

Abstract

The addition of azides to acylnitroso hetero Diels-Alder cycloadducts derived from cyclopentadiene affords exo triazolines in excellent yield. The reaction is greatly affected by reducing the level of alkene strain, while sterically demanding azides do not hinder the reaction. Conversion of the triazolines to aziridines is also described.
The nitroso hetero Diels-Alder (HDA) reaction provides a useful method for incorporating 1,4-aminoalcohol moieties into a carbon framework in a diastereoselective fashion.1 Stereoselective nitroso HDA reactions2 have also provided access to new classes of synthetically useful molecules. While a variety of nitroso species have been investigated for use in HDA reactions,3 our research has largely focused on acyl- and carboxylnitroso species due to the ease of their synthesis and high reactivity. Hydroxamic acids and N-hydroxy carbamates, when oxidized in the presence of cyclic dienes, afford hetero Diels-Alder adducts 1.1a,4 Reduction of the N-O bond of 1 results in amino alcohols,5 which are suitable intermediates for the synthesis of natural products,6 carbocyclic nucleosides,7 and other important biologically active molecules.8 The C-O bond can also be cleaved through metal-mediated reactions in the presence of nucleophiles9 or electrophiles,10 yielding 1,4-benzodiazepines11 and other useful synthetic intermediates. The N-acyl group can also be cleaved under relatively mild conditions (where R = alkyl or aryl).12 While extensive chemistry has been developed that capitalizes on the strained nature of 1, modifications of the olefin have been mainly limited to epoxidation,6c dihydroxylation,13 and oxidative cleavage.14
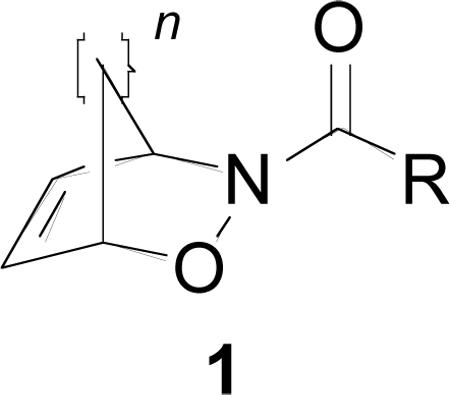
In an effort to expand upon the versatility of 1, we were interested in selective functionalization of the olefin to produce new structural features. Encouraged by a recent report highlighting the addition of nitrile oxides to 1,15 we wish to report on studies regarding the reactivity of 1 with azides to form triazolines and their subsequent transformation to aziridines.
Intermolecular [3+2] cycloaddition reactions of azides to strained bicyclic alkenes are well-documented in the literature.16 Examples include additions to norbornene, 2-azabicyclo[2.2.1]hept-5-en-3-one (ABH) 2 to afford 2’-3’-epimino-carbocyclic nucleosides,17 2,3-diazabicyclo[2.2.1]hept-5-enes 3 to afford 1,4-dihydropyridines,18 as well as many other strained systems.16h,19 However, the addition of azides to bicyclic oxazines such as 1 has not been disclosed in the literature.

Since we wished to probe the role of olefin strain on reactivity with azides, we chose to subject the olefins of 1a-c as well as protected amino alcohols 4a-c to reactions with various azides (Scheme 1). Oxazines 1a-c can be prepared on a large scale from hydroxylamine hydrochloride in two steps in moderate to high yields.20 N-O bond reduction with Mo(CO)621 followed by protection of the resulting alcohol yielded monocyclic olefins 4a-c.22
Scheme 1.

Synthesis of cycloadducts and protected amino alcohols.
We were very pleased to find that when 1a was treated with benzyl azide,23 the regioisomeric exo-triazolines 5a and 6a were obtained in quantitative yield after stirring for 2 days neat at room temperature (eq 1). Similar reactions of 2-azabicyclo[2.2.2]hept-5-en-3-ones reportedly required high pressures for the cycloaddition reaction to occur.17c The exo- specificity of the reaction is in agreement with what has been reported in the literature in related reactions of bicyclo[2.2.1]hept-5-ene-2,3-dicarboximides,16h ABH (2 ) and derivatives,24 7-oxabicyclo[2.2.1]hept-5-en-2-yl derivatives, and 3,18,25 and was confirmed by single crystal x-ray diffraction of triazolines 5a and 6a.
 |
(1) |
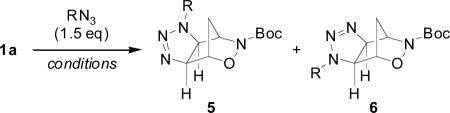
In an effort to ascertain the limitations of this chemistry, we treated alkene 1a with various organic azides under a number of reaction conditions (Table 1). The reaction proceeded equally well refluxing in toluene or chloroform (entries 4 and 8), however it was sluggish when stirred in solution at room temperature (entry 2). The steric bulk of the azide had little effect on either the regioselectivity or yield of the reaction, with primary, secondary, tertiary, and aryl azides affording triazolines 5 and 6 in excellent yield (entries 8-12).
Table 1.
Reaction of 1a with various azides
 | ||||
|---|---|---|---|---|
| Entry | R | Conditionsa | Productsb | Yieldc |
| 1 | Bn | A | 5a/6a | 99% |
| 2 | Bn | B | 5a/6a | 99% |
| 3 | Add | B | 5b/6b | 95% |
| 4 | Bn | C | 5a/6a | 88% |
| 5 | n-octyl | C | 5c/6c | 99% |
| 6 | cyclopentyl | C | 5d/6d | 97% |
| 7 | Ph | C | 5e/6e | 97% |
| 8 | Bn | D | 5a/6a | 88% |
| 9 | n-octyl | D | 5c/6c | 81% |
| 10 | cyclopentyl | D | 5d/6d | 86% |
| 11 | Add | D | 5b/6b | 85% |
| 12 | Ph | D | 5e/6e | 99% |
A = neat, RT, 2 days; B = CHCl3, RT, 4 weeks; C = CHCl3, reflux, 3 days; D = PhCH3, reflux, 4 h.
Ratio of 5:6 = 1.0 : 1.1 in all cases as determined by 1H NMR.
Isolated yield.
Ad = 1-adamantyl.
The structures of 5 and 6 were assigned based on single crystal x-ray diffraction for 5a and 6a, and by 2D-NMR experiments for 5b-e and 6b-e. The exo stereochemistry was confirmed from the observed 4J-coupling (“W-coupling”) of H2 and H3 with H5’, but not with H5. The position of the Boc group relative to the R group was confirmed by HMBC experiments based on the observed 3J-coupling between H4 (or H1) and the carbonyl (Figure 1).26
Figure 1.

Observed W-coupling and 3JCH-coupling of exo triazolines
Trimethylsilyl azide has been reported to add to norbornene systems and other bicyclic olefins to yield triazolines and/or aziridines directly.18,27 Since we were interested in obtaining the unsubstituted aziridine 7, 1a was subjected to azidotrimethylsilane under various conditions (Scheme 2). Under a variety of reaction conditions (neat, 25-80°C, PhCH3, 25-110°C), only decomposition of the alkene was observed. Tosyl azide28 has also been reported to afford aziridines directly,17b and upon treatment of 1a with tosyl azide, we were able to obtain aziridine 8 in good yield.
Scheme 2.

Reaction of 1a to yield aziridines directly.
Substantial rate increases were reported for [3+2] cycloadditions (especially “Click chemistry”29 reactions) when both reactants were “floated” on water compared to reactions that were stirred with or without solvent.30 We found no significant increase in the rate of reaction when 1a was treated with benzyl azide “on water” or in a concentrated solution of toluene “on water” when compared to stirring the two reagents neat at room temperature.
The effect of the strain of the alkene on the formation of triazolines was also studied. When the bicyclo[2.2.2] cycloadduct 1b was treated with 1.4 equivalents of benzyl azide neat at room temperature, no reaction was observed. When 1b was treated with a large excess of benzyl azide in refluxing toluene, however, a mixture of triazolines 9-12 was obtained in moderate yield (Scheme 3). In contrast to bicyclo[2.2.1] cycloadduct 1a, endo triazolines 11 and 12 were formed along with the exo triazolines 9 and 10. The influence of ring strain on reactivity was especially evident when bicyclo[2.2.4] cycloadduct 1c and monocyclic alkenes 4a-c were subjected to the same conditions. When heated neat or in toluene, only trace amounts of triazoline products were observed.
Scheme 3.

Reaction of other alkenes with benzyl azide.
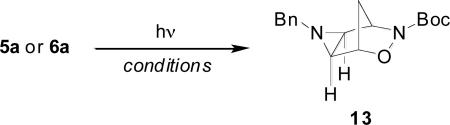
In the course of this study, we found that triazolines 5a and 6a were stable to temperatures up to 120°C and did not decompose readily in ambient light. When irradiated below ~300 nm, 5a and 6a readily underwent photolytic conversion to aziridine 13 in 2-4 h (Table 2). At wavelengths higher than 300 nm, the time required for the reaction to proceed to 100% conversion increased greatly (entry 5).
Table 2.
Photolysis of triazolines 5a and 6a
 | |||||
|---|---|---|---|---|---|
| Entry | Isomer | Solvent | Filter | Timea | Yieldb |
| 1 | 5a | CHCl3 | quartz | 1.5 h | 65% |
| 2 | 6a | CHCl3 | quartz | 3.5 h | 78% |
| 3 | 5a | CH3CN | vycor | 2 h | 75% |
| 4 | 6a | CH3CN | vycor | 3 h | 67% |
| 5 | 5a | CH3CN | pyrex | 20 h | 93% |
Reactions were monitored by TLC and/or 1H NMR.
Isolated yield.
Surprisingly, N-O bond reduction of triazolines 5a and 6a proceeded cleanly to afford triazolines 14 and 15 without decomposition of the triazoline ring (Scheme 4). Triazoline 14 was cleanly transformed to aziridine 16 upon irradiation in acetonitrile; however, a complex mixture of products was observed when triazoline 15 was subjected to the same conditions. The reason for this result is unclear at this time and will be investigated further.
Scheme 4.

Structural elaboration of triazolines 5a and 6a.
In summary, the addition of azides to the electron-neutral alkene of cycloadduct 1a affords a mixure of exo triazolines in good to excellent yield. The steric bulk of the azide appears not to play a significant role in the course of the reaction; however, the reactivity of the alkene was diminished significantly with cycloadducts derived from larger cyclic alkenes (1b and 1c), or monocyclic alkenes (4a-c).
Experimental Section
General Procedure for the Synthesis of Triazolines Using Method A
The alkene (1 mmol) and azide (1.5 mmol) were combined and stirred neat at 25 °C in a single-necked round-bottomed flask. The progress of the reaction was monitored by TLC or 1H NMR for the disappearance of the alkene, and the crude material was chromatographed through silica gel 60 (230-400 mesh).
General Procedure for the Synthesis of Triazolines Using Method B
The alkene (1 mmol) and azide (1.5 mmol) were dissolved in 10 mL of CHCl3 in a single-necked round-bottomed flask and stirred at 25 °C. The progress of the reaction was monitored by TLC or 1H NMR for the disappearance of the alkene (typically 4 weeks), and the crude material was chromatographed through silica.
General Procedure for the Synthesis of Triazolines Using Method C
The alkene (1 mmol) and azide (1.5 mmol) were dissolved in 10 mL of CHCl3 in a single-necked round-bottomed flask fitted with a condensor and heated to reflux in an oil bath (oil temp. = 80 °C). The progress of the reaction was monitored by TLC or 1H NMR for the disappearance of the alkene (typically 3 days), and the crude material was chromatographed through silica.
General Procedure for the Synthesis of Triazolines Using Method D
The alkene (1 mmol) and azide (1.5 mmol) were dissolved in 10 mL of PhCH3 in a single-necked round-bottomed flask fitted with a condensor and heated to reflux in an oil bath (oil temp. = 125 °C). The progress of the reaction was monitored by TLC or 1H NMR for the disappearance of the alkene (typically 3-4 h), and the crude material was chromatographed through silica.
tert - butyl (3aα, 4β,7β,7aα)-1-benzyl-3a,4,7,7a-tetrahydro-4,7-methano[1,2,3]triazole[4,5-d][1,2]oxazine-6(1H)-carboxylate (5a) and tert-butyl (3aα,4β,7β,7aα)-3-benzyl-3a,4,7,7a-tetrahydro-4,7-methano[1,2,3]triazole[4,5-d][1,2]oxazine-6(3H)-carboxylate (6a)
Prepared following the general procedure for the synthesis of triazolines using Method A. Cycloadduct 1a (198 mg, 1.01 mmol) and benzyl azide (211 mg, 1.58 mmol) were reacted for 48 h. The brown crude material was chromatographed through 15 g of silica using a solvent gradient of 100% CH2Cl2 to 98% CH2Cl2/EtOAc to afford triazoline 6a (159 mg, 48% yield) as a white solid, then 85% CH2Cl2/EtOAc to afford triazoline 5a (170 mg, 51% yield) as a white solid (99% total combined yield). Analytical and x-ray crystallographic samples of 5a and 6a were prepared by recrystallization from EtOAc/hexanes. 5a: mp = 104-105 °C; λmax = 255 nm; 1H NMR (500 MHz, CDCl3) δ 7.37–7.27 (m, 5H), 4.92 (s, 1H), 4.90 (d, J = 14.5 Hz, 1H), 4.82 (d, J = 9.8 Hz, 1H), 4.72 (d, J = 14.5 Hz, 1H), 4.09 (s, 1H), 3.55 (d, J = 9.8 Hz, 1H), 1.70 (dt, J = 11.4, 1.5 Hz, 1H), 1.45 (d, J = 11.5 Hz, 1H), 1.39 (s, 9H) ppm; 13C NMR (125 MHz, CDCl3) δ 156.4, 135.6, 128.8, 128.24, 128.17, 84.6, 82.6, 79.9, 61.2, 59.3, 53.4, 32.4, 27.9 ppm; HRMS (FAB), m/z (M+H): calcd for C17H23N4O3+, 331.1770; obsd, 331.1745. 6a: mp = 100-101 °C; λmax = 255 nm; 1H NMR (500 MHz, CDCl3) δ 7.35-7.24 (m, 5H), 4.92 (d, J = 14.7 Hz, 1H), 4.85 (s, 1H), 4.84 (d, J = 9.9 Hz, 1H), 4.61 (d, J = 14.7 Hz, 1H), 4.09 (s, 1H), 3.50 (d, J = 9.9 Hz, 1H), 1.68 (d, J = 11.5 Hz, 1H), 1.45-1.43 (m, 10H) ppm; 13C NMR (125 MHz, CDCl3) δ 156.4, 135.6, 128.8, 128.3, 128.2, 83.7, 82.7, 79.6, 61.5, 59.9, 53.6, 32.3, 27.9 ppm; HRMS (FAB), m/z (M+H): calcd for C17H23N4O3+, 331.1770; obsd, 331.1753.
General Procedure for Photolysis of Triazolines. tert-butyl (1α,2β,4β,5α)-3-benzyl-6-oxa-3,7-diazatricyclo[3.2.1.02,4]octane-7-carboxylate (13)
5a (333 mg, 1.01 mmol) was dissolved in 300 mL of degassed CH3CN and transferred to a 450-mL photochemical reaction vessel. The solution was irradiated in an immersion-well reactor under a stream of Ar with a Hanovia 450W mercury lamp equipped with a Vycor filter sleeve. Reaction progress was monitored by TLC and 1H NMR for the disappearance of 5a. After 3 h, the reaction was concentrated and the crude material was chromatographed through 30 g of silica using a gradient consisting of 100% CH2Cl2 to 95% CH2Cl2/EtOAc to afford 13 as a colorless oil (205 mg, 67% yield). 1H NMR (500 MHz, CDCl3) δ 7.35-7.27 (m, 5H), 4.79 (m, 1H), 4.62 (m, 1H), 3.40 (d, J = 13.5 Hz, 1H), 3.35 (d, J = 14.0 Hz, 1H), 2.29-2.22 (m, 3H), 1.50 (s, 9H), 1.39 (d, J = 10.5 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ 157.5, 138.6, 128.4, 127.7, 127.3, 82.3, 79.9, 60.7, 59.1, 37.1, 36.7, 29.4, 28.2 ppm; MS (FAB) m/z 303 (M+H), 247, 203, 171 (100%); HRMS (FAB) m/z (M+H): calcd for C17H23N2O3+, 303.1709; obsd, 303.1712.
Supplementary Material
Acknowledgements
Support from the NIH (AI054193 and GM 68012) is gratefully acknowledged. The authors would also like to thank Jaroslav Zajicek for assistance with 2D-NMR experiments, Bill Boggess and Nonka Sevov for mass spectroscopic analyses, and Bruce Noll for X-ray crystallographic data. MJM gratefully acknowledges the Hans-Köll Institute and the University of Notre Dame for support of a sabbatical leave.
Footnotes
Supporting Information Available: Experimental details and characterization data for triazolines 5a-e, 6a-e, 9-12, and 14-16, including 1D and 2D-NMR spectra and x-ray crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Footnotes
- 1.a Kirby GW. Chem. Soc. Rev. 1977;6:1–24. [Google Scholar]; b Corrie JET, Kirby GW, Mackinnon JWM. J. Chem. Soc., Perkin Trans. 1. 1985:883–886. [Google Scholar]; c Leach AG, Houk KN. J. Org. Chem. 2001;66:5192–5200. doi: 10.1021/jo0104126. [DOI] [PubMed] [Google Scholar]
- 2.a Yamamoto Y, Yamamoto H. J. Am. Chem. Soc. 2004;126:4128–4129. doi: 10.1021/ja049849w. [DOI] [PubMed] [Google Scholar]; b Kirby GW, Nazeer M. Tetrahedron Lett. 1988;29:6173–6174. [Google Scholar]; c Ritter AR, Miller MJ. J. Org. Chem. 1994;59:4602–4611. [Google Scholar]
- 3.a Miller CA, Batey RA. Org. Lett. 2004;6:699–702. doi: 10.1021/ol0363117. [DOI] [PubMed] [Google Scholar]; b Calvet G, Dussaussois M, Blanchard N, Kouklovsky C. Org. Lett. 2004;6:2449–2451. doi: 10.1021/ol0491336. [DOI] [PubMed] [Google Scholar]; c Ware RW, Day CS, King SB. J. Org. Chem. 2002;67:6174–6180. doi: 10.1021/jo0202839. [DOI] [PubMed] [Google Scholar]; d Ware RW, Jr., King SB. J. Org. Chem. 2000;65:8725–8729. doi: 10.1021/jo001230z. [DOI] [PubMed] [Google Scholar]
- 4.Vogt PF, Miller MJ. Tetrahedron. 1998;54:1317–1348. [Google Scholar]
- 5.a Shireman BT, Miller MJ. Tetrahedron Lett. 2000;41:9537–9540. [Google Scholar]; b Keck GE, Wager TT, McHardy SF. Tetrahedron. 1999;55:11755–11772. [Google Scholar]; c Keck GE, McHardy SF, Wager TT. Tetrahedron Lett. 1995;36:7419–7422. [Google Scholar]
- 6.a Li F, Warshakoon NC, Miller MJ. J. Org. Chem. 2004;69:8836–8841. doi: 10.1021/jo048606j. [DOI] [PubMed] [Google Scholar]; b Malpass JR, Hemmings DA, Wallis AL, Fletcher SR, Patel S. J. Chem. Soc., Perkin Trans. 1. 2001:1044–1050. [Google Scholar]; c Justice DE, Malpass JR. Tetrahedron. 1996;52:11977–11994. [Google Scholar]
- 7.a Li F, Brogan John B, Gage Jennifer L, Zhang D, Miller Marvin J. J. Org. Chem. 2004;69:4538–4540. doi: 10.1021/jo0496796. [DOI] [PubMed] [Google Scholar]; b Kim K-H, Miller MJ. Tetrahedron Lett. 2003;44:4571–4573. [Google Scholar]; c Li H, Miller MJ. J. Org. Chem. 1999;64:9289–9293. [Google Scholar]
- 8.a Jiang MX-W, Warshakoon NC, Miller MJ. J. Org. Chem. 2005;70:2824–2827. doi: 10.1021/jo0484070. [DOI] [PubMed] [Google Scholar]; b Lee W, Miller MJ. J. Org. Chem. 2004;69:4516–4519. doi: 10.1021/jo0495034. [DOI] [PubMed] [Google Scholar]
- 9.a Surman MD, Mulvihill MJ, Miller MJ. J. Org. Chem. 2002;67:4115–4121. doi: 10.1021/jo016275u. [DOI] [PubMed] [Google Scholar]; b Surman MD, Mulvihill MJ, Miller MJ. Tetrahedron Lett. 2002;43:1131–1134. [Google Scholar]; c Surman MD, Miller MJ. J. Org. Chem. 2001;66:2466–2469. doi: 10.1021/jo010094a. [DOI] [PubMed] [Google Scholar]; d Mulvihill MJ, Surman MD, Miller MJ. J. Org. Chem. 1998;63:4874–4875. [Google Scholar]
- 10.Lee W, Kim K-H, Surman MD, Miller MJ. J. Org. Chem. 2003;68:139–149. doi: 10.1021/jo026488z. [DOI] [PubMed] [Google Scholar]
- 11.Surman MD, Mulvihill MJ, Miller MJ. Org. Lett. 2002;4:139–141. doi: 10.1021/ol017036w. [DOI] [PubMed] [Google Scholar]
- 12.Keck GE, Webb RR, Yates JB. Tetrahedron. 1981;37:4007–4016. [Google Scholar]
- 13.Keck GE, Romer DR. J. Org. Chem. 1993;58:6083–6089. [Google Scholar]
- 14.a Shireman BT, Miller MJ. J. Org. Chem. 2001;66:4809–4813. doi: 10.1021/jo015544d. [DOI] [PubMed] [Google Scholar]; b Nora GP, Miller MJ, Moellmann U. Bioorg. Med. Chem. Lett. 2006;16:3966–3970. doi: 10.1016/j.bmcl.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 15.Quadrelli P, Mella M, Paganoni P, Caramella P. Eur. J. Org. Chem. 2000;14:2613–2620. [Google Scholar]
- 16.a Braese S, Gil C, Knepper K, Zimmermann V. Angew. Chem., Int. Ed. 2005;44:5188–5240. doi: 10.1002/anie.200400657. [DOI] [PubMed] [Google Scholar]; b Shea KJ, Kim JS. J. Am. Chem. Soc. 1992;114:4846–4855. [Google Scholar]; c Becker KB, Hohermuth MK. Helv. Chim. Acta. 1979;62:2025–36. [Google Scholar]; d Taniguchi H, Ikeda T, Imoto E. Bull. Chem. Soc. Jpn. 1978;51:1859–65. [Google Scholar]; e Scheiner P. Tetrahedron. 1968;24:2757–66. [Google Scholar]; f Scheiner P. J. Am. Chem. Soc. 1968;90:988–992. [Google Scholar]; g Rolf H. Angew. Chem., Int. Ed. Engl. 1963;2:565–598. [Google Scholar]; h Tarabara IN, Kas'yan AO, Yarovoi MY, Shishkina SV, Shishkin OV, Kas'yan LI. Russ. J. Org. Chem. 2004;40:992–998. [Google Scholar]
- 17.a Ishikura M, Katagiri N. Recent Res. Devel. Organic Chem. 2004;8:197–205. [Google Scholar]; b Ishikura M, Murakami A, Katagiri N. Org. Biomol. Chem. 2003;1:452–453. doi: 10.1039/b210963h. [DOI] [PubMed] [Google Scholar]; c Ishikura M, Kudo S, Hino A, Ohnuki N, Katagiri N. Heterocycles. 2000;53:1499–1504. [Google Scholar]
- 18.Stout DM, Takaya T, Meyers AI. J. Org. Chem. 1975;40:563–569. [Google Scholar]
- 19.Huenig S, Kraft P. Heterocycles. 1995;40:639–652. [Google Scholar]
- 20.Zhang D, Sueling C, Miller MJ. J. Org. Chem. 1998;63:885–888. doi: 10.1021/jo971696q. [DOI] [PubMed] [Google Scholar]
- 21.Cicchi S, Goti A, Brandi A, Guarna A, De Sarlo F. Tetrahedron Lett. 1990;31:3351–3354. [Google Scholar]
- 22.a Li F, Miller MJ. J. Org. Chem. 2006;71:5221–5227. doi: 10.1021/jo060555y. [DOI] [PubMed] [Google Scholar]; b Sirisoma NS, Woster PM. Tetrahedron Lett. 1998;39:1489–1492. [Google Scholar]
- 23.Alvarez SG, Alvarez MT. Synthesis. 1997:413–414. [Google Scholar]
- 24.Malpass JR, Belkacemi D, Griffith GA, Robertson MD. ARKIVOC. 2002:164–174. [Google Scholar]
- 25.Reymond J-L, Vogel P. Tetrahedron Lett. 1988;29:3695–3698. [Google Scholar]
- 26.For a full description of the assignment of stereochemical relationships and the position of the R and Boc groups using 2D-NMR experiments, see the supporting information.
- 27.Peterson WR, Jr., Arkles B, Washburne SS. J. Organomet. Chem. 1976;121:285–291. [Google Scholar]
- 28.Pollex A, Hiersemann M. Org. Lett. 2005;7:5705–5708. doi: 10.1021/ol052462t. [DOI] [PubMed] [Google Scholar]
- 29.a Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; b Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 30.a Narayan S, Muldoon J, Finn MG, Fokin VV, Kolb HC, Sharpless KB. Angew. Chem., Int. Ed. 2005;44:3275–3279. doi: 10.1002/anie.200462883. [DOI] [PubMed] [Google Scholar]; b Klijn JE, Engberts JBFN. Nature. 2005;435:746–747. doi: 10.1038/435746a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.