

Abstract
Homology-directed repair of DNA damage has recently emerged as a major mechanism for the maintenance of genomic integrity in mammalian cells. The highly conserved strand transferase, Rad51, is expected to be critical for this process. XRCC3 possesses a limited sequence similarity to Rad51 and interacts with it. Using a novel fluorescence-based assay, we demonstrate here that error-free homology-directed repair of DNA double-strand breaks is decreased 25-fold in an XRCC3-deficient hamster cell line and can be restored to wild-type levels through XRCC3 expression. These results establish that XRCC3-mediated homologous recombination can reverse DNA damage that would otherwise be mutagenic or lethal.
Keywords: Homologous recombination, double-strand break repair, gene conversion, mammalian cells, XRCC3, Rad51-related proteins
Cells treated with X-rays suffer potentially lethal DNA double-strand breaks (DSBs), in addition to a variety of other lesions (Ward 1995). As in cells from other organisms, cycling mammalian cells possess a robust capacity to repair DSBs by homologous recombination (Liang et al. 1998). One type of homologous repair is gene conversion, a process in which a donor DNA sequence with homology to both sides of the DSB supplies genetic information to repair the DSB (Taghian and Nickoloff 1997; Elliott et al. 1998; Liang et al. 1998). Effectively, the information in the homologous sequence is copied into the broken locus, making the repaired locus an exact copy of the donor sequence, without altering the donor sequence. The error-free nature of gene conversion suggests a particularly important contribution in maintaining genetic integrity. To further understand both the mechanism of homologous recombination in mammalian cells, as well as the role of homologous recombination in mammalian cell growth and genome integrity, it will be critical to identify genetic loci that influence homologous recombination.
Several such genetic loci have been identified in Saccharomyces cerevisiae, but mutations of the homologous genetic loci in mammalian cells do not always result in the same phenotypes. Mice with a disrupted Rad51 gene die early in embryogenesis and cells recovered from mutant embryos do not proliferate (Lim and Hasty 1996; Tsuzuki et al. 1996). The severity of the Rad51 mutant phenotype is surprising, considering that yeast Rad51 mutants are viable in both S. cerevisiae (Shinohara et al. 1992) and Schizosaccharomyces pombe (Muris et al. 1993). The phenotype of Rad52 mutants also differs greatly between yeast and mouse. Mutation of Rad52 in S. cerevisiae confers extreme sensitivity to ionizing radiation due to a defect in recombinational repair. In contrast, mutation of the Rad52 homolog in mice leads to no discernible radiation sensitivity and a mild defect in homologous recombination (Rijkers et al. 1998). In the case of Rad54 mutation, however, the yeast and mammalian phenotypes are more comparable (Essers et al. 1997).
Because many yeast genes that are involved in homologous recombination were originally identified as mutations that conferred sensitivity to ionizing radiation (Petes et al. 1991), ionizing radiation-sensitive mammalian cells represent good candidates for genes with similar functions. Nine such complementation groups have been isolated from hamster cells (Thompson and Jeggo 1995; Jeggo 1998; Thompson and Schild 1999). Several of these cell lines have been studied genetically for defects in DSB repair. The XRCC4-7 groups, which are defective for a DNA ligase IV-interacting protein (XRCC4), Ku86, Ku70, and DNA-PKcat, respectively, are involved in nonhomologous repair of DSBs as demonstrated in assays for both V(D)J recombination (Taccioli et al. 1993) and chromosomal DSB repair (Liang et al. 1996). XRCC1 interacts with DNA ligase III and is implicated in DNA base excision repair (Caldecott et al. 1994). The XRCC2 and XRCC3 mutants were found to have high levels of chromosomal instability and extreme sensitivity to cross-linking agents (Tebbs et al. 1995; Liu et al. 1998).
The irs1SF cell line, which is defective in XRCC3 (Tebbs et al. 1995; Fuller and Painter 1988), has not been characterized at the molecular level for its role in DSB repair. The XRCC3 protein shares limited homology with the Rad51 strand transferase in a presumed ATP-binding domain and also interacts with Rad51 (Tebbs et al. 1995; Liu et al. 1998). In this report we have examined directly the irs1SF mutant to determine whether XRCC3 is involved in homologous repair of DNA damage. For this endeavor, we designed a new recombination reporter system to efficiently assay chromosomal gene conversion events in response to DSBs. The system utilizes a modified gene for green fluorescent protein (GFP) as a recombination reporter and the I-Sce I endonuclease for the introduction of DSBs (Jasin 1996). The 18-bp I-Sce I site (Colleaux et al. 1988) is inserted within one copy of a GFP gene, inactivating it, but when cleaved, a homologous repair event with a linked donor GFP gene fragment restores functional GFP expression. The gene conversion events are readily detected by flow cytometry in a 2-day assay. Using this system, we have determined that the irs1SF mutant has a homologous repair defect that can be restored by expression of XRCC3, but not other proteins predicted to be involved in recombinational repair.
Results
XRCC3-deficient cells have impaired homologous repair of DSBs
To test whether XRCC3-deficient mammalian cells are defective for homologous repair of DSBs, we constructed the recombination substrate DR–GFP. A gene conversion event within this substrate results in the expression of intact GFP protein (Fig. 1A), which can be assayed as cellular green fluorescence. DR–GFP is composed of two differentially mutated GFP genes oriented as direct repeats and separated by a drug selection marker, the puromycin N-acetyltransferase gene (Fig. 1B). One of the GFP genes, SceGFP, is mutated to contain the recognition site for the rare-cutting I-Sce I endonuclease and, as a result, will undergo a DSB when I-Sce I is expressed in vivo. The I-Sce I site was incorporated at a BcgI restriction site by substituting 11 bp of wild-type GFP sequences with those of the I-Sce I site (Fig. 1A). These substituted base pairs also supply two in-frame stop-codons, which terminate translation and inactivate the protein. Downstream of the SceGFP gene is an 812-bp internal GFP fragment termed iGFP. Homologous sequences in the two mutated GFP genes are separated by 3.7 kb.
Figure 1.

GFP expression plasmids. (A) Modified GFP gene. The modified GFP gene encodes the EGFP protein fused to a nuclear localization signal (N) and zinc finger domain (Z). It is expressed from a hCMV enhancer/chicken β-actin promoter (arrow) on a spliced message (not shown). GFP is modified to SceGFP so as to contain an I-SceI site and in-frame termination codons (underline). (B) DR–GFP recombination substrate. Downstream of the SceGFP gene is iGFP, a 5′ and 3′-truncated GFP gene. (C) (STGC) product. In a STGC, a DSB at the I-Sce I site is repaired from the iGFP gene on the same chromatid or sister chromatid, to result in a functional GFP gene. (D) Homology-mediated deletion product.
Two homologous recombination products are possible with DR–GFP, a short tract gene conversion (STGC) product (Fig. 1C) or a deletion product (Fig. 1D). The STGC product results from a noncross-over gene conversion within the limited amount of homology (≤812 bp), whereby the iGFP sequence acts as a donor of wild-type sequence information to the broken SceGFP gene. The deletion product could result from a conservative recombination event with an associated cross-over, a long tract gene conversion, or from the nonconservative single-strand annealing pathway in which the sequence between the two GFP repeats is degraded. Whereas the STGC event restores an intact GFP gene, a deletional event retains only the 5′ portion of the GFP gene that would encode a carboxy-terminal truncation. To determine whether this truncation product would be functional, the gene was constructed and transiently transfected into cells. Fluorescent cells were not detected above background levels (data not shown), demonstrating that only events that restore an intact GFP gene would be scored with the DR–GFP substrate.
The DR–GFP substrate was stably transfected into both wild-type AA8 hamster cells and the derivative XRCC3-deficient irs1SF cell line. Puromycin-resistant cell lines were isolated and screened by Southern blotting for those that had integrated one to two copies of the DR–GFP substrate and flow cytometric analysis was performed (Fig. 2A). A small number of GFP+ cells were detected in some of the DR–GFP-containing cell lines (≤10−4). Because the parental AA8 and irs1SF cells did not contain any positive cells (data not shown), this result suggests that the GFP+ cells in DR–GFP-containing cell lines may be spontaneous recombinants, although this has not been verified. In control experiments in which the cell lines were electroporated with a lacZ expression vector and then incubated for 48 hr in nonselective medium, there was no change in the number of GFP+ cells.
Figure 2.

The irs1SF cell line is defective for DSB-induced homologous recombination because of XRCC3 deficiency. Wild-type (DRaa) and XRCC3-deficient (DRirs) cell lines were electroporated with each of the indicated expression vectors (100 μg of the I-Sce I vector; 20 μg of the XRCC3 vector), and 5 × 104 cells were analyzed by flow cytometry. (A) Two-color fluorescence analysis for cell lines DRaa-40 and DRirs-10. The percentage of green fluorescent cells falling above the diagonal for each transfection are indicated. (FL1) Green fluorescence; (FL2) orange fluorescence. (B) Summary of results from several cell lines.
To determine whether DSBs will promote STGC within the DR–GFP substrate, cells were electroporated with the pCβASce vector (100 μg) to transiently express the I-Sce I endonuclease (Fig. 2A). I-Sce I expression in the wild-type cell line DRaa-40 led to >5% of cells being GFP+, a >700-fold induction over untransfected cells. This large induction of gene conversion with a DSB is similar to what has been observed previously when DSB-induced gene conversion was measured with drug selection markers (e.g., Liang et al. 1998). In contrast, I-Sce I expression in the mutant DRirs-10 cell line led to only 0.22% of cells being GFP+, that is, 25-fold lower than in the wild-type cell line, demonstrating that the irs1SF cell line has a defect in DSB-promoted homologous recombination.
The recombination defect of irs1SF can be complemented by expression of XRCC3
The irs1SF cell line was derived from a mutagenized culture of AA8 cells, raising the possibility that the recombination defect observed above may not be the direct result of mutation of the XRCC3 gene. To test whether the recombination defect in this cell line is due to XRCC3 deficiency, cotransfections were carried out in which the I-Sce I expression vector was electroporated with an XRCC3 expression vector. Coexpression of XRCC3 with I-Sce I resulted in a 25-fold increase in the number of GFP+ cells in the mutant cell line, restoring recombination to wild-type levels (Fig. 2A). No such increase was observed in wild-type cells. Transfection of the XRCC3 expression vector without I-Sce I did not detectably increase recombination over spontaneous levels in either the mutant or wild-type cell lines (data not shown). These findings indicate that the DSB-promoted recombination defect in the irs1SF cell line is the direct result of its XRCC3 deficiency.
We performed this analysis on multiple independently isolated DR–GFP-containing wild-type and irs1SF cell lines (Fig. 2B). In the three wild-type cell lines, ∼5% of the cells transfected with the I-Sce I expression vector were GFP+, whether or not they were cotransfected with the XRCC3 expression vector. In the two irs1SF-derived cell lines, recombination was much lower, but increased 25- to 30-fold when XRCC3 was expressed with I-Sce I. The absolute level of GFP+ cells was higher in the complemented DRirs-14 cell line, likely due to the presence of a second copy of the SceGFP gene. Thus, the recombination defect of irs1SF cells has been observed in independently isolated DR–GFP-containing cell lines.
Fluorescent cells have a functional GFP gene arising from gene conversion
To prove that fluorescent cells had undergone gene conversion within the DR–GFP reporter, we sorted GFP+ cells by flow cytometry. Sorted populations were collected from both the wild-type and mutant cells after transfection of the I-Sce I expression vector (20 μg) either with or without the XRCC3 expression vector. We used a lower amount of the I-SceI expression vector in these transfections to conserve DNA. Prior to sorting, ∼0.5% to 1.5% of the I-Sce I-transfected cells were GFP+ with the exception of the uncomplemented mutant, in which only 0.04% of the cells were GFP+ (Fig. 3). On expansion, the sorted populations from the wild-type cell line and the complemented mutant were 93% to 96% fluorescent, whereas the population derived from the uncomplemented mutant was 81% fluorescent. The decreased purity is likely due to the low number of GFP+ cells in the starting uncomplemented mutant cell population. Each cell population maintained fluorescence at this level during continued growth in culture (data not shown).
Figure 3.

Green fluorescence is stably expressed after I-Sce I expression. Wild-type DRaa-40 cells and XRCC3-deficient DRirs-14 cells were electroporated with the I-Sce I expression vector or both the I-Sce I and XRCC3 expression vectors, sorted on the basis of green fluorescence, and then reanalyzed by flow cytometry. Percent GFP+ cells before sorting (unshaded peak) and after sorting (shaded peaks) are indicated. Not shown is the DRaa-40 cell line transfected with both the I-Sce I and XRCC3 expression vectors, in which the presorted population was 0.66% GFP+ and the post-sorted population was 93.5% GFP+. Also not shown are the lacZ-transfected cells that were not sorted; 0.01% GFP+ for DRaa-40 and <0.01% GFP+ for DRirs-14. (The frequency of GFP+ cells is lower in this experiment than the one shown in Fig. 2, because only 20 μg of each of the expression vectors was electroporated.)
Genomic DNA was prepared from the sorted populations and analyzed by Southern blotting with the GFP-coding sequence as probe (Fig. 4). A STGC at the I-Sce I site restores the 11 bp of wild-type GFP sequence at the BcgI site, which is readily detectable by enzyme digestion in conjunction with SalI and HindIII. The nonrecombinant SceGFP gene gives bands of 2140 and 881 bp when the SalI/HindIII band is digested by I-Sce I but maintains the 3021-bp band when digested by BcgI. Conversely, a STGC that restores an intact GFP gene results in a SalI/HindIII band of 3021-bp that is not cleavable by I-Sce I, but that is cleaved by BcgI to 2140 and 881 bp. The banding pattern indicative of STGC was found in each of the GFP+ sorted populations. These results indicate that the fluorescence assay is a direct measure of the percentage of cells that have undergone STGC.
Figure 4.
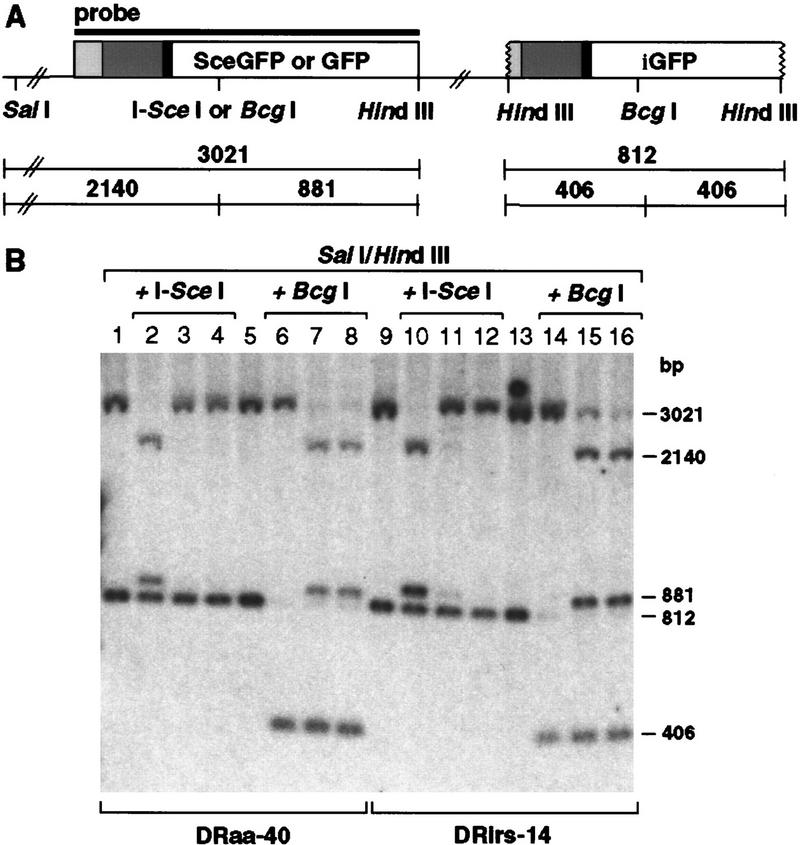
Green fluorescence is a measure of STGC. (A) Map of the parental or recombined DR–GFP substrate containing SceGFP or GFP, respectively. Sizes of expected fragments are indicated in base pairs. (B) Southern blot analysis of sorted cell lines. Genomic DNA was digested with the indicated endonucleases. (Lanes 1,2,5,6) Untransfected wild-type cell line DRaa-40; (lanes 3,7) GFP+ DRaa-40 cells from transfection of the I-Sce I expression vector; (lanes 4,8) GFP+ DRaa-40 cells from cotransfection of the I-Sce I and XRCC3 expression vectors; (lanes 9,10,13,14) untransfected mutant cell line DRirs-14; (lanes 11,15) GFP+ DRirs-14 cells from transfection of the I-Sce I expression vector; (lanes 12,16) GFP+ DRirs-14 cells from cotransfection of the I-Sce I and XRCC3 expression vectors. The residual 3021-bp band after BcgI digestion in lanes 7,8,15, and 16 is likely due to GFP-negative cells in the population (see Fig. 3).
Transient expression of proteins other than XRCC3 does not suppress the recombination defect
In yeast, expression of Rad51 or Rad52 from a high-copy number plasmid suppresses the repair defect of strains deficient in Rad51-related proteins (Hays et al. 1995; Johnson and Symington 1995). To determine whether transient expression of other proteins in addition to XRCC3 would suppress the gene conversion defect in the irs1SF cells, we transiently expressed three other proteins predicted to be involved in recombinational repair in mammalian cells. Unlike XRCC3 expression, none of XRCC2, Rad51, nor Rad52, singly or in combination, was capable of complementing the recombination defect in the irs1SF cells (Fig. 5).
Figure 5.

DSB-induced recombination is restored in the irs1SF cell line by transient expression of XRCC3 but not other recombination proteins. The DRirs-14 cell line was electroporated with each of the indicated expression vectors (20 μg). The total amount of DNA electroporated was maintained at a constant level by the addition of pCMV–lacZ. A total of 5 × 105 cells were analyzed by flow cytometry. (hRad51 or 51) Human Rad51; (mRad52 or 52) mouse Rad52; (X2) XRCC2; (X3) XRCC3.
Discussion
In this report we demonstrated that XRCC3 promotes the repair of DSBs by homologous recombination using a novel fluorescence-based assay. The GFP reporter allows homology-directed repair to be scored by flow cytometry within 24–48 hr after transfection of the endonuclease expression vector, with a sensitivity for detection of recombinants at a level of 10−4 or less. Levels of homologous repair observed in wild-type cells with this system are comparable to those with drug selection markers. With this GFP reporter system, we determined that the irs1SF mutant cell line has markedly reduced levels of homologous repair, measured to be 25-fold lower than the parental cell line. The recombination defect can be corrected by expression of XRCC3 but not other proteins predicted to be involved in recombinational repair in mammalian cells.
The irs1SF mutant has a broad spectrum of sensitivities to DNA damaging agents. It has modest sensitivity to both ionizing and UV radiation (2- to 3-fold), modest sensitivity to alkylating agents (2- to 10-fold), and extreme sensitivity to DNA cross-linking agents (60- to 100-fold) (Fuller and Painter 1988; Caldecott and Jeggo 1991). One other hamster mutant, the irs1 cell line, is known to have a similar spectrum of sensitivity to DNA-damaging agents (Jones et al. 1987; Caldecott and Jeggo 1991). This mutant is deficient in XRCC2 and, similar to irs1SF cells, is also defective in DSB-promoted recombination (Johnson et al. 1999). Both mutant cell lines, in contrast to a nonhomologous DNA end-joining repair mutant (xrs-6; Kemp and Jeggo 1986), show a highly elevated frequency of spontaneous chromosomal aberrations (Tebbs et al. 1995; Fuller and Painter 1988; Liu et al. 1998). Thus, homologous recombination may play a particularly important role in mammalian cells in the repair of spontaneously arising DNA damage, as may occur during DNA replication when a sister chromatid would be available to act as a template for homology-directed repair. The maintenance of genomic integrity by homologous repair processes is likely to be necessary not only for cell survival, as is surmised from the lethality of Rad51 knockout mice (Lim and Hasty 1996; Tsuzuki et al. 1996), but also for prevention of tumorigenesis, as the tumor suppressor genes BRCA1 and BRCA2 have been linked to these processes (Kinzler and Vogelstein 1997). Recently, BRCA1-deficient cells have been directly demonstrated to have a homologous repair defect using an I-Sce I assay system (Moynahan et al. 1999).
The specific defect in homology-directed repair that has been observed in the irs1SF cell line is in gene conversion, a process that is thought to require homologous strand invasion. The human Rad51 protein, like its Escherichia coli RecA and yeast Rad51 homologs, is a strand transferase, promoting the invasion of a single-stranded molecule into a homologous duplex (Sung 1994; Baumann and West 1998). Human XRCC3 is only 22% identical to human Rad51, yet key residues in the Walker A and B motifs, which are known to be involved in ATP binding, are conserved (Liu et al. 1998). Similarly, XRCC2 is 20% identical to Rad51, with the highest identity in the same motifs (Liu et al. 1998). Although it is possible that XRCC3 (and XRCC2) has strand transferase activity like Rad51, a more likely alternative is that XRCC3 is an accessory factor for Rad51. In this way, XRCC3 would act more like the yeast Rad51-related proteins Rad55 and Rad57, which have been shown to promote the strand exchange activity of the S. cerevisae Rad51 protein, rather than having strand exchange activity themselves (Sung 1997b). Supporting this, XRCC3 shares more sequence identity with yeast Rad57 (28%) than with human Rad51 (Liu et al. 1998) and it also interacts with Rad51 (Liu et al. 1998), as does Rad55 with the yeast Rad51 protein (Hays et al. 1995; Johnson and Symington 1995).
A notable difference between XRCC3 and Rad55/Rad57 is that Rad51 overexpression in yeast will suppress the DNA repair defect of rad55 or rad57 mutants (Hays et al. 1995; Johnson and Symington 1995), whereas we see no suppression of the recombination defect in the irs1SF mutant by transient expression of exogenous Rad51. Similarly, overexpression of Rad52 in yeast, a protein that also stimulates the Rad51 strand exchange activity in vitro (Sung 1997a), will suppress the X-ray sensitivity of rad55 or rad57 mutants (Hays et al. 1995). Yet, as with Rad51, we see no suppression of the recombination defect in the irs1SF cell line by expression of exogenous mouse Rad52. In our assays, only XRCC3 expression suppressed the defect in the irs1SF mutant, although we cannot rule out that higher levels of Rad51 or Rad52 would have an effect. Interestingly, XRCC3 is required for the assembly of Rad51 complexes that are formed in vivo in response to DNA-damaging agents (Bishop et al. 1998), raising the possibility that the defect in the irs1SF cell line will not be suppressible by any protein except XRCC3. In summary, our results provide further evidence for a role for the Rad51 pathway in maintaining genomic stability in mammalian cells by promoting homologous repair of DNA damage.
Materials and methods
DNA manipulations
The modified GFP gene (NZ-EGFP) was constructed from the enhanced GFP gene (EGFP; Clontech) and contains a threefold repeated nuclear localization signal (Donoho et al. 1998) and a zinc finger DNA-binding domain from the GAGA2 transcription factor (Gly-310 to Val-372; Pedone et al. 1996) with preferred mammalian codons. This modified GFP-coding region was cloned into the vector pCAGGS (Niwa et al. 1991). The SceGFP gene was constructed by replacing the 34-bp BcgI fragment from the GFP gene with 34 bp that include the 18 bp I-Sce I site. The pgk–pur gene is from pHA262pur (a kind gift of Hein te Riele, Netherlands Cancer Institute, Amsterdam, The Netherlands). I-Sce I is expressed in pCβASce from a hCMV enhancer/chicken β-actin promoter (Richardson et al. 1998). In each of the other expression vectors, the protein is expressed from a hCMV enhancer/promoter. The human XRCC3 expression vector, pXR3-10, is a kind gift of K. Brookman (Livermore). The human XRCC2 expression vector has been described previously (Johnson et al. 1999), as has the Rad51 expression vector, pCDNA–HA-51 [a kind gift of D. Weaver; (Scully et al. 1997)]. Both of these vectors have been shown to express functional proteins. The Rad52 expression vector, pFlag:mR52 (R.D. Johnson and M. Jasin, unpubl.) was derived from pKSMmRad52cDNA (a kind gift of R. Rothstein, Columbia University, New York, NY).
Transfections and recombination assays
Stable integrants were constructed by electroporating cells in tissue culture medium with circular pDR–GFP at 625 V/cm, 960 μF, and plating in puromycin at 12 μg/ml (AA8) or 5 μg/ml (irs1SF). Puromycin-resistant colonies were screened by Southern blots for an intact DR–GFP reporter with SalI and HindIII, and subsequently with BcgI, I-SceI, and other enzymes to estimate DR–GFP copy number. To assay GFP expression, cells were electroporated as described above (Fig. 5) or under modified conditions found to result in a higher transfection efficiency (Figs. 2–4). The modified conditions consist of adding fresh tissue culture medium to cells 4 hr prior to electroporation and electroporating cells in phosphate-buffered saline and then diluting cells immediately in tissue culture medium. After electroporation, cells were plated in nonselective medium for 48 hr, rinsed twice with phosphate-buffered saline, recovered by treatment with trypsin/EDTA, concentrated by centrifugation, and resuspended in tissue culture medium prior to flow cytometry. Cells were analyzed in a Becton-Dickinson FACScan on a green (FL1) versus orange (FL2) fluorescence plot with 20% or 25% orange minus green compensation to aid in the detection of faint positives. GFP-negative cells possess autofluorescence and fall along the green/orange diagonal, whereas GFP+ cells appear in a separate population shifted off the diagonal of cells and toward FL1. Expression of a functional GFP gene in DR–GFP recombinants appears to confer no selective advantage or disadvantage, because the ratio of GFP positive to negative cells in a mixed population remains stable during expansion of cells for 2 weeks or longer.
Acknowledgments
We thank Dr. Jeremy Stark for helpful comments on the manuscript. This work was supported by the National Research Service Award fellowship (GM18640) to R.D.J. and Human Frontiers Science Program (RG-270/97) and National Institutes of Health (GM54688) grants to M.J. A portion of this work was done under the auspices of the U.S. Department of Energy under contract no. W-7405-ENG-48.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL m-jasin@ski.mskcc.org; FAX (212) 717-3317.
References
- Baumann P, West SC. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem Sci. 1998;23:247–251. doi: 10.1016/s0968-0004(98)01232-8. [DOI] [PubMed] [Google Scholar]
- Bishop DK, Ear U, Bhattacharyya A, Calderone C, Beckett M, Weichselbaum RR, Shinohara A. Xrcc3 is required for assembly of Rad51 complexes in vivo. J Biol Chem. 1998;273:21482–21488. doi: 10.1074/jbc.273.34.21482. [DOI] [PubMed] [Google Scholar]
- Caldecott K, Jeggo P. Cross-sensitivity of gamma-ray-sensitive hamster mutants to cross-linking agents. Mutat Res. 1991;255:111–121. doi: 10.1016/0921-8777(91)90046-r. [DOI] [PubMed] [Google Scholar]
- Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, Thompson LH. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol. 1994;14:68–76. doi: 10.1128/mcb.14.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colleaux L, d'Auriol L, Gailbert F, Dujon B. Recognition and cleavage site of the intron-encoded omega transposase. Proc Natl Acad Sci. 1988;85:6022–6026. doi: 10.1073/pnas.85.16.6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoho G, Jasin M, Berg P. Analysis of gene targeting and intrachromosomal homologous recombination stimulated by genomic double-strand breaks in mouse embryonic stem cells. Mol Cell Biol. 1998;18:4070–4078. doi: 10.1128/mcb.18.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Winderbaum J, Nickoloff JA, Jasin M. Gene conversion tracts from double-strand break repair in mammalian cells. Mol Cell Biol. 1998;18:93–101. doi: 10.1128/mcb.18.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers J, Hendriks RW, Swagemakers SMA, Troelstra C, de Wit J, Bootsma D, Hoeijmakers JHJ, Kanaar R. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell. 1997;89:195–204. doi: 10.1016/s0092-8674(00)80199-3. [DOI] [PubMed] [Google Scholar]
- Fuller LF, Painter RB. A Chinese hamster ovary cell line hypersensitive to ionizing radiation and deficient in repair replication. Mutat Res. 1988;193:109–121. doi: 10.1016/0167-8817(88)90041-7. [DOI] [PubMed] [Google Scholar]
- Hays SL, Firmenich AA, Berg P. Complex formation in yeast double-strand break repair: Participation of Rad51, Rad52, Rad55, and Rad57 proteins. Proc Natl Acad Sci. 1995;92:6925–6929. doi: 10.1073/pnas.92.15.6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- Jeggo PA. DNA breakage and repair. Adv Genet. 1998;38:185–218. doi: 10.1016/s0065-2660(08)60144-3. [DOI] [PubMed] [Google Scholar]
- Johnson RD, Symington LS. Functional differences and interactions among the putative RecA homologs Rad51, Rad55, and Rad57. Mol Cell Biol. 1995;15:4843–4850. doi: 10.1128/mcb.15.9.4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RD, Liu N, Jasin M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature. 1999;401:397–399. doi: 10.1038/43932. [DOI] [PubMed] [Google Scholar]
- Jones NJ, Cox R, Thacker J. Isolation and cross-sensitivity of X-ray-sensitive mutants of V79-4 hamster cells. Mutat Res. 1987;183:279–286. doi: 10.1016/0167-8817(87)90011-3. [DOI] [PubMed] [Google Scholar]
- Kemp LM, Jeggo PA. Radiation-induced chromosome damage in X-ray-sensitive mutants (xrs) of the Chinese hamster ovary cell line. Mutat Res. 1986;166:255–263. doi: 10.1016/0167-8817(86)90025-8. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761. doi: 10.1038/386761a0. , 763. [DOI] [PubMed] [Google Scholar]
- Liang F, Romanienko PJ, Weaver DT, Jeggo PA, Jasin M. Chromosomal double-strand break repair in Ku80 deficient cells. Proc Natl Acad Sci. 1996;93:8929–8933. doi: 10.1073/pnas.93.17.8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Han M, Romanienko PJ, Jasin M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc Natl Acad Sci. 1998;95:5172–5177. doi: 10.1073/pnas.95.9.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Lamerdin JE, Tebbs RS, Schild D, Tucker JD, Shen MR, Brookman KW, Siciliano MJ, Walter CA, Fan W, Narayana LS, et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell. 1998;1:783–793. doi: 10.1016/s1097-2765(00)80078-7. [DOI] [PubMed] [Google Scholar]
- Moynahan, M.E., J.W. Chiu, B.H. Koller, and M. Jasin. 1999. Brca1 controls homology-directed repair. Mol. Cell. (In press). [DOI] [PubMed]
- Muris DF, Vreeken K, Carr AM, Broughton BC, Lehmann AR, Lohman PH, Pastink A. Cloning the RAD51 homologue of Schizosaccharomyces pombe. Nucleic Acids Res. 1993;21:4586–4591. doi: 10.1093/nar/21.19.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J-i. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–200. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Pedone PV, Ghirlando R, Clore GM, Gronenborn AM, Felsenfeld G, Omichinski JG. The single Cys2-His2 zinc finger domain of the GAGA protein flanked by basic residues is sufficient for high-affinity specific DNA binding. Proc Natl Acad Sci. 1996;93:2822–2826. doi: 10.1073/pnas.93.7.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes TD, Malone RE, Symington LS. Recombination in yeast. In: Broach JR, Pringle JR, Jones EW, editors. The molecular and cellular biology of the yeast Saccharomyces: Genome dynamics, protein synthesis, and energetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1991. pp. 407–521. [Google Scholar]
- Richardson C, Moynahan ME, Jasin M. Double-strand break repair by interchromosomal recombination: Suppression of chromosomal translocations. Genes & Dev. 1998;12:3831–3842. doi: 10.1101/gad.12.24.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijkers T, Van Den Ouweland J, Morolli B, Rolink AG, Baarends WM, Van Sloun PP, Lohman PH, Pastink A. Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol Cell Biol. 1998;18:6423–6429. doi: 10.1128/mcb.18.11.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265:1241–1243. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- ————— Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem. 1997a;272:28194–28197. doi: 10.1074/jbc.272.45.28194. [DOI] [PubMed] [Google Scholar]
- ————— Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes & Dev. 1997b;11:1111–1121. doi: 10.1101/gad.11.9.1111. [DOI] [PubMed] [Google Scholar]
- Taccioli GE, Rathbun G, Oltz E, Stamato T, Jeggo PA, Alt FW. Impairment of V(D)J recombination in double-strand break repair mutants. Science. 1993;260:207–210. doi: 10.1126/science.8469973. [DOI] [PubMed] [Google Scholar]
- Taghian DG, Nickoloff JA. Chromosomal double-strand breaks induce gene conversion at high frequency in mammalian cells. Mol Cell Biol. 1997;17:6386–6393. doi: 10.1128/mcb.17.11.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbs RS, Zhao Y, Tucker JD, Scheerer JB, Siciliano MJ, Hwang M, Liu N, Legerski RJ, Thompson LH. Correction of chromosomal instability and sensitivity to diverse mutagens by a cloned cDNA of the XRCC3 DNA repair gene. Proc Natl Acad Sci. 1995;92:6354–6358. doi: 10.1073/pnas.92.14.6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LH, Jeggo PA. Nomenclature of human genes involved in ionizing radiation sensitivity. Mutat Res. 1995;337:131–134. doi: 10.1016/0921-8777(95)00018-f. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Schild D. The contribution of homologous recombination in preserving genome integrity in mammalian cells. Biochimie. 1999;81:87–105. doi: 10.1016/s0300-9084(99)80042-x. [DOI] [PubMed] [Google Scholar]
- Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci. 1996;93:6236–6340. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JF. Radiation mutagenesis: The initial DNA lesions responsible. Radiat Res. 1995;142:362–368. [PubMed] [Google Scholar]