). These results further support that the only requirement for protein p6 to form a stable nucleoprotein complex at PA2c is that protein p4 be able to bind to its recognition site at PA3.
Figure 5.

Effect of p6 on expression of PA2c in the absence of the p4-binding site at PA3. All of the transcription reactions included a 258-bp DNA fragment containing the C2 promoter (0.5 nm) and B. subtilis ςA–RNA polymerase at 20 nm. Reactions 1–4 also included the 366-bp φ29 DNA fragment (1 nm) used in the above DNase I footprinting experiments. In reactions 5–8, the 366 bp was substituted by a shorter DNA fragment (198 bp) that lacks PA3 and the associated p4-binding site. Proteins p4 and p6 were present at 60 nm and 14.4 μm, respectively.
Figure 6.
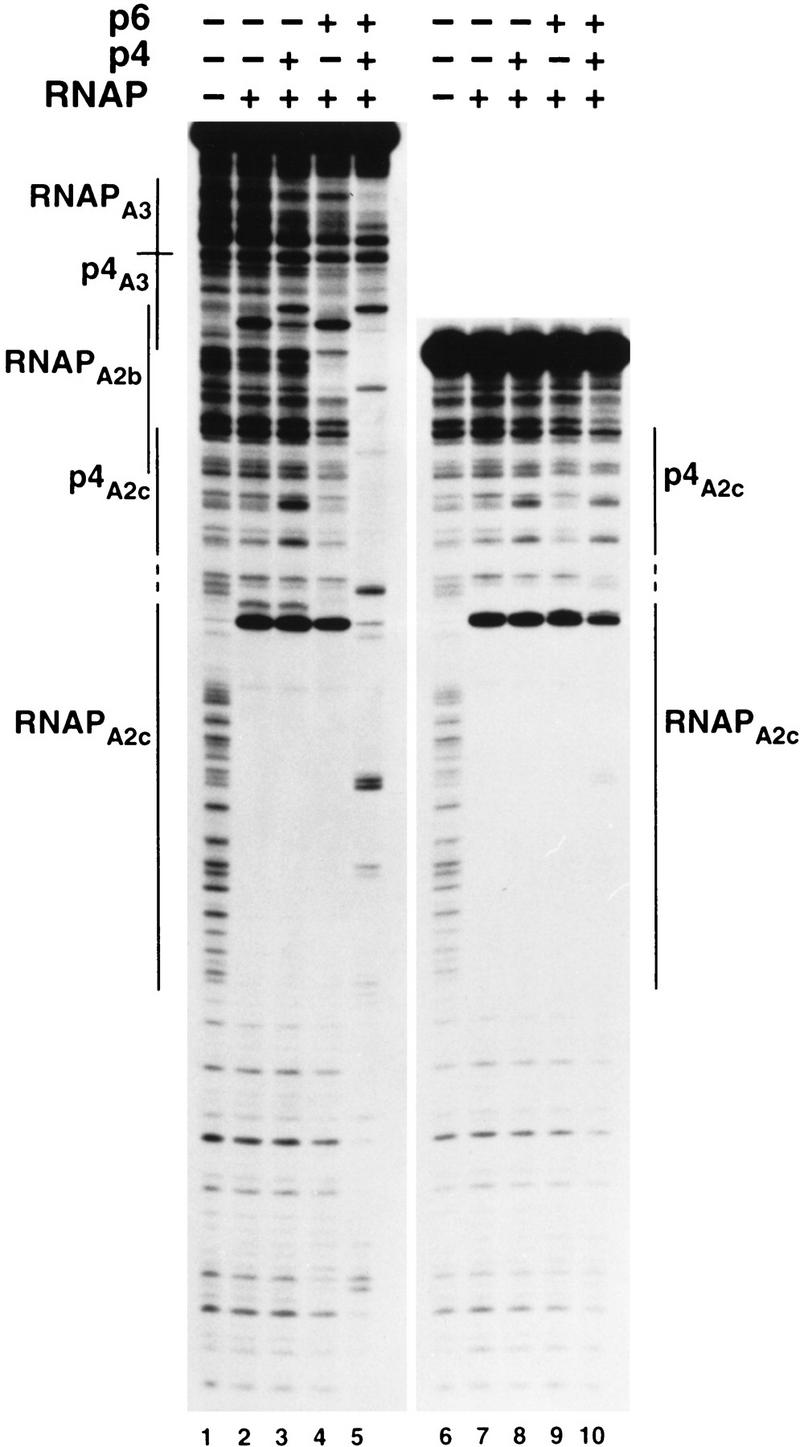
Complex formation by p6 in the absence of the p4-binding site at PA3. DNase I footprinting reactions 1–5 were performed with the 366-bp DNA fragment used in experiments above. In reactions 6–10, the 366-bp fragment was substituted by the 198-bp DNA fragment lacking PA3 and its associated p4-binding site. RNAP, protein p4, and protein p6 were present at 20 nm, 60 nm, and 14.4 μm, respectively. The locations of the p4- and RNAP-binding sites are indicated by lines.
Specificity of the functional interactions between proteins p6 and p4
Nf and GA-1 are two φ29-related phages whose protein p6 counterparts have recently been purified (Freire et al. 1996). Nf is evolutionarily more related to φ29 than is GA-1, as inferred from a number of relevant properties (see Freire et al. 1996): (1) the p6 proteins of φ29 and Nf show a higher sequence homology (73%) than do the φ29 p6 and its GA-1 counterpart (58%); (2) the nucleoprotein complexes formed at the genome ends by the φ29 and Nf p6 proteins are structurally very similar but different from that formed by the GA-1 p6. We analyzed the ability of the two heterologous proteins to form nucleoprotein complexes with the 366-bp φ29 DNA fragment in the presence and in the absence of the φ29 protein p4 (Fig. 7). The experiments performed with the Nf p6 gave results very similar to those discussed above for its counterpart in φ29. Thus, when both φ29 protein p4 and the Nf p6 were present, the p4–RNAP complex at PA2c was displaced by p6. By contrast, the binding of the GA-1 p6 to the φ29 DNA fragment was equally inefficient in the presence or absence of the φ29 p4. In agreement with the DNase I footprinting results, primer extension analysis showed that the Nf p6 protein enhanced PA2c repression and PA3 activation by the φ29 protein p4 as efficiently as its φ29 counterpart (data not shown). These observations lead to the conclusion that the mutually cooperative effects observed between the φ29 proteins p6 and p4 require that p6 and, consequently, its nucleoprotein complex, be specific.
Figure 7.
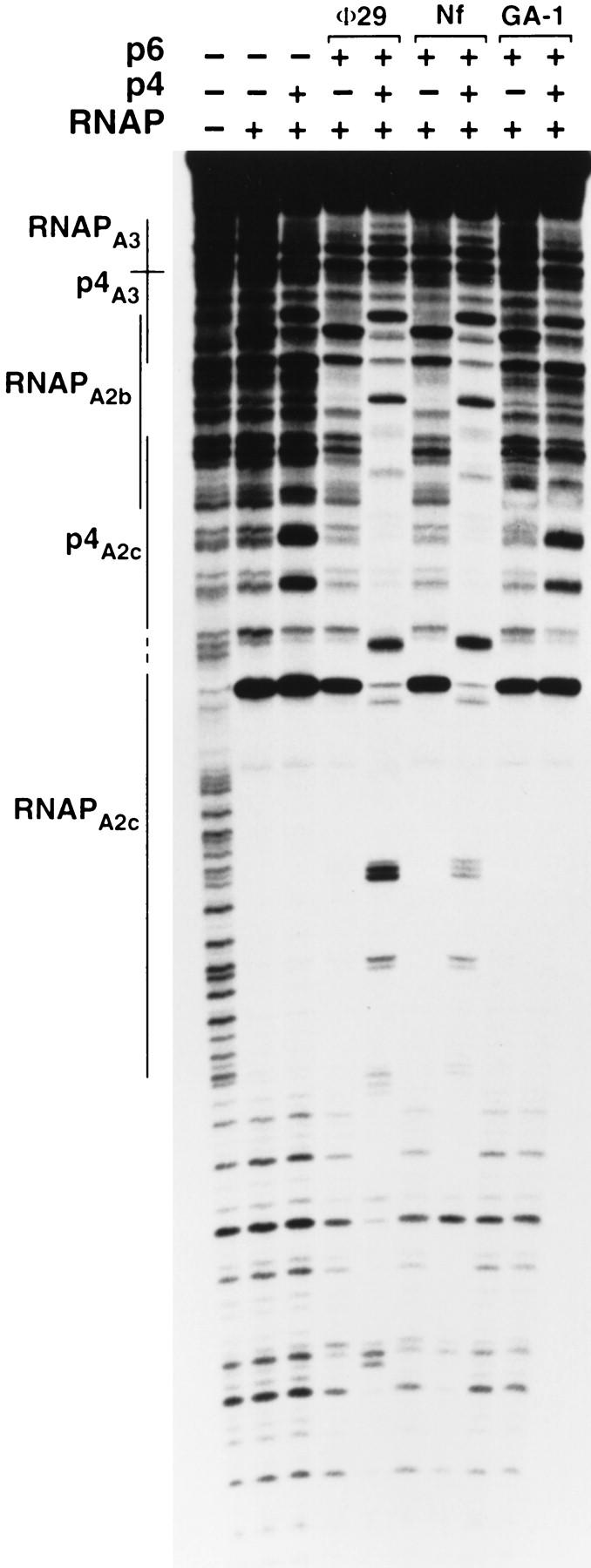
Complex formation by p6 proteins from related phages in the absence and presence of φ29 p4 protein. The locations of the p4- and RNAP-binding sites are marked with lines. DNase I footprinting reactions included the 366-bp φ29 DNA fragment, B. subtilis ςA–RNA polymerase (20 nm), protein p4 from φ29 (330 nm), and the p6 proteins (14.4 μm) from φ29, Nf, or GA-1, as indicated.
Discussion
Protein p6 belongs to a class of dsDNA-binding proteins that, like the prokaryotic HU protein and the HMG-1/HMG-2 protein family, bind DNA through the minor groove with low sequence specificity and act as architectural elements in various DNA processes. Protein p6 is involved in the initiation of φ29 DNA replication, in the repression of the early C2 promoter, and in the compaction of the viral genome (see Salas and Rojo 1993; Gutiérrez et al. 1994). Here we provide evidence for yet another crucial role for p6: its collaboration with the regulatory protein p4 to repress the two main early promoters, PA2b and PA2c, and to activate the late promoter, PA3, thereby regulating the early-to-late transcriptional switch of the viral life cycle.
The interplay between p6 and p4 in DNA binding and transcriptional regulation
The binding of p4 to its site at PA3 is essential for p6 to exert its effect on PA2c transcription because p6 alone binds with a low affinity to this DNA region. With p4 bound to its PA3 site, p6 binds to the upstream DNA region forming a nucleoprotein complex stable enough to block PA2c–RNAP binding and thereby, to repress transcription from PA2c. This complex, in turn, promotes PA2b repression and PA3 activation by increasing the binding affinity of p4 for its PA3 site.
The observed cooperativity between p6 and p4 in DNA binding and transcriptional regulation is probably based on their DNA architectural properties. Protein p6 bends the DNA on binding, and DNA sequence bendability influences p6–DNA complex formation (see Salas and Rojo 1993). Protein p4, on the other hand, requires the specific DNA conformation adopted by A tracts (Koo et al. 1986) for efficient binding (Nuez et al. 1994) and also curves DNA on binding (see Rojo and Salas 1995; Rojo et al. 1998). Transient flexing of DNA by p6 may help p4 in creating curvature both by promoting interactions between two p4 dimers (each bound to one inverted recognition sequence at the target site), and between these and the DNA backbone, thereby enhancing p4–PA3 binding affinity. The p4-induced DNA curvature could then allow stable binding by the first p6 dimer, followed by additional p6 dimers propagating along the DNA in a cooperative fashion, as occurs at the genomic ends (Prieto et al. 1988; Gutiérrez et al. 1994). Unlike other DNA-bending proteins, p6 does not appear to bind preferentially to prebent DNA. This, and the fact that the footprint of p4 bound to PA3 barely changes in the presence of p6, makes it unlikely that the latter colocalizes with p4 at its PA3 site in the final nucleoprotein complex. The footprints generated by p4 and p6 are contiguous, consistent with a model in which p4 would promote binding of the first p6 dimer immediately upstream of its binding site at PA3 (see Fig. 8). Our proposed model, where p4 and p6 localize next to one another, does not exclude the possibility that interactions between the two proteins also play a role in stabilizing the DNA–protein complexes.
Figure 8.

Two different strategies for regulating the early–late transcriptional switch of phage φ29 in vitro. (A) In the absence of protein p6, repression of the early A2b and A2c promoters and activation of the late A3 promoter is directly mediated by protein p4 and its physical interaction with RNAP. Protein p4 activates PA3 by binding to its site at this promoter and stabilizing RNAP at PA3 as a closed complex. Binding of p4 to this site also entails repression of PA2b, whose −35 region overlaps with the p4-binding site at PA3. Repression of PA2c, on the other hand, stems from p4 binding to its site at PA2c and holding the RNAP at the promoter (for review, see Rojo et al. 1998). (B) In the presence of p6 and p4, p6 appears as the direct mediator of PA2c repression by forming a nucleoprotein complex that precludes RNAP binding to this promoter. Moreover, in the presence of p6, binding of p4 to its site at PA3 is enhanced, as is PA3 activation and PA2b repression. The cartoon shows RNAP as a large light gray protein; the small dark gray spheres depict protein p4 dimers, and the unshaded oblate ellipses represent protein p6 dimers. For simplicity, the complexed DNA is drawn as a straight rod.
The transcriptional consequences of the cooperativity between p4 and p6 coincide with those observed previously in the presence of p4 alone (see Rojo et al. 1998). However, whereas PA2b repression and PA3 activation occur by the same p4-mediated mechanism regardless of whether only p4 or both p4 and p6 are present, the mechanism underlying PA2c repression is different in each case (see Fig. 8). When p6 is absent, binding of p4 to its PA2c site causes repression by preventing RNAP promoter clearance (Monsalve et al. 1996a). However, in the presence of both p4 and p6, it is p6 that directly represses PA2c by blocking access to RNAP. The opposite regulatory outcomes at PA2c and PA3 by p4 alone are explained in terms of the strength of the RNAP–promoter interactions involved, strong at PA2c and weak at PA3 for lack of a distinct −35 box (Monsalve et al. 1997). When both p4 and p6 are present, the same opposite regulatory outcomes at PA2c and PA3 could be more related to the differences between the two p4-binding sites and, in particular, the affinity of p4 for each of the binding sites. First, the formation of a stable p6–nucleoprotein complex is driven by the binding of p4 to its high-affinity site at PA3, a process unaffected by the presence of RNAP. Second, although RNAP by itself binds more strongly to PA2c than to PA3, p6 displaces a p4–RNAP complex from PA2c while enhancing its binding to PA3. It is thus remarkable how the phage exploits two alternative mechanisms for regulating the early–late transcriptional switch, by combining a high-affinity binding site for p4 with a low-affinity binding site for RNAP at PA3, and vice versa at PA2c.
Protein p6 as a mediator of the switch between the early and late stages of φ29 transcription in vivo
A direct assessment of our results in vivo is difficult because protein p6 is essential in viral DNA replication (for review, see Salas and Rojo 1993). However, several lines of evidence provide support for p6 involvement in regulating the phage early–late transcriptional switch in vivo. Protein p6 is very abundant in φ29-infected cells, its intracellular concentration reaching values as high as 1 mm at late times of infection, an amount estimated to be almost twice that required to complex all the φ29 DNA molecules present at that time (Abril et al. 1997). The p6/DNA molar ratios at which p6 affects transcription from PA2c, PA2b, and PA3 in vitro are in this same range. Various observations indicate that p4 is far less abundant in φ29-infected cells (Monsalve et al. 1995). The phage would economize on p4 levels by exploiting the high abundance of p6 and its DNA-binding properties, in a process that physically links binding of p4 to PA3 with the stable binding of p6 to PA2c. It seems likely that the p6-independent mechanism of PA2c repression will prevail at intermediate times of infection, when the p6 concentration is still not very high. As infection proceeds and p6 levels build up, the p6-dependent repression mechanism would become increasingly important, eventually becoming the predominant one. Nevertheless, given the dynamic nature of the p6–nucleoprotein complexes, it is conceivable that both mechanisms of PA2c repression coexist in the cell, acting on different DNA molecules.
Comparison with other systems
In most cases where HU and HMG-1 and HMG-2 promote assembly of higher-order structures, the nonspecific architectural protein appears to be recruited to the nucleoprotein complex whose formation it stimulates. Thus, binding of HU within the final complex has been observed in the Mu transpososome (Lavoie and Chaconas 1993, 1994), the λ intasome (Segall et al. 1994), the Mu early operator–Mu repressor complex (Betermier et al. 1995), the replication origin of pKYM (Yasukawa et al. 1997), and GalR bound to gal operators (Aki and Adhya 1997). A model has been proposed to reconcile the nonspecific DNA binding and bending properties of HU with its site-specific binding in these higher-order complexes. First, random binding of HU to DNA would promote association of spatially separated specific elements by bending the intervening DNA region; once the control protein forms the right protein–protein or protein–DNA contacts, HU would be specifically directed to the intervening loop through its high affinity for bent DNA. The nonspecific bending protein is thus ‘trapped’ and contributes to the overall stabilization of the higher-order complex (see Grosschedl et al. 1994; Lavoie and Chaconas 1994; Segall et al. 1994; Betermier et al. 1995; Aki and Adhya 1997; Yasukawa et al. 1997). In the case of HMG-1 and HMG-2, both DNA-mediated and protein–protein interactions have been implicated in the synergistic stimulation between HMG-1 and HMG-2 and the specific control protein (Zappavigna et al. 1996; Boonyaratanakornkit 1998; Jayaraman et al. 1998). The p6–p4 protein pair resembles the above systems in the reciprocal nature of their functional interactions. However, the φ29 system would be novel in at least two respects. First, the nonspecific protein in φ29 (p6) is not trapped in the complex formed by the control protein (p4) and its specific recognition site but, rather, it is specifically targeted to the adjacent DNA region. Second, the functional consequence of the mutual stabilization between the two proteins is to regulate not one but three different processes simultaneously, namely, transcriptional activation of one promoter and repression of two neighboring promoters.
Materials and methods
DNA substrates
The 366-bp end-labeled DNA fragment containing PA2c, PA2b, and PA3 was obtained by PCR amplification from φ29 DNA with primers 5′-GATTTCTCTCTGCATCA-3′ (primer 1) and 5′-CAAAATATCTTCGTGTTC-3′ (primer 2). The shorter DNA fragment (198 bp) containing only PA2c and part of PA2b was generated by PCR amplification using primers 1 and 3 (5′-ATACAGGTGTGGTTAAA-3′). To label these two DNA fragments at only one of the ends (the 5′ end) for DNase I footprinting, primer 1 was treated with polynucleotide kinase and [γ-32P]ATP prior to the amplification reaction. A DNA fragment (258 bp) containing the C2 promoter was obtained by PCR using oligonucleotides 5′-GTGTTTGTGTTGATGATG-3′ and 5′-AAAGTAGGGTACAGCGACAAC-3′. DNA templates for in vitro transcription reactions were full-length φ29 DNA with the terminal protein covalently attached to each 5′ end (TP-DNA) or any of the PCR-generated DNA fragments described above (amplified using unlabeled primers).
Proteins and nucleotides
B. subtilis ςA RNA polymerase was purified as described previously (Sogo et al. 1979). Protein p6 was purified essentially as described by Pastrana et al. (1985). Purification of protein p4 was as described (Mencía et al. 1993). Unlabeled nucleoside triphosphates (ultrapure) and deoxynucleoside triphosphates were purchased from Pharmacia, [α-32P]dATP (3000 Ci/mmole) and [γ-32P] ATP (3000 Ci/mmole) were obtained from Amersham International.
In vitro transcription assays
Transcription reactions were performed in a total volume of 25 μl containing 1 nm of template DNA, 50 mm ammonium sulfate, 25 mm Tris-HCl at pH 7.5, 10 mm MgCl2, 1 μg of poly[d(I-C)], 2 mm dithiothreitol, RNasin ribonuclease inhibitor (7.5 units), 0.2 mm each ATP, CTP, GTP, and UTP, and 20 nm B. subtilis ςA RNA polymerase; where indicated, different amounts of protein p6 (1.8–14.4 μm) and/or protein p4 (660–20 nm) were also included. Unless otherwise stated, in reactions containing p6 the template DNA was preincubated with the protein at 37°C for 10 min prior to addition of RNA polymerase and/or p4. Mixtures were incubated in the absence of NTPs for 10 min at 37°C, and transcription was started by addition of the NTPs. After 15 min at 37°C, the reactions were stopped with EDTA (20 mm final concentration) and the transcripts were analyzed by primer extension as described (Monsalve et al. 1995). The primers were selected to produce a 68-, 78-, 98-, and 184-nucleotide product for PA3, PA2c, PC2, and PA2b, respectively.
DNase I footprinting
DNase I footprinting (Galas and Schmitz 1978) was performed with the end-labeled DNA fragments described above. In a total volume of 25 μl, the DNA fragment (in 25 mm Tris-HCl at pH 7.5, 10 mm MgCl2, 1 μg of poly[d(I-C)] as nonspecific competitor DNA) was incubated in the absence or presence of the following proteins: B. subtilis ςA RNA polymerase (20 nm), protein p4 (660–20 nm), protein p6 (1.8–14.4 μm). In reactions containing p6, the DNA fragment was preincubated with p6 at 37°C for 10 min prior to the addition of DNase I or any other protein, unless otherwise stated. Incubation of DNA with p4 and/or RNAP was for 10 min at 37°C. The footprinting reaction was initiated by the addition of 2 ng of DNase I, the digestion was then allowed to proceed for 2 min at 37° C and finally stopped by the addition of EDTA (20 mm final concentration). The DNA was precipitated with ethanol in the presence of 10 μg of carrier tRNA and analyzed by 8 m urea–6% PAGE. Chemical sequencing reactions of the same DNA fragment used to obtain the DNase I footprint were run in parallel as size standards.
Acknowledgments
We thank Drs. S. Padmanabhan, F. Rojo, and J.M. Hermoso for critical reading of the manuscript, J.M. Lázaro for protein purification, J. Saturno for help with figure preparation, and L. Villar for technical assistance. This investigation was funded by research grants 2R01 GM27242-19 from the National Institutes of Health, PB93-0173 from the Dirección General de Investigación Científica y Técnica, and an Institutional grant from Fundación Ramón Areces. M.E.-A. was supported by the Ministerio de Educación y Cultura (Spain).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
This work is dedicated to the memory of Professor Eladio Viñuela.
E-MAIL msalas@cbm.uam.es; FAX 34-91 397 84 90.
References
- Abril AM, Salas M, Andreu JM, Hermoso JM, Rivas G. Phage φ29 protein p6 is in a monomer-dimer equilibrium that shifts to higher association states at the millimolar concentrations found in vivo. Biochemistry. 1997;36:11901–11908. doi: 10.1021/bi970994e. [DOI] [PubMed] [Google Scholar]
- Aki T, Adhya S. Repressor induced site-specific binding of HU for transcriptional regulation. EMBO J. 1997;16:3666–3674. doi: 10.1093/emboj/16.12.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlung T, Ingmer H. H-NS: A modulator of environmentally regulated gene expression. Mol Microbiol. 1997;24:7–17. doi: 10.1046/j.1365-2958.1997.3151679.x. [DOI] [PubMed] [Google Scholar]
- Barthelemy I, Mellado RP, Salas M. In vitro transcription of bacteriophage φ29 DNA: Inhibition of early promoters by the viral replication protein p6. J Virol. 1989;63:460–462. doi: 10.1128/jvi.63.1.460-462.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betermier M, Rousseau P, Alazard R, Chandler M. Mutual stabilization of bacteriophage Mu repressor and histone-like proteins in nucleoprotein structure. J Mol Biol. 1995;249:332–341. doi: 10.1006/jmbi.1995.0300. [DOI] [PubMed] [Google Scholar]
- Bianchi ME, Beltrame M, Paonessa G. Specific recombination of cruciform DNA by nuclear protein HMG-1. Science. 1989;243:1053–1059. doi: 10.1126/science.2922595. [DOI] [PubMed] [Google Scholar]
- Boonyaratanakornkit V, Melvin V, Prendegast P, Altmann M, Ronfani L, Bianchi ME, Taraseviciene L, Nordeen SK, Allegretto EA, Edwards DP. High-mobility group chromatin proteins 1 and 2 functionally interact with steroid hormone receptors to enhance their DNA binding in vitro and transcriptional activity in mammalian cells. Mol Cell Biol. 1998;18:4471–4487. doi: 10.1128/mcb.18.8.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracco L, Kotlarz D, Kolb A, Diekmann S, Buc H. Synthetic curved DNA sequences can act as transcriptional activators in Escherichia coli. EMBO J. 1989;8:4289–4296. doi: 10.1002/j.1460-2075.1989.tb08615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin M, Reeves R. High mobility group proteins. Prog Nucleic Acids Res Mol Biol. 1996;54:35–100. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- Falconi M, Higgins NP, Spurio R, Pon CL, Gualerzi CO. Expression of the gene encoding the major bacterial nucleoid protein H-NS is subject to transcriptional autorepression. Mol Microbiol. 1993;10:273–282. doi: 10.1111/j.1365-2958.1993.tb01953.x. [DOI] [PubMed] [Google Scholar]
- Flashner Y, Gralla JD. DNA dynamic flexibility and protein recognition: Differential stimulation by bacterial histone-like protein HU. Cell. 1988;54:713–721. doi: 10.1016/s0092-8674(88)80016-3. [DOI] [PubMed] [Google Scholar]
- Freire R, Salas M, Hermoso JM. A new protein domain for binding to DNA through the minor groove. EMBO J. 1994;13:4353–4360. doi: 10.1002/j.1460-2075.1994.tb06755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire R, Serrano M, Salas M, Hermoso JM. Activation of replication origins in φ29-related phages requires the recognition of initiation proteins to specific nucleoprotein complexes. J Biol Chem. 1996;271:31000–31007. doi: 10.1074/jbc.271.48.31000. [DOI] [PubMed] [Google Scholar]
- Galas DJ, Schmitz A. DNase footprinting: A simple method for detection of protein-DNA binding specificity. Nucleic Acids Res. 1978;5:3157–3170. doi: 10.1093/nar/5.9.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosschedl RK, Giese K, Pagel J. HMG domain proteins: Architectural elements in the asssembly of nucleoprotein structures. Trends Genet. 1994;10:94–100. doi: 10.1016/0168-9525(94)90232-1. [DOI] [PubMed] [Google Scholar]
- Gutiérrez C, Freire R, Salas M, Hermoso JM. Assembly of phage φ29 genome with viral protein p6 into a compact complex. EMBO J. 1994;13:269–276. doi: 10.1002/j.1460-2075.1994.tb06257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges-Garcia Y, Hagerman PJ, Pettijohn DE. DNA ring closure mediated by protein HU. J Biol Chem. 1989;264:14621–14623. [PubMed] [Google Scholar]
- Jayaraman L, Moorthy NC, Murthy KGK, Manley JL, Bustin M, Prives C. High mobility group protein-1 (HMG-1) is a unique activator of p53. Genes & Dev. 1998;12:462–472. doi: 10.1101/gad.12.4.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo H, Wu H, Crothers DM. DNA bending at adenine-thymine tracts. Nature. 1986;320:501–506. doi: 10.1038/320501a0. [DOI] [PubMed] [Google Scholar]
- Lavoie BD, Chaconas G. Site-specific HU binding in the Mu transpososome: Conversion of a sequence-independent DNA-binding protein into a chemical nuclease. Genes & Dev. 1993;7:2510–2519. doi: 10.1101/gad.7.12b.2510. [DOI] [PubMed] [Google Scholar]
- ————— A second high affinity HU binding site in the phage Mu transpososome. J Biol Chem. 1994;269:15571–15576. [PubMed] [Google Scholar]
- Mencía M, Salas M, Rojo F. Residues of the Bacillus subtilis phage φ29 transcriptional activator required both to interact with RNA polymerase and to activate transcription. J Mol Biol. 1993;233:695–704. doi: 10.1006/jmbi.1993.1546. [DOI] [PubMed] [Google Scholar]
- Mencía M, Monsalve M, Rojo F, Salas M. Transcription activation by phage φ29 protein p4 is mediated by interaction with the α subunit of Bacillus subtilis RNA polymerase. Proc Natl Acad Sci. 1996a;93:6616–6620. doi: 10.1073/pnas.93.13.6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencía M, Monsalve M, Salas M, Rojo F. Transcriptional activator of phage φ29 late promoter: Mapping of residues involved in interaction with RNA polymerase and in DNA bending. Mol Microbiol. 1996b;20:273–282. doi: 10.1111/j.1365-2958.1996.tb02616.x. [DOI] [PubMed] [Google Scholar]
- Monsalve M, Mencía M, Rojo F, Salas M. Transcription regulation in bacteriophage φ29: Expression of the viral promoters throughout the infection cycle. Virology. 1995;207:23–31. doi: 10.1006/viro.1995.1048. [DOI] [PubMed] [Google Scholar]
- ————— Activation and repression of transcription at two different phage φ29 promoters are mediated by interaction of the same residues of regulatory protein p4 with RNA polymerase. EMBO J. 1996a;15:383–391. [PMC free article] [PubMed] [Google Scholar]
- Monsalve M, Mencía M, Salas M, Rojo F. Protein p4 represses phage φ29 A2c promoter by interacting with the α subunit of RNA polymerase. Proc Natl Acad Sci. 1996b;93:8913–8918. doi: 10.1073/pnas.93.17.8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsalve M, Calles B, Mencía M, Salas M, Rojo F. Transcription activation or repression by phage φ29 protein p4 depends on the strength of the RNA polymerase-promoter interactions. Mol Cell. 1997;1:99–107. doi: 10.1016/s1097-2765(00)80011-8. [DOI] [PubMed] [Google Scholar]
- Monsalve M, Calles B, Mencía M, Rojo F, Salas M. Binding of phage φ29 protein p4 to the early A2c promoter: Recruitment of a repressor by the RNA polymerase. J Mol Biol. 1998;283:559–569. doi: 10.1006/jmbi.1998.2084. [DOI] [PubMed] [Google Scholar]
- Nash HA. Bending and supercoiling of DNA at the attachment site of bacteriophage lambda. Trends Biochem Sci. 1990;15:222–227. doi: 10.1016/0968-0004(90)90034-9. [DOI] [PubMed] [Google Scholar]
- ————— . The E. coli HU and IHF proteins; accessory factors for complex protein-DNA assemblies. In: Lin ECC, Lynch AS, editors. Regulation of gene expression in E. coli. Austin, TX: R.G. Landes Co.; 1996. pp. 149–179. [Google Scholar]
- Nuez B, Rojo F, Salas M. Requirement for an Abstract structure at the binding site of phage φ29 transcriptional activator. J Mol Biol. 1994;237:175–181. doi: 10.1006/jmbi.1994.1219. [DOI] [PubMed] [Google Scholar]
- Oñate S, Prendergast P, Wagner JP, Nissen M, Reeves R, Pettijohn DE, Edwards DP. The DNA-bending protein HMG-1 enhances progesterone receptor binding to its target DNA sequences. Mol Cell Biol. 1994;14:3376–3391. doi: 10.1128/mcb.14.5.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana R, Lázaro JM, Blanco L, García JA, Méndez E, Salas M. Overproduction and purification of protein p6 of Bacillus subtilis phage φ29: Role in the initiation of DNA replication. Nucleic Acids Res. 1985;13:3083–3100. doi: 10.1093/nar/13.9.3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontiggia A, Negri A, Beltrame M, Bianchi ME. Protein HU binds specifically to kinked DNA. Mol Microbiol. 1993;7:343–350. doi: 10.1111/j.1365-2958.1993.tb01126.x. [DOI] [PubMed] [Google Scholar]
- Preobrajenskaya O, Boullard A, Boubrick F, Schnarr M, Rouvière-Yaniv J. The protein HU can displace the LexA repressor from its DNA-binding sites. Mol Microbiol. 1994;13:459–467. doi: 10.1111/j.1365-2958.1994.tb00440.x. [DOI] [PubMed] [Google Scholar]
- Prieto I, Serrano M, Lázaro JM, Salas M, Hermoso JM. Interaction of the bacteriophage φ29 protein p6 with double-stranded DNA. Proc Natl Acad Sci. 1988;85:314–318. doi: 10.1073/pnas.85.2.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo F, Salas M. Transcriptional regulators: Protein-DNA complexes and regulatory mechanisms. Methods Mol Genet. 1995;6:421–438. [Google Scholar]
- Rojo F, Mencía M, Monsalve M, Salas M. Transcription activation and repression by interaction of a regulator with the α subunit of RNA polymerase: The model of phage φ29 protein p4. Prog Nucleic Acid Res Mol Biol. 1998;60:29–46. doi: 10.1016/s0079-6603(08)60888-0. [DOI] [PubMed] [Google Scholar]
- Salas M, Rojo F. Replication and transcription of bacteriophage φ29 DNA. In: Hoch JA, Losick R, editors. Bacillus subtilis and other Gram-positive bacteria: Biochemistry, physiology, and molecular genetics. Washington, D.C.: American Society for Microbiology; 1993. pp. 843–857. [Google Scholar]
- Segall AM, Goodman SD, Nash HA. Architectural elements in nucleoprotein complexes: Interchangeability of specific and nonspecific DNA binding proteins. EMBO J. 1994;13:4536–4548. doi: 10.1002/j.1460-2075.1994.tb06775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Gutiérrez C, Salas M, Hermoso JM. The superhelical path of the DNA in the nucleoprotein complex that activates the initiation of phage φ29 DNA replication. J Mol Biol. 1993;230:248–259. doi: 10.1006/jmbi.1993.1140. [DOI] [PubMed] [Google Scholar]
- Shore D, Baldwin RL. Energetics of DNA twisting. J Mol Biol. 1983;170:957–981. doi: 10.1016/s0022-2836(83)80198-3. [DOI] [PubMed] [Google Scholar]
- Sogo JM, Inciarte MR, Corral J, Viñuela E, Salas M. RNA polymerase binding sites and transcription of the DNA of Bacillus subtilis phage φ29. J Mol Biol. 1979;127:411–436. doi: 10.1016/0022-2836(79)90230-4. [DOI] [PubMed] [Google Scholar]
- Spurio R, Falconi M, Brandi A, Pon CL, Gualerzi CO. The oligomeric structure of nucleoid protein H-NS is necessary for recognition of intrinsically curved DNA and for DNA bending. EMBO J. 1997;16:1795–1805. doi: 10.1093/emboj/16.7.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrier CS, Roodi N, Yee CJ, Bailey LR, Jensen RA, Bustin M, Parl FF. High mobility group (HMG) protein and TATA-binding protein-associated factor TAF(II)30 affect estrogen receptor-mediated transcriptional activation. Mol Endocrinol. 1997;11:1009–1019. doi: 10.1210/mend.11.8.9962. [DOI] [PubMed] [Google Scholar]
- Wang JC, Giaever GN. Action at a distance along DNA. Science. 1988;240:300–304. doi: 10.1126/science.3281259. [DOI] [PubMed] [Google Scholar]
- Werner MH, Burley SK. Architectural transcription factors: Proteins that remodel DNA. Cell. 1997;88:733–736. doi: 10.1016/s0092-8674(00)81917-0. [DOI] [PubMed] [Google Scholar]
- Whiteley HR, Ramey WD, Spiegelman GB, Holder RD. Modulation of in vivo and in vitro transcription of bacteriophage φ29 early genes. Virology. 1986;155:392–401. doi: 10.1016/0042-6822(86)90202-3. [DOI] [PubMed] [Google Scholar]
- Williams RM, Rimsky S. Molecular aspects of the E. coli nucleoid protein, H-NS: A central controller of gene regulatory networks. FEMS Microbiol Lett. 1997;156:175–185. doi: 10.1111/j.1574-6968.1997.tb12724.x. [DOI] [PubMed] [Google Scholar]
- Yamada H, Yoshida T, Tanaka K, Sasakawa C, Mizuno T. Molecular analysis of the Escherichia coli hns gene encoding a DNA binding protein, which preferentially recognises curved DNA sequences. Mol & Gen Genet. 1991;230:332–336. doi: 10.1007/BF00290685. [DOI] [PubMed] [Google Scholar]
- Yasukawa H, Ozaki E, Nakahama K, Masamune Y. HU protein binding to the replication origin of the rolling-circle plasmid pKYM enhances DNA replication. Mol & Gen Genet. 1997;254:548–554. doi: 10.1007/s004380050450. [DOI] [PubMed] [Google Scholar]
- Zappavigna V, Falciola L, Citterich M, Mavilio F, Bianchi ME. HMG-1 interacts with HOX proteins and enhances their DNA binding and transcriptional activation. EMBO J. 1996;15:4981–4991. [PMC free article] [PubMed] [Google Scholar]
- Zuber F, Kotlarz D, Rimsky S, Buc H. Modulated expression of promoters containing upstream curved DNA sequences by the Escherichia coli nucleoid protein H-NS. Mol Microbiol. 1994;12:231–240. doi: 10.1111/j.1365-2958.1994.tb01012.x. [DOI] [PubMed] [Google Scholar]
- Zwilling S, Konig H, Wirth T. High mobility group protein 2 functionally interacts with the POU domains of octamer transcription factors. EMBO J. 1995;14:1198–1208. doi: 10.1002/j.1460-2075.1995.tb07103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]