

Abstract
Cyclin D1 is part of a cell cycle control node consistently deregulated in most human cancers. However, studies with cyclin D1-null mice indicate that it is dispensable for normal mouse development as well as cell growth in culture. Here, we provide evidence that ras-mediated tumorigenesis depends on signaling pathways that act preferentially through cyclin D1. Cyclin D1 expression and the activity of its associated kinase are up-regulated in keratinocytes in response to oncogenic ras. Furthermore, cyclin D1 deficiency results in up to an 80% decrease in the development of squamous tumors generated through either grafting of retroviral ras-transduced keratinocytes, phorbol ester treatment of ras transgenic mice, or two-stage carcinogenesis.
Keywords: Knockout, Tg.AC, cell cycle, keratinocyte, two-stage carcinogenesis
Research carried out over the last several years has revealed a distinct biochemical pathway regulating G1 progression in mammalian cells. In this pathway, a cyclin D–cyclin-dependent kinase (CDK)4/6 protein complex phosphorylates the retinoblastoma protein (pRb) to promote progression through a late G1 checkpoint (Weinberg 1995). Such process may be negatively regulated by a series of CDK inhibitors (CDKIs), including p15, p16, p21, and p27. Many of the proteins that participate in this regulatory circuit have been implicated in the causation of various types of human tumors (Sherr 1996). For example, several laboratories have reported that subsets of tumors of a particular type show loss of pRb or, alternatively, overexpression of cyclin D1 (Jiang et al. 1993; Bartkova et al. 1994; Schauer et al. 1994). Similarly, in yet other tumor types, loss of p16 and pRb appears to be mutually exclusive (Otterson et al. 1994; Aagaard et al. 1995; Shapiro et al. 1995). These observations have led to the hypothesis that inactivation of cyclin D1–CDK4/6–p16–pRb by means of either loss of suppressor activity of pRb or p16, or overexpression of cyclin D1, can promote tumor development (Weinberg 1995; Sherr 1996).
Several lines of evidence indicate that the cyclin D1–CDK4/6–p16–pRb regulatory circuit lies downstream of oncogenic ras. The tumor suppressor activity of p16 was demonstrated through its ability to inhibit ras-induced proliferation and cellular transformation (Serrano et al. 1995), and it was recently observed that cell cycle arrest following ras inactivation is dependent on functional pRb (Peeper et al. 1997). Moreover, cyclin D1 expression can be stimulated in vitro by oncogenic ras in epithelial (Filmus et al. 1994) as well as fibroblast (Liu et al. 1995) cell lines, apparently through activation of the MAP kinase cascade (Albanese et al. 1995; Liu et al. 1995). Data from experimental and human cancers also indicate that cyclin D1 mediates the proliferative response to oncogenic ras. Thus, mouse skin tumors induced by a two-stage carcinogenesis protocol, which contain a characteristic oncogenic mutation in codon 61 of the Ha-ras gene, overexpress cyclin D1 (Robles and Conti 1995), although they have a very low frequency of cyclin D1 gene amplification (Bianchi et al. 1993). Such amplification represents a common mechanism of cyclin D1 up-regulation in human tumors (Peters 1994). Similar results have been observed in carcinogen-induced rat mammary tumors (Sgambato et al. 1995), which also carry characteristic ras mutations (Zarbl et al. 1985). Recently, a strong positive correlation between cyclin D1 and c-Ki–ras immunoexpression was reported in human epithelial ovarian tumors (Hung et al. 1996). However, none of these reports demonstrates that cyclin D1 is necessary for oncogenic ras-mediated growth.
Here, we provide biochemical and genetic evidence that cyclin D1 is a critical target for oncogenic ras in mouse skin. Cyclin D1, its association with CDK4, and CDK4 kinase activity are up-regulated in response to retroviral ras. In addition, ras-mediated skin tumorigenesis is substantially reduced in a cyclin D1-deficient background.
Results
Retroviral ras-associated expression of cyclin D1 and cyclin E
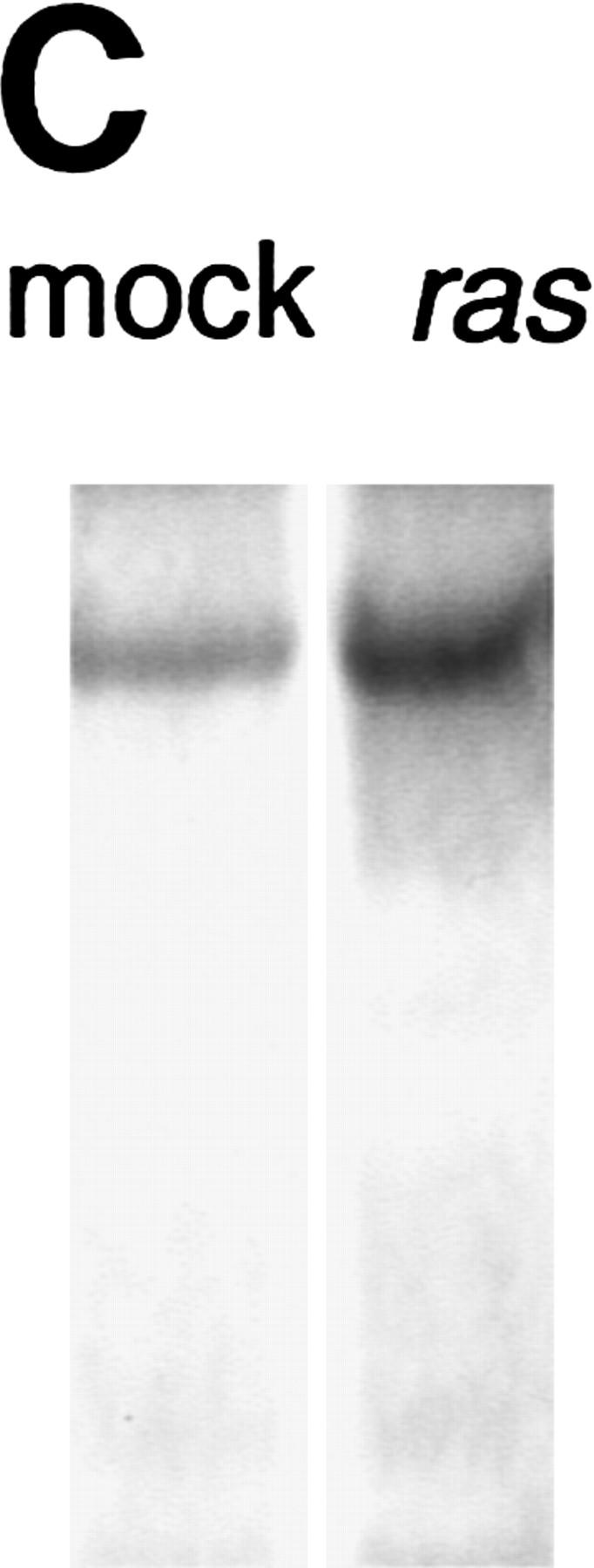
Retroviral introduction of a mutant ras oncogene into cultured murine keratinocytes results in an increased proliferation rate, resistance to Ca2+-induced terminal differentiation, and development of benign tumors when grafted onto athymic nude mice (Roop et al. 1986). Because of their role as positive regulators of the cell cycle, G1 cyclins were likely to mediate this response. Through total RNA Northern analysis, we found that among the G1 cyclins, cyclins D1 and E were induced by v-Ha-ras, cyclin D2 remained unchanged, and cyclin D3 mRNA was almost undetectable and was not induced by ras (Fig. 1A; data not shown). Western blot analysis of whole cell extracts showed a predominant induction of cyclin D1, a modest increase in cyclin E, and no change in cyclin D3 (Fig. 1B). A polyclonal cyclin D1 antibody weakly cross-reactive with cyclin D2 confirmed cyclin D1 induction and showed an apparent increase in cyclin D2 level (Fig. 1B).
Figure 1.

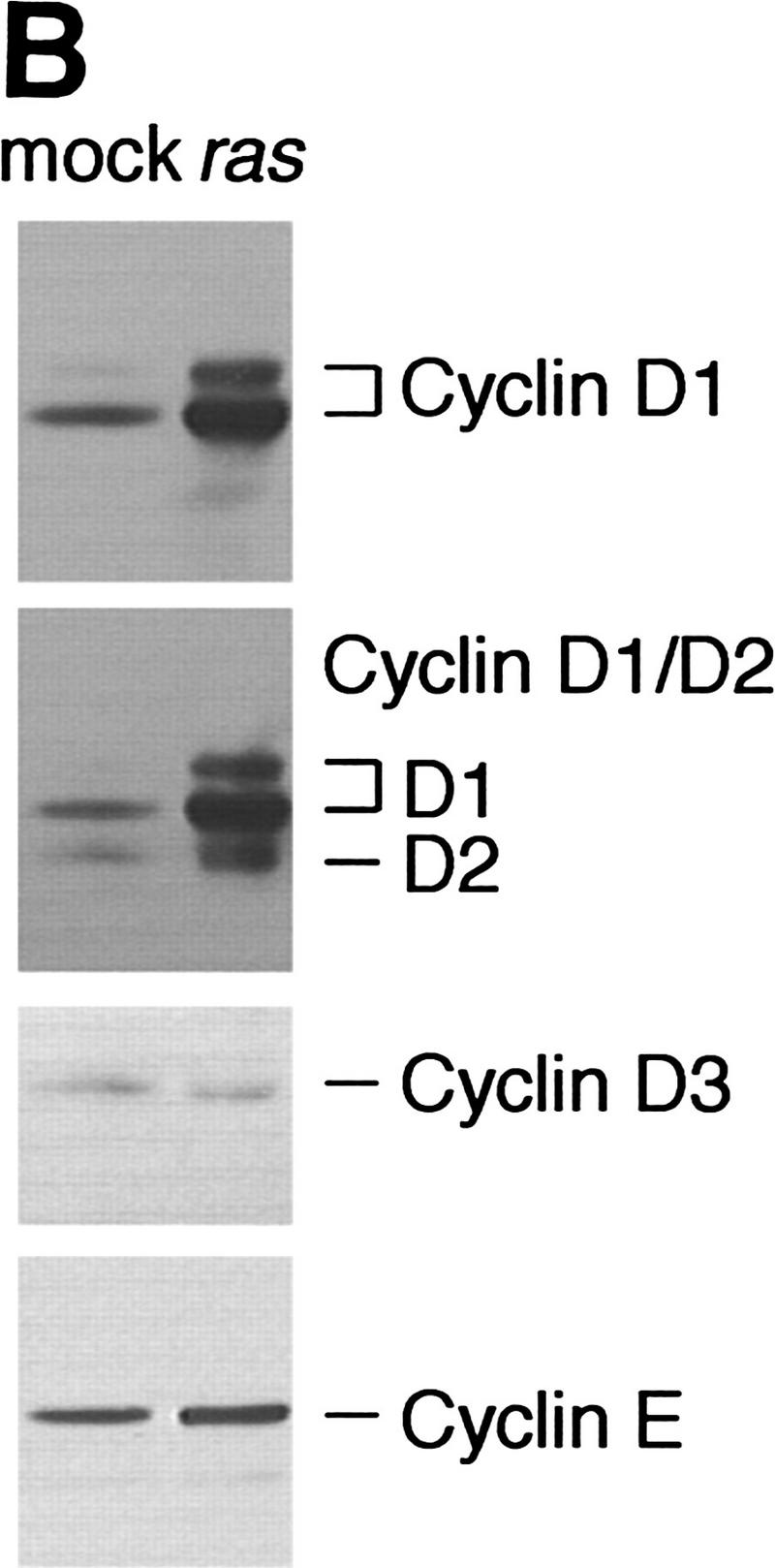
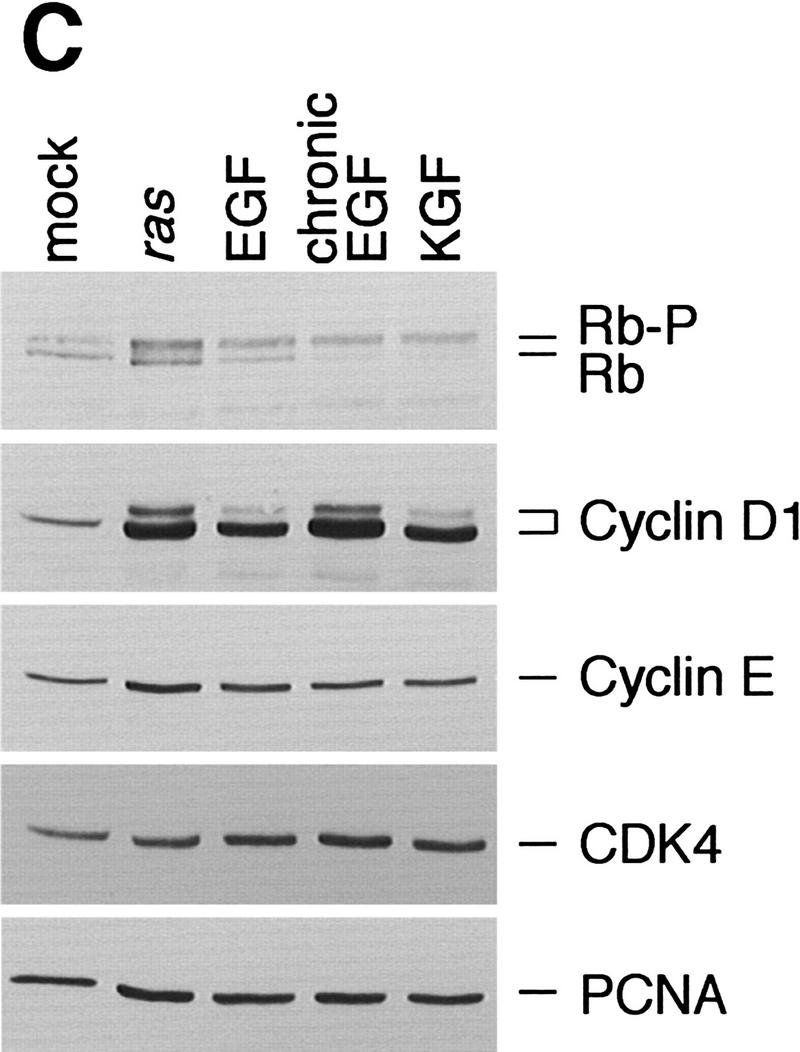
Expression of G1 cyclins mRNA (A) and proteins (B) in mock and v-Ha-ras-infected murine primary keratinocytes. (C) Comparative induction of cyclin D1 protein expression and pRb hyperphosphorylation in mock-infected (mock), v-Ha-ras-infected (ras), and after single-dose EGF (EGF), continuous presence of EGF in the media (chronic EGF), and single-dose KGF (KGF) treatments of primary murine keratinocytes.
Certain growth factors such as epidermal growth factor (EGF) or keratinocyte growth factor (KGF) are known to induce a proliferative response in cultured keratinocytes (Dlugosz et al. 1994). Consistent with extensive data regarding the role of cyclin D1 in the regulation of G1-phase transit in response to growth factors, EGF and KGF also stimulated cyclin D1 expression (Fig. 1C). The magnitude of the increase in cyclin D1 expression obtained by oncogenic ras was comparable to that induced by the continuous presence of EGF in the culture media (Fig. 1C). Other cell cycle proteins were affected only minimally by either v-Ha-ras infection or growth factor treatment, and p16 was undetectable (Fig. 1C; data not shown).
In many and perhaps all types of normal cells, inactivation of pRb by hyperphosphorylation during late G1 phase of the cell cycle is a prerequisite for S-phase entry (Weinberg 1995). Upon infection with retroviral ras, cultured keratinocytes showed an increase in the proportion of high-molecular-weight bands that are characteristic of the hyperphosphorylated forms of pRb (Fig. 1C). This band shift indicative of a higher degree of phosphorylation is concomitant with cyclin D1 induction and coincides with the increase in keratinocyte proliferation that is observed after ras infection. As expected, EGF and KGF also stimulated pRb hyperphosphorylation (Fig. 1C).
Association of cyclin D1 with CDK4 and CDK4 kinase activity are stimulated by v-Ha-ras
In CDK immunoprecipitates, cyclin D1 was found associated exclusively with CDK4 and not with CDK2 or CDK6 in normal and ras-infected keratinocytes (Fig. 2A), although these three CDKs are expressed in murine keratinocytes (data not shown). This interaction was confirmed by immunoprecipitation of cyclin D1 followed by CDK4 immunoblotting (Fig. 2B). In both cases, we observed that the amount of CDK4-bound cyclin D1 increased after ras infection proportionally to the increase in cyclin D1 in whole cell extracts. As expected, cyclin E was found associated with CDK2 in both normal and ras-infected keratinocytes (data not shown). Binding of the CDKI p21 and p27 to cyclin–CDK complexes is known to regulate their catalytic activity in response to antimitogens (Sherr and Roberts 1995). However, a possible function of these CDKIs in the assembly of active cyclin–CDK complexes has been suggested recently (LaBaer et al. 1997). We therefore studied the presence of these CDKIs in cyclin and CDK immunoprecipitates to determine if the pattern of binding changed after v-Ha-ras infection. We found that p21 and p27 were associated with CDK2 and CDK4 even after v-Ha-ras infection (Fig. 2A). Both inhibitors were also present in cyclin D1 immunoprecipitates (Fig. 2B). Surprisingly, the expression of p21 increased after ras transduction; however, the in vitro kinase activity of CDK4 toward pRb was clearly enhanced by retroviral ras (Fig. 2C), confirming the association among oncogenic ras, cyclin D1 expression, and pRb phosphorylation.
Figure 2.


Analysis of G1 cyclin–CDK complex formation in mock and v-Ha-ras-infected murine primary keratinocytes. Whole cell extracts and CDK (A) or cyclin D1 (B) immunoprecipitates were subjected to immunoblot analysis with cyclin D1, p21, and p27 (A), or CDK4, p21, and p27 (B). (C) Phosphorylation of pRb by CDK4 immunocomplex.
Absence of cyclin D1 impairs ras-associated tumor development
Immunohistochemical analysis of cyclin D1 expression in archival tumor samples generated by grafting in vitro-initiated primary murine keratinocytes into nude mice showed that in all cases, cyclin D1 was strongly expressed in basal undifferentiated keratinocytes from tumors and absent in adjacent normal skin (data not shown). This indicated a correlation between oncogenic ras and cyclin D1 expression in vivo. However, the question remained whether cyclin D1 is an essential mediator of keratinocyte transformation by oncogenic ras or is independently induced as a nonessential consequence of the proliferative response generated by ras transformation. To address these possibilities, we studied ras-mediated tumorigenesis in cyclin D1-deficient mice.
Primary keratinocytes were isolated from newborn cyclin D1 knockout, heterozygous, and wild-type siblings and transduced with retroviral v-Ha-ras. In vitro growth and morphology of cyclin D1-deficient keratinocytes was similar to those of their wild-type counterparts before and after retroviral infection (data not shown). This observation is consistent with that of normal in vitro growth of cyclin D1-deficient mouse embryo fibroblasts (Fantl et al. 1995). The growth of tumors generated by grafting v-Ha-ras infected keratinocytes was followed for several weeks after removal of the grafting chamber, and the combined results of two independent experiments are shown in Figure 3A. Tumors obtained by chamber grafting of ras-infected cyclin D1-deficient primary keratinocytes were at least 50% smaller throughout the experiment than those from isogenic wild-type and heterozygous controls. However, cyclin D1-deficient papillomas presented a morphology and histopathology indistinguishable from that of the controls, except for expression of cyclin D1 (Fig. 3B,C).
Figure 3.

(A) Primary keratinocytes from each cyclin D1 genotype were infected with v-Ha-ras retrovirus and grafted onto nude mice. Approximate tumor volume was calculated as tumor height × length × width in mm. Error bars, s.e.m. Immunohistochemical staining of cyclin D1 in a cyclin D1 heterozygous (B) and a cyclin D1 knockout (C) graft.
A second confirmatory study of tumor susceptibility in the presence of exogenous ras involved the development of benign skin tumors in the v-Ha-ras transgenic Tg.AC mouse strain in response to treatment with the phorbol ester TPA (Spalding et al. 1993). Upon treatment with TPA, the ras transgene is expressed in preneoplastic focal follicular hyperplasias and in papillomas, but not in non-tumor-bearing Tg.AC mouse skin (Hansen and Tennant 1994a,b). Consistently, we found that cyclin D1 expression was up-regulated in papillomas and even in follicular hyperplasias of Tg.AC mice but not in TPA-treated uninvolved skin (data not shown). Genetically matched female mice were generated that carried a v-Ha-ras transgene (Tg.AC+/−) and were cyclin D1−/− (n = 7) or cyclin D1+/− (n = 5). This cross also yielded v-Ha-ras transgene-negative (Tg.AC−/−) cyclin D1−/− (n = 10) or cyclin D1+/− (n = 5) females that were used as controls (Fig. 4A). As expected, in the absence of v-Ha-ras transgene no tumors appeared in response to TPA treatment, regardless of cyclin D1 status. A multitude of papillomas developed on the backs of Tg.AC+/− mice as a result of TPA application. Papilloma multiplicity was up to sixfold higher in Tg.AC+/− cyclin D1+/− mice compared to Tg.AC+/− cyclin D1−/− siblings. Tumor appearance was delayed by 1 week in cyclin D1-deficient Tg.AC mice, but the fraction of mice with papillomas at the end of the experiment was similar in both groups (3/5 for Tg.AC+/− cyclin D1+/− and 5/7 for Tg.AC+/− cyclin D1−/−).
Figure 4.

(A) Papilloma development in isogenic female mice generated through backcrossing of the Tg.AC transgene onto cyclin D1 knockout background and treated with TPA. (B) Papilloma development in isogenic cyclin D1 knockout (−/−), heterozygous (+/−), and wild-type (+/+) mice subjected to a two-stage carcinogenesis protocol using DMBA as initiator and TPA as promoter.
To determine if skin tumor development mediated by mutation of endogenous Ha-ras was also dependent on cyclin D1 expression, a third experimental approach consisting of a two-stage protocol of skin carcinogenesis was undertaken next. Such protocol involves the application of a single dose of the genotoxic compound 9,10-dimethyl-1,2-benz[a]anthracene (DMBA), followed by multiple applications of TPA, and results in the development of benign skin lesions. These papillomas contain a characteristic oncogenic mutation in codon 61 of the Ha-ras gene and overexpress cyclin D1 (Robles and Conti 1995). In this experiment tumor appearance was delayed by 2 weeks in cyclin D1−/− mice, and the fraction of mice with papillomas at the end of the experiment (20 weeks) was 56% (5/9) in cyclin D1−/−, 96% (23/24) in cyclin D1+/−, and 100% (11/11) in cyclin D1+/+ mice. Papilloma multiplicity in cyclin D1−/− mice was 25% of that of cyclin D1+/− mice and 22% of that of cyclin D1+/+ mice (Fig. 4B).q
Taken together, the results of these three protocols of experimental carcinogenesis indicate that cyclin D1 has a unique role in promoting ras-mediated growth in mouse skin and that ras-mediated tumor development is dependent on pathways that favor cyclin D1 expression.
Possible mechanism of v-Ha-ras stimulation of cyclin D1 expression in keratinocytes
Cyclin D1 induction by activated ras genes has been reported previously in a number of established fibroblast cell lines (Albanese et al. 1995; Liu et al. 1995; Winston et al. 1996) and in intestinal epithelial cells (Filmus et al. 1994). In fibroblasts, cyclin D1 seems to be directly induced by the ras-signaling cascade. Because this induction is insensitive to growth factor receptor blockade, it appears that it is not dependent on an extracellular autocrine-signaling loop (Winston et al. 1996). However, autocrine stimulation has proven to be necessary for development of oncogenic ras-associated epithelial tumors, as indicated by the fact that EGF receptor (EGFR)-deficient keratinocytes form smaller tumors than their wild-type counterparts when transformed by retroviral ras and subsequently grafted onto nude mice (Dlugosz et al. 1997). In view of our data from tumorigenic assays indicating a role for cyclin D1 in the proliferation of ras-transformed keratinocytes, we hypothesized that in epithelial cells, cyclin D1 induction by oncogenic ras might also be dependent on autocrine stimulation. Consistently, we found that exposure of cultured keratinocytes to the protein tyrosine kinase inhibitor Tyrphostin 47, which is known to block EGFR signaling (Dvir et al. 1991), resulted in down-regulation of cyclin D1 in both EGF-treated as well as v-Ha-ras-infected keratinocytes (Fig. 5A). It has been demonstrated previously that at the dosage used in this experiment (50 μm), Tyrphostin 47 is not cytotoxic but cytostatic for cultured keratinocytes (Dvir et al. 1991), and its effect can be reversed by culturing in fresh media devoid of tyrphostin (data not shown). However, although tyrphostin preferentially inhibits the EGFR-associated kinase, it may inhibit other kinases as well. Therefore, to test our hypothesis in a system that was specifically deficient in EGFR, we analyzed cyclin D1 protein expression before and after ras transduction in keratinocytes derived from EGFR wild-type and null newborn mice and grown in basal media (Dlugosz et al. 1997). With equal total protein loading and equal expression of v-Ha-ras, expression of cyclin D1 was four-fold higher in EGFR wild-type keratinocytes than in their EGFR null siblings upon retroviral transduction of v-Ha-ras (Fig. 5B).
Figure 5.


(A) v-Ha-ras-infected primary murine keratinocytes were treated with 50 μm of tyrphostin 47 or DMSO for 18 hr. As a control, primary murine keratinocytes were treated with 10 ng/ml EGF for 18 hr in addition to tyrphostin. (B) Expression of cyclin D1 and v-Ha-ras in mock- and v-Ha-ras-infected primary murine keratinocytes isolated from EGFR knockout mice and wild type. Expression of actin was used for equal loading correction.
Discussion
In view of the critical role of cyclin D1 in G1 progression it was surprising that cyclin D1-deficient mice developed with few very specific tissue defects (Fantl et al. 1995; Sicinski et al. 1995). Nonetheless, the observation of defective proliferation of cells from the retina and mammary gland confirmed the functional importance of cyclin D1 in vivo and was consistent with the role of cyclin D1 in mammary gland proliferation and carcinogenesis (Peters 1994).
Using three diverse models of ras-mediated tumorigenesis of mouse skin in a cyclin D1-deficient background, we have provided genetic evidence of the role of cyclin D1 as a downstream mediator of oncogenic ras. This is in agreement with previous biochemical evidence of signaling between ras and cyclin D1 and ras and pRb (Peeper et al. 1997). We found that although lack of cyclin D1 did not completely inhibit cellular transformation, it significantly reduced the efficiency of tumor development. During preliminary studies of protein expression we have observed no compensatory up-regulation of either cyclins D2 or D3 in the tumors that developed in a cyclin D1-deficient background (M.L. Rodríguez-Puebla and C.J. Conti, in prep.). This indicates that cyclin D1 is a preferential target of oncogenic ras and underscores the functional uniqueness of each D-type cyclin. It is also consistent with previous observations in vitro using cyclin D1-deficient fibroblasts (Fantl et al. 1995). Other evidence supporting this hypothesis is that cyclin D1 did not behave as an oncogene when expressed in stratified epithelia (Robles et al. 1996) and did not synergize with oncogenic ras in the development of squamous tumors (A.I. Robles, M.L. Rodríquez-Puebla, and C.J. Conti, unpubl.).
We have extended our observations to show that oncogenic ras-mediated expression of cyclin D1 is also dependent on autocrine stimulation through the EGFR. Our data are consistent with previous reports on the importance of autocrine stimulation through the EGFR in epithelial tumors (Dlugosz et al. 1997; Gangarosa et al. 1997) and is in conflict with the observation that in ras-transformed fibroblasts cyclin D1 induction is insensitive to growth factor receptor blockade (Winston et al. 1996). This difference might be explained by a response that is cell-type specific. Both v-H-ras and v-H-raf can transform NIH-3T3 fibroblasts, and cyclin D1 is up-regulated in each case (Liu et al. 1995). In contrast, in rat intestinal epithelial cells a raf-independent EGFR autocrine loop is necessary for ras transformation (Gangarosa et al. 1997). It would appear that the crosstalk between cyclin D1 and autocrine stimulation through the EGFR is unique to oncogenic ras in epithelial cells, and its implications warrant further investigation.
Materials and methods
Cell culture
Primary keratinocytes were isolated from newborn mice, cultured, and infected with retroviral v-Ha-ras as described previously (Dlugosz et al. 1995). All cultures were harvested at day 8 of culture for RNA or protein extraction. Growth factors were added to the basic media at 10 ng/ml where indicated. In one set of experiments, 50 μm of the protein tyrosine kinase inhibitor Tyrphostin 47 (RG-50864 or AG 213, Sigma) or its vehicle (DMSO) alone were added to the cultures for 18 hr before harvesting for protein extraction.
Northern blotting
Total RNA was extracted from mock- and v-Ha-ras-infected primary keratinocytes with Trizol (GIBCO BRL). Total RNA (20 μg) was separated on a 0.8% agarose gel, blotted onto a nylon membrane (Amersham), and hybridized with 32P-labeled cDNAs. Human cyclin D1 cDNA was provided by A. Arnold (Massachusetts General Hospital, Boston, MA), murine cyclin D2 and murine cyclin D3 cDNAs were provided by C. Sherr (St. Jude Children’s Research Hospital, Memphis, TN), and murine cyclin E cDNA was provided by J.A. DeLoia (University of Pittsburgh, PA). GAPD (ATCC) was used as a control for equal loading. Hybridizations were performed with QuickHyb (Stratagene) followed by autoradiography.
Immunoblotting and immunoprecipitation
Cultures were washed in ice-cold PBS and lysed in 50 mm Tris (pH 7.4) containing 150 mm NaCl, 0.5% NP-40, 50 mm NaF, 2 mm Na3VO4, 1 mm DTT, 1 mm PMSF, and 25 μg/ml aprotinin. One hundred micrograms was electrophoresed through 10% or 7.5% SDS-PAGE and transferred to Hybond-ECL membranes (Amersham). Immunoblots were incubated for 1 hr at room temperature in blocking solution (TBS with 5% milk and 0.05% Tween 20) followed by the primary antibody diluted in blocking solution for 1–2 hr. The following primary antibodies were used: mouse monoclonal antibodies against cyclin D1 (Zymed), Rb (Pharmingen), Ha-ras (Transduction Labs), and actin (clone C4, Boehringer Mannheim), and rabbit polyclonal antibodies against cyclin D1 (Upstate Biotechnology), weakly cross-reactive with cyclin D2, and against cyclin D3 (C-16), cyclin E (M-20), PCNA, CDK2, CDK4, CDK6, p21, and p27 (Santa Cruz Biotechnology). The immunoblots were developed with a horseradish peroxidase-conjugated secondary antibody followed by chemiluminescence (Amersham).
One milligram of total cell extract was used for coimmunoprecipitation followed by immunoblotting. Extracts were incubated with 1 μg of antibody, and the immunocomplexes were precipitated with protein G–Sepharose (GIBCO BRL), washed, separated through 10% SDS-PAGE, and transferred to nitrocellulose. Immunoblots were treated as above.
Primary keratinocytes derived from EGFR wild-type and null newborns (Dlugosz et al. 1997) were lysed in 50 mm Tris (pH 7.4) containing 1% NaDOC, 1% Triton X-100, 0.1% SDS, 150 mm NaCl, 0.5 mm EDTA, 1 mm EGTA, 10 mm NaF, 200 μm Na3VO4, 1 mm DTT, 1 mm PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Twenty-five micrograms of protein was electrophoresed through a 12% SDS–polyacrylamide gel followed by Western blotting.
CDK4 kinase assay
Mock- and v-Ha-ras-infected newborn primary keratinocyte cultures were lysed in 50 mm HEPES (pH 7.5) containing 150 mm NaCl, 2.5 mm EGTA, 1 mm EDTA, 0.1 % Tween 20, 1 mm DTT, 0.1 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 mm NaF, 0.1 mm Na3VO4, and 10 mm β-glycerophosphate. The cell lysate was frozen and stored at −70°C. CDK4-associated kinase activity was measured as described (Matsushime et al. 1994) against recombinant pRb using polyclonal CDK4 antibody (Santa Cruz Biotechnology) to pull down the active kinase complex.
Tumorigenicity assays
Grafting of v-H-ras-infected keratinocytes Crossing cyclin D1 heterozygous siblings generated genetically matched cyclin D1 knockout, heterozygous, and wild-type newborn mice. After genotyping (Sicinski et al. 1995) skins with equal genotype were pooled for keratinocyte isolation, plated, and infected with the v-Ha-ras-encoding retrovirus. Five days after infection they were trypsinized, combined with wild-type primary fibroblasts, and transferred to a grafting chamber surgically implanted on the back of nude mice (Dlugosz et al. 1995). The chamber was removed 1 week after grafting, and tumor development was measured as tumor height × length × width.
TPA treatment of v-Ha-ras transgenic Tg.AC mice
Female homozygous v-Ha-ras transgenic Tg.AC mice were mated with a cyclin D1 knockout male, and the F1 offspring was backcrossed with the cyclin D1-deficient parent. Female (2–5 weeks old) mice were initially treated with 2.5 μg of TPA (Sigma) applied topically in 200 μl of acetone twice a week for 5 weeks. Treatment was then reduced to 1 μg of TPA twice a week for 13 weeks. Mice were housed in cages independent of genotype, and the number of lesions was scored weekly by palpation.
Two-stage carcinogenesis
For chemical carcinogenesis studies, cyclin D1-deficient mice were backcrossed into a Sencar background. Genetically matched cyclin D1 knockout, heterozygous, and wild-type newborns were initiated at day 1 after birth by application of 50 μg of DMBA in 100 μl of acetone. At day 7, animals received 2.5 μg of TPA in 100 μl of acetone twice a week for 2 weeks. The TPA regimen was subsequently changed to 5 μg of TPA in 200 μl of acetone twice a week for 8 weeks and back to 2 μg of TPA in 200 μl of acetone for a total of 20 weeks.
Histology and immunohistochemistry
Tissues were fixed in 10% buffered formalin or 4% paraformaldehyde, and embedded in paraffin. Sections of all tissues were stained with hematoxylin and eosin for histopathological evaluation. Detection of cyclin D1 expression by immunostaining of formalin-fixed paraffin-embedded tissues has been described previously (Robles and Conti 1995), as well as detection of ras transgene by mRNA in situ hybridization using 35S-labeled riboprobe (Hansen and Tennant 1994a).
Acknowledgments
We thank Brent Whalin, Margaret LaCava, April Ott, Jimmy Lynn Rosborough, and David Morgan for technical assistance, and the Histology Core Facility at SPRD for tissue processing. We also thank Dr. Jean C. Zenklusen for helpful discussions. This work was supported by National Institutes of Health grant CA42157.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL SA83125@odin.mdacc.tmc.edu; FAX (512) 237-2444.
References
- Aagaard L, Lukas J, Bartkova J, Kjerulff AA, Strauss M, Bartek J. Aberrations of p16Ink4 and retinoblastoma tumour-suppressor genes occur in distinct sub-sets of human cancer cell lines. Int J Cancer. 1995;61:115–120. doi: 10.1002/ijc.2910610120. [DOI] [PubMed] [Google Scholar]
- Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Lukas J, Strauss M, Bartek J. The PRAD-1/cyclin D1 oncogene product accumulates aberrantly in a subset of colorectal carcinomas. Int J Cancer. 1994;58:568–573. doi: 10.1002/ijc.2910580420. [DOI] [PubMed] [Google Scholar]
- Bianchi AB, Fischer SM, Robles AI, Rinchik EM, Conti CJ. Overexpression of cyclin D1 in mouse skin carcinogenesis. Oncogene. 1993;8:1127–1133. [PubMed] [Google Scholar]
- Dlugosz AA, Cheng C, Denning MF, Dempsey PJ, Coffey RJ, Jr, Yuspa SH. Keratinocyte growth factor-receptor ligands induce TGFα expression and activate the epidermal growth factor-receptor signaling pathway in cultured epidermal keratinocytes. Cell Growth & Differ. 1994;5:1283–1292. [PubMed] [Google Scholar]
- Dlugosz AA, Glick AB, Tennenbaum T, Weinberg WC, Yuspa SH. Isolation and utilization of epidermal keratinocytes for oncogene research. Methods Enzymol. 1995;254:3–20. doi: 10.1016/0076-6879(95)54003-2. [DOI] [PubMed] [Google Scholar]
- Dlugosz AA, Hansen L, Cheng C, Alexander N, Denning MF, Threadgill DW, Magnuson T, Coffey RJ, Jr, Yuspa SH. Targeted disruption of the epidermal growth factor receptor impairs growth of squamous papillomas expressing the v-rasHa oncogene but does not block in vitro keratinocyte responses to oncogenic ras. Cancer Res. 1997;57:3180–3188. [PubMed] [Google Scholar]
- Dvir A, Milner Y, Chomsky O, Gilon C, Gazit A, Levitzki A. The inhibition of EGF-dependent proliferation of keratinocytes by tyrphostin tyrosine kinase blockers. J Cell Biol. 1991;113:857–865. doi: 10.1083/jcb.113.4.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes & Dev. 1995;9:2364–2372. doi: 10.1101/gad.9.19.2364. [DOI] [PubMed] [Google Scholar]
- Filmus J, Robles AI, Shi W, Wong MJ, Colombo LL, Conti CJ. Induction of cyclin D1 overexpression by activated ras. Oncogene. 1994;9:3627–3633. [PubMed] [Google Scholar]
- Gangarosa LM, Sizemore N, Graves-Deal R, Oldham SM, Der CJ, Coffey RJ. A Raf-independent epidermal growth factor receptor autocrine loop is necessary for ras transformation of rat intestinal epithelial cells. J Biol Chem. 1997;272:18926–18931. doi: 10.1074/jbc.272.30.18926. [DOI] [PubMed] [Google Scholar]
- Hansen LA, Tennant RW. Focal transgene expression associated with papilloma development in v-Ha-ras-transgenic Tg.AC mice. Mol Carcinog. 1994a;9:143–156. doi: 10.1002/mc.2940090306. [DOI] [PubMed] [Google Scholar]
- ————— Follicular origin of epidermal papillomas in v-Ha-ras transgenic Tg.AC mouse skin. Proc Natl Acad Sci. 1994b;91:7822–7826. doi: 10.1073/pnas.91.16.7822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung WC, Chai CY, Huang JS, Chuang LY. Expression of cyclin D1 and c-Ki-ras gene product in human epithelial ovarian tumors. Hum Pathol. 1996;27:1324–1328. doi: 10.1016/s0046-8177(96)90345-7. [DOI] [PubMed] [Google Scholar]
- Jiang W, Zhang Y-J, Khan SM, Hollstein MC, Santella RM, Lu S-H, Harris CC, Montesano R, Weinstein IB. Altered expression of the cyclin D1 and retinoblastoma genes in human esophageal cancer. Proc Natl Acad Sci. 1993;90:9026–9030. doi: 10.1073/pnas.90.19.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes & Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Chao JR, Jiang MC, Ng SY, Yen JJ, Yang YH. Ras transformation results in an elevated level of cyclin D1 and acceleration of G1 progression in NIH3T3 cells. Mol Cell Biol. 1995;15:3654–3663. doi: 10.1128/mcb.15.7.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otterson GA, Kratze RA, Coxon A, Kim YW, Kaye FJ. Absence of p16ink4 protein is restricted to a subset of lung cancer cell lines that retains wildtype RB. Oncogene. 1994;9:3375–3378. [PubMed] [Google Scholar]
- Peeper DS, Upton TM, Ladha MH, Neuman E, Zalvide J, Bernards R, DeCaprio JA, Ewen ME. Ras signalling linked to the cell–cycle machinery by the retinoblastoma protein. Nature. 1997;386:177–181. doi: 10.1038/386177a0. [DOI] [PubMed] [Google Scholar]
- Peters G. The D-type cyclins and their role in tumorigenesis. J Cell Sci. 1994;S18:89–96. doi: 10.1242/jcs.1994.supplement_18.13. [DOI] [PubMed] [Google Scholar]
- Robles AI, Conti CJ. Early overexpression of cyclin D1 protein in mouse skin carcinogenesis. Carcinogenesis. 1995;16:781–786. doi: 10.1093/carcin/16.4.781. [DOI] [PubMed] [Google Scholar]
- Robles AI, Larcher F, Whalin RB, Murillas R, Richie E, Gimenez-Conti IB, Jorcano JL, Conti CJ. Expression of cyclin D1 in epithelial tissues of transgenic mice results in epidermal hyperproliferation and severe thymic hyperplasia. Proc Natl Acad. 1996;93:7634–7638. doi: 10.1073/pnas.93.15.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roop DR, Lowy DR, Tambourin PE, Strickland J, Harper JR, Balaschak M, Spangler EF, Yuspa SH. An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature. 1986;323:822–824. doi: 10.1038/323822a0. [DOI] [PubMed] [Google Scholar]
- Schauer IE, Siriwardana S, Langan TA, Sclafani RA. Cyclin D1 overexpression vs. retinoblastoma inactivation: Implications for growth control evasion in non-small cell and small cell lung cancer. Proc Natl Acad Sci. 1994;91:7827–7831. doi: 10.1073/pnas.91.16.7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Gomez-Lahoz E, DePinho RA, Beach D, Bar-Sagi D. Inhibition of ras-induced proliferation and cellular transformation by p16INK4. Science. 1995;267:249–252. doi: 10.1126/science.7809631. [DOI] [PubMed] [Google Scholar]
- Sgambato A, Han EK-H, Zhang Y-J, Moon RC, Santella RM, Weinstein IB. Carcinogenesis. 1995;16:2193–2198. doi: 10.1093/carcin/16.9.2193. [DOI] [PubMed] [Google Scholar]
- Shapiro GI, Edwards CD, Kobzik L, Godleski J, Richards W, Sugarbaker DJ, Rollins BJ. Reciprocal Rb inactivation and p16INK4 expression in primary lung cancers and cell lines. Cancer Res. 1995;55:505–509. [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes & Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 1995;82:621–630. doi: 10.1016/0092-8674(95)90034-9. [DOI] [PubMed] [Google Scholar]
- Spalding JW, Momma J, Elwell MR, Tennant RW. Chemically induced skin carcinogenesis in a transgenic mouse line (Tg.AC) carrying a v-Ha-ras gene. Carcinogenesis. 1993;14:1335–1341. doi: 10.1093/carcin/14.7.1335. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;31:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Winston JT, Coats SR, Wang Y-Z, Pledger W. Regulation of the cell cycle machinery by oncogenic ras. Oncogene. 1996;12:127–134. [PubMed] [Google Scholar]
- Zarbl H, Sukumar S, Arthur AV, Martin-Zanca D, Barbacid M. Direct mutagenesis of Ha-ras-1 oncogenes by N-nitroso-N-methylurea during initiation of mammary carcinogenesis in rats. Nature. 1985;315:382–385. doi: 10.1038/315382a0. [DOI] [PubMed] [Google Scholar]