

Abstract
Osteogenesis imperfecta, discussed in Baldridge et al. 2008 is an inherited bone fragility disorder with a wide range of clinical severity that in the majority of cases is caused by mutations in COL1A1 or COL1A2, the genes that encode the two collagen type I alpha chains. Here we describe genotype-phenotype correlations in OI patients who have mutations affecting collagen type I. This paper is based on findings in a large single-centre OI population and a review of the literature.
1. Introduction
Osteogenesis imperfecta [1] is a heritable skeletal disorder characterized by bone fragility and often short stature. Extraskeletal features include blue sclerae, dentinogenesis imperfecta (DI), and a variable degree of postpubertal hearing loss.
The spectrum of clinical severity is wide, ranging from nearly asymptomatic individuals with a mild predisposition to fractures, normal stature and normal lifespan, to severe bone deformities, mobility impairments, and very short stature to perinatal lethality [2].
Classically four types of OI are distinguished, as described by Sillence et al. in 1979 [3]. This classification is based on clinical features. OI type I is the mildest form. Patients usually have normal or slightly short stature with straight long bones and blue sclerae. DI is infrequent in this type. The main clinical issue in OI type I concerns the spine, where vertebral compression fractures can lead to mild scoliosis. OI type II is usually lethal during the perinatal period, with in utero onset of severe long-bone deformities and rib fractures that lead to respiratory compromise and eventually death. OI type III is the most severe form of OI that is compatible with life. Patients have severe short stature and severe long bone deformities, with reduced mobility. They typically have DI and blue sclera. Scoliosis can lead to fatal respiratory compromise. OI type IV is a moderately severe form of OI with multiple fractures, mild to moderate bone deformities, and variable short stature. Sclerae in this type are white or grey and DI is not a constant feature.
Even though this classification is widely used, it is somewhat subjective in nature and depends on the amount of information that is available at any given time. For example, it can occur that a fetus is diagnosed with OI type II, the lethal form, on prenatal ultrasound, but after birth turns out to be less severely affected than initially thought and is eventually reclassified as OI type III or even type VI, if adequate management is initiated early on. In fact, the aim of current multidisciplinary treatment approaches including physiotherapy, occupational therapy, bisphosphonate treatment, and corrective surgical interventions is to shift the severity of the skeletal phenotype from severe (corresponding to OI type III) to moderate (type IV) and from moderate to mild (type I). Thus, an initial OI type classification for a specific patient can change if treatments or other factors decrease the severity of the phenotype.
The majority of OI patients have a mutation in either the COL1A1 or the COL1A2 gene, the two genes coding for alpha 1 and alpha 2 chains of collagen type I. Collagen type I is a heterotrimer consisting of two alpha 1 chains and one alpha 2 chain. It is initially synthesized as a pro-alpha chain with a propeptide at each end (N-propeptide and C-propeptide). The propeptides are necessary for pro-alpha chain association and triple helix formation that starts at the carboxy-terminal propeptide and extends to the aminoterminal propeptide in a zipper-like manner. The triple helical domains are composed of uninterrupted repeats of the Gly-X-Y tripeptide. Triple helix formation requires a glycine residue in every third position of the chains because glycine is the only residue small enough to fit into the restricted space at the inside of the helix.
Two general classes of mutations in collagen type I are associated with OI. The first class of mutation leads to haploinsufficiency, typically resulting in mild OI type I [4]. Haploinsufficiency is usually the consequence of a nonsense mutation or a frame shift mutation that introduces a premature termination codon in the coding sequence of one COL1A1 allele. This initiates nonsense-mediated decay of the mRNA [5]. The result of these mutations is a quantitative defect in collagen type I, as only about half of the normal amount of the protein is produced. However, the collagen type I protein that is produced has a normal structure. Some splice-site mutations and C-propeptide mutations in COL1A1 can also lead to haploinsufficiency.
The second class of mutation consists of sequence abnormalities that lead to structural mutations in collagen type I. This includes helical glycine mutations, which are the most frequent type of mutations leading to OI. Such mutations usually replace glycine by another amino acid. Any mutation that disrupts the Gly-X-Y sequence of the collagen triple helix will result in impaired collagen function. Such mutations impair triple helix formation, its stability, and its interactions with the extracellular matrix. Very few mutant collagen monomers are needed to disrupt the structure of the various multitrimer assemblies. Glycine mutations in collagen type I thus exert what is known as a dominant negative effect.
Many studies on individual OI patients or small patient groups have examined the molecular and cellular consequences of collagen type I mutations, especially in skin fibroblasts, but less information is available on osteoblasts. So far, very little is known about how a specific collagen type I mutation leads to a particular phenotype. As a general rule, the phenotypic severity depends on the affected alpha chain, the position of the mutation, the substituting amino acid, or the combination of these three variables.
As of June 2011, more than 2000 different collagen type I mutations have been identified in OI patients and are listed in a database (http://www.le.ac.uk/ge/collagen/) [6, 7]. With the constant increase in the number of identified mutations in COL1A1 and COL1A2, genotype-phenotype correlation becomes more and more pertinent.
In this paper, we provide an overview of genotype-phenotype correlations in OI patients who have mutations affecting collagen type I.
2. Lethality
A 2007 compilation of 832 triple helical collagen type I mutations distinguished two main classes of mutations [6–8]: point mutations that cause the substitution of a triple helical glycine residue by another amino acid (n = 682) and mutations that alter splice sites (n = 150). The majority of these mutations caused OI types II, III, or IV.
The only phenotypic characteristic that could be evaluated in this report was whether the individuals carrying each mutation had survived the immediate postnatal period or not. Glycine substitutions in the alpha 1 chain of collagen type I were more frequent than mutations in the alpha 2 chain (57% versus 43%) and more likely to have a lethal outcome (36% of alpha 1 and 19% of alpha 2 mutations were lethal). Substitutions in the first 200 residues of the alpha 1 chain were found to be usually nonlethal.
Two exclusively lethal regions in the alpha 1 chain (helix positions 691–823 and 910–964), and eight clusters of lethality in the alpha 2 chain were identified. These lethal areas align with major ligand binding regions for extracellular matrix proteins, which might explain the lethal outcome of mutations occurring at these sites. Even though the identification of these “lethal regions” provides a useful rule of thumb, there are certainly many exceptions to the rule. For example, a recent study identified seven individuals who had mutations in the putative lethal regions of alpha 1 (n = 1) and alpha 2 (n = 6) and who did not have a lethal outcome [9]. This observation is important for genetic counseling, especially in the context of prenatal diagnosis.
Glycine substitutions are more likely to have a lethal outcome when the substituting amino acid in the alpha 1 chain is charged (like aspartate or glutamate acid or arginine) or branched (valine) and affects a glycine at an amino acid position that is C-terminal of position 200 [8]. No such relationship was found for the alpha 2 chain. Glycine substitutions in the alpha 2 chain are more likely to be concordant for the outcome than glycine substitutions in the alpha 1 chain.
3. Deformities and Fractures at Birth
In a study on 117 OI patients, no significant relationship was found between the presence of limb deformities and fractures at birth and either the type of affected collagen type I alpha chain or the position of the glycine mutation within the alpha chain [9]. A smaller percentage of patients with arginine substitutions in alpha 1 had fractures or deformities at birth than patients with serine substitutions (44% versus 83%; P < 0.05). There was no relationship between the substituting amino acid and the prevalence of fractures or deformities at birth in the alpha 2 chain.
4. Anthropometry
In a study of 86 OI patients, mean standing height was reduced in all groups of patients but height reduction was significantly more marked in patients with qualitative collagen defects than in those with quantitative defects [10]. The same pattern was observed for truncal height and arm span z-score. Arm span to height ratio was increased in children with qualitative collagen defects (P < 0.05), but not in those with a quantitative collagen defect. Head circumference was increased in all OI types, but significantly more so in patients with qualitative collagen defects. Head circumference was disproportionate to height. The disproportion was again more pronounced in patients with qualitative collagen defects.
A recent study in 161 OI patients (median age: 13 years) who had glycine mutations in the triple helical domain of collagen type I found that mutations in COL1A1 and in COL1A2 led to similar average height z-scores [9]. An inverse relationship was found between the height and the location of the mutation in the triple helical domain of the alpha 2 chain (Figure 1(b)). Thus, a mutation closer to the carboxy-terminal end of the triple helical domain of alpha 2 had a more detrimental effect on height. No such correlation was observed in the alpha 1 chain (Figure 1(a)). Results in the alpha 2 chain are consistent with the gradient model of disease severity, which posits that mutations at the carboxy-terminal end of the alpha 2 chain are more disruptive to triple helix formation and therefore cause a more severe phenotype.
Figure 1.
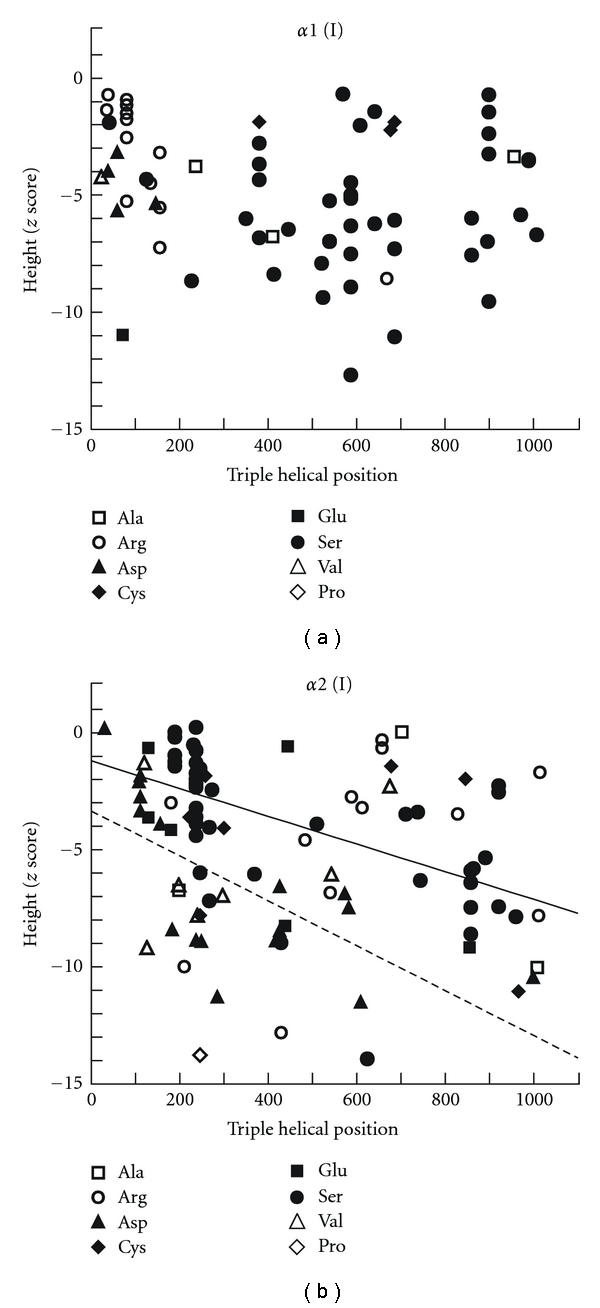
Relationship between the positions of glycine substitutions in the triple helical domain of type I collagen and height z-scores in α1 chain of collagen I (a) and α2 chain of collagen I (b). The oblique solid line in B represents the regression line between the position of serine substitutions in α2 chain of collagen I and height (r = −0.73, P < 0.001; Spearman's rank correlation). The oblique dashed line in (b) represents the regression line between the position of aspartate substitutions in α2 chain of collagen I and height (r = −0.75, P < 0.001). A mutation closer to the carboxy-terminal end of the triple helical domain of alpha 2 chain had a more detrimental effect on height (b). No such correlation was observed in the alpha 1 chain (a), adapted with some modification from [11].
Height is not only affected by the location of the glycine mutation but also by the specific substituting amino acid [9]. Serine substitutions are the most common type of mutations in both the alpha 1 and the alpha 2 chain and seem to lead to a shorter average stature when they affect the alpha 1 chain [9]. Arginine substitutions are the second most frequent mutations in the alpha 1 chain. Compared to patients with serine substitutions, patients with arginine mutations on average had less severe short stature [9]. Aspartate substitutions are the second most frequent mutations in the alpha 2 chain and on average lead to a severe short stature. Other substituting amino acids in either chain were not frequent enough for statistical analysis.
In a study on the relationship between genotype and skeletal phenotype in 192 OI patients (age range 3 weeks to 16.9 years), it was noted that patients with haploinsufficiency mutations on average were taller (height z-score −1.3 (SD: 1.1) than patients with helical mutations in the alpha 1 (mean height z-score −5.5 (SD: 3.1)) or alpha 2 chains (mean height z-score −5.3 (SD: 3.3)) [11].
5. Dental Phenotype
DI is histologically characterized by an abnormal dentine structure [12]. Clinically it is defined by discoloration of the teeth that ranges from grey-brown to opalescent blue. In a study of 88 OI patients, the prevalence of DI was 28% [13]. The majority of patients with DI had a qualitative collagen defect (96%), whereas only a minority of patients (4%) had a quantitative collagen defect. Lund et al. did not observe an association between DI and any specific structural abnormality or any particular position of the abnormality along the collagen I alpha chains [13].
In an analysis based on genomic DNA sequencing data, Rauch et al. found that the majority of OI patients with a glycine mutation have clinically recognizable DI [9]. The prevalence of DI was similar for mutations affecting the alpha 1 or the alpha 2 chain (71% versus 66%, resp., P = 0.53). Among mutations affecting the alpha 1 chain, arginine substitutions were less often associated with DI (25%) than serine substitutions (89%). In the same study it was observed that glycine mutations affecting the first 120 amino acids of the collagen type I triple helical domain did not lead to DI (Figure 2). Although at this point there is no mechanistic link between collagen type I mutations and DI, a high degree of concordance for DI was observed for OI patients who shared the same glycine mutation in the helical domain. It thus appears that the dental phenotype is directly linked to the collagen abnormality and is not significantly influenced by other genetic or environmental factors.
Figure 2.
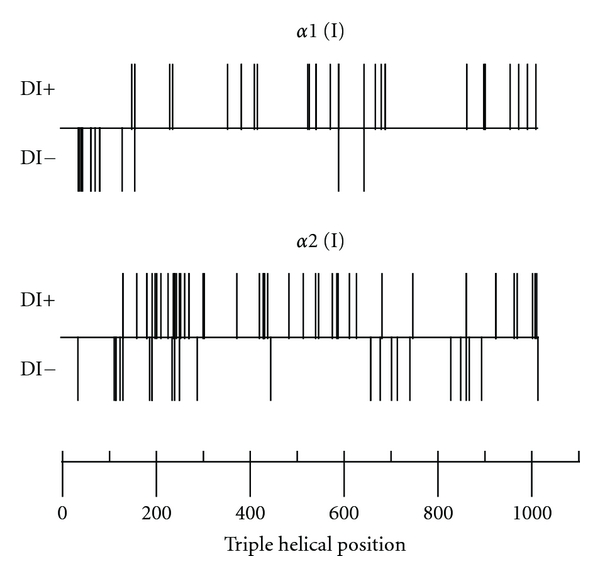
Relationship between the triple helical position of glycine mutations in collagen type I α chains and the presence (+) or absence (−) of dentinogenesis imperfecta (DI), adapted from [9].
6. Scleral Hue
Patients with mutations in the helical domain of the alpha 1 chain tend to have blue sclera more often (75%) than those with mutations in the alpha 2 chain (57%) [9]. It was also noticed that mutation affecting the first 120 amino acids of the triple helical domain consistently led to blue sclerae (Figure 3). Among patients who shared the same collagen type I mutation the concordance for the scleral phenotype was less than for the dental phenotype. This may be due to the fact that blue sclera is a more variable feature than DI and in some patients tends to disappear with age.
Figure 3.
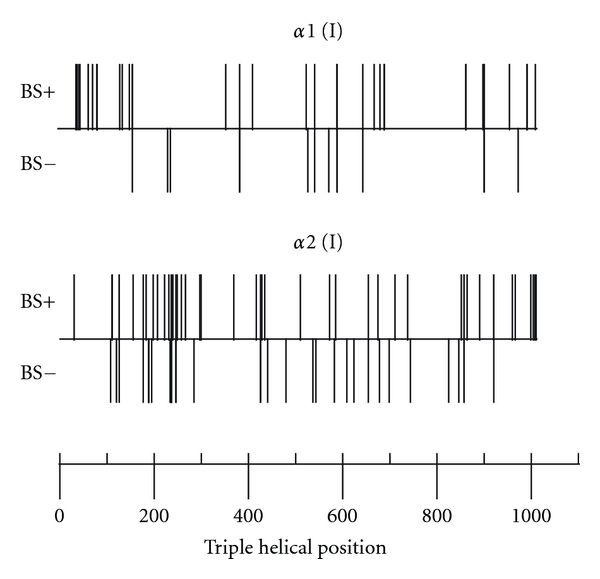
Relationship between the triple helical position of glycine mutations in collagen type I α chains and the presence (+) or absence (−) of blue sclera, adapted from [9].
7. Skeletal Phenotype
7.1. Bone Mineral Density
In a study on 192 OI patients, the mean lumbar spine areal bone mineral density (aBMD) z-score was higher in patients with haploinsufficiency mutations (−3.4 (SD: 1.0)) than in patients with helical mutations in the alpha 1 chain (−4.9 (SD: 1.5)) or the alpha 2 chain (−5.0 (SD: 1.3)) [11]. After adjustment for age, sex, and height z-scores, the mean lumbar spine aBMD z-scores were higher by 0.7 in the haploinsufficiency group compared to both helical mutation groups. The surprisingly small difference in aBMD between these two mutation groups is not reflective of the marked difference in clinical phenotype, suggesting that factors other than aBMD are likely to influence the severity of the bone disease. The same study found no significant association between the lumbar spine aBMD z-scores and the affected alpha chain or the substituting amino acid or the position of the mutation on the triple helical mutations [11].
7.2. Iliac Bone Histomorphometry
A histomorphometric study in 96 patients showed that bone size, cortical width, and the amount of trabecular bone were all significantly lower in OI patients than in healthy controls, but no significant difference in bone size and the amount of trabecular bone was found between patients with haploinsufficiency mutations and those with helical glycine mutations [11] (Figure 4(a)). However, cortical width was higher in the haploinsufficiency group, suggesting that the tissue-level differences between the two mutation groups was related mainly to differences in bone modeling on periosteal and endocortical bone surfaces (Figures 4(a) and 5).
Figure 4.
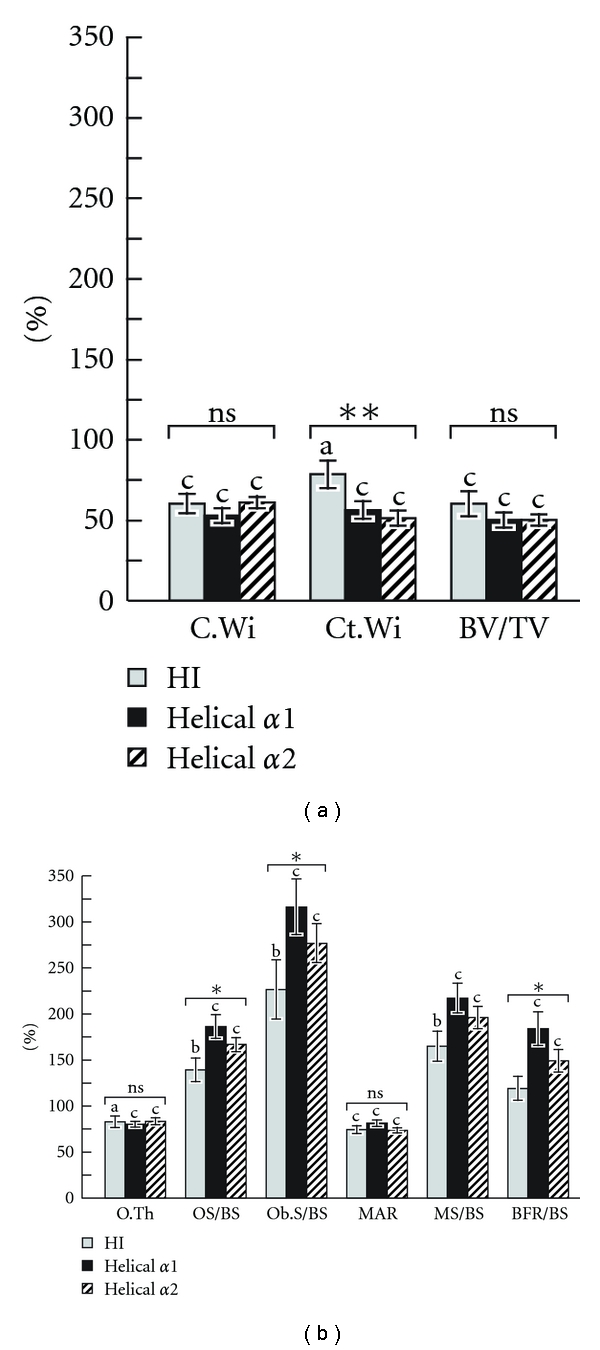
Iliac bone histomorphometric results in patients with COL1A1 haploinsufficiency mutations (HI), as well as with COL1A1 and COL1A2 mutations leading to glycine substitutions in the helical domain of collagen type I. Results are expressed as a percentage of the average result in the age-specific reference range [14]. The letters above the bars indicate the significance of the difference from 100% (i.e., the average result in healthy controls): a: P < 0.05; b: P < 0.01; c: P < 0.001. The significance of the variation between genotype groups is indicated by the symbols above the bar groups: ns = not significant; *P < 0.05; **P < 0.01; (a) C.Wi: core width; Ct.Wi: cortical width; BV/TV: bone volume per tissue volume; (b) O.Th: osteoid thickness; OS/BS: osteoid surface per bone surface, Ob.S/BS: osteoblast surface per bone surface; MAR: mineral apposition rate; MS/BS: mineralizing surface per bone surface; BFR/BS: bone-formation rate per bone surface, adapted with modification from [11].
Figure 5.
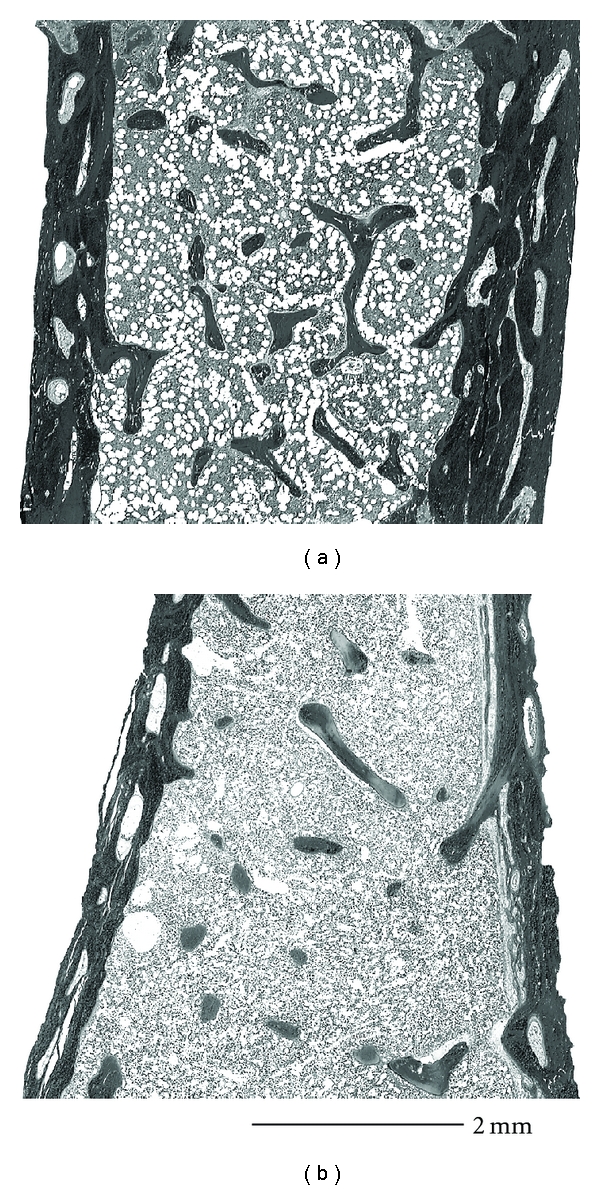
Representative examples of iliac bone samples. (a) Haploinsufficiency mutation, in a 4-year-old boy with a c.1981C > T nucleotide change in COL1A1 that creates a stop codon (p.Gln661X). Core width 4.8 mm, cortical width 765 μm, bone volume per tissue volume 12.9%. (b) Helical glycine mutation, 9-year-old boy with a c.1090G > A nucleotide change in COL1A2 that creates an amino acid change (p.Gly364Ser). Core width 4.2 mm, cortical width 449 μm, and bone volume per tissue volume 8.9%, adapted from [11].
The genotypic groups differed little in trabecular bone metabolism. Bone turnover was generally elevated in OI patients, but this abnormality was more marked in patient with helical glycine mutations (Figure 4(b)). There was no obvious correlation between the affected alpha chain, the position of the mutation, or the substituting amino acid and the histomorphometric parameters in the helical mutation group.
In summary, patients with haploinsufficiency mutations have a milder skeletal phenotype and so far there is no obvious correlation between the genotype and the skeletal phenotype in the helical glycine mutation group.
7.3. Wormian Bones
Wormian bones on skull radiography are a characteristic but not pathognomonic finding of OI and can sometimes help to differentiate between OI and child abuse. Wormian bones are accessory skull bones that are surrounded by suture lines. An analysis of 195 OI patients with a median age of 11.8 years found a significant number of Wormian bones in 59% of cases [15]. A smaller study in adults had found a similar prevalence of Wormian bones [16].
The prevalence of Wormian bones strongly depended on genotype, as they were present in 29% of patients with haploinsufficiency mutations, 96% of patients with helical glycine mutation in the alpha 1 chain, 72% of patients with helical glycine mutation in the alpha 2 chain, 48% of patients with splice site mutations, 33% of patients with C-propeptide mutations [15]. Regression analysis showed that height z-score and the presence of glycine substitutions were the main patient characteristics that were associated with the presence of Wormian bones. Wormian bones were observed in patients as young as 2 weeks old, suggesting that they are likely to develop in utero. Evaluation of serial skull X-rays showed that Wormian bones do not “disappear” with time but tend to persist.
7.4. Cranial Base Abnormalities
Several cranial base abnormalities can be distinguished: Basilar invagination is a primary developmental anomaly and is radiologically defined by the upward migration of the top of C2 (the odontoid process) into the foramen magnum (Figure 6). Basilar impression is the acquired form of the basilar invagination and is diagnosed when the odontoid process is positioned far above the caudal borders of the skull secondary to an upward displacement of basilar and condylar regions of the occipital bone leading to an infolding of the foramen magnum and a translocation of the upper cervical spine into the brainstem [17]. Platybasia is defined by the presence of a flat anterior cranial base angle (the nasion-sella-basion angle) (Figure 7).
Figure 6.
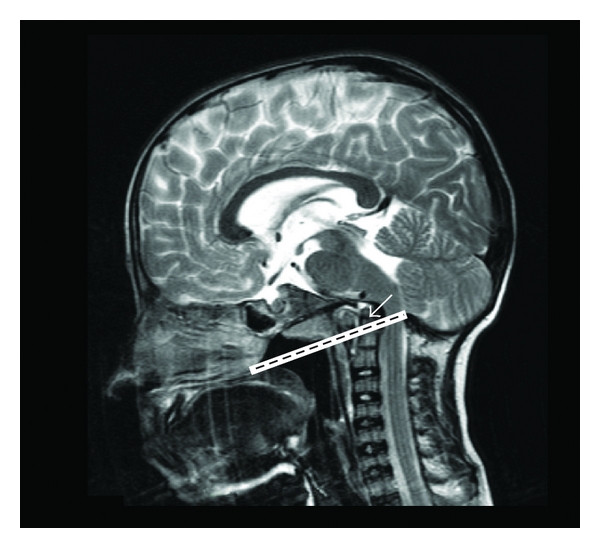
Basilar impression on brain MRI. The tip of the odontoid process (arrow) projects > 5 mm above the Chamberlain line (line between the hard palate and the opisthion which is the midpoint on the posterior margin of the foramen magnum).
Figure 7.
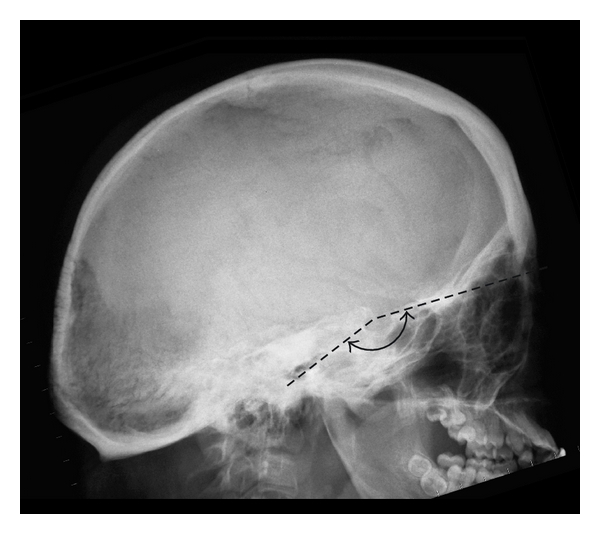
Platybasia: a flat cranial base as a consequence of a flat nasion-sella-basion angle (Welcher basal angle or sphenoid angle > 140°).
A cephalometric analysis of 187 mostly pediatric OI patients found at least one of these cranial base abnormalities in 22% of patients [19]. Platybasia was the most common finding (16%), followed by basilar impression (6%) and basilar invagination (4%). A similar prevalence of cranial base abnormalities had been observed in a smaller study of adult patients [16].
Cranial base abnormalities appeared to depend on genotype, as they were present in 6% of patients with haploinsufficiency mutations, in 43% of patients with helical glycine substitutions in alpha 1, in 32% of patients with helical glycine substitutions in alpha 2, and in 17% of patients with splice-site mutations in either COL1A1 or COL1A2 [19]. However, regression analyses found that only the height z-score but not genotype was a significant independent predictive factor of cranial base abnormalities in OI patients. This suggests that cranial base abnormalities do not directly result from collagen type I mutations, but rather depend on the severity of the skeletal phenotype that is caused by these mutations.
8. Other Genotype-Phenotype Correlations
8.1. Intracranial Hemorrhage and Limb Anomalies
A specific phenotype seems to be associated with mutations located in exon 49 of the COL1A2 gene, which codes for the carboxy-terminal end of the alpha 2 triple helical domain [18] (Figure 8). Three unrelated patients with OI type III, two girls and one boy, with mutations in this area had small hands and feet, brachydactyly with marked shortening of the distal phalanges, hypoplastic nails and loose skin around fingers (Figure 9). In addition, these three patients developed spontaneous intracranial hemorrhage at different ages (between 4 months to 15 years old). So far, there is no mechanistic explanation for this association.
Figure 8.
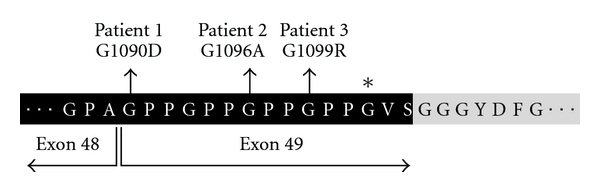
Amino acid sequence of collagen α2 (I) between residues 1087 and 1111. Residues that are part of the triple-helical domain are shown on black background. The first residues of the C-telopeptide are shown in italics and on gray background. The asterisk indicates the glycine residue that is affected in a boy with OI type IV who has normal height and a normal appearance of hands and feet, adapted from [18].
Figure 9.
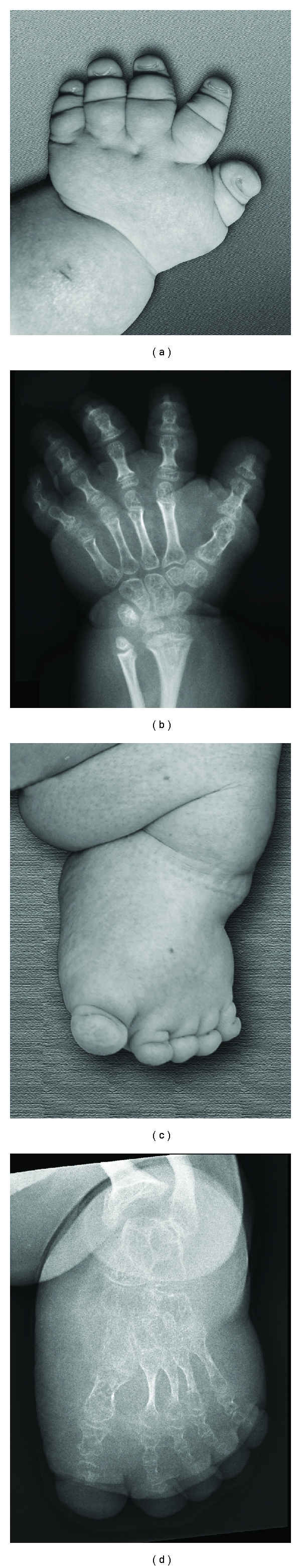
Radiographs of hands and feet in a 15-year-old female. Small stubby hands and feet with hypoplastic nails (a, c). Severe shortening and thinning of all tubular bones including metacarpal and metatarsal bones, especially marked at the distal phalanges (b, d), adapted with modification from [18].
8.2. Ehlers-Danlos Syndrome (EDS) Association
Nonglycine substitutions are rarely disease causing in the triple helical domain of collagen type I. A specific arginine to cysteine substitution at position 888 of the alpha 1 triple helical domain was reported to lead to a phenotype that combined features of OI and EDS [20]. Other nonglycine substitutions in alpha 1 or alpha 2 have been reported to be associated with osteopenia, and vascular rupture [21], Caffey disease with joint laxity [22], and Marfan variant [23].
8.3. Hearing
The relationship between genotype and hearing loss in OI has not been studied in great detail. In one study on adults with OI, 60% of patients with glycine substitutions and 70% of patients with haploinsufficiency mutations had hearing loss [24]. The different mutations resulted in overlapping hearing phenotypes and there was a marked intrafamilial variability in hearing phenotype between family members with the same mutation. However there was no statistically significant correlation between the mutated gene or the mutation type and hearing pattern. The basis of hearing loss in OI is thus likely to be complex and multifactorial.
9. Summary of Main Points
Glycine-to-serine substitutions are the most frequent type of mutation in the triple helix domains of the collagen type I alpha 1 and alpha 2 chains.
Serine substitutions tend to lead to a more severe phenotype in the alpha 1 than in the alpha 2 chain.
The clinical severity of serine substitutions correlates with the position of the mutation in the alpha 2 chain, but not in the alpha 1 chain.
Substitutions by arginine, aspartate, glutamate, and valine beyond the first 200 amino acid residues of alpha 1 are generally lethal, but have variable outcome in alpha 2.
Mutations affecting the first 120 amino acids of the collagen type I triple helix lead to blue sclera but do not cause dentinogenesis imperfect.
Glycine substitutions in alpha 1 almost always are associated with the presence of Wormian bones, whereas about three quarters of patients with glycine substitutions in alpha 2 and one quarter of patients with haploinsufficiency mutations have Wormian bones.
More than a third of patients with glycine substitutions in the alpha 1 or alpha 2 chain but only 1 in 20 patients with haploinsufficiency mutations have cranial base abnormalities.
Mutations at the C-terminal end of the alpha 2 chain are associated with limb anomalies and intracranial hemorrhage.
No correlation was found between the mutated gene or mutation type and the hearing pattern in OI.
It is essential to keep in mind that these are general rules for genotype phenotype correlation in autosomal dominant OI patients but exceptions do arise and it is particularly important to keep this in mind in the context of genetic counseling specifically in prenatal cases.
Acknowledgments
This study was supported by the Shriners of North America and the Fonds de la Recherche en Santé du Québec.
References
- 1.Baldridge D, Schwarze U, Morello R, et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Human Mutation. 2008;29(12):1435–1442. doi: 10.1002/humu.20799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rauch F, Glorieux FH. Osteogenesis imperfecta. The Lancet. 2004;363(9418):1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 3.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. Journal of Medical Genetics. 1979;16(2):101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byers PH. Collagens: building blocks at the end of the development line. Clinical Genetics. 2000;58(4):270–279. doi: 10.1034/j.1399-0004.2000.580404.x. [DOI] [PubMed] [Google Scholar]
- 5.Byers PH. Killing the messenger: new insights into nonsense-mediated mRNA decay. Journal of Clinical Investigation. 2002;109(1):3–6. doi: 10.1172/JCI14841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalgleish R. The human type I collagen mutation database. Nucleic Acids Research. 1997;25(1):181–187. doi: 10.1093/nar/25.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalgleish R. The Human Collagen Mutation Database 1998. Nucleic Acids Research. 1998;26(1):253–255. doi: 10.1093/nar/26.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marini JC, Forlino A, Cabral WA, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Human Mutation. 2007;28(3):209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rauch F, Lalic L, Roughley P, Glorieux FH. Genotype-phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I. European Journal of Human Genetics. 2010;18(6):642–647. doi: 10.1038/ejhg.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lund AM, Müller J, Skovby F. Anthropometry of patients with osteogenesis imperfecta. Archives of Disease in Childhood. 1999;80(6):524–528. doi: 10.1136/adc.80.6.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rauch F, Lalic L, Roughley P, Glorieux FH. Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. Journal of Bone and Mineral Research. 2010;25(6):1367–1374. doi: 10.1359/jbmr.091109. [DOI] [PubMed] [Google Scholar]
- 12.Majorana A, Bardellini E, Brunelli PC, Lacaita M, Cazzolla AP, Favia G. Dentinogenesis imperfecta in children with osteogenesis imperfecta: a clinical and ultrastructural study. International Journal of Paediatric Dentistry. 2010;20(2):112–118. doi: 10.1111/j.1365-263X.2010.01033.x. [DOI] [PubMed] [Google Scholar]
- 13.Lund AM, Jensen BL, Nielsen LA, Skovby F. Dental manifestations of osteogenesis imperfecta and abnormalities of collagen I metabolism. Journal of Craniofacial Genetics and Developmental Biology. 1998;18(1):30–37. [PubMed] [Google Scholar]
- 14.Glorieux FH, Travers R, Taylor A, et al. Normative data for iliac bone histomorphometry in growing children. Bone. 2000;26(2):103–109. doi: 10.1016/s8756-3282(99)00257-4. [DOI] [PubMed] [Google Scholar]
- 15.Semler O, Cheung MS, Glorieux FH, Rauch F. Wormian bones in osteogenesis imperfecta: correlation to clinical findings and genotype. American Journal of Medical Genetics A. 2010;152(7):1681–1687. doi: 10.1002/ajmg.a.33448. [DOI] [PubMed] [Google Scholar]
- 16.Kovero O, Pynnönen S, Kuurila-Svahn K, Kaitila I, Waltimo-Sirén J. Skull base abnormalities in osteogenesis imperfecta: a cephalometric evaluation of 54 patients and 108 control volunteers. Journal of Neurosurgery. 2006;105(3):361–370. doi: 10.3171/jns.2006.105.3.361. [DOI] [PubMed] [Google Scholar]
- 17.Hayes M, Parker G, Ell J, Sillence D. Basilar impression complicating osteogenesis imperfecta type IV: the clinical and neuroradiological findings in four cases. Journal of Neurology Neurosurgery and Psychiatry. 1999;66(3):357–364. doi: 10.1136/jnnp.66.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faqeih E, Roughley P, Glorieux FH, Rauch F. Osteogenesis imperfecta type III with intracranial hemorrhage and brachydactyly associated with mutations in exon 49 of COL1A2. American Journal of Medical Genetics A. 2009;149(3):461–465. doi: 10.1002/ajmg.a.32653. [DOI] [PubMed] [Google Scholar]
- 19.Cheung MS, Arponen H, Roughley P, et al. Cranial base abnormalities in osteogenesis imperfecta: phenotypic and genotypic determinants. Journal of Bone and Mineral Research. 2011;26(2):405–413. doi: 10.1002/jbmr.220. [DOI] [PubMed] [Google Scholar]
- 20.Cabral WA, Makareeva E, Letocha AD, et al. Y-position cysteine substitution in type I collagen (α1(I) R888C/p.R1066C) is associated with osteogenesis imperfecta/Ehlers-Danlos Syndrome phenotype. Human Mutation. 2007;28(4):396–405. doi: 10.1002/humu.20456. [DOI] [PubMed] [Google Scholar]
- 21.Malfait F, Symoens S, De Backer J, et al. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Human Mutation. 2007;28(4):387–395. doi: 10.1002/humu.20455. [DOI] [PubMed] [Google Scholar]
- 22.Gensure RC, Mäkitie O, Barclay C, et al. A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. Journal of Clinical Investigation. 2005;115(5):1250–1257. doi: 10.1172/JCI22760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips CL, Shrago-Howe AW, Pinnell SR, Wenstrup RJ. A subsitution at a non-glycine position in the triple-helical domain of proα2(I) collagen chains present in an individual with a variant of the Marfan syndrome. Journal of Clinical Investigation. 1990;86(5):1723–1728. doi: 10.1172/JCI114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartikka H, Kuurila K, Körkkö J, et al. Lack of correlation between the type of COL1A1 or COL1A2 mutation and hearing loss in osteogenesis imperfecta patients. Human Mutation. 2004;24(2):147–154. doi: 10.1002/humu.20071. [DOI] [PubMed] [Google Scholar]