

Abstract
The iodination of chlorinated aromatic compounds using Ag2SO4/I2, AgSbF6/I2, AgBF4/I2 and AgPF6/I2 offers access to iodoarenes that are valuable intermediates in organic synthesis. Specifically, iodination of phenols, anisoles and anilines with a 3,5-dichloro substitution pattern preferentially yielded the ortho, para and para iodinated product, respectively. In the case of chlorobenzene and 3-chlorotoluene, AgSbF6/I2, AgBF4/I2 and AgPF6/I2, but not Ag2SO4/I2, selectively introduced the iodine in para position to the chlorine substituent.
Keywords: Phenol, anisole, aniline, chlorobenzene, 3-chlorotoluene, non-coordinating ions, silver sulfate, silver hexafluoroantimonate, silver tetrafluoroborate, silver hexafluorophosphate
1. Introduction
The iodoarene moiety is an important structural motif in biologically active molecules (e.g. thyroid hormone) and a synthetic intermediate for a variety of fine chemistry products (e.g. isovanillyl sweeteners1), radiopharmaceuticals,2 environmental contaminants3,4 and numerous bioactive compounds, such as camptothecin,5 cephalosporin derivatives,6 dehydrotubifoline,7 morphine,8 sangliferine A,9 ecteinascidine,10 and berkelic acid methyl ester.11 One example of a prescription drug synthesized from an iodoarene intermediate is galanthamine, an acetylcholinesterase inhibitor for the symptomatic treatment of senile dementia of Alzheimer patients.12 The usefulness of iodoarenes as synthetic intermediates is partly due to the fact that the iodo substituent can undergo a multitude of transition metal-catalyzed cross-coupling reactions.13,14
In particular the electrophilic iodination of phenols, anisoles and anilines provides straightforward access to a range of valuable iodoarene intermediates.15,16 A variety of iodine atom donating reagents, such as N-iodosuccinimide/p-toluenesulfonic acid17 and iodine monochloride (ICl),18 have been used successfully for the iodination of aromatic compounds. In addition, elemental iodine (I2) is a particularly attractive source of iodine atoms.15,16 Iodination reactions using I2 require activation by protons, metal ions or a suitable solvent and trapping of the hydriodic acid formed during the reaction to prevent cleavage of carbon-iodide bonds. Finally, oxidative activation strategies have been employed to generate reactive iodonium species or to oxidize the released iodide to iodine, thus allowing a stochiometric use of the iodine atoms present in the reaction.15,16 Most iodination reagents give good-to-excellent yields of iodinated phenols, anisoles and anilines and display a high para regioselectivity. In para-substituted aromatic compounds, iodination typically results in mono- or even di-iodination in ortho positions.
Iodinated phenols, anisoles and anilines with chlorine substituents in the meta position are of interest as starting materials for a variety of drug molecules19–21 and environmental contaminants.3,4 These compounds are frequently synthesized via the reduction of a suitable nitrobenzene followed by a Sandmeyer reaction to introduce the iodo substituent.3,4,22–24 Although a direct iodination of a suitable chlorinated precursor would greatly improve access to these building blocks, the regioselectivity of the iodination of chlorinated aromatic compounds has been poorly characterized. For example, 3,5-dichloro-2-iodophenol, a starting material for the synthesis of heat shock protein-90 (HSP-90) inhibitors, can only be synthesized in moderate yield by iodination of 3,5-dichlorophenol with NaH/I2.19 2,5-Dichloro-4-iodophenol, a precursor of cephalosporin derivatives with activity against methicillin-resistant Staphylococcus aureus, was synthesized from 2,5-dichlorophenol with Ag2SO4/I2.6 Several chlorinated iodo- and diiodoanilines have been prepared by iodination of the corresponding chlorinated aniline with iodine monochloride.20,21,25,26 For example, 2-iodo-3,4-dichloroaniline, a starting material for preparation of indolyl substituted benzoic acids for the treatment of urinary tract disorders, has been synthesized by ICl/AcOH in only 35% yield.26
One reason for the lack of direct iodination procedures for chlorinated aromatic compounds is the challenging separation of different iodinated regioisomers (Scheme 1) and the formation of by-products resulting from dehalogenation, polysubstitution and other side-reactions, which considerably complicates the product isolation and purification. Here, we systematically investigate the regioselective iodination of a series of chlorinated phenols, anisoles, anilines and other aromatic compounds using a series of iodination reagents, with a special emphasis on iodination reactions using I2 and silver salts with non-coordinating anions.
Scheme 1.

Regioselective iodination of chlorinated phenols, anisoles, anilines, chlorobenzenes and chlorotoluenes using different silver salts as iodination reagents.
2. Results and discussion
2.1. Exploratory iodination of phenol (1a), 3,5-dichlorophenol (1b) and 3,5-dichloroanisole (1c)
2.1.1. Conventional iodination reagents
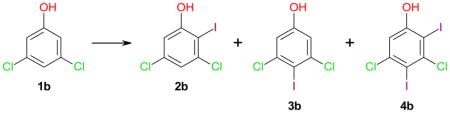
The iodination of phenol (1a) with different iodination reagents has been investigated extensively and typically results in good yields and para selectivity.15 Building on published iodination approaches for 1a, this study initially investigated the regioselectivity of the iodination of 3,5-dichlorophenol 1b (Table 1). The corresponding iodides 2b and 3b are useful starting materials for the synthesis of HSP-90 inhibitors19 or metabolites of polychlorinated biphenyls (PCBs).3,4 Iodination with I2 in ethanol resulted in complete conversion of 1b within 16 hours and displayed ortho selectivity; however, the yield of the ortho iodinated product 2b was only 16% (entry 1–1). N-Iodosuccinimide (NIS)/p-toluenesulfonic acid (PTSA) as the iodine atom donating reagent17 resulted in almost complete conversion of 1b within 24 h, with a 3b: 2b ratio of approximately 3: 1 (entry 1–2). A more pronounced regioselectivity has been reported previously for the iodination of phenol (1a) with NIS/PTSA (3a: 2a > 14: 1).17
Table 1.
Effect of iodinating reagents, solvents and temperature on the iodination of 3,5-dichlorophenol (1b).*
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Reaction Conditionsa | Reaction time (h) | Conversion (%) | Yield
|
||
| 2b (%) | 3b (%) | 4b (%) | ||||
| 1–1 | I2 (1.5 eq.), C2H5OH | 16 | 100 | 16 | 1 | T |
| 1–2 | N-Iodosuccinimide, PTSA, CH3CN | 24 | < 100 | 18 | 57 | T |
| 1–3 | BTMACl2I, ZnCl2, AcOH, RTb | 24 | < 100 | 5 | 4 | nd |
| 1–4 | BTMACl2I, ZnCl2, AcOH, 90 °Cc | 2 | < 100 | 46 | 39 | T |
| 1–5 | CAN, I2, CH3CN | 24 | > 0 | 2 | 2 | nd |
| 1–6 | Ag2SO4, I2, CH3CNd | 16 | > 0 | 11 | 3 | T |
| 1–7 | Ag2SO4, I2, DCM-MeOH-H2O (1:1:1, v/v)d | 2 | 100 | 9 | 2 | T |
| 1–8 | Ag2SO4, I2, β-cyclodextrine | 1 | 100 | 8 | nd | nd |
| 1–9 | Ag2SO4, I2, n-hexaned | 16 | < 100 | 49 | 41 | T |
| 1–10 | Ag(OCOCF3)2, I2, C2H5OH | 16 | < 100 | 28 | 4 | T |
Percent conversion and yields were determined by GC-MS;
one equivalent (eq.) of each key reagent was employed if not mentioned otherwise;
BTMACl2I (1.5 eq.) and ZnCl2 (1.5 eq.);
BTMACl2I (1.1 eq.) and ZnCl2 (1.5 eq.);
I2 (1.5 eq.) and Ag2SO4 (1.1 eq.);
β-cyclodextrin in DMSO was added to a solution containing 1b and Ag2SO4/I2 (1 eq.: 1 eq.) in DCM (DMSO: DCM = 1: 1, v/v);
T = traces were detected by GC-MS; nd = not detected; BTMACl2I = benzyltrimethylammonium dichloroiodinate; RT = room temperature; PTSA = p-toluenesulfonic acid.
Although nearly complete conversion was observed within 24 h for the iodination of 1b with benzyltrimethylammonium dichloroiodinate (BTMACl2I)/ZnCl23 at room temperature, the total yield of iodides 2b and 3b was poor and no diiodinated products were detected (entry 1–3). BTMACl2I/ZnCl23 at 90 °C also resulted in almost complete conversion of 1b and the formation of essentially a 1:1 mixture of 2b and 3b (entry 1–4). Only 4% conversion and no regioselectivity was observed when 1b was iodinated CAN/I2 in acetonitrile (entry 1–5).27,28 In contrast, the iodination of phenol with CAN/I2 has been reported to give 70% yield of the 2- and 4-iodinated products, with a ratio of 2a: 3a of 7: 3.28 Overall, the yields and/or regioselectivity with the conventional iodination reagents were unsatisfactory (yields < 41%), with only NIS/PTSA resulting in a reasonable yield of 3b (57%).
2.1.2. Iodinations of 3,5-dichlorophenol 1b using Ag2SO4/I2 and related silver reagents
Considering the poor yield and regioselectivity of more conventional iodination reagents (Table 1, entries 1–1 to 1–5), a series of silver salt/I2 reagents was studied as iodination reagents for 1b. Silver salts, such as Ag2SO4/I26,29–31 and Ag(OCOCF3)/I232,33, have been used extensively for the iodination of aromatic compounds. They activate I2 by forming insoluble silver iodide, thus generating an electrophilic iodine species. The reactive iodine species appears to be identical in many of these reactions and is thought to react with the respective aromatic compound via a σ-complex.34 As shown in Table 1, only a small percentage of 1b was iodinated with Ag2SO4/I2 in acetonitrile (entry 1–6), whereas complete or almost complete conversion of 1b was observed with all other silver salts investigated (entries 1–7 to 1–10). However, several reagents displayed poor yields, possibly due to the high reactivity of the respective reagent (entries 1–7 and 1–8).
β-Cyclodextrin has been shown to improve the regioselectivity of bromination reactions in organic solvents due to complexation of the aromatic phenol or aniline,35,36 but to decrease the ortho-to-para ratio for the ortho-iodination of phenol (1a) in aqueous solution.37 In this study, β-cyclodextrin had no advantageous effect on the regioselectivity of the iodination of 1b with Ag2SO4/I2 in DMSO/DCM (entry 1–8). Iodination of 1b with Ag2SO4/I2 in n-hexane resulted in good yields (total yield of 2b + 3b is 90%), but displayed poor regioselectivity (2b: 3b ~ 1: 1; entry 1–9). The iodination with Ag(OCOCF3)/I2 in ethanol resulted in an almost complete conversion of 1b and gave unsatisfactory yields after 16 hours, with a 7-times higher yield of the ortho iodinated product 2b (entry 1–10).
2.1.3. Iodination of 3,5-dichloroanisole 1c using Ag2SO4/I2
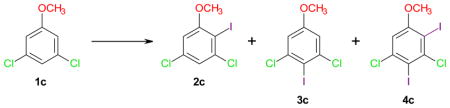
The iodination of 3,5-dichloroanisole (1c) was investigated as a structural analog to 3,5-dichlorophenol (1b) (Table 2). The structures of the iodination products 3c and 4c were confirmed by crystal structure analysis to ensure a correct interpretation of the product ratios (Figure S1). The iodination of 1c with NIS/PTSA, which gave the best iodination results with phenol 1b, yielded the 4-substituted product 3c in 68% yield (complete conversion) (entry 2-1). However, considerable quantities of 2c and 4c were also formed (2c: 3c ~ 1: 5 and 4c: 3c ~ 1: 23). Subsequent experiments investigated the yield and regioselectivity of the iodination of anisole 1c with Ag2SO4/I2 in different solvents. Iodination of 1c in DCM resulted in poor yields of 2c and 3c, possibly due to the formation of multi-iodinated products, and limited regioselectivity (entry 2–2). While the yields of the iodination reaction in hexane were excellent (87% total yield), the regioselectivity was relatively poor, with 3c being the major product (entry 2–3). This is comparable with the iodination of 1b in hexane, which also resulted in poor regioselectivity (entry 1–9). Significantly improved para regioselectivity was observed for reactions performed in acetonitrile (entries 2–4 and 2–5). In particular iodination with 1.5 equivalents Ag2SO4 and 1.1 equivalents I2 gave 3c in 65% yield, with 2c: 3c ~ 1: 16 (complete conversion) (entry 2–4). Increasing the molar ratios of Ag2SO4 and I2 gave a somewhat lower yield of 3c and a decreased regioselectivity (2c: 3c ~ 1: 12) (entry 2–5). A reasonable para selectivity was also observed in DMSO; however, the yields of 3c were only moderate (35% yield; 94% conversion) (entry 2–6).
Table 2.
Effect of solvents and molar ratio of the starting materials on the iodination of 3,5-dichloroanisole (1c) with Ag2SO4/I2.*
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Reaction Conditions | Reaction time (h) | Conversion (%) | Yield
|
||
| 2c (%) | 3c (%) | 4c (%) | ||||
| 2-1 | NIS, PTSA, CH3CNa | 18 | 100 | 14 | 68 | 3 |
| 2–2 | Ag2SO4, I2, DCMb | 60 | 100 | 4 | 7 | 9 |
| 2–3 | Ag2SO4, I2, n-hexaneb | 16 | 100 | 28 | 48 | 11 |
| 2–4 | Ag2SO4, I2, CH3CNc | 42 | 100 | 4 | 65 | T |
| 2–5 | Ag2SO4, I2, CH3CNd | 19 | 90 | 4 | 49 | T |
| 2–6 | Ag2SO4, I2, DMSOa | 4 | 94 | 4 | 35 | nd |
Percent conversion and yields were determined by GC-MS;
(1.0 eq.) and Ag2SO4 (1.0 eq.);
I2 (1.5 eq.) and Ag2SO4 (1.1 eq.);
I2 (1.1 eq.) and Ag2SO4 (1.5 eq.);
I2 (2.0 eq.) and Ag2SO4 (2.0 eq.);
T = traces were detected by GC-MS; nd = not detected.
2.2. Iodination with silver salt with non-coordinating anions and I2 (AgX/I2)
Since neither the conventional nor the silver-based iodination reagents offered a clear advantage for the regioselective iodination of phenol 1b or anisole 1c (Tables 1 and 2), the present study investigated the hypothesis that anions with different ligand binding strength may modulate the reactivity and, thus, regioselectivity of silver salt/I2 reagents. In particular non-coordinating anions SbF6−, BF4− and PF6− are of interest in this context because their ligand binding strengths decrease in the order SbF6− > BF4− > PF6−.38 Although AgBF4/I2 has been used for the synthesis of iodoarenes from aryltrimethylsilanes, this reagent has not been investigated for the direct electrophilic iodination of aromatic compounds.39,40 Furthermore, several other iodinating reagents, such as bis(sym-collidine)iodine(I) hexafluorophosphate41 or HgO/HBF4/I2 on SiO3,42 contain non-coordinating anions. However, to the best of our knowledge iodination reactions with I2 and AgSbF6, AgBF4 or AgPF6 have not been employed in aromatic iodination reactions.
2.2.1. Iodination of phenol 1a and 3,5-dichorophenol 1b with AgX/I2
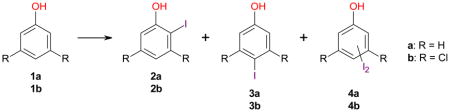
As mentioned above, the iodination of phenol (1a) with a range of reagents, for example KI/H2O2/AcOH,43 KI/KClO3/HCl,44 CAN/I2,28 NaBO3·4H2O/I2 in ionic liquids,45 H5PV2Mo10O40 polyoxometalate/I2,46 ICl/DDQ/ferrocenium tetrakis(3,5-bis(trifluoromethyl)phenyl)borate47 or NIS/PTSA,17 typically results in good yields and para selectivity; however, ortho iodination of 1a reportedly occurs with a number of silver salts and iodine, for example Ag2SO4/I2 and AgNO3/I2 in DCM.48 In this study, conversion of 79% and 100% were observed for iodinations of 1a with AgSbF6/I2 and AgBF4/I2, respectively, and the yields of 2a and 3a were poor (Table 3; entries 3-1 and 3-2). One possible explanation for the poor yields is the formation of poly-iodinated and other byproducts that cannot be detected by GC-MS. An intriguing observation is that the para substituted product 3a was formed in 46% yield (91% conversion) with AgPF6/I2 (entry 3–3). This suggests that the side reactions responsible for the low yield with AgSbF6/I2 and AgBF4/I2 did not play a role in the iodination of 1a with AgPF6/I2, possibly due to its lower reactivity. However, this reagent does not offer an apparent advantage compared to conventional iodination reagents.
Table 3.
Iodination of phenol (1a) and 3,5-dichlorophenol (1b) using different iodination reagents.*
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Reaction Conditionsa | Reaction time (h) | Conversion (%) | Yield
|
||
| 2 (%) | 3 (%) | 4 (%) | ||||
| (A) Phenol (1a) | ||||||
| 3-1 | AgSbF6, I2, DCM | 23 | 79 | 2 | 1 | T |
| 3-2 | AgBF4, I2, DCM | 1.5 | 100 | 7 | 3 | T |
| 3–3 | AgPF6, I2, DCM | 23 | 91 | 4 | 46 | T |
| (B) 3,5-Dichlorophenol (1b) | ||||||
| 3–4 | Ag2SO4, I2, DCMb | 16 | 100 | 53 | 2 | T |
| 3–5 | AgSbF6, I2, DCM | 16 | <100 | 82 | T | nd |
| 3–6 | AgBF4, I2, DCMc | 1 | <100 | >90 | 5 | nd |
| 3–7 | AgPF6, I2, DCM | 16 | <100 | 57 | 10 | nd |
Percent conversion and yields were determined by GC-MS;
one equivalent (eq.) of each reagent was employed if not mentioned otherwise;
I2 (1.5 eq.) and Ag2SO4 (1.5 eq.);
I2 (1.1 eq.) and Ag2SO4 (1.1 eq.);
T = traces were detected by GC-MS; nd = not detected.
Compared to 1a, significantly improved yields and regioselectivities were observed for iodinations of 1b with Ag2SO4/I2, AgSbF6/I2, AgBF4/I2 and AgPF6/I2 in DCM (Table 3). These reactions gave moderate-to-good yields of the ortho product 2b (Table 3, entries 3–4 to 3–7). Iodination of 1b with Ag2SO4/I2 in DCM gave 2b in 53% yield (entry 3–4). In contrast, iodination of 2,5-dichlorophenol under comparable conditions has been reported to yield the corresponding para substituted product, 2,5-dichloro-4-iodophenol, in 86% yield.6 AgBF4/I2 was the most reactive reagent among the silver salts investigated, with complete conversion of 1b after only 1 h (entry 3–6). The highest 2b: 3b ratio was obtained with AgSbF6/I2, which afforded 2b in 82% yield (entry 3–5). In this reaction, only traces of the para product 3b were detected by GC-MS. A relatively poor regioselectivity was observed for AgPF6/I2, with a 2b: 3b ratio of approximately 6: 1. The opposite regioselectivity was observed for NIS/PTSA, with 2b: 3b ~ 1: 3 (entry 1–2).
2.2.2. Iodination of anilines 1d-g with AgX/I2
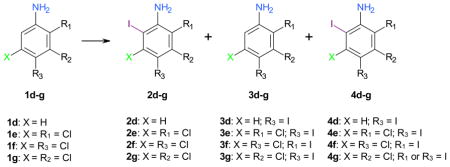
The iodination of aniline (1d) with Ag2SO4/I2 in ethanol has been reported to result in the formation of 3d in 46% yield.31 Similarly, the direct iodination of aniline (1d) with different reagents, for example KI/H2O2/AcOH,43 KI/KClO3/HCl,44 KI/KIO3/HCl,49 CAN/I2,28 NaBO3·4H2O/I2 in ionic liquids,45 H5PV2Mo10O40 polyoxometalate/I2,46 ICl/DDQ/ferrocenium tetrakis(3,5-bis(trifluoromethyl)phenyl)borate47 or bis(sym-collidine)iodine(I) hexafluorophosphate,41 yields 3d as the major product. The only reported selective synthesis of 2d (46% yield) by direct iodination of 1d employs Ag2SO4/I2 in 1,2-ethanediol as iodinating reagent.50 In this study, the iodination of aniline (1d) with AgSbF6/I2 and AgPF6/I2 resulted in the formation of 4-iodoaniline (3d) in 25% (57% conversion) and 22% (69% conversion) yield, respectively (Table 4, entries 4-1). While no 2- and 3-iodoanilines were detected with either reagent, significant amounts of a diiodo- and, in the case of AgSbF6/I2, a triiodo-aniline were detected by GC-MS. Therefore, AgSbF6/I2 and AgPF6/I2 do not offer a more straightforward access to para iodinated aniline 3d.
Table 4.
Percent conversion (C) and yields of mono and diiodinated products from selected chlorinated anilines using different iodination reagents (R1 to R3 = H if not mentioned otherwise).*,#
 | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Starting Material 1 | Reaction Time (t) and Reaction Conditions | |||||||||||||||||||||
| Ag2SO4, I2, DCM | AgSbF6, I2, DCM | AgBF4, I2, DCM | AgPF6, I 2, DCM | ||||||||||||||||||||
| Time (h) | C | 2 | 3 | 4 | Time (h) | C | 2 | 3 | 4 | Time (h) | C | 2 | 3 | 4 | Time (h) | C | 2 | 3 | 4 | ||||
| Yield (%) | Yield (%) | Yield (%) | Yield (%) | ||||||||||||||||||||
| 4-1 |
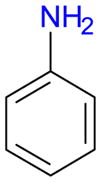
|
d | 15 | 57 | -a | 25 | T | 15 | 69 | -a | 22 | T | |||||||||||
| 4-2ab |
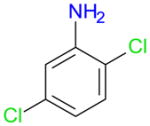
|
ed | 18 | 84 | T | 47 | 2 | 18 | 83 | nd | 59 | 3 | 18 | 49 | T | 22 | 1 | 18 | 79 | T | 14 | 2 | |
| 4-2bc | 17e | 99 | 3 | ~94 | 3 | 3 | 76 | 5 | 48 | 6 | 3 | 88 | nd | 55 | 7 | 3 | 68 | 1 | 48 | 9 | |||
| 4-3af |
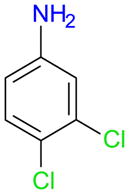
|
fg | 17 | 96 | 8 | 77 | 8 | 17 | 52 | 5 | 6 | 5 | 15 | 48 | 1 | T | 1 | ||||||
| 4-3bb | 15 | 58 | 2 | 9 | 5 | 15 | 53 | 3 | 6 | 5 | |||||||||||||
| 4–4 |
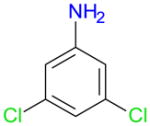
|
g | 18 | 87 | 22 | 66 | T | 18 | 65 | T | 36 | T | 19 | 56 | 1 | 24 | T | 18 | 59 | T | 19 | T | |
Percent conversion and yields were determined by GC-MS;
no traces of 2- and 3-iodoaniline were detected by GC-MS;
the reaction scheme for the iodination of aniline (1d) is shown in Scheme 1;
the silver salt and I2 were stirred for 30 minutes before addition of the respective starting material;
the silver salt and β-cyclodextrin were stirred in the respective solvent for 30 minutes, followed by addition of I2; the starting material was added after stirring for another 15 minutes;
2e = 3,6-dichloro-2-iodoaniline; 3e = 2,5-dichloro-4-iodoaniline; 4e = 3,6-dichloro-2,4-diiodoaniline;
methanol was used as reaction solvent;
the silver salt and the respective starting material were stirred for 30 minutes before addition of I2;
2f = 3,4-dichloro-2-iodoaniline; 3f = 4,5-dichloro-2-iodoaniline; 4f = 3,4-dichloro-2,6-diiodoaniline;
T = traces were detected by GC-MS; nd = not detected; ND = not determined, but considerable quantities were detected according to GC-MS.
2,5-Dichloroaniline (1e) was iodinated in para position to yield 3e in 47% (84% conversion) with Ag2SO4/I2 and 59% (83% conversion) with AgSbF6/I2 (entries 4-2a). Small quantities of diiodoaniline 4e were detected by GC-MS with both reagents. Under similar reaction conditions, AgBF4/I2 and AgPF6/I2 gave only poor yields of 3e plus small quantities of the diiodoaniline 4e, which suggests that both reagents may be too reactive for the selective mono-iodination of 1e.
Ag2SO4/I2 also appeared to be a good iodination reagent for 3,4-dichloroaniline (1f), resulting in the formation of a 77% yield of 4,5-dichloro-2-iodoaniline (3f) (entries 4-3a). The other reagents investigated gave poor conversions of approximately 50% and overall yields of the possible mono- and di-iodination ≤ 16%. In the case of 1f, the order of the addition of the starting material and I2 did not alter the percent conversion or the regioselectivity of the reaction (entries 4-3a versus 4-3b), a finding that most likely applies to this type of iodination reaction in general.
All four reagents showed some para selectivity for the iodination of 3,5-dichloroaniline (1g), which is the structural analog of 3,5-dichlorophenol (1b) and 2,5-dichloroanisole (1c). However, only Ag2SO4/I2 resulted in a good conversion (87%) and a reasonable yield (66%) of 3g (entries 4–4). According to GC-MS analysis, all four iodination reagents resulted in the formation of two diiodinated anilines. Compared to the other three reagents, iodination with Ag2SO4/I2 appeared to yield a larger amount of diiodinated products.
2,5-Dichloroaniline (1e) was selected to investigate the potential role of β-cyclodextrin on the yield and selectivity of the iodination reactions (entries 4-2b). Addition of β-cyclodextrin has been shown to improve the regioselectivity of bromination reactions in CCl4.35,36 Iodination of 1e resulted in improved yields of the para iodinated aniline 3e for all reagents, with exception of AgSbF6/I2 (entries 4-2a versus 4-2b). However, the yield of the diiodoaniline 4e also increased, thus resulting in less favorable ratios of 3e: 4e for all reagents. The only exception was the reaction with Ag2SO4/I2/β-cyclodextrin in methanol, where 3e was the major product with a yield of ~94% (99% conversion). These reaction conditions suggest that the iodination of chlorinated anilines in the presence of β-cyclodextrin may offer an excellent access to iodinated anilines, such as 3e, especially if the reaction is performed in a protic solvent. These observations are in contrast to the fact that the addition of β-cyclodextrin (see entry 1–8) did not offer an obvious advantage compared to other silver salts/I2 reagents investigated for the iodination of 1b (Table 1). This is most likely due to the different reaction conditions employed.
Overall, Ag2SO4/I2 and AgSbF6/I2 appeared to be the best reagents for the iodination of chlorinated anilines by providing a reasonable regioselectivity; however, the yields are typically moderate. One possible explanation for the relatively moderate yields of the iodination of anilines 1e–g is the use of DCM as solvent. Significantly better yields have been reported for the iodination of various chloro and nitro anilines with Ag2SO4/I2 in ethanol31 and 1,2-ethanediol.50 However, the regioselectivity of reactions using ethanol as solvent are relatively poor.31 For example, iodination of 3-nitroaniline with Ag2SO4/I2 in ethanol has a reported yield 90% of the corresponding 4- and 6-iodinated anilines in a 3: 1 ratio.31 In the present study, iodination of 1e–g typically occurred with much more pronounced regioselectivity, with product ratios frequently > 20: 1 (entries 4-2 to 4–4). This improved regioselectivity of iodination reactions with silver salts/I2 in non-polar solvents may be advantageous compared to the higher yielding reactions in protic solvents.
2.2.3. Iodination of miscellaneous aromatic compounds with AgX/I2
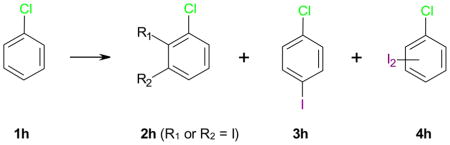
In addition to chlorinated phenols, anisoles and anilines 1, the present study also investigated the iodination of several other aromatic compounds with the four silver salt/I2 reagents (Tables 5 and 6). Chlorobenzene (1h), a deactivated aromatic compound, did not react with Ag2SO4/I2 (Table 5; entry 5-1). AgSbF6/I2 and AgPF6/I2 iodinated 1h preferentially in the para position; however the conversion was relatively low for both reagents (entries 5-2 and 5-4). The best iodination results were obtained with AgBF4/I2, which yielded the para iodinated product 3h in 87% (93% conversion) (entry 5-3). Only traces of a diiodinated chlorobenzene were detected in the case of AgSbF6/I2 and AgBF4/I2. The largest relative amount of the diiodinated product was observed with AgBF4/I2. The iodination of chlorobenzene with other silver salts/I2, such as AgOTf/I2, has been reported to yield 3h only in moderate yield.33,51 In contrast, several other conventional reagents have given good-to-excellent yields of 3h;52–56 however, the respective reaction conditions required the use of concentrated sulfuric acid (e.g., NaI/conc. H2SO4 at 60 °C52), strong oxidizers (e.g., NaI/oxone in water,53 NaI/H2O2/CeCl3·7H2O54 or NaI/Ce(OH)3O2H/SDS55) or elemental fluorine56. Therefore, AgBF4/I2 may offer a mild approach to para iodinated chlorobenzenes.
Table 5.
Iodination of chlorobenzene (1h) using different iodination reagents.*
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Reaction Conditionsa | Reaction time (h) | Conversion (%) | Yield
|
||
| 2h (%) | 3h (%) | 4h (%) | ||||
| 5-1 | Ag2SO4, I2, DCM | 18 | 0 | - | - | - |
| 5-2 | AgSbF6, I2, DCM | 17 | 73 | 8b | 59 | T |
| 5-3 | AgBF4, I2, DCM | 18 | 93 | 1b | 87 | T |
| 5-4 | AgPF6, I2, DCM | 12 | 56 | 6b | 47 | - |
Percent conversion and yields were determined by GC-MS;
one equivalent (eq.) of each reagent was employed;
unidentified monoiodinated chlorobenzene; the yield was estimated using the relative response factor of the corresponding 4-chloro-iodobenzene;
T = traces were detected by GC-MS.
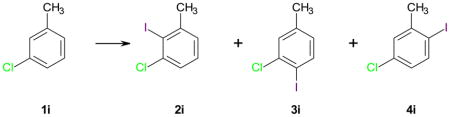
Table 6.
Iodination of 3-chlorotoluene (1i) using different iodination reagents.*
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Reaction Conditionsa | Reaction time (h) | Conversion (%) | Yield
|
||
| 2i (%) | 3i (%) | 4i (%) | ||||
| 6-1 | Ag2SO4, I2, DCM | 16 | 10 | - | 2 | 8 |
| 6-2 | AgSbF6, I2, DCM | 16 | 100 | 3 | 7 | >90 |
| 6-3 | AgBF4, I2, DCM | 16 | 100 | 3 | 8 | 70 |
| 6-4 | AgPF6, I2, DCM | 16 | 100 | 3 | 15 | 80 |
Percent conversion and yields were determined by GC-MS;
one equivalent (eq.) of each reagent was employed.
Similar to chlorobenzene (1h), iodination of 3-chlorotoluene (1i) with Ag2SO4/I2 only yielded traces of iodinated products (Table 6, entry 6-1). In contrast, the other three reagents resulted in the formation of good yields of 5-chloro-2-iodotoluene (4i), with yields > 90 being observed for AgSbF6/I2 (entries 6-2 to 6-4). In comparison, the only other reported direct iodination of 1i with KI/NaNO3 result in a mixture of 3i and 4i.57 Although the present study does not provide a clear rank order for the different silver salt/I2 reagents, the iodination experiments with 1h and 1i demonstrate that, as expected, the iodination reagents with the non-coordinating anions SbF6−, BF4− and PF6− are more reactive compared to Ag2SO4/I2, with AgBF4/I2 being the most reactive iodination reagent. One possible explanation for this observation is that there are fewer interactions between the reactive iodonium intermediate and the respective anion, which results in a more electrophilic iodinating species.
2.3. Synthesis of hydroxylated polychlorinated biphenyls
Selected hydroxylated metabolites of two PCB congeners were synthesized to demonstrate the usefulness of the iodination reactions described above. In short, the respective iodoanisoles 2c or 3c were synthesized by iodination of 1b with BTMACl2I/ZnCl2/AcOH at room temperature (25% yield) followed by methylation with dimethyl sulfate (99% yield) or directly from 1c with Ag2SO4/I2 (44% yield), respectively, and coupled with the respective benzene boronic acid 5 to yield the desired methoxylated PCB 6 (Scheme 2). Subsequent demethylation with BBr3 in DCM yielded the desired hydroxylated PCB metabolite 7. The structure of the two PCB derivatives 6a and 6b was verified by crystal structure analysis, thus providing additional evidence for the structure of the respective iodoanisoles 2c and 3c (Figure S2).
Scheme 2.

Synthesis of hydroxylated polychlorinated biphenyl 7a using the ortho iodinated 3,5-dichloroanisole 2c.
3. Conclusion
Although the iodination of phenol (1a) and aniline (1d) typically proceeds with good yield and regioselectivity, conventional iodination reagents do not necessarily allow a convenient and regioselective iodination of chlorinated phenols, anisoles and anilines 1. The present study demonstrates that iodination reactions with Ag2SO4/I2 and AgX/I2, where X is a non-cooordinating anion SbF6−, BF4− or PF6−, provides a convenient access to selected iodoarenes. Specifically, the iodination of 3,5-dichlorophenol (1b) with Ag2SO4/I2 and all three AgX/I2 in DCM gave moderate-to-good yields of the ortho product 2b. In contrast, iodination of the corresponding anisole 1c with Ag2SO4/I2 in acetonitrile yielded the para product 3c. All silver salt/I2 reagents iodinated the chlorinated anilines 1e–g preferentially in para position, with Ag2SO4/I2/β-cyclodextrin being the best reagent for this reaction. In the case of chlorobenzene (1h) and 3-chlorotoluene (1i), the three AgX/I2 reagents, but not Ag2SO4/I2, yielded iodinated products in good yields and regioselectivity. These findings suggest that silver salt-based iodination reagents may offer straightforward access to select iodinated aromatic compounds. In particular, the three AgX/I2 systems may offer access to iodinated intermediates that are difficult to synthesize with other reagents, including Ag2SO4/I2.
4. Experimental
All chemicals were purchased from commercial suppliers and used without further purification. Column chromatography was carried out on silica gel (100–200 mesh) from Sorbent Technologies (Atlanta, GA, USA). Melting points were determined on a Mel-Temp melting point apparatus and are uncorrected. NMR spectra were measured at room temperature on a Bruker Avance-300 or a Bruker Avance DRX-400 spectrometer in the University of Iowa Central NMR Research Facility (Iowa City, IA, USA) using CDCl3 as solvent. Chemical shifts are reported in parts per million relative to CDCl3 (1H, δ 7.24; 13C, δ 77.00). GC-MS analysis of all compounds was performed in the electron impact (EI) mode on an Agilent 6890N Gas Chromatograph coupled with an Agilent 5975 Mass Selective Detector (Agilent Technologies, CA, USA) using a HP-1 (Methyl Silicone Gum) column (Hewlett Packard, PA, USA). The following conditions were used for the GC-MS analysis: injector: 250 °C, starting temperature: 50 °C, final temperature: 250 °C, heating rate: 20 °C/min, hold 5 min. For all compounds investigated, the retention time followed the order ortho < para iodinated product. Only the isotopic ion with the lowest mass is reported for all fragments observed in the MS spectra. HRMS were recorded by the High Resolution Mass Spectrometry Facility of the University of California Riverside (Riverside, CA, USA).
4.1. General procedure for the iodination of chlorinated benzene derivatives 1a–i
The respective silver salt (0.32 g, 1 mmol) and iodine (0.25 g, 1 mmol) were typically added to a stirred solution of the benzene derivative 1a–i (1 mmol) in dichloromethane (3 mL). The reaction mixture was allowed to stir at room temperature for approximately 16 h (see Tables 1–6). The reaction mixture was cooled with ice-cold water, quenched with an aqueous solution of sodium metabisulfite (0.2 mL) and, in the case of anilines, 2 M NaOH (0.2 mL). The mixture was filtered through Celite® and the residue was washed with dichloromethane (3 × 3 mL). The combined filtrate was washed with aq. sodium bicarbonate (3 mL), water (3 mL) and brine (3 mL). The combined organic phases were dried over Na2SO4 and the solvent was removed under reduced pressure. The residue was redissolved in dichloromethane (10 mL) and the percent conversion of the starting material and the yields of the iodination products were determined by GC-MS using diethylene glycol di-n-butyl ether as internal standard. The relative response factor for the respective analyte (RRFA) was calculated from a calibration standard containing known amounts of the internal standard and the respective analytes using the formula RRFA = AIS · MA/(AA · MIS), where AIS is the peak area of the internal standard, AA is the area of an analyte (i.e., starting material or iodination product), MA is the mass of the analyte and MIS is the mass of the internal standard. The mass of the analyte in the reaction mixture was determined as MA = (RRFA · MIS · AA)/AIS. All samples were analyzed at least in duplicate. The iodination products of selected reactions were separated by column chromatography to obtain milligram quantities for their characterization and use as analytical standards. In the case of 3g, the isolated quantities were not sufficient for 13C NMR analysis.
4.1.1. 3,5-Dichloro-2-iodophenol 2b19
White solid; Mp: 81–83 °C; 1H NMR (400 MHz, CDCl3): δ/ppm 7.07 (m, 1 H), 6.90 (m, 1 H), 5.69 (s, 1 H); 13C NMR (100 MHz, CDCl3): δ/ppm 156.9, 139.0, 135.9, 121.6, 113.4, 89.0; mass spectrum m/z (relative abundance %): 288 (M·+, 60), 252 (10), 133 (10), 97 (10), 62 (10); HRMS m/z : calculated for C6H2OCl2I [M-H] 286.8533; Found 286.8533.
4.1.2. 3,5-Dichloro-4-iodophenol 3b
White solid; Mp: 134–135 °C (hexane); 1H NMR (300 MHz, CDCl3): δ/ppm 6.92 (s, 2 H), 5.17 (s, 1 H); 13C NMR (75 MHz, CDCl3): δ/ppm 156.1, 140.8, 115.2, 92.6; mass spectrum m/z (relative abundance%): 288 (M·+, 80), 133 (10), 97 (10); HRMS m/z: calculated for C6H2OCl2I [M-H] 286.8533; Found 286.8532.
4.1.3. 3,5-Dichloro-2-iodoanisole 2c
White solid; 1H NMR (300 MHz, CDCl3): δ/ppm 7.12 (d, J = 2.1 Hz, 1 H), 6.67 (d, J = 2.1 Hz, 1 H), 3.88 (s, 3 H); 13C NMR (75 MHz, CDCl3): δ/ppm 160.2, 140.3, 135.5, 121.6, 109.4, 89.1, 57.0; mass spectrum m/z (relative abundance %): 302 (M·+, 60), 287 (10), 259 (10), 160 (20), 97 (10); HRMS m/z: calculated for C7H5OCl2I [M] 301.8757; Found 301.8760.
4.1.4. 3,5-Dichloro-4-iodoanisole 3c3,58
White solid; Mp: 49–50 °C (Lit.: 62 °C58); 1H NMR (300 MHz, CDCl3): δ/ppm 6.94 (s, 2 H), 3.78 (s, 3 H); 13C NMR (75 MHz, CDCl3): δ/ppm 160.2, 140.7, 113.8, 92.1, 55.8; mass spectrum m/z (relative abundance %): 302 (M·+, 60), 287 (10), 259 (10), 160 (10), 97 (10); HRMS m/z: calculated for C7H5OCl2I [M] 301.8757; Found 301.8763.
4.1.5. 3,5-Dichloro-2,4-diiodoanisole 4c
White solid; Mp: 143–144 °C; 1H NMR (400 MHz, CDCl3): δ/ppm 6.85 (s, 1 H), 3.89 (s, 3 H); 13C NMR (100 MHz, CDCl3): δ/ppm 160.0, 144.5, 140.8, 109.3, 91.6, 88.5, 57.2; mass spectrum m/z (relative abundance %): 428 (M·+, 70), 413 (15), 286 (15); HRMS m/z: calculated for C7H4OCl2I2 [M] 427.7723; Found 427.7718.
4.1.6. 3,6-Dichloro-2-iodoaniline 2e23
Brown solid; Mp: 98 °C (Lit.: 68 °C23); 1H NMR (400 MHz, CDCl3): δ/ppm 7.16 (d, J = 8.4 Hz, 1 H), 6.80 (d, J = 8.4 Hz, 1 H), 4.77 (br s, 2 H); 13C NMR (100 MHz, CDCl3): δ/ppm 145.2, 137.7, 129.4, 118.3, 115.3, 87.9; mass spectrum m/z (relative abundance %): 287 (M·+, 60), 160 (20), 1245 (20); HRMS m/z: calculated for C6H4NCl2I [M] 286.8766; Found 286.8770.
4.1.7. 2,5-Dichloro-4-iodoaniline 3e23
Brown solid; Mp: 53 °C (Lit.: 57 °C23); 1H NMR (400 MHz, CDCl3): δ/ppm 7.59 (s, 1 H), 6.82 (s, 1 H), 4.11 (br s, 2 H); 13C NMR (100 MHz, CDCl3): δ/ppm 143.7, 138.9, 137.1, 118.1, 115.3, 81.6; mass spectrum m/z (relative abundance %): 287 (M·+, 50), 160 (20), 135 (10), 124 (10), 97 (10); HRMS m/z: calculated for C6H5NCl2I [M+H] 287.8838; Found 287.8826.
4.1.8. 3,6-Dichloro-2,4-diiodoaniline 4e25
Brown solid; Mp: 110 °C (Lit.: 111–112 °C25); 1H NMR (400 MHz, CDCl3): δ/ppm 7.71 (s, 1 H), 4.82 (br s, 2 H); 13C NMR (100 MHz, CDCl3): δ/ppm 145.4, 140.7, 138.6, 115.8, 86.0, 79.0; mass spectrum m/z (relative abundance %): 413 (M·+, 70), 286 (20), 159 (10); HRMS m/z: calculated for C6H4NCl2I2 [M+H] 413.7805; Found 413.7787.
4.1.9. 3,4-Dichloro-2-iodoaniline 2f26
Brown solid; Mp: 40 °C; 1H NMR (400 MHz, CDCl3): δ/ppm 7.20 (d, J = 8.8 Hz, 1 H), 6.58 (d, J = 8.8 Hz, 1 H), 4.31 (br s, 2 H); 13C NMR (100 MHz, CDCl3): δ/ppm 147.7, 136.7, 130.1, 120.4, 112.9, 88.8; mass spectrum m/z (relative abundance %): 287 (M·+, 70), 160 (15), 124 (15); HRMS m/z: calculated for C6H5NCl2I [M+H] 287.8838; Found 287.8836.
4.1.10. 4,5-Dichloro-2-iodoaniline 3f20,21
Brown solid; Mp: 67 °C; 1H NMR (400 MHz, CDCl3): δ/ppm 7.64 (s, 1 H), 6.78 (s, 1 H), 4.12 (br s, 2 H); 13C NMR (100 MHz, CDCl3): δ/ppm 146.4, 139.0, 133.1, 121.5, 115.0, 81.0; mass spectrum m/z (relative abundance %): 287 (M·+, 60), 160 (20), 133 (20); HRMS m/z: calculated for C6H5NCl2I [M+H] 287.8838; Found 287.8830.
4.1.11. 3,4-Dichloro-2,6-diiodoaniline 4f25
Brown solid; Mp: 116 °C (Lit.: 120–121 °C25); 1H NMR (400 MHz, CDCl3): δ/ppm 7.73 (s, 1 H), 4.85 (s, 2 H); 13C NMR (100 MHz, CDCl3): δ/ppm 147.1, 138.7, 137.3, 120.3, 86.2, 77.7; mass spectrum m/z (relative abundance %): 413 (M·+, 70), 286 (15), 159 (15); HRMS m/z: calculated for C6H4NCl2I2 ([M+H] 413.7805; Found 413.7785.
4.1.12. 3,5-Dichloro-2-iodoaniline 2g24
Brown solid; Mp: 46 °C; 1H NMR (400 MHz, CDCl3): δ/ppm 6.84 (d, J = 2.4 Hz, 1 H), 6.57 (d, J = 2.4 Hz, 1 H), 4.39 (br s, 2 H); 13C NMR (100 MHz, CDCl3): δ/ppm 149.5, 139.8, 135.2, 118.5, 117.7, 85.8; mass spectrum m/z (relative abundance %): 287 (M·+, 70), 160 (15), 124 (15); HRMS m/z: calculated for C6H5NCl2I [M+H] 287.8838; Found 287.8833.
4.1.13. 3,5-Dichloro-4-iodoaniline 3g22
Brown solid; Mp: 143 °C; 1H NMR (400 MHz, CDCl3): δ/ppm 6.68 (s, 2 H), 3.76 (br s, 2 H); mass spectrum m/z (relative abundance %): 287 (M·+, 60), 160 (20), 133 (20); HRMS m/z: calculated for C6H5NCl2I [M+H] 287.8838; Found 287.8824.
4.1.14. 3,5-Dichloro-2,6-diiodoaniline and 3,5-dichloro-2,4-diiodoaniline 4g
Brown solid; Mp: 110 °C; 1H NMR (400 MHz, CDCl3): δ/ppm 6.78 (s, 1 H), 4.44 (br s, 2 H); 13C NMR (100 MHz, CDCl3): δ/ppm 149.1, 143.8, 140.3, 118.5, 111.74, 111.66, 86.2, 84.8; mass spectrum m/z (relative abundance %): 413 (M·+, 70), 286 (15), 159 (15); HRMS m/z: calculated for C6H4NCl2I2 [M+H] 413.7805; Found 413.7774.
4.2. Synthesis of PCB derivatives
4.2.1. Synthesis of 4,4′,6-trichloro-2-methoxybiphenyl 6a
A mixture of 2c (0.45 g, 1.5 mmol), 4-chlorophenylboronic acid (5a) (0.47 g, 3.0 mmol), bis(dibenzylideneacetone) palladium (20 mg, 22.5 μmol), 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (DPDB) (40 mg, 0.1 mmol) and powdered K3PO4 (0.95 mg) in toluene (3.5 mL) were heated at 100 °C in a sealed tube under a nitrogen atmosphere as described previously.4 The tube was allowed to cool to room temperature and the reaction mixture was passed through a Celite® bed. The residue was washed with dichloromethane (2 × 25 mL) and the combined filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel with n-hexane as eluent and the pure compound was crystallized from methanol-dichloromethane to yield 4,4′,6-trichloro-2-methoxybiphenyl (6a) as a colorless solid in 18% yield. Mp: 58–59 °C (chloroform-methanol); 1H NMR (300 MHz, CDCl3): δ/ppm 7.41 (AAXX′ system, 2 H), 7.20 (AA′XX′ system, 2 H), 7.13 (d, J = 1.8 Hz, 1 H), 6.87 (d, J = 1.8 Hz, 1 H), 3.73 (s, 3 H, −OCH3); 13C NMR (100 MHz, CDCl3): δ/ppm 158.1, 134.7, 134.3, 133.7, 132.8, 131.7, 128.3, 127.3, 121.6, 110.2, 56.2; Anal. Calcd for C13H9Cl3O: C, 54.30; H, 3.15; Found: C, 54.39; H, 3.13; mass spectrum m/z (relative abundance %): 286 (M·+, 100), 249 (6), 236 (82), 216 (20), 173 (40).
4.2.2. 2,2′,5′,6-Tetrachloro-4-methoxybiphenyl 6b
Synthesized as described above by the Suzuki coupling of 3c (0.50 g, 1.66 mmol) and 2,5-dichlorophenylboronic acid (5b) (0.48 g, 2.5 mmol) to afford 6b as a colorless solid in 77% yield. Mp: 87 °C (chloroform-methanol); 1H NMR (400 MHz, CDCl3): δ/ppm 7.41 (d, J = 8.8 Hz, 1 H), 7.32 (dd, J = 2.4 & 8.8 Hz, 1 H), 7.21 (d, J = 2.4 Hz, 1 H), 6.97 (s, 2 H), 3.84 (s, 3 H, −OCH3); 13C NMR (100 MHz, CDCl3): δ/ppm 159.9, 137.3, 135.2, 132.7, 132.4, 131.5, 130.5, 129.7, 128.2, 113.9, 55.8; Anal. Calcd for C13H8Cl4O: C, 48.49; H, 2.48; Found: C, 48.73; H, 2.37; HRMS m/z: calculated for C13H8OCl4 (M·+) 319.9324, found 319.9325.
4.2.3. 4,4′,6-Trichlorobiphenyl-2-ol 7a
BBr3 (1.2 mL, 1.2 mmol, 1M solution in heptane) was added to a stirred solution of 6a (70 mg, 0.24 mmol) in anhydrous CH2Cl2 (5 mL) under a nitrogen atmosphere.3 The reaction was stirred at room temperature for 5 days, quenched by pouring onto crushed ice and extracted with dichloromethane (5 mL). The organic layer was washed with 2 M NaOH solution (5 mL), the aqueous layer was acidified with 2 N HCl (5 mL) and extracted with dichloromethane (3 × 5 mL). The combined organic layer was washed with water (25 mL), brine (25 mL), dried over (Na2SO4) and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel using a hexane-chloroform gradient (100% to 90% hexane) to yield 4,4′,6-trichlorobiphenyl-2-ol (7a) as a colorless oil in 29% yield. 1H NMR (400 MHz, CDCl3): δ/ppm 7.50 (AA′XX′ system, 2 H), 7.26 (AA′XX′ system, 2 H), 7.08 (d, J = 2.0 Hz, 1 H), 6.93 (d, J = 2.0 Hz, 1 H), 4.95 (s, 1 H, −OH); 13C NMR (100 MHz, CDCl3): δ/ppm 154.2, 135.4, 134.7, 134.3, 131.9, 130.8, 129.8, 124.9, 121.9, 114.8; mass spectrum m/z (relative abundance %): 272 (M·+, 47), 236 (18), 237 (38), 202 (100), 173 (42), 139 (46), 118 (27), 86 (82); HRMS m/z: calculated for C12H6OCl3 [M-H] 270.9484, found 270.9481.
4.2.4. 2,2′,5′,6-Tetrachlorobiphenyl-4-ol 7b
Prepared from 2,2′,5′,6-tetrachloro-4-methoxybiphenyl (6b) (0.31 g, 1 mmol) as described above to afford 7b as a colorless solid in 87% yield. Mp: 101 °C (chloroform-methanol); 1H NMR (400 MHz, CDCl3): δ/ppm 7.42 (d, J = 8.4 Hz, 1 H), 7.33 (dd, J = 2.4 & 8.4 Hz, 1 H), 7.21 (d, J = 2.4 Hz, 1 H), 6.94 (s, 2 H), 5.57 (s, 1 H, -OH); 13C NMR (100 MHz, CDCl3): δ/ppm 156.0, 137.1, 135.3, 132.7, 132.4, 131.4, 130.5, 129.7, 128.5, 115.4; mass spectrum m/z (relative abundance %): 306 (M·+, 75), 270 (5), 235 (5); HRMS m/z: calculated for C12H6OCl4 [M] 305.9167, found 305.9177.
4.3. X-ray crystal structure analysis
X-ray diffraction data were collected at 90.0(2) K on either a Nonius KappaCCD or a Bruker-Nonius X8 Proteum diffractometer with graded-multilayer focusing optics as described previously.59 Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 827884 to 827887. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Supplementary Material
Scheme 3.

Synthesis of hydroxylated polychlorinated biphenyl 7a using the para iodinated 3,5-dichloroanisole 3c.
Acknowledgments
This research was supported by grants ES05605, ES013661 and ES017425 from the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH). Contents of this manuscript are solely the reponsibility of the authors and do not necessarily represent the official views of the NIEHS/NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Naidu AB, Ganapathy D, Sekar G. Synthesis. 2010:3509. [Google Scholar]
- 2.Pacuszka T, Panasiewicz M. J Labelled Compd Radiopharm. 2000;43:1255. [Google Scholar]
- 3.Waller SC, He YA, Harlow GR, He YQ, Mash EA, Halpert JR. Chem Res Toxicol. 1999;12:690. doi: 10.1021/tx990030j. [DOI] [PubMed] [Google Scholar]
- 4.Joshi SN, Vyas SM, Duffel MW, Parkin S, Lehmler HJ. Synthesis. 2011:1045. doi: 10.1055/s-0030-1258454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comins DL, Baevsky MF, Hong H. J Am Chem Soc. 1992;114:10971. [Google Scholar]
- 6.Springer DM, Luh BY, Goodrich J, Bronson JJ. Bioorg Med Chem. 2003;11:265. doi: 10.1016/s0968-0896(02)00336-x. [DOI] [PubMed] [Google Scholar]
- 7.Rawal VH, Michoud C, Monestel R. J Am Chem Soc. 1993;115:3030. [Google Scholar]
- 8.Hong CY, Overman LE. Tetrahedron Lett. 1994;35:3453. [Google Scholar]
- 9.Nicolaou KC, Xu J, Murphy F, Barluenga S, Baudoin O, Wei HX, Gray DLF, Ohshima T. Angew Chem, Int Ed. 1999;38:2447. [PubMed] [Google Scholar]
- 10.Endo A, Yanagisawa A, Abe M, Tohma S, Kan T, Fukuyama T. J Am Chem Soc. 2002;124:6552. doi: 10.1021/ja026216d. [DOI] [PubMed] [Google Scholar]
- 11.Buchgraber P, Snaddon TN, Wirtz C, Mynott R, Goddard R, Fuerstner A. Angew Chem, Int Ed. 2008;47:8450. doi: 10.1002/anie.200803339. [DOI] [PubMed] [Google Scholar]
- 12.Chang JH, Kang HU, Jung IH, Cho CG. Org Lett. 2010;12:2016. doi: 10.1021/ol100617u. [DOI] [PubMed] [Google Scholar]
- 13.Tsuji J. Innovations in organic synthesis. John Wiley & Sons, Ltd; Chichester: 2000. Transition metal reagents and catalysts. [Google Scholar]
- 14.Diederich F, Stang PJ. Metal-catalyzed cross-coupling reactions. Wiley-VCH Verlag GmbH; Weinheim: 1998. [Google Scholar]
- 15.Stavber S, Jereb M, Zupan M. Synthesis. 2008. p. 1487. [Google Scholar]
- 16.Hanson JR. J Chem Res. 2006:277. [Google Scholar]
- 17.Bovonsombat P, Leykajarakul J, Khan C, Pla-on K, Krause MM, Khanthapura P, Ali R, Doowa N. Tetrahedron Lett. 2009;50:2664. [Google Scholar]
- 18.Shashidhar GVS, Satyanarayana N, Sundaram EV. Indian J Chem, Sect A. 1987;26A:333. [Google Scholar]
- 19.Kung P-P, Meng JJ. 2010018481. International patent WO. 2010
- 20.Yu MS, Lopez De Leon L, McGguire MA, Botha G. Tetrahedron Lett. 1998;39:9347. [Google Scholar]
- 21.Li X, Yin W, Sarma PVVS, Zhou H, Ma J, Cook JM. Tetrahedron Lett. 2004;45:8569. [Google Scholar]
- 22.Cooper CB, McFarland JW, Blair KT, Fontaine EH, Jones CS, Muzzi ML. Bioorg Med Chem Lett. 1994;4:835. [Google Scholar]
- 23.Rodighiero G. Ann Chim. 1951;41:43. [Google Scholar]
- 24.Di Fabio R, Giacobbe S, Bertani B, Micheli F. 9712870. World patent WO. 1997
- 25.Waring WS. 895395. Great Britain patent GB. 1962
- 26.Lee D, Marino JP, Zhao Y. 2005009993. World patent WO. 2005
- 27.Sugiyama T. Bull Chem Soc Jpn. 1981;54:2847. [Google Scholar]
- 28.Das B, Krishnaiah M, Venkateswarlu K, Reddy VS. Tetrahedron Lett. 2007;48:81. [Google Scholar]
- 29.Sy WW. Tetrahedron Lett. 1993;34:6223. [Google Scholar]
- 30.Sy WW, Lodge BA, By AW. Synth Commun. 1990;20:877. [Google Scholar]
- 31.Sy WW. Synth Commun. 1992;22:3215. [Google Scholar]
- 32.Glennon RA, Young R, Benington F, Morin RD. J Med Chem. 1982;25:1163. doi: 10.1021/jm00352a013. [DOI] [PubMed] [Google Scholar]
- 33.Haszeldine RN, Sharpe AG. J Chem Soc. 1952:993. [Google Scholar]
- 34.Galli C. J Org Chem. 1991;56:3238. [Google Scholar]
- 35.Suresh P, Annalakshmi S, Pitchumani K. Tetrahedron. 2007;63:4959. [Google Scholar]
- 36.Velusamy P, Pitchumani K, Srinivasan C. Tetrahedron. 1996;52:3487. [Google Scholar]
- 37.Veglia AV, de Rossi RH. J Org Chem. 1988;53:5281. [Google Scholar]
- 38.Honeychuck RV, Hersh WH. Inorg Chem. 1989;28:2869. [Google Scholar]
- 39.Wilson SR, Jacob LA. J Org Chem. 1986;51:4833. [Google Scholar]
- 40.Jacob LA, Chen BL, Stec D. Synthesis. 1993:611. [Google Scholar]
- 41.Brunel Y, Rousseau G. Tetrahedron Lett. 1995;36:8217. [Google Scholar]
- 42.Barluenga J, Campos PJ, Gonzalez JM, Asensio G. J Chem Soc, Perkin Trans 1. 1984:2623. [Google Scholar]
- 43.Reddy KSK, Narender N, Rohitha CN, Kulkarni SJ. Synth Commun. 2008;38:3894. [Google Scholar]
- 44.Sathiyapriya R, Karunakaran RJ. Synth Commun. 2006;36:1915. [Google Scholar]
- 45.Bhilare SV, Deorukhkar AR, Darvatkar NB, Salunkhe MM. Synth Commun. 2008;38:2881. [Google Scholar]
- 46.Branytska OV, Neumann R. J Org Chem. 2003;68:9510. doi: 10.1021/jo035271h. [DOI] [PubMed] [Google Scholar]
- 47.Mukaiyama T, Kitagawa H, Matsuo J-i. Tetrahedron Lett. 2000;41:9383. [Google Scholar]
- 48.Al-Lohedan HA. Orient J Chem. 1990;6:251. [Google Scholar]
- 49.Adimurthy S, Ramachandraiah G, Ghosh PK, Bedekar AV. Tetrahedron Lett. 2003;44:5099. [Google Scholar]
- 50.Zhang Y, Ren T, Zhu W, Xie Y. Org Prep Proced Int. 2007;39:90. [Google Scholar]
- 51.Mulholland GK, Zheng QH. Synth Commun. 2001;31:3059. [Google Scholar]
- 52.Pasha MA, Myint YY. Synth Commun. 2004;34:2829. [Google Scholar]
- 53.Firouzabadi H, Iranpoor N, Kazemi S. Can J Chem. 2009;87:1675. [Google Scholar]
- 54.Firouzabadi H, Iranpoor N, Kazemi S, Ghaderi A, Garzan A. Adv Synth Catal. 2009;351:1925. [Google Scholar]
- 55.Firouzabadi H, Iranpoor N, Garzan A. Adv Synth Catal. 2005;347:1925. [Google Scholar]
- 56.Rozen S, Zamir D. J Org Chem. 1990;55:3552. [Google Scholar]
- 57.Makhon’kov DI, Cheprakov AV, Beletskaya IP. Zh Org Khim. 1988;24:2251. [Google Scholar]
- 58.Goldschmidt S, Suchanek L. Chem Ber. 1957;90:19. [Google Scholar]
- 59.Lehmler HJ, Parkin S, Robertson LW. Chemosphere. 2002;46:485. doi: 10.1016/s0045-6535(01)00177-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.