

Abstract
Ubiquitin-mediated proteolysis controls the abundance of many cell cycle regulatory proteins. Recent work in Saccharomyces cerevisiae suggests that a complex consisting of Cdc53, Skp1, and a third component known as an F-box protein (termed SCF) in combination with Cdc34 specifically targets regulatory proteins for degradation, and that substrate specificity is likely to be mediated by the F-box subunit. A screen for genetic interactions with a cdc34 mutation yielded MET30, which encodes an F-box protein. MET30 is an essential gene required for cell cycle progression and met30 mutations interact genetically with mutations in SCF components. Furthermore, physical interactions between Met30, Cdc53, Cdc34, and Skp1 in vivo provide evidence for an SCFMet30 complex. We demonstrate the involvement of Met30 in the degradation of the Cdk-inhibitory kinase Swe1. Swe1 is stabilized in met30 mutants and GST-Met30 pull-down experiments reveal that Met30 specifically binds Swe1 in vivo. Furthermore, extracts prepared from cdc34 or met30 mutants are defective in polyubiquitination of Swe1. Taken together, these data suggest that SCF-mediated proteolysis may contribute to the regulation of entry into mitosis. Our data, in combination with previously published results, also provide evidence for distinct SCF complexes in vivo and support the idea that their F-box subunits mediate SCF substrate specificity.
Keywords: Met30, F-box, Swe1, SCF, ubiquitination
Proteolysis is one of the principal mechanisms whereby cellular protein levels are regulated. Therefore, it is not surprising, that cell cycle transitions are frequently controlled by degradation of regulatory proteins. Proteolysis is particularly suitable for cell cycle regulation, because it is a rapid irreversible process and, thus, ensures unidirectional progression through cell cycle phase transitions.
In eukaryotes, most regulated proteolysis is initiated by polyubiquitination of target proteins. Ubiquitination requires ATP-dependent activation of ubiquitin by ubiquitin-activating enzymes (E1), transfer to a conserved cystein residue on ubiquitin-conjugating enzymes (E2 or Ubc) and, finally, conjugation of ubiquitin to target proteins. Ubiquitin conjugation often requires specificity factors (E3 or ubiquitin ligase) that are thought to recognize and directly interact with substrate proteins, and, in some cases, serve as thiol–ubiquitin intermediates. The E1–E2–E3 reaction cascade results in polyubiquitination of substrate proteins on lysine residues. These polyubiquitinated proteins are subsequently recognized and degraded by the 26S proteasome, a complex protease-containing organelle (Hochstrasser 1996).
Eukaryotes express a family of distinct E2 enzymes. All share characteristic motifs that have allowed the identification of the complete set of E2 enzymes in Saccharomyces cerevisiae (Ubc1–Ubc13) by sequence analysis of the S. cerevisiae genome. Although E2 enzymes confer some substrate specificity, the general view is that E3 enzymes are responsible for the majority of substrate recognition and targeting, and are therefore the components subjected to regulation. Despite their central role, E3 enzymes are the least well-characterized factors involved in ubiquitination, probably because they generally do not share a single identifying sequence motif or mode of action. However, several families of proteins with E3 function have been identified, including the HECT domain proteins (Huibregtse et al. 1995), Ubr1-related ubiquitin ligases (Varshavsky 1996), and the anaphase-promoting complex (APC, also called the cyclosome) (Lamb et al. 1994; Irniger et al. 1995; King et al. 1995; Sudakin et al. 1995).
Recent work in S. cerevisiae suggests that a complex consisting of Cdc53, Skp1, and Cdc4 functions as an E3 ubiquitin ligase for the Cdk-inhibitor Sic1 (Bai et al. 1996; Feldmann et al. 1997; Skowyra et al. 1997). This complex has been termed SCFCdc4 (Skp1-Cullin (or Cdc53)-F-box protein) indicating that the F-box protein in this particular complex is Cdc4. F-box proteins share a short sequence motif, the F-box, which has been shown to mediate binding to Skp1 (Bai et al. 1996). Data base searches have revealed that F-box proteins exist as a small family. Interestingly, the yeast F-box protein Grr1 has been linked to G1-cyclin proteolysis (Barral et al. 1995) and a physical interaction with the G1 cyclins Cln1 and Cln2 has been demonstrated in vitro (Skowyra et al. 1997) suggesting the existence of an SCFGrr1 complex. These observations have led to the proposal of a model in which different F-box proteins associated with Skp1 and Cdc53 define the substrate specificity of SCF ubiquitin ligase complexes (Bai et al. 1996; Feldmann et al. 1997; Skowyra et al. 1997). SCF-dependent ubiquitin conjugation in budding yeast has been shown to be catalyzed by the E2 enzyme Cdc34, which interacts with Cdc53 and can ubiquitinate Sic1 and Cln2 (Deshaies et al. 1995; Mathias et al. 1996; Willems et al. 1996; Verma et al. 1997). Currently, however, it cannot be ruled out that other E2 enzymes function in conjunction with SCF.
In a genetic screen designed to identify limiting components of the Cdc34-dependent ubiquitination pathway, we isolated the gene encoding the F-box protein Met30. MET30 is an essential gene and has been recently implicated in repression of genes required for methionine metabolism. Mutants harboring the dominant MET30-1 allele were shown to fail to repress expression of MET25, a gene required for methionine biosynthesis, in response to S-adenosylmethionine (Thomas et al. 1995). However, this defect cannot explain the lethality confered by met30 loss-of-function mutations.
Here, we report that Met30 is required for proteolysis of the Cdk-inhibitory kinase Swe1. Swe1 is the S. cerevisiae homolog of fission yeast Wee1 and phosphorylates the cyclin-dependent kinase Cdc28 on tyrosine 19 (Booher et al. 1993). Phosphorylation of specific tyrosines by Wee1-like kinases renders cyclin-dependent kinases inactive. Entry into mitosis in many organisms is largely regulated by Wee1-dependent tyrosine phosphorylation followed by dephosphorylation of Cdks (Dunphy 1994). Although the enzymatic machinery that controls tyrosine phosphorylation and dephosphorylation of Cdks is conserved in budding yeast, phosphorylation on tyrosine 19 of Cdc28 is not required for proper timing of mitosis during the unperturbed cell cycle (Russell et al. 1989; Amon et al. 1992; Booher et al. 1993). However, it is critical for the inhibition of mitosis in response to activation of the morphogenesis checkpoint (Lew and Reed 1995a,b) and participates in adaptation from other checkpoint arrests (Rudner and Murray 1996).
In this paper we show that the F-box protein Met30 is part of an SCFMet30 complex in vivo and specifically targets Swe1 to the Cdc34-dependent proteolysis pathway. These results link regulation of tyrosine phosphorylation of Cdks with SCF-regulated proteolysis. Because highly conserved Met30 homologs are found in other eukaryotes, including mammals, SCF-mediated ubiquitination of Wee1-like kinases may play a significant role in control of entry into mitosis.
Results
Isolation of MET30 as an enhancer of cdc34-3 mutants
The S. cerevisiae ubiquitin-conjugating enzyme Cdc34 has been implicated in the proteolysis of several key regulatory proteins, including the Cdk inhibitors Sic1 and Far1 (McKinney et al. 1993; Feldmann et al. 1997; Skowyra et al. 1997), the G1 cyclins Cln1, Cln2, Cln3 (Deshaies et al. 1995; Yaglom et al. 1995; Lanker et al. 1996), and the DNA replication factor Cdc6 (Piatti et al. 1996; Drury et al. 1997). The current model predicts that different E3 ligases mediate substrate specificity. To identify such E3 enzymes, we used a haploinsufficiency screen with a temperature-sensitive cdc34 mutant. We reasoned that a 50% reduction of a limiting component in the Cdc34 pathway should result in enhancement of the temperature sensitivity of diploid cdc34-3 cells (for description of the rationale of the screen, see Marini et al. 1996). Therefore, we mutagenized diploid cdc34-3 mutants by random heterozygous transposon-mediated gene disruptions (see Materials and Methods), and selected mutants that were unable to grow at the semipermissive temperature of the parental strain (32°C). Three such mutants exhibited a 2:2 segregation of lethality linked to the inserted URA3 marker associated with the transposon. Sequence analysis of the DNA flanking the transposon insertion sites revealed that the same ORF was disrupted in all three mutants and that the site of integration for two mutants was identical. Analysis of the disruption sites by use of the Yeast Genome Data Base identified the mutated gene as MET30. To verify that mutation of MET30 caused the enhancement of the temperature sensitivity in the three mutants, we replaced one copy of the entire MET30 ORF in a diploid cdc34-3 mutant with a gene conferring resistance to the drug G418. This heterozygous gene disruption resulted in the same reduced permissive temperature for cdc34-3 mutants that we observed with the original enhancer mutants (Fig. 1A). A disruption of one copy of MET30 in a diploid wild-type strain did not affect growth at any temperature (data not shown). Tetrad analysis of the heterozygously disrupted wild-type strain revealed a 2:2 segregation of lethality with all viable spores being sensitive to G418, confirming that MET30 is an essential gene, as has been reported previously (Thomas et al. 1995).
Figure 1.

Genetic interactions between MET30, CDC34, and SCF components. (A) Herterozygous disruption of MET30 enhances the temperature sensitivity of diploid cdc34-3 mutants. Cells were grown at 23°C, sequentially replica plated to four new plates, and incubated at the temperatures indicated. (B) High-copy suppression. met30-6, cdc53-1, and cdc4-3 mutants were transformed with 2μ-based plasmids harboring no insert (vector), MET30 (2μ-MET30), or CDC34 (2μ-CDC34). Transformants were grown at 23°C and replicas were incubated at 34°C (met30-6 and cdc53-1) or 30°C (cdc4-3). To analyze genetic interactions between MET30 and SKP1, met30-6 mutants expressing SKP1 under control of the GAL1 promoter were grown on glucose plates at 23°C, replica plated to galactose plates to induce expression of SKP1 and incubated at the restrictive temperature of 34°C.
Met30 interacts genetically with Cdc53, Cdc34, and Skp1
To analyze the functional relationship between Met30 and proteins involved in the Cdc34-dependent ubiquitination pathway, we next tested for genetic interactions with other SCF components. To carry out a detailed genetic analysis, we constructed temperature-sensitive met30 mutants. For this purpose, the coding region of MET30 was mutagenized in vitro by PCR and mutants unable to grow at 37°C were selected. One of these, met30-6, was chosen for further analysis.
We had already demonstrated a genetic interaction between cdc34 temperature sensitivity and MET30 gene dosage (Fig. 1A). To provide additional support for this interaction, we transformed haploid met30-6 mutants with CDC34 cloned into a high-copy plasmid. Increased dosage of Cdc34 suppressed the lethality of met30-6 mutants at a normally nonpermissive temperature (34°C). As expected, MET30 on a high-copy plasmid rescued the mutant as well (Fig. 1B). We then extended the same kind of analysis to cdc53-1 and cdc4-3 mutants. The growth defect of cdc53-1 mutants at restrictive temperature was suppressed by both high-copy MET30 and high-copy CDC34. However, increased levels of Met30 or Cdc34 did not rescue cdc4-3 mutants. Finally, a genetic link between SKP1 and MET30 was established, because overexpression of Skp1 suppressed the temperature sensitivity of met30-6 mutants (Fig. 1B).
Met30 physically interacts with Cdc34, Cdc53, and Skp1
These genetic data suggest that Cdc34, Cdc53, and Skp1 interact with Met30. However, genetic interactions can result from indirect physiological interactions. Therefore, we sought direct evidence for a physical association between Met30 and components of the Cdc34-dependent ubiquitination machinery. Proteins from cells expressing biologically active GST-tagged Skp1 and epitope-tagged Met30(HA)3 were purified on glutathione beads. Proteins bound to the beads were eluted with glutathione and analyzed by immunoblotting. Met30 specifically copurified with Skp1 (Fig. 2A). We then performed coimmunoprecipitation experiments with strains expressing Met30 tagged with the RGS6H epitope and Cdc34(HA)3 or Cdc53(HA)3, respectively. Met30 was found in immune complexes with Cdc34 and Cdc53 (Fig. 2B). The two mobility species of Cdc53 detected are consistent with a report suggesting that the slower migrating isoform of Cdc53 is modified with the ubiquitin-related protein Rub1 (Lammer et al. 1998). Whereas the stoichiometry of the Met30/Cdc53 association was unchanged in different experiments, the amount of Met30 associated with Cdc34 was very variable but always significant.
Figure 2.

Physical interaction of Met30 with Skp1, Cdc34, and Cdc53. (A) Met30 forms a complex with Skp1 in vivo. Cells harboring an endogenously expressed (HA)3-tagged MET30 and expressing GST–SKP1 or GST under the control of the CUP1 promotor were grown in the presence of 100 μm copper to an OD600 ≅ 0.5. Protein extracts were incubated with glutathione beads. After several washes with binding buffer the beads were washed with binding buffer, including 0.8 m of sodium chloride (wash). Bound proteins were eluted with glutathione and subjected to Western blot analysis with an antibody directed against the HA epitope. Probing the blot with antibodies directed against GST revealed a two- to threefold excess of GST compared with GST–Skp1 (data not shown). (B) Met30 is in a complex with Cdc34 and Cdc53 in vivo. Cells expressing a RGS6H epitope-tagged MET30 under control of the GAL1 promotor and harboring either endogenously expressed (HA)3-tagged CDC34 or CDC53 were grown in galactose to an OD600 ≅ 0.5. Immunocomplexes were purified with 12CA5 antibodies (directed against the HA epitope) coupled to protein A, separated by SDS-PAGE, and analyzed by immunoblotting with antibodies directed against the HA or the RGS6H epitopes to detect Cdc53 and Cdc34 or Met30, respectively. Cells expressing Met30(RGS6H) but no HA-tagged protein were used as a control (no HA tag).
Taken together, both the genetic and the biochemical results suggest that Met30 is part of a multiprotein complex with Cdc34, Cdc53, and Skp1. In accordance with the previously proposed nomenclature, we refer to the Skp1–Cdc53–Met30 complex as SCFMet30. While this paper was being revised, a similar analysis that arrived at the same conclusion was published (Patton et al. 1998).
Swe1 is a potential target of SCFMet30
The arrest phenotype of met30-6 mutants at 37°C was heterogeneous, including both budded and unbudded cells with either G1 or G2/M DNA content. The majority of the population arrested as round unbudded cells. We noticed that a fraction of the budded met30-6 cells displayed an elongated bud phenotype (Fig. 3C), which is characteristic of cells in which activation of Clb1-4/Cdc28 kinase has been delayed or prevented (Lew and Reed 1993). One mechanism whereby Clb1-4/Cdc28 activation can be blocked is inhibitory phosphorylation of Cdc28 at tyrosine 19 by the kinase Swe1 (Booher et al. 1993). This phosphorylation is reversed by the phosphatase Mih1 leading to the activation of the Cdc28 kinase (Russell et al. 1989). To determine whether this regulatory pathway might be affected in met30 mutants, we generated met30-6 mih1::LEU2 and met30-6 mih1::LEU2 swe1::URA3 strains. At 37°C, the met30-6 mih1::LEU2 cells arrested with a much higher proportion of cells exhibiting the elongated bud phenotype (Fig. 3D), whereas elongated buds were almost absent in the met30-6 mih1::LEU2 swe1::URA3 cells (Fig. 3E). As described previously, deletion of MIH1 in an otherwise wild-type strain did not cause a significant cell cycle delay or lead to generation of cells with elongated buds (Fig. 3A,B). This result suggested that Swe1 might be more abundant or more active in met30-6 cells than in wild-type cells.
Figure 3.
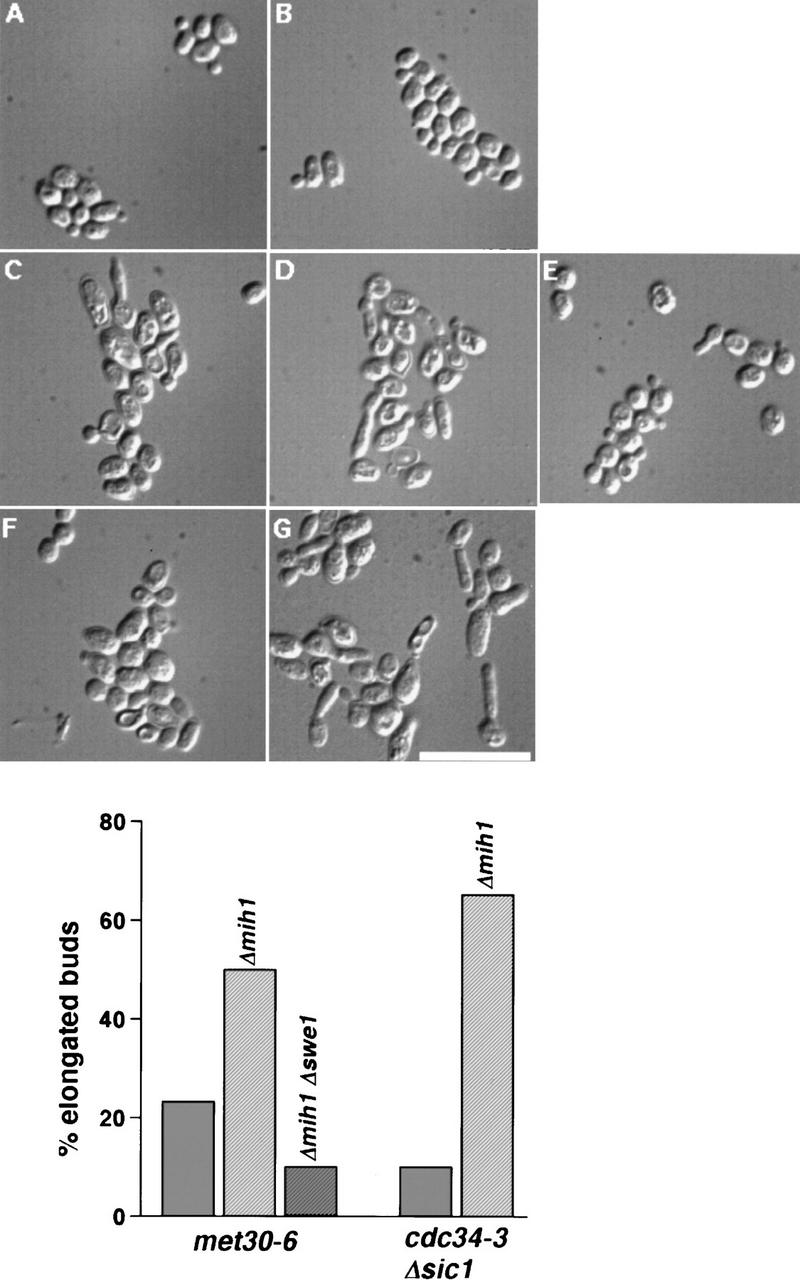
Hyperactivation of the Swe1 pathway in met30 and cdc34 mutants. Cells were grown at 23°C in YEPD and shifted to 37°C for 5 hr, followed by fixation in 70% ethanol and visualization by DIC microscopy. Strains were as follows (percent of elongated cells or buds in the population are indicated in parentheses): (A) wild type (0%), (B) mih1::LEU2 (0%), (C) met30-6 (23%), (D) met30-6 mih1::LEU2 (50%), (E) met30-6 mih1::LEU2 swe1::URA3 (10%—these were smaller and less straight than the elongated buds in C and D), (F) cdc34-3 sic1::URA3 (10%), (G) cdc34-3 sic1::URA3 mih1::LEU2 (65%). Two hundred cells were counted in each sample. Bar, 10 μm. The graph illustrates the percentage of cells with elongated buds.
A role for Cdc34 in control of the Swe1/Mih1 pathway has not been reported, but such a role might have been missed because the failure of cdc34 mutants to degrade the Cdk inhibitor Sic1 leads to a cell cycle arrest prior to G2, when Swe1 and Mih1 become relevant to cell cycle progression. cdc34-3 sic1::URA3 cells arrested in G2/M with a predominantly large round bud phenotype (Fig. 3F), as reported previously (Schwob et al. 1994). To determine whether an increase in Swe1 activity occurred in cdc34-3 mutants, we generated a cdc34-3 sic1::URA3 mih1::LEU2 strain. At 37°C, a large proportion (65%) of these cells arrested with elongated buds (Fig. 3G). Deletion of SWE1 in cdc34-3sic1::URA mutants did not supress the G2/M arrest phenotype, suggesting that accumulation of another Cdc34 target is responsible for this cell cycle arrest (data not shown). However, these cells no longer had elongated buds (data not shown), indicating that this component of the phenotype is caused by Swe1.
In combination, these findings suggest that one defect in both met30 and cdc34 mutants is a higher level of Swe1 protein or activity. This does not make a major contribution to the arrest phenotype of either mutant alone because Mih1 suffices to keep Cdc28 sufficiently active; deletion of MIH1 uncovers a role for these proteins in controlling Cdc28 tyrosine phosphorylation.
Given the expectation that Met30 and Cdc34 act by targeting proteins for ubiquitination and subsequent degradation, the simplest explanation of the above findings would be that Swe1 is itself a target of SCFMet30. Consistent with this explanation, analysis of the Swe1 sequence revealed two regions with PEST motifs characteristic of unstable proteins (Rogers et al. 1986) that are often targets of the Cdc34-dependent ubiquitination pathway. To examine the stability of Swe1 protein, a 12myc epitope tag was fused to the carboxyl terminus of Swe1, and expression of this construct was placed under the control of the inducible GAL1 promoter. Wild-type and met30-6 mutant cells containing this construct were incubated at 37°C for 2 hr, induced to express Swe1–myc12 with galactose for 10 min, and then labeled with [35S]methionine/cysteine for 10 min. Swe1–myc12 expression was subsequently turned off by transferring the cells to dextrose medium, and an excess of cold methionine and casamino acids was added. Degradation of labeled Swe1–myc12 was significantly inhibited in the met30-6 mutant relative to wild type (Fig. 4A,C). In contrast, Swe1–myc12 degradation in cdc4-3 mutants proceeded at wild-type rates (Fig. 4A,C). To exclude possible cell cycle position effects, we analyzed Swe1 turnover in cells synchronized in S phase. Cells were arrested by addition of hydroxyurea and pulse-chase analysis was performed as described above. Again, Swe1–myc12 degradation was significantly inhibited in met30-6 mutants relative to wild type (Fig. 4B,C). The G1 cyclin, Cln2, was not stabilized in a met30-6 mutant, confirming that Swe1 stabilization is not caused by a general proteolysis defect associated with the met30-6 mutation (data not shown).
Figure 4.

Swe1 is stabilized in met30 but not cdc4 mutants. (A) Wild-type (top), cdc4-3 (middle), or met30-6 (bottom) cells containing the GAL1–SWE1–myc12 construct were shifted to 37°C for 2 hr. Galactose was then added to 2% and the cells were incubated for a further 10 min to induce Swe1–myc12 expression. Cells were then labeled 10 min at 37°C followed by termination of Swe1 synthesis and labeling during a 4-hr time course. The immunoprecipitated Swe1 was analyzed by SDS-PAGE and PhosphorImaging. (Left lane) Immunoprecipitates from cells lacking the Swe1–myc construct. The band running below Swe1–myc12 (asterisk) is a background band. (B) A similar experiment was conducted with synchronized cells. Wild-type and met30-6 cells were arrested by addition of 200 mm hydroxyurea (HU) for 3 hr at 23°C prior to the temperature shift to 37°C. Cell cycle arrest was monitored visually (>80% large budded cells). Pulse-chase analysis was performed as described in A but in the continuous presence of 200 mm HU. (C) The 35S-labeled Swe1–myc12 signal was quantitated by PhosphorImager and ImageQuant v1.2 software, normalized to the background band and expressed as percent of signal at t = 0.
Met30 physically interacts with Swe1 in vivo
In the current model for SCF-mediated ubiquitination, F-box proteins specifically recognize and bind target proteins. A physical interaction between Cdc4 and its target Sic1, as well as binding of Grr1 to Cln1, has been demonstrated in vitro (Feldmann et al. 1997; Skowyra et al. 1997). We asked whether a complex of Met30 and its putative target Swe1 could be detected in vivo. Extracts were prepared from yeast cells expressing an epitope-tagged allele of Swe1 and a GST–Met30 fusion, and Met30-associated complexes were purified on glutathione beads. Swe1 specifically copurified with GST–Met30, whereas no binding to GST was detected (Fig. 5). This physical interaction is most likely required to target Swe1 to the Cdc34-degradation pathway.
Figure 5.

Met30 interacts physically with Swe1. Cells were grown in YEP raffinose to an OD600 ≅ 0.3 and expression of Swe1(myc)12, Clb2 and GST–Met30 was induced by addition of galactose for 3 hr. As a control, a strain expressing GST instead of GST–Met30 was processed in parallel. Protein complexes were purified on glutathione beads and parallel samples were separated on two SDS–polyacrylamide gels. After immunoblotting, one of the blots was probed with a polyclonal antibody directed against GST to detect GST–Met30 and GST, respectively. The other blot was analyzed with the 9E10 antibody against the myc epitope on Swe1. (Asterisk) A protein crossreacting with the anti-GST antibody.
In the experiment described above, Swe1 was expressed under the control of the GAL1 promoter that leads to cell cycle arrest as a result of inhibition of Cdc28 kinase activity. However, simultaneous overexpression of the B-type cyclin Clb2 suppresses this cell cycle arrest, presumably because excess Clb2 hyperactivated the Cdc28 kinase. Interestingly, overexpression of Clb2 increased the amount of Swe1 that copurified with Met30 (data not shown), suggesting a potential link between Clb2/Cdc28 mediated phosphorylation of Swe1 and SCFMet30 mediated ubiquitination.
Extracts prepared from cdc34 or met30 mutants are defective in Swe1 polyubiquitination
Our data suggest that Met30 is required for Swe1 ubiquitination. However, to establish a more direct link between Met30 function and Swe1 ubiquitination, we analyzed the ability of wild-type and mutant yeast cell extracts to ubiquitinate Swe1. To this end, we incubated radiolabeled Swe1, produced by in vitro translation, with whole yeast cell extracts and analyzed the reaction products by SDS-PAGE. Incubation with extracts prepared from wild-type cells resulted in accumulation of more slowly migrating forms of Swe1 (Fig. 6A). This reaction was dependent on the addition of ATP (data not shown). To ask whether the slower migrating forms of Swe1 were a result of polyubiquitination, we added mutant ubiquitin (Ubi-R48) to the reaction. Mutation of lysine 48 to arginine impairs chain elongation in polyubiquitination reactions because lysine 48 is the major site of isopeptide linkage between ubiquitin molecules (Chau et al. 1989). Addition of Ubi-R48 to the in vitro assay significantly reduced the mobility shift of Swe1 in response to incubation with wild-type extract, indicating that the more slowly migrating Swe1 forms correspond to polyubiquitinated species (Fig. 6A). To address the question of a requirement of Cdc34 and Met30 function for Swe1 polyubiquitination, we prepared extracts from cdc34 and met30 mutants. Whereas incubation of Swe1 with wild-type cell extracts resulted in the accumulation of polyubiquitinated species of Swe1, no change in Swe1 mobility was observed after incubation with either mutant extract (Fig. 6B). This deficiency to catalyze Swe1 polyubiquitination is a result of impaired Cdc34 and Met30 activity, respectively, rather than the result of a general inactivity of the extracts, because Swe1 ubiquitination is restored when both mutant extracts are combined. Cdc34 and Met30 functions are therefore required for polyubiquitination of Swe1 in vitro.
Figure 6.

Swe1 in vitro ubiquitination. (A) Swe1 is ubiquitinated in whole cell extracts. 35S-labeled Swe1 was incubated with whole yeast cell extracts at 23°C (lanes 2,3) or on ice (lane 1) in the presence of an ATP-regenerating system and ubiquitin (lanes 1,2) or mutant R48-ubiquitin (lane 3), respectively. The reaction mix was separated by SDS-PAGE and analyzed with a PhosphorImager. (B) Swe1 ubiquitination is defective in cdc34 and met30 mutants. In vitro ubiquitination activity of extracts prepared from cdc34-3 or met30-6 mutants was compared with wild-type extracts. (Lane 5) Complementation experiment in which both mutant extracts were mixed (ratio, 2:1 = cdc34-3:met30-6). Extracts were preincubated at 23°C for 5 min to allow complex formation. 35S-labeled Swe1, ATP-regenerating system and ubiquitin were then added and reactions incubated at 23°C (lanes 2– 6) or on ice (lane 1). No extract was added to the reaction shown in lane 6. Reaction products were analyzed as described in A.
Discussion
The F-box components of SCF target distinct substrates to the Cdc34-dependent ubiquitination pathway
We isolated MET30 in a genetic screen on the basis of its strong specific interaction with a temperature-sensitive cdc34 mutation. The presence of an F-box motif in Met30 immediately suggested that Met30 might be part of an E3 complex targeting selected proteins to the Cdc34-dependent ubiquitination pathway. In this paper, we demonstrate the existence of such an E3 complex in vivo by showing that Met30 physically associates with Skp1 and Cdc53 and refer to this complex as SCFMet30. While this paper was being revised, similar results were reported by Patton et al. (1998). Whereas the F-box motif in Met30 suggests a direct interaction with Skp1 on the basis of previous work, we have no evidence for a direct physical contact between Met30 and Cdc53 or Cdc34. By use of insect cells expressing various combinations of Cdc4, Cdc53, Skp1, and Cdc34, a direct contact between Cdc4/Skp1, Skp1/Cdc53, and Cdc53/Cdc34, respectively, was proposed (Skowyra et al. 1997). We envision a similar situation, with Met30 replacing Cdc4 as the F-box protein in this complex. This model suggests a rather nonspecific role for Skp1, Cdc53, and Cdc34 in SCF-mediated proteolysis, with Cdc53 and Skp1 serving principally as a bridge between F-box proteins and the ubiquitin-conjugating enzyme Cdc34. The F-box proteins may represent the specificity factors that physically interact with substrate proteins and enable their ubiquitination by Cdc34. On the basis of current data, the precise molecular organization of SCF complexes in vivo is not clear. For example, it has not yet been established whether individual SCF complexes consist of one molecule each of Cdc53, Skp1, and one F-box protein, or whether complexes of higher stoichiometry exist.
Little is known about the regulation of SCF activity. Two-hybrid experiments with the F-box protein Grr1 have shown that the Grr1/Skp1 interaction is modulated by carbon source (Li and Johnston 1997), suggesting that complex formation between Skp1 and the F-box component might be subjected to regulation, consequently influencing the rate of substrate proteolysis.
On the basis of the current model of SCF-mediated ubiquitination, it cannot be ruled out that ubiquitin conjugation to some substrates can be catalyzed by E2 enzymes other than Cdc34. It has been shown that glucose-induced HXT1 expression depends on functional Cdc53 and Skp1, but not on Cdc34 (Li and Johnston 1997).
Targets of Met30
MET30 was originally identified in a screen for mutants defective in transcriptional repression of MET25 (Thomas et al. 1995). This screen resulted in the isolation of a dominant allele, MET30-1, that was shown to impair adenosylmethionine-mediated transcriptional regulation. Furthermore, a two-hybrid interaction could be demonstrated between Met30 and the transcriptional activator of MET25, Met4 (Thomas et al. 1995). These results, together with our experiments that link Met30 with ubiquitin-dependent proteolysis, suggest Met4 as a possible target of Met30-mediated degradation. The constitutive transcription of MET25 observed may result from the hyperaccumulation of Met4 linked to a defect in Met30-dependent proteolysis. However, Met4 stabilization in met30 mutants remains to be shown.
Recent work on the Hedgehog and Wingless signaling pathway in Drosophila suggests that the F-box protein Slimb is required for the degradation of β-catenin and other proteins. It has been proposed that Slimb is a homolog of S. cerevisiae Cdc4 (Jiang and Struhl 1998), but sequence comparison revealed that Met30 is, in fact, the closest budding yeast relative of Slimb (Fig. 7). Although budding yeast does not have a close homolog of β-catenin, it is conceivable that Met30 and Slimb target other proteins that have been conserved through evolution in both metazoa and yeast. It remains to be determined, for example, whether the metazoan homologs of Swe1 are targeted by an SCF complex containing Slimb (see below).
Figure 7.

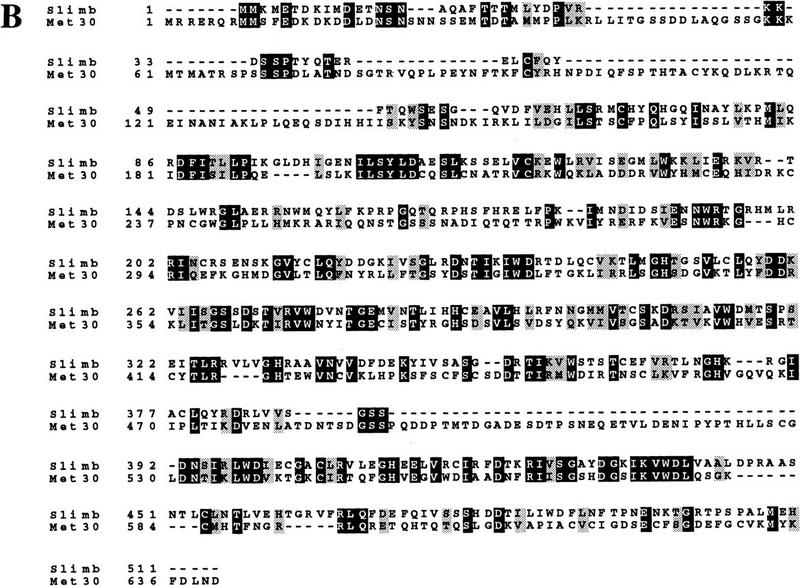
Met30 is closely related to Drosophila Slimb. (A) Dendrogram illustrating that Met30 is more closely related to Slimb and a human homolog h-βTrCP (Margottin et al. 1998) than Cdc4. The dendrogram was generated with GeneWorks 2.5 software. (B) Met30 and Slimb sequences were aligned with Clustalw. Identical amino acids are shown as white letters on black background, and related residues as black letters on gray background.
We report here that Swe1 is stabilized in met30 mutants, suggesting that the Cdc28-inhibitory kinase is degraded in a Met30-dependent manner. However, MET30 is an essential gene, with both deletion mutants and several different temperature-sensitive mutants arresting predominantly in G1 as unbudded cells. Defects in repression of genes involved in methionine biosynthesis or failure to degrade Swe1 cannot explain this cell cycle arrest phenotype, suggesting that other proteins are subjected to Cdc34-dependent ubiquitination by Met30.
All of the budding yeast F-box proteins analyzed genetically to date appear to be involved in the proteolysis of at least several proteins. Remarkably, they seem to target simultaneously both proteins implicated in cell cycle regulation and control of general metabolism. For example, Cdc4 has been implicated in ubiquitination of the cell cycle regulatory proteins Sic1, Far1, and Cdc6 (McKinney et al. 1993; Drury et al. 1997; Feldmann et al. 1997; Henchoz et al. 1997; Skowyra et al. 1997) as well as the transcriptional activator Gcn4 (D. Kornitzer, pers. comm.). Similarly, Grr1 is required for proteolysis of the G1 cyclins Cln1 and Cln2 (Barral et al. 1995) as well as a yet unknown target that mediates glucose repression. Thus, recognition and targeting by a particular F-box protein appears to depend more on protein structure than on function. Surprisingly though, there are no obvious sequence homologies relating substrates of a particular F-box protein. Thus, the mechanism whereby substrate specificity is achieved remains to be determined.
SCF-mediated proteolysis and entry into mitosis
In most eukaryotes, entry into mitosis is controlled by phosphorylation and dephosphorylation of cyclin-dependent kinases. Wee1-like kinases phosphorylate Cdks on conserved tyrosine residues, thus inhibiting their activity. Dephosphorylation by Cdc25 phosphatases results in activation of the Cdk/cyclin complexes and triggers entry into mitosis. Our results demonstrate that Swe1, the budding yeast homolog of Wee1, is degraded in a Met30-dependent manner. Wee1 might be regulated by Met30 homologs in other eukaryotes, because proteins highly homologous to Met30 are found in many other eukaryotes, including mammals. Furthermore, analyzing Wee1 protein sequences from various organisms (human, Xenopus, Drosophila, and fission yeast), we identified at least one PEST motif in each protein, suggesting that they are degraded by the Cdc34–SCF pathway. In fact, synchrony experiments in Schizosaccharomyces pombe have shown that Wee1 protein levels are greatly reduced at the onset of mitoses despite constant transcript levels (Aligue et al. 1997).
The abruptness of the G2/M transition is thought to be the result of positive feedback loops, with cyclin B/Cdk activation leading to Wee1 inactivation and Cdc25 activation. It is possible that cyclin B/Cdk-mediated phosphorylation of Wee1 creates a signal for ubiquitination and leads to Wee1 degradation. Thus, SCF-mediated proteolysis may play a significant role in the regulation of the phosphorylation state of cyclin-dependent kinases, contributing to control of entry into mitosis.
In budding yeast, the presence of Swe1 does not affect cell cycle progression in the unperturbed cell cycle. However, when cell polarity is impaired, Swe1 introduces a prolonged G2 delay, which has been ascribed to a checkpoint monitoring cell morphogenesis (Lew and Reed 1995a,b; Sia et al. 1996). Our findings suggest that Met30-dependent ubiquitination contributes to maintaining low Swe1 levels in unperturbed cells, and it will be interesting to determine whether this pathway may be blocked in response to the morphogenesis checkpoint.
Materials and methods
Yeast strains and culture
All yeast strains were isogenic to 15DaubΔ, a bar1Δ ura3Δns derivative of BF264-15D (MATa ade1 his2 leu2-3,112 trp1-1a) (Reed et al. 1985). All strains were grown in standard culture medium and standard yeast genetic methods were used (Guthrie and Fink 1991).
Enhancer screen and cloning of MET30
A diploid cdc34-3 strain was transformed with three different pools of a mTn-3xHA/lacZ transposon mutagenized yeast genomic library resulting in random heterozygous gene disruptions marked with the URA3 gene (Burns et al. 1994, Ross et al. 1997). A total of 8000 uracil prototrophic transformants were replica plated to YEPD plates and incubated at semipermissive temperature (32°C). Colonies that failed to grow at 32°C were picked from the master plate, retested, and sporulated. The disrupted genes from mutants that showed cosegregation of lethality and uracil prototrophy were identified by rescue of the mTn-3xHA/lacZ insertion including flanking regions (see http://ycmi.med.yale.edu/YGAC/home.html). The MET30 ORF including 1000 bp up- and downstream was amplified by PCR (primer PK55, 5′-GTCTGGTATACCGACACAG-3′, and PK56, 5′-CCTCGGATTCAT ACTGGAA-3′) and a 3.2-kb XbaI–SphI fragment was subcloned into puc19 (resulting in plasmid Pp70) and YCplac33 (resulting in plasmid Pp88; Gietz and Sugino 1988). To disrupt MET30, a HincII–BglII fragment containing the entire coding region was replaced by a DNA fragment conferring kanamycin and G418 resistance [HincII–HpaI fragment from plasmid kanMX2 (Wach et al. 1994)].
Construction of temperature-sensitive met30 mutants
Temperature-sensitive met30 alleles were isolated following a procedure similar to the one described by Muhlrad et al. (1992). Briefly, primers PK55 and PK56 were used to amplify MET30 under mutagenic conditions (1 mm dCTP, dTTP, 0.2 mm dGTP, dATP, 30 pmole of each primer, 7 mm MgCl2, 0.5 mm MnCl2, 20 mm Tris-HCl at pH 8.4, 50 mm KCl, and 0.5 units of Taq DNA polymerase in a total volume of 100 μl). The PCR product was cotransformed with PstI-cut Pp88 into a strain disrupted for MET30 and kept alive by a GAL1–MET30 inducible allele. The PstI digestion removes most of the MET30 coding region and the gap was repaired in vivo by homologous recombination with the mutagenized MET30 PCR fragment. Transformants were grown on glucose plates at 25°C, replica plated and colonies unable to grow at 37°C were selected. Plasmids containing temperature-sensitive met30 alleles were rescued, a XbaI–SphI fragment containing the gene was subcloned into YIplac204 (Gietz and Sugino 1988), and the met30 mutant alleles were then integrated at the TRP1 locus.
Immunoprecipitations
Strains expressing carboxy-terminal epitope-tagged proteins were constructed by inserting the DNA sequence encoding the epitope tag in front of the stop codon. All tagged proteins were functional because they rescued a strain deleted for the corresponding gene. Amino-terminal GST-tagged Skp1 was expressed under the control of the CUP1 promoter on a single copy plasmid. An XbaI–KpnI fragment (from plasmid pYEXTM4T, Amrad Biotech) containing the CUP1 promotor in front of the GST-coding region was ligated with XbaI–KpnI-digested YCplac33 (resulting in plasmid Pp87; Gietz and Sugino 1988). The Skp1 coding region was amplified with PCR primers that added a BamHI site to each end and cloned into the single BamHI site of Pp87. Expression of GST–Skp1 was induced by addition of CuSO4 to a final concentration of 100 μm. For immunoprecipitations cells were broken with glass beads in buffer A (50 mm Tris-HCl at pH 7.5, 150 mm NaCl, 0.1% NP40, 10 mm sodium pyrophosphate, 5 mm EDTA, 5 mm EGTA, 0.1 mm orthovanadate, 1 mm PMSF, and 2 μg/ml aprotinin, leupeptin, and pepstatin) by 5 × 1 min vortexing with 1-min cooling intervals on ice. Cell debris was sedimented for 15 min at 14000g and protein concentration was determined by absorption at 280 nm. A total of 500 μg of protein was incubated with 12CA5 antibodies (ascites fluid) directed against the HA epitope (covalently coupled to protein A beads) or glutathione beads, respectively. Unbound proteins were removed by four washes in 1 ml of buffer A, and bound proteins were eluted by boiling in 100 μl of gel-loading buffer. Proteins were separated by SDS-PAGE and analyzed by immunoblotting with anti-HA (12CA5, BAbCO) anti-RGS6H (Qiagen), or anti-GST antibodies (prepared by immunizing rabbits with purified GST purified from Escherichia coli), respectively.
Construction of the GAL–SWE1–myc12 plasmid
A 3.7-kb XbaI–BamHI fragment from pSWE1-HA (Booher et al.1993) containing GAL1–SWE1–HA was cloned into an altered version of pRS306 that had the polylinker sites from KpnI to SmaI deleted. The NheI site in the HA tag sequence was converted into a SalI site, the DNA was digested with NheI and ligated with a 100-fold molar excess of the synthetic oligonucleotide 5′-CTAGCGTCGACG-3′, which anneals to itself to generate a SalI site that ligates into the NheI site. A 400-bp XhoI–SalI fragment containing 12 tandem copies of the myc epitope (P. Russell, TSRI) was cloned into the generated SalI site. The construct was digested with StuI for integration at URA3.
Determination of Swe1 stability
Cells containing the GAL1–SWE1–myc12 construct were grown in YEPS (1% yeast extract, 2% Bacto-peptone, 2% sucrose) at 23°C and shifted to 37°C for 2 hr. Galactose was then added to 2% and the cells were incubated for a further 10 min to induce Swe1–myc12 expression. Cells were then harvested by centrifugation, resuspended at 108 cells/ml in labeling medium [yeast nitrogen base minus methionine, 2% sucrose, and 2% galactose with 0.25 mCi/ml (0.183 mm) Trans35S-Label (ICN)], and incubated for a further 10 min at 37°C. Labeled cells were collected by filtration, washed with prewarmed YEPD (1% yeast extract, 2% Bacto-peptone, 2% dextrose), and resuspended at 3 × 107 cells/ml in fresh YEPD supplemented with 3 mm methionine and 0.5% casamino acids to prevent further labeling. Incubation was continued at 37°C for 4 hr. Aliquots of cells were diluted into ice-cold 10 mm NaN3, harvested by centrifugation, washed with icecold 10 mm NaN3, and frozen at −80°C. To determine Swe1 stability in synchronized cultures, cells were grown in YEPS at 23°C, arrested by addition of 200 mm hydroxyurea for 3 hr (>80% accumulation of large budded cells), and then shifted to 37°C for an additional hour to inactivate the temperature-sensitive alleles. Cells were processed as described above but in the presence of 200 mm hydroxyurea. Cell pellets were lysed in ice-cold 50 mm Tris (pH 7.5), 5 mm EDTA, 1 mm sodium pyrophosphate, 150 mm sodium chloride, 1% NP-40, 1 mm sodium orthovanadate, 1 mm PMSF, 1 μg/ml leupeptin, and 1 μg/ml pepstatin. Swe1–myc12 was immunoprecipitated with the anti-c-Myc (9E10) antibody (Santa Cruz Biotechnology) from lysates containing equal amounts of radioactive label, and the immunoprecipitates were washed three times with lysis buffer, boiled 10 min in 1× SDS sample loading buffer, and separated by SDS-PAGE in 8% polyacrylamide gels. Dried gels were exposed to Molecular Dynamics storage phosphor screen for 24–48 hr, scanned on a Molecular Dynamics 445 SI PhosphorImager, and analyzed with ImageQuant version 1.2 software.
Preparation of ubiquitination-competent yeast extracts
All extracts were prepared from cells deleted for the PEP4 gene. Cells were grown to an OD600 ≅ 1, washed twice with ice cold water, and the cell pellet was resuspended in YEB [30 mm HEPES at pH 7.3, 100 mm sodium acetate, 1 mm EDTA, 1 mm EGTA, 10% glycerol, 2 mm DTT, and proteinase inhibitor cocktail (Complete minus EDTA, Boehringer)]. Sodium acetate was added to a final concentration of 200 mm and cells were lysed with a French press. Extracts were centrifuged 10 min at 27000g and 15 min at 136000g. After filtration through a 45-μm filter, extracts were desalted on a G25 column (8 ml) that was pre-equlibrated with YEB and concentrated with an amicon ultrafiltration cell (membrane YM10). Alliquots of extracts (30–45 μg/μl) were flash frozen in liquid nitrogen and stored at −70°C.
In vitro ubiquitination assay
35S-Labeled Swe1 was produced by in vitro transcription/translation of a PCR fragment containing the SWE1 coding region and the T7 promoter (TNT T7 Quick Coupled Transcription/Translation System, Promega). Mutant ubiquitin (R48) was expressed in E. coli and purified as described (Beers and Callis 1993).
Ubiquitination reactions (total volume 10 μl) contained 50 μg extract or 75 μg in case of the complementation experiment (50 μg cdc34-3 + 25 μg met30-6), 5 μg ubiquitin (Sigma), 1 μl 10× ATP regenerating system (10 mm ATP, 600 mm creatine phosphate, 10 mm magnesium acetate, 1.5 mg/ml creatine kinase in YEB), 2 μl 5× SCF-buffer (25 mm magnesium acetate, 5 mm DTT, 5 μm okadaic acid, 150 nm MG132 (Calbiochem), and proteinase inhibitor cocktail (Complete minus EDTA, Boehringer) and 1.5 μl of 35S-labeled Swe1. Reactions were incubated at 23°C for 5–15 min and terminated by addition of gel-loading buffer. Samples were separated on a 7.5% SDS–polyacrylamide gel and analyzed by exposing the gel to a PhosphorImaging screen.
Acknowledgments
We are grateful to C. Wittenberg for pointing out the F-box in Met30, D. Finley and S. Sadis for the GST–MET30 and 6xHIS-ubiquitin(R48) expression plasmids and helpful discussions during the course of this work. We thank M. Snyder and P. Ross-Mcdonald for the generous gift of the transposon mutagenized yeast library. We thank K. Flick, S. Haase, and C. Wittenberg for helpful suggestions on the manuscript and all members of the Scripps-cell cycle group in the McGowan, Reed, Russell, and Wittenberg laboratories for a stimulating environment and many constructive discussions. P.K. was supported by a Max Kade Fellowship. This work was supported by National Institutes of Health grant GM-38328 to S.I.R. and U.S. Public Health Service grant GM-53050 and by funds from the Searle Scholars Program/The Chicago Community Trust to D.J.L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL sreed@scripps.edu; FAX (619) 784-2781.
References
- Aligue R, Wu L, Russell P. Regulation of Schizosaccharomyces pombe Wee1 tyrosine kinase. J Biol Chem. 1997;272:13320–13325. doi: 10.1074/jbc.272.20.13320. [DOI] [PubMed] [Google Scholar]
- Amon A, Surana U, Muroff I, Nasmyth K. Regulation of p34CDC28 tyrosine phosphorylation is not required for entry into mitosis in S. cerevisiae. Nature. 1992;355:368–371. doi: 10.1038/355368a0. [DOI] [PubMed] [Google Scholar]
- Bai C, Sen P, Hofman K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Barral Y, Jentsch S, Mann C. G1 cyclin turnover and nutrient uptake are controlled by a common pathway in yeast. Genes & Dev. 1995;9:399–409. doi: 10.1101/gad.9.4.399. [DOI] [PubMed] [Google Scholar]
- Beers EP, Callis J. Utility of polyhistidine-tagged ubiquitin in the purification of ubiquitin-protein conjugates and as an affinity ligand for the purification of ubiquitin-specific hydrolases. J Biol Chem. 1993;268:21645–21649. [PubMed] [Google Scholar]
- Booher RN, Deshaies RJ, Kirschner MW. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993;2:3417–3426. doi: 10.1002/j.1460-2075.1993.tb06016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns N, Grimwald B, Ross-Macdonald PB, Choi EY, Finberg K, Roeder SG, Snyder M. Large-scale analysis of gene expression, protein localization, and gene disruption in Saccharomyces cerevisiae. Genes & Dev. 1994;8:1087–1105. doi: 10.1101/gad.8.9.1087. [DOI] [PubMed] [Google Scholar]
- Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Chau V, Kirschner MW. Ubiquitination of the G1 cyclin Cln2p by the Cdc34p-dependent pathway. EMBO J. 1995;14:303–312. doi: 10.1002/j.1460-2075.1995.tb07004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury LS, Perkins G, Diffley J. The Cdc4/34/53 pathway targets Cdc6p for proteolysis in budding yeast. EMBO J. 1997;16:5966–5976. doi: 10.1093/emboj/16.19.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy WG. The decision to enter mitosis. Trends Cell Biol. 1994;4:202–207. doi: 10.1016/0962-8924(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Feldmann RMR, Correll CC, Kaplan KB, Deshaies RJ. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to yeast genetics and molecular biology, methods in enzymology. San Diego, CA: Academic Press; 1991. [PubMed] [Google Scholar]
- Henchoz S, Chi Y, Catarin B, Herskowitz I, Deshaies RJ, Peter M. Phosphorylation- and ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor far1p in budding yeast. Genes & Dev. 1997;11:3046–3060. doi: 10.1101/gad.11.22.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Gen. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Beaudenon S, Howley PM. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci. 1995;92:5249. doi: 10.1073/pnas.92.11.5249-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irniger S, Piatti S, Michaelis C, Nasmyth K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 1995;81:269–277. doi: 10.1016/0092-8674(95)90337-2. [DOI] [PubMed] [Google Scholar]
- Jiang J, Struhl G. Regulation of the Hedgehog and Wingless signalling pathways by the F- box/WD40-repeat protein Slimb. Nature. 1998;391:493–496. doi: 10.1038/35154. [DOI] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20s complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Lamb JR, Michaud WA, Sikorski RS, Hieter PA. Cdc16p, Cdc23p and Cdc27p form a complex essential for mitosis. EMBO J. 1994;13:4321–4328. doi: 10.1002/j.1460-2075.1994.tb06752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammer D, Mathias N, Laplaza JM, Jiang W, Liu Y, Callis J, Goebl M, Estelle M. Modification of yeast Cdc53p by the ubiquitin-related protein Rub1p affects function of the SCFCdc4 complex. Genes & Dev. 1998;12:914–926. doi: 10.1101/gad.12.7.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanker S, Valdivieso MH, Wittenberg C. Rapid degradation of the G1 cyclin Cln2 induced by CDK-dependent phosphorylation. Science. 1996;271:1597–1601. doi: 10.1126/science.271.5255.1597. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Morphogenesis in the yeast cell cycle: Regulation by Cdc28 and cyclins. J Cell Biol. 1993;120:1305–1320. doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J Cell Biol. 1995a;129:739–749. doi: 10.1083/jcb.129.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Cell cycle control of morphogenesis in budding yeast. Curr Opin Genet Dev. 1995b;5:17–23. doi: 10.1016/s0959-437x(95)90048-9. [DOI] [PubMed] [Google Scholar]
- Li FN, Johnston M. Grr1 of Saccharomyces cerevisiae is connected to the ubiquitin proteolysis machinery through Skp1: Coupling glucose sensing to gene expression and the cell cycle. EMBO J. 1997;16:5629–5638. doi: 10.1093/emboj/16.18.5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. A novel human WD protein, h-βTrCP, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998;4:565–574. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- Marini NJ, Meldrum E, Buehrer B, Hubberstey AV, Stone DE, Traynor-Kaplan A, Reed SI. A pathway in the yeast cell division cycle linking protein kinase C (Pkc1) to activation of Cdc28 at START. EMBO J. 1996;15:3040–3052. [PMC free article] [PubMed] [Google Scholar]
- Mathias N, Johnson SL, Winey M, Adams AEM, Goetsch L, Pringle JR, Byers B, Goebl MG. Cdc53p acts in concert with Cdc4p and Cdc34p to control the G1-to-S-phase transition and identifies a conserved family of proteins. Mol Cell Biol. 1996;16:6634–6643. doi: 10.1128/mcb.16.12.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney JD, Chang F, Heintz N, Cross FR. Negative regulation of FAR1 at the start of the cell cycle. Genes & Dev. 1993;7:833–843. doi: 10.1101/gad.7.5.833. [DOI] [PubMed] [Google Scholar]
- Muhlrad D, Hunter R, Parker R. A rapid method for localized mutagenesis of yeast genes. Yeast. 1992;8:79–82. doi: 10.1002/yea.320080202. [DOI] [PubMed] [Google Scholar]
- Patton EE, Willems AR, Sa D, Kuras L, Thomas D, Craig KL, Tyers M. Cdc53 is a scaffold protein for multiple Cdc34/Skp1/F-box protein complexes that regulate cell division and methionine biosynthesis in yeast. Genes & Dev. 1998;12:692–705. doi: 10.1101/gad.12.5.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatti S, Böhm T, Cocker JH, Diffley JFX, Nasmyth K. Activation of S-phase-promoting CDKs in late G1 defines a “point of no return” after which Cdc6 synthesis cannot promote DNA replication in yeast. Genes & Dev. 1996;10:1516–1531. doi: 10.1101/gad.10.12.1516. [DOI] [PubMed] [Google Scholar]
- Reed SI, Hadwiger JA, Lorincz AT. Protein kinase activity associated with the product of the yeast cell division cycle gene CDC28. Proc Natl Acad Sci. 1985;82:4055–4059. doi: 10.1073/pnas.82.12.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: The PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- Ross MP, Sheehan A, Roeder GS, Snyder M. A multipurpose transposon system for analyzing protein production, localization, and function in Saccharomyces cerevisiae. Proc Natl Acad Sci. 1997;94:190–195. doi: 10.1073/pnas.94.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner AD, Murray AW. The spindle assembly checkpoint. Curr Opin Cell Biol. 1996;8:773–780. doi: 10.1016/s0955-0674(96)80077-9. [DOI] [PubMed] [Google Scholar]
- Russell P, Moreno S, Reed SI. Conservation of mitotic controls in fission and budding yeasts. Cell. 1989;57:295–303. doi: 10.1016/0092-8674(89)90967-7. [DOI] [PubMed] [Google Scholar]
- Schwob E, Böhm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Sia RA, Herald HA, Lew DJ. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol Biol Cell. 1996;7:1657–1666. doi: 10.1091/mbc.7.11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–198. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Kuras L, Barbery R, Cherest H, Blaiseau PL, Surdin-Kerjan Y. Met30p, a yeast transcriptional inhibitor that responds to S-adenosylmethionine, is an essential protein with WD40 repeats. Mol Cell Biol. 1995;15:6526–6534. doi: 10.1128/mcb.15.12.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. The N-end rule: Function, mysteries, uses. Proc Natl Acad Sci. 1996;93:12142–12149. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Feldman RM, Deshaies RJ. SIC1 is ubiquitinated in vitro by a pathway that requires CDC4, CDC34, and cyclin/CDK activities. Mol Biol Cell. 1997;8:1427–1437. doi: 10.1091/mbc.8.8.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Willems AR, Lanker S, Patton EE, Craig KL, Nason TF, Mathias N, Kobayashi R, Wittenberg C, Tyers M. Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell. 1996;86:453–463. doi: 10.1016/s0092-8674(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Yaglom J, Linskens MHK, Sadis S, Rubim DM, Futcher B, Finley D. p34Cdc28-mediated control of Cln3 cyclin degradation. Mol Cell Biol. 1995;15:731–741. doi: 10.1128/mcb.15.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]