

Abstract
The periodic fever syndromes are disorders of innate immunity. They may be inherited or acquired and present as recurrent attacks of apparently spontaneous self-limiting inflammation without evidence of autoantibodies or infection. Over the past decade-and-a-half there has been significant progress in their understanding and treatment.
Keywords: human studies, inflammation/inflammatory mediators including eicosanoids, tumour antigens
Introduction
The periodic fever syndromes are disorders of innate immunity characterized by recurring episodes of fever and systemic inflammatory symptoms, affecting the serosal surfaces, joints, skin and eyes. They include the hereditary syndromes: familial Mediterranean fever (FMF), tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS), mevalonate kinase deficiency [MKD – previously known as hyperimmunoglobulin D and periodic fever syndrome (HIDS)], the cryopyrin-associated periodic syndromes (CAPS), Blau syndrome, deficiency of the interleukin (IL)-1-receptor antagonist (DIRA), pyogenic arthritis, pyoderma gangrenosum and acne syndrome (PAPA syndrome): diseases of uncertain genetic aetiology including periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome and acquired diseases such as Schnizler's syndrome.
Despite similarities in their symptoms, they have differing aetiologies, inheritance, duration and frequency of ‘attacks’, and the overall clinical picture of the various disorders is often distinct (Table 1). These diseases are compatible with near-normal life expectancy, bar the serious risk of developing reactive systemic (AA) amyloidosis. Recent advances in unravelling their molecular pathogenesis have led to radically improved diagnosis and therapies, and have provided advances into the understanding of aspects of innate immunity in general. The aim of this paper is to provide an up-to-date overview of these syndromes and their management.
Table 1.
The major autoinflammatory conditions of known genetic aetiology.
 |
FMF
The term ‘familial Mediterranean fever’ was coined in 1958 and the associated gene, MEFV, was identified by positional cloning in 1997 [1,2]. MEFV is expressed constitutively in neutrophils, eosinophils, monocytes, dendritic cells and synovial fibroblasts and is up-regulated in response to inflammatory cytokines [3]. The more than 80 MEFV mutations associated with FMF encode either single amino acid substitutions or deletions (Infevers registry database http://fmf.igh.cnrs.fr/ISSAID/infevers/) [4–6]. Disease-causing mutations occur mainly in exon 10, but also within exons 1, 2, 3, 5 and 9. Mutations in each of the two MEFV alleles are found in 85% of patients with FMF; although 14–25% of patients with a clinical diagnosis of FMF have only one identified mutation, the great majority of individuals with a single mutated allele are healthy carriers [7,8].
Neither the structure nor function of pyrin (the protein product of MEFV) have been characterized completely, although it is a member of the death domain superfamily and likely to be involved in homotypic protein–protein interactions [9,10] which potentiate production of proinflammatory cytokines [11].
FMF is most common in Middle Eastern populations, but occurs worldwide [12]. The sexes are affected equally and the disease usually presents in childhood. The prevalence of FMF is estimated to be one in 250 to one in 500 among Sephardic Jews and one in 1000 in the Turkish population. Carrier frequency exceeds one in four in some eastern Mediterranean populations, prompting speculation that the FMF trait may have conferred survival benefit, possibly through enhanced resistance to infection.
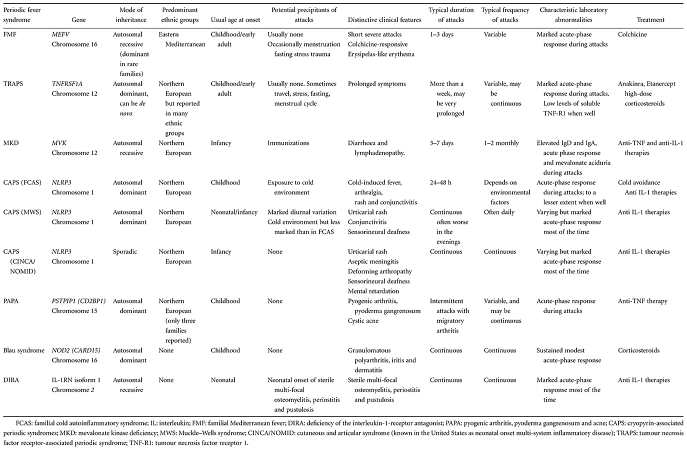
Apparently spontaneous attacks of FMF occur irregularly, although precipitants such as minor physical or emotional stress, the menstrual cycle or diet are well recognized, and resolve within 72 h. Peritonitis occurs in 85% of cases, and 40% of patients will undergo exploratory surgery before diagnosis is made. Pleuritic chest pain occurs in 40% of patients, characteristically unilaterally. Meningitic headache occurs particularly in children, but the nervous system is not usually involved. Orchitis occurs in fewer than 5%, again most commonly in early childhood. Joint involvement is generally mild and affects the lower limbs: arthralgia is common in acute attacks and a chronic destructive arthritis is rare. A degree of myalgia can occur during acute attacks, and 20% of patients have persistent muscle pain on exertion, usually affecting the calves. Protracted febrile myalgia is a very rare complication, characterized by severe pain in the lower limb or abdominal musculature that lasts for weeks and can be accompanied by a vasculitic rash; it usually responds to high-dose corticosteroid therapy. A characteristic erysipelas-like rash occurs in 20% of patients, usually around the ankles (Fig. 1).
Fig. 1.
Erysipelas-like erythema during an attack of familial Mediterranean fever (FMF). This rare but highly characteristic manifestation of FMF is usually seen on the lower legs or feet. It is accompanied by systemic inflammation and resolves with 2–3 days.
Acute attacks are accompanied by a neutrophilic leucocytosis and a dramatic acute-phase response. Investigations may be required to exclude other diagnoses, but X-ray, ultrasound or echocardiography during attacks is usually unrewarding.
Diagnosis is supported by DNA analysis, but remains clinical and centres on the history of recurrent self-limiting attacks of fever and serositis that are prevented by colchicine [13]. Genetic results must be interpreted cautiously, given that not all individuals with paired pathogenic MEFV mutations develop FMF, and others with heterozygous carrier status can do so. Furthermore, most diagnostic laboratories offer only limited analysis, as MEFV is a large gene.
Supportive measures, particularly analgesia, are often required during acute attacks, but the mainstay of management is long-term prophylactic treatment with low-dose colchicine. This serendipitous discovery in 1972 [14] has entirely transformed the prognosis of this disease. Continuous treatment with colchicine at a dose of 1–2 mg daily in adults prevents or substantially reduces symptoms of FMF in at least 95% of cases, and almost completely eliminates the risk of AA amyloidosis (see below). The mechanism of action of colchicine remains incompletely understood, but it modulates neutrophil adhesion, mobility and cytokine release in a presumably somewhat specific manner, possibly by competing with pyrin as a substrate for caspase 1, in patients with defective pyrin variants [15,16].
Long-term colchicine is advisable in every patient with FMF and mandatory in those who already have AA amyloidosis. Although colchicine is very toxic in acute overdose, the low daily doses required for treatment of FMF are generally very well tolerated. Diarrhoea is the most common side effect and can usually be avoided by gradual introduction of the drug and, if necessary, temporary withdrawal of lactose from the diet. Despite theoretical concerns, there is no evidence that colchicine causes infertility or birth defects, and it can be taken safely by nursing mothers [17]. Colchicine has now been licensed by the US Food and Drug administration for the prophylactic treatment of FMF from the age of 4 years upwards. Genuine resistance to colchicine is rare, although poor compliance is frequent. In treatment-refractory patients there is emerging evidence that etanercept or anakinra can be efffective [18,19].
TRAPS
TRAPS is very rare, with an estimated prevalence of about 1 per million in Europe. It is an autosomal dominant disease associated with mutations in TNF receptor superfamily 1A (TNFRSF1A), a 10-exon gene located on chromosome 12p13 [20]. TNF is a key mediator of inflammation with actions including pyrexia, cachexia, leucocyte activation, induction of cytokine secretion, expression of adhesion molecules and resistance to intracellular pathogens. TNF receptor 1 (TNF-R1) is a member of the death domain superfamily. It has a cysteine-rich extracellular domain, a transmembrane domain and an intracellular death domain. Binding of soluble circulating TNF causes receptor trimers to form with downstream induction of inflammation. The mechanism(s) by which heterozygous TNFRSF1A mutations cause TRAPS remains unclear and may well be mutation-specific [21,22]. Most TRAPS-associated mutations are within exons 2–4, and about half are substitutions affecting highly conserved cysteine residues in the extracellular domain. Under normal circumstances, TNF signalling is terminated by metalloproteinase-dependent cleavage of the extracellular domain, thereby releasing soluble TNF-R1 which competes with the cell surface receptor in binding circulating TNF. While cleavage of certain TNFR1 variants is impaired, producing a ‘shedding defect’, this is not the case with other TRAPS-causing mutations, which must exert their pathogenic effect by different means. It is thought that mutant misfolded receptors may give rise to enhanced or prolonged signalling, possibly through retention within the endoplasmic reticulum [21–24].
TRAPS was described in 1982 as familial Hibernian fever, reflecting the Irish/Scottish ancestry of patients in early reports, but TRAPS has been reported subsequently in many ethnic groups. Both sexes are affected and presentation is usually before the age of 4 years. Most mutations are associated with high penetrance but two variants, P46L and R92Q, which can be associated with mild TRAPS, are present in approximately 10% of healthy West Africans [25] and 2% of healthy Caucasians, respectively.
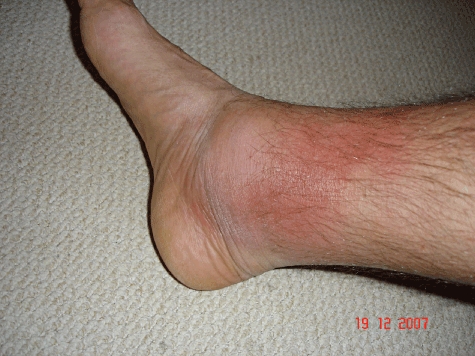
TRAPS attacks are often far less distinct than in FMF. Febrile episodes typically last weeks and symptoms are near-continuous in a third of patients. Approximately half of patients give no clear family history, many of whom have the P46L or R92Q variants, which are associated with milder disease and later onset [26]. The clinical picture varies: more than 95% of patients experience fever, and 80% have arthralgia or myalgia that classically displays centripetal migration; abdominal pain occurs in 80%, and rash which may be erythematous, oedematous plaques (often overlying areas of mylagia) and discrete reticulate or serpiginous lesions occur in 70% of patients (Fig. 2). Other features include headache, pleuritic pain, lymphadenopathy, conjunctivitis and periorbital oedema. There are also reports of central nervous system (CNS) manifestations and magnetic resonance imaging (MRI) findings resembling multiple sclerosis [27]. Symptoms are accompanied almost universally by a marked acute-phase response. Between attacks, the plasma concentration of soluble TNF-R1 may be abnormally low in patients with decreased receptor shedding. Genetic testing is central to diagnosis.
Fig. 2.
Typical rash accompanying an attack of tumour necrosis factor receptor-associated periodic syndrome (TRAPS). Rashes are seen in about 80% of TRAPS attacks and tend to last for 10 days–3 weeks. They may be very varied both between and within individuals; there can be discrete erythematous macules or papules or large erythematous plaques and both localized and generalized annular, reticulate and serpiginous lesions are also seen.
Despite high initial hopes for response to anti-TNF biologics, treatment of TRAPS often remains disappointing. Acute attacks respond to high-dose corticosteroids, and etanercept but interestingly, not infliximab, is useful as long-term treatment in some patients, although response may gradually decline [28]. More recent reports suggest that IL-1 blockade with anakinra can be very effective in preventing attacks [29].
MKD
MKD, also known as hyperimmunoglobulin D and periodic fever syndrome (HIDS), is an autosomal recessive disease caused by mutations in the mevalonate kinase (MVK) gene [30]. About 60 mutations have been described, the most common of which encode MVK variants V377I and I268T. MVK is the enzyme after 3-hydroxy-3-methylglutaryl-co-enzyme A (HMG CoA) reductase in the cholesterol, farnasyl and isoprenoid biosynthesis pathway. Most disease-causing MVK mutations are missense variants that reduce enzyme activity by >90% [31]. Other mutations resulting in near complete absence of enzyme activity cause a much more severe inflammatory disease known as mevalonic aciduria (MVA), features of which include stillbirth, congenital malformations, severe psychomotor retardation and early death.
It is not yet known how MVK deficiency causes inflammation, although reduction in prenylation due to failure of flux through the isoprenoid pathway currently seems more likely to be responsible than accumulation of the enzyme's substrate [32,33]. A variety of effects of statins on caspase-1 activation and IL-1 secretion have been postulated, but a clinical study of simvastatin in six patients with MKD suggested only minor benefit [34], and two children with MVA were reported to develop severe inflammatory disease following statin treatment [35].
MKD is extremely rare and is predominantly a disease of north-western Europe, probably through a founder effect, although it has been reported in many other countries and other ethnic groups. It was described in the Netherlands in 1984 and the international registry in Nijmegen has data on just over 200 patients (http://www.hids.net). The carriage rate of MVK V337I is one in 350 in the Dutch population [36].
The disease occurs equally in both sexes and usually presents in the first year of life [37]. Attacks are irregular, typically lasting 3–7 days, and are provoked characteristically by vaccination, minor trauma, surgery or stress, perhaps triggered by a reduction in MVK enzyme associated with increased body temperature [38]. Attacks typically comprise fever, cervical lymphadenopathy, splenomegaly and abdominal pain with vomiting and diarrhoea. Headache, arthralgia, large joint arthritis, erythematous macules and papules and aphthous ulcers are also common. MKD often ameliorates partially in adult life.
Diagnosis of MKD is supported by a high serum immunoglobulin (Ig)D concentration, although this is not specific and not always present [39]. More accessibly, serum IgA concentration is also elevated in 80% patients. Attacks are accompanied by an acute-phase response, leucocytosis and the transient presence of mevalonic acid in the urine. A mutation in both alleles of the MVK gene can be identified in most patients. Treatment is largely supportive, and responses to etanercept [40,41] and anakinra have been reported.
CAPS
CAPS comprises an overlapping severity spectrum, ranging from mild to severe, otherwise known as familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome (MWS) and chronic infantile neurological, cutaneous and articular syndrome (CINCA), which is known in the United States as neonatal onset multi-system inflammatory disease (NOMID). CAPS is associated with mutations in NLRP3/CIAS1 on chromosome 1q44, a gene that encodes the death domain protein NOD-like receptor protein 3 (NLRP3) [42]. Dominant inheritance occurs in about 75% of patients with FCAS and MWS, whereas CINCA, at the most severe end of the clinical spectrum, is usually due to de novo mutation. More than 60, mainly missense, mutations have been reported, with the vast majority in exon 3. The genotype–phenotype relationship can differ markedly between individuals, even within a family.
NLRP3 encodes a death domain protein that has a pyrin domain, a nucleotide-binding site (NBS) domain and a leucine-rich repeat (LRR) motif. Recognition via the LRR of a variety of danger signals results in the association of NLRP3 with other members of the death domain superfamily to form a multimeric cytosolic protein complex, known as the inflammasome [43,44]. This results in activation of caspase 1, which cleaves pro-IL-1 to produce active IL-1 beta; it also up-regulates nuclear factor kappa B (NF-κB) expression and thereby increases IL-1 gene expression. IL-1 is a major proinflammatory cytokine that mediates responses to infection and tissue injury and, as proved by the complete response of CAPS to IL-1 receptor blockade, is pivotal in causing the clinical features of this disease [45].
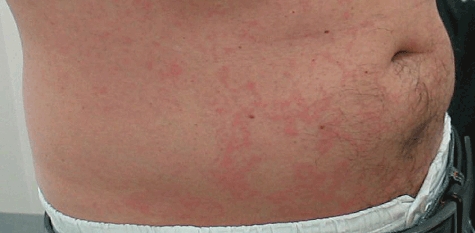
Most reported patients with CAPS have European ancestry, but cases have been described from South Asia and elsewhere [46]. Onset of disease is usually in early infancy, often from birth, and there is no sex bias. FCAS is characterized by recurrent episodes of cold-induced fever, arthralgia, conjunctivitis and rash (Fig. 3). MWS was described in 1962 [47] as a similar syndrome but with often daily attacks complicated by progressive sensorineural deafness in 40% of patients, and a high risk of AA amyloidosis. CINCA is a sporadic, severe inflammatory disorder that presents in the neonatal period with multi-system involvement including the skin, skeletal system and central nervous system [48]. Bony overgrowth and premature ossification may occur, particularly in the skull and knees; chronic aseptic meningitis results in developmental retardation, blindness and deafness.
Fig. 3.
Classical diffuse urticarial rash of cryopyrin-associated periodic syndromes (CAPS). The lesions are only slightly itchy and are often remarkably asymptomatic.
Clinical disease is accompanied by an acute-phase response, and often anaemia of chronic disease. Fundoscopy and brain imaging may show features consistent with elevated intracranial pressure. A mutation in NLRP3 can be identified in almost all patients with clinical FCAS or MWS, although mutations are only found in about 50% of children with classical CINCA; it is possible that ‘mutation-negative’ cases of FCAS and MWS may also exist but are simply not being recognized.
Daily injections of anakinra (recombinant IL-1 receptor antagonist) produces rapid and complete clinical and serological remission in CAPS [45]. Various new IL-1 inhibitors are also proving to be very effective [49]. It is hoped that early anti-IL-1 therapy may prevent developmental abnormalities.
DIRA
This autosomal recessive disease was first described in 2009 [50]. It is due to mutations in IL-1RN that result in a total deficiency of IL-1 receptor antagonist. The disease has been reported in only a handful of families of varying ethnic origins. The disease presents in the immediate neonatal period with a pustular rash, joint swelling, osteolytic lesions and periosteitis typically affecting the distal ribs and the long bones. Characteristic heterotopic bone formation around the proximal femur appears almost universal. Treatment is anakinra, with good responses seen in all cases treated so far.
PAPA syndrome
This exceptionally rare autosomal dominant disease is caused by mutations in the proline serine threonine phosphatase-interacting protein 1 (PTSTPIP) gene encoding a protein also known as CD2 binding protein 1 (CB2BP1) [51]. The underlying pathogenesis remains poorly understood, although there is evidence that CD2BP1, which interacts with actin and is an important component of cytoskeletal organization, interacts with pyrin and disease-associated mutations appear to potentiate this. PAPA is characterized clinically by severe acne and recurrent pustular sterile arthritis that occurs typically after minor trauma. Early reports suggest that therapy with anakinra may be effective.
Blau syndrome
This sarcoid-like syndrome is an autosomal dominant syndrome of granulomatous infiltration of the joints (causing camptodactyly), skin and sometimes viscera, associated with uveitis [52]. It is associated with missense mutations in NOD2/CARD15. This is another member of the death domain superfamily [53] which has also been implicated in familial Crohn's disease, also a granulomatous disease. Treatment is with corticosteroids and there is one case report of response to anakinra [54].
PFAPA
This was first described in 1987 in a dozen otherwise healthy American children [55]. The diagnosis is clinical and is suggested by the presence of a recurrent fever of early onset (aged less than 5 years) and one of the following associated symptoms: oral apthous ulcers, cervical lymphadenopathy or pharyngitis, in the absence of evidence of recurrent upper respiratory tract infections or cyclic neutropenia. Characteristically, the children are entirely well between attacks and their parents frequently report that they seem less susceptible than other children to minor viral infections. The largest case-series to date has been published recently by Feder and Salazar [56]. They describe the phenotype in more than 100 children seen in a single centre in Connecticut. The median age at presentation was 2·5 years and 83% of children presented before their fifth birthday. There was a slight male preponderance of 62%. The patients were of European ancestry in 81% of cases, but large case-series have also been described in Israel, suggesting that this is by no means a disease limited to those of northern European descent [57,58]. The ‘acronym symptoms’ of apthous oral ulcers, pharyngitis and cervical lymphadenopathy were definitely not universal and are frequently not all present during a single attack. The diagnosis is clinical and there is no diagnostic test. During attacks the acute phase response is often strikingly elevated. In general the prognosis is good and most children will outgrow their symptoms within a decade.
For many clinicians the strongest diagnostic pointers are the extreme regularity of attacks (although there may be some skipping of attacks, particularly in the summer months) and the response to a small dose of corticosteroids. None the less, before making a diagnosis of PFAPA other causes must be excluded including recurrent viral or bacterial infections and cyclic neutropenia. In addition the inherited fever syndromes should be considered; indeed, a recent paper has shown that 16% of patients who fulfilled the diagnostic criteria for PFAPA in fact had an inherited periodic fever syndrome and another 18% carried a known fever gene mutation or polymorphism [59].
Most children have received repeated courses of antibiotics without benefit for presumed recurrent pharyngitis before the diagnosis is made. The first-line treatment of PFAPA is a single dose of corticosteroid given at the start of the attack. Padeh et al. suggested that the dramatic response to a single oral dose of corticosteroids is sufficiently unique to PFAPA syndrome that it could be used as a diagnostic criterion [57]. More than 70% of patients who responded to steroids were given it for recurrent attacks with good effect, although attack-free interval did appear to shorten in 50% of treated children.
The H2 receptor antagonist cimetidine has been also reported to be effective in PFAPA. Feder and Salazar describe treatment of 26 children using 150 mg twice daily for a total of 6–12 months. Prophylactic cimetidine stopped all attacks in 27%, with only one patient having a recurrence of disease when treatment was stopped. The only major side effect encountered was that four children could not tolerate the taste. Colchicine has been tried in PFAPA with variable reports of success. Tasher et al. reported that eight of the nine children had a good clinical response to colchicine prophylaxis in PFAPA at doses of 0·5–1 mg daily, but other series have reported less benefit [57]. Our unreported experience has also been favourable, and in patients who require prophylactic treatment it is our first choice of agent.
Tonsillectomy can be curative in PFAPA: a randomized study in 39 children followed-up for 18 months demonstrated a remission of symptoms in 62% of the children who had a tonsillectomy and only 5% of controls [60]. Curiously, the histology of the resected material in children who underwent tonsillectomy for PFAPA showed typical appearances of chronic inflammation with no specific features [61]. In general, more than 50% of children appear to have excellent long-term outcomes after tonsillectomy, but this may be biased data as many centres do not refer children for surgery unless their tonsils are persistently enlarged. Therefore, these very high responses may be seen only in the subgroup of cases with abnormal tonsils whose symptoms are driven by persistent tonsillar inflammation.
Schnitzler's syndrome
This was first reported in 1974 and is characterized by a chronic urticarial rash, a monoclonal IgM gammopathy and systemic inflammation usually presenting as fever [62]. The median age at onset is 51 years [63] and the youngest reported case was aged 13. There is a slight male preponderance, and the majority of reported cases to date are of Caucasian ancestry [64]. Clinically, Schnitzler's syndrome resembles CAPS very closely, except that it is clearly an acquired syndrome presenting in mid- to late adulthood (Fig. 4). The monoclonal protein appears central to the pathogenesis, although the mechanism remains unclear [65]. About a fifth of patients eventually progress to overt plasma cell malignancy. Chemotherapy has been tried in the past, but does not appear to relieve the syndrome and should only be used for conventional haematological indications. The treatment of choice of Schnitzler's syndrome is IL-1 blockade. Anakinra has been reported in a number of small series to abolish symptoms completely, although it has no effect on the paraprotein concentration (lipsker).
Fig. 4.
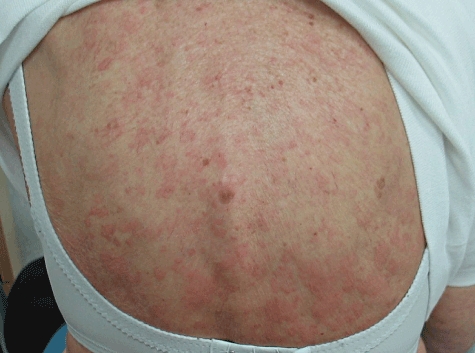
Diffuse trunk urticarial rash due to Schnizler's syndrome associated with an immunoglobulin M kappa paraprotein. The rash and symptoms disappeared within hours of starting anakinra.
Long-term outcomes
Although CINCA/NOMID and DIRA can be sufficiently severe to cause death within the first few decades, life expectancy among many patients with autoinflammatory disorders is good, and is expected to be excellent in those for whom there is now effective therapy. The most serious and life-threatening complication of these diseases generally is AA amyloidosis.
AA amyloidosis
AA amyloidosis is an often fatal disorder, predominantly affecting the kidneys, which occurs in a small proportion of patients with chronic inflammatory diseases [66]. AA amyloid fibrils are derived from the circulating acute-phase reactant serum amyloid A protein (SAA). SAA is synthesized by the liver under transcriptional regulation of IL-1, IL-6 and TNF-α[67], and its plasma concentration may rise a thousand-fold in the presence of inflammation [68]. AA amyloidosis is much more common among patients with inherited periodic fever syndromes than other chronic inflammatory diseases. The median duration of inflammatory disease in patients who develop amyloidosis is about 20 years. Up to 60% of patients with FMF died of renal failure due to AA amyloidosis before prophylactic colchicine was widely available, and even recently it was reported in 13% of a large Turkish series. The incidence of AA amyloidosis in TRAPS and CAPS is approximately 25%, but is less than 5% in MKD, perhaps because the disease often ameliorates spontaneously in early adulthood.
The natural history of untreated AA amyloidosis is of renal failure and early death, but this can be prevented completely by effective treatment of the underlying inflammatory disorder and long-term suppression of SAA levels to near-normal values.
Conclusion
The periodic fever syndromes are a group of autoinflammatory diseases which present largely, but not exclusively, in childhood. Diagnosis is often dependent upon genetic testing, which is available from the National Amyloidosis Centre in the UK and from a variety of international centres (list available at http://www.printo.it/eurofever/genetic_analysys.asp). Recent advances in diagnosis have been accompanied by improvements in treatment, particularly in FMF, CAPS, TRAPS and Schnizler's syndrome, and for the majority of affected individuals the long-term prognosis is good.
Disclosure
Supported in part by the European Community Seventh Framewok Programme (Coordination Theme 1 [Health] grant HEALTH-F2-2008-200923[EUROTRAPS]).
References
- 1.Consortium TFF. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 2.Consortium TIF. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90:797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 3.Centola M, Wood G, Frucht DM, et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 2000;95:3223–31. [PubMed] [Google Scholar]
- 4.Sarrauste de Menthiere C, Terriere S, Pugnere D, Ruiz M, Demaille J, Touitou I. INFEVERS: the Registry for FMF and hereditary inflammatory disorders mutations. Nucleic Acids Res. 2003;31:282–5. doi: 10.1093/nar/gkg031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Touitou I, Lesage S, McDermott M, et al. Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat. 2004;24:194–8. doi: 10.1002/humu.20080. [DOI] [PubMed] [Google Scholar]
- 6.Milhavet F, Cuisset L, Hoffman HM, et al. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat. 2008;29:803–8. doi: 10.1002/humu.20720. [DOI] [PubMed] [Google Scholar]
- 7.Lachmann HJ, Sengul B, Yavuzsen TU, et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology (Oxf) 2006;45:746–50. doi: 10.1093/rheumatology/kei279. [DOI] [PubMed] [Google Scholar]
- 8.Kone-Paut I, Hentgen V, Guillaume-Czitrom S, Compeyrot-Lacassagne S, Tran TA, Touitou I. The clinical spectrum of 94 patients carrying a single mutated MEFV allele. Rheumatology. 2009;48:840–2. doi: 10.1093/rheumatology/kep121. [DOI] [PubMed] [Google Scholar]
- 9.Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–86. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tidow N, Chen X, Müller C, et al. Hematopoietic-specific expression of MEFV, the gene mutated in familial Mediterranean fever, and subcellular localization of its corresponding protein, pyrin. Blood. 2000;95:1451–5. [PubMed] [Google Scholar]
- 11.Chae JJ, Wood G, Masters SL, et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci USA. 2006;103:9982–7. doi: 10.1073/pnas.0602081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mikula M, Buller A, Sun W, Strom CM. Prevalence of known mutations in the familial Mediterranean fever gene (MEFV) in various carrier screening populations. Genet Med. 2008;10:349–52. doi: 10.1097/GIM.0b013e3181723cc8. [DOI] [PubMed] [Google Scholar]
- 13.Livneh A, Langevitz P, Zemer D, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40:1879–85. doi: 10.1002/art.1780401023. [DOI] [PubMed] [Google Scholar]
- 14.Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med. 1972;287:1302. doi: 10.1056/NEJM197212212872514. [DOI] [PubMed] [Google Scholar]
- 15.Rigante D, La Torraca I, Avallone L, Pugliese AL, Gaspari S, Stabile A. The pharmacologic basis of treatment with colchicine in children with familial Mediterranean fever. Eur Rev Med Pharmacol Sci. 2006;10:173–8. [PubMed] [Google Scholar]
- 16.Chia EW, Grainger R, Harper JL. Colchicine suppresses neutrophil superoxide production in a murine model of gouty arthritis: a rationale for use of low-dose colchicine. Br J Pharmacol. 2008;153:1288–95. doi: 10.1038/bjp.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben-Chetrit E, Levy M. Reproductive system in familial Mediterranean fever: an overview. Ann Rheum Dis. 2003;62:916–19. doi: 10.1136/ard.62.10.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mor A, Pillinger MH, Kishimoto M, Abeles AM, Livneh A. Familial Mediterranean fever successfully treated with etanercept. J Clin Rheumatol. 2007;13:38–40. doi: 10.1097/01.rhu.0000255772.25658.7c. [DOI] [PubMed] [Google Scholar]
- 19.Roldan R, Ruiz AM, Miranda MD, Collantes E. Anakinra: new therapeutic approach in children with familial Mediterranean fever resistant to colchicine. Joint Bone Spine. 2008;75:504–5. doi: 10.1016/j.jbspin.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 20.McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–44. doi: 10.1016/s0092-8674(00)80721-7. [DOI] [PubMed] [Google Scholar]
- 21.Todd I, Radford PM, Daffa N, Bainbridge SE, Powell RJ, Tighe PJ. Mutant tumor necrosis factor receptor associated with tumor necrosis factor receptor-associated periodic syndrome is altered antigenically and is retained within patients' leukocytes. Arthritis Rheum. 2007;56:2765–73. doi: 10.1002/art.22740. [DOI] [PubMed] [Google Scholar]
- 22.Rebelo SL, Bainbridge SE, Amel-Kashipaz MR, et al. Modeling of tumor necrosis factor receptor superfamily 1A mutants associated with tumor necrosis factor receptor-associated periodic syndrome indicates misfolding consistent with abnormal function. Arthritis Rheum. 2006;54:2674–87. doi: 10.1002/art.21964. [DOI] [PubMed] [Google Scholar]
- 23.D'Osualdo A, Ferlito F, Prigione I, et al. Neutrophils from patients with TNFRSF1A mutations display resistance to tumor necrosis factor-induced apoptosis: pathogenetic and clinical implications. Arthritis Rheum. 2006;54:998–1008. doi: 10.1002/art.21657. [DOI] [PubMed] [Google Scholar]
- 24.Nedjai B, Hitman GA, Yousaf N, et al. Abnormal tumor necrosis factor receptor I cell surface expression and NF-κB activation in tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008;58:273–83. doi: 10.1002/art.23123. [DOI] [PubMed] [Google Scholar]
- 25.Tchernitchko D, Chiminqgi M, Galacteros F, et al. Unexpected high frequency of P46L TNFRSF1A allele in sub-Saharan West African populations. Eur J Hum Genet. 2005;13:513–15. doi: 10.1038/sj.ejhg.5201344. [DOI] [PubMed] [Google Scholar]
- 26.Ravet N, Rouaghe S, Dode C, et al. Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann Rheum Dis. 2006;65:1158–62. doi: 10.1136/ard.2005.048611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmann LA, Lohse P, Konig FB, Feneberg W, Hohlfeld R, Kumpfel T. TNFRSF1A R92Q mutation in association with a multiple sclerosis-like demyelinating syndrome. Neurology. 2008;70:1155–6. doi: 10.1212/01.wnl.0000296279.98236.8a. [DOI] [PubMed] [Google Scholar]
- 28.Nowlan ML, Drewe E, Bulsara H, et al. Systemic cytokine levels and the effects of etanercept in TNF receptor-associated periodic syndrome (TRAPS) involving a C33Y mutation in TNFRSF1A. Rheumatology (Oxf) 2006;45:31–7. doi: 10.1093/rheumatology/kei090. [DOI] [PubMed] [Google Scholar]
- 29.Gattorno M, Pelagatti MA, Meini A, et al. Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008;58:1516–20. doi: 10.1002/art.23475. [DOI] [PubMed] [Google Scholar]
- 30.van der Meer JW, Vossen JM, Radl J, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet. 1984;1:1087–90. doi: 10.1016/s0140-6736(84)92505-4. [DOI] [PubMed] [Google Scholar]
- 31.Cuisset L, Drenth JP, Simon A, et al. Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur J Hum Genet. 2001;9:260–6. doi: 10.1038/sj.ejhg.5200614. [DOI] [PubMed] [Google Scholar]
- 32.Houten SM, Frenkel J, Waterham HR. Isoprenoid biosynthesis in hereditary periodic fever syndromes and inflammation. Cell Mol Life Sci. 2003;60:1118–34. doi: 10.1007/s00018-003-2296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneiders MS, Houten SM, Turkenburg M, Wanders RJ, Waterham HR. Manipulation of isoprenoid biosynthesis as a possible therapeutic option in mevalonate kinase deficiency. Arthritis Rheum. 2006;54:2306–13. doi: 10.1002/art.21960. [DOI] [PubMed] [Google Scholar]
- 34.Simon A, Drewe E, van der Meer JW, et al. Simvastatin treatment for inflammatory attacks of the hyperimmunoglobulinemia D and periodic fever syndrome. Clin Pharmacol Ther. 2004;75:476–83. doi: 10.1016/j.clpt.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 35.Hoffmann GF, Charpentier C, Mayatepek E, et al. Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics. 1993;91:915–21. [PubMed] [Google Scholar]
- 36.Houten SM, van Woerden CS, Wijburg FA, Wanders RJ, Waterham HR. Carrier frequency of the V377I (1129G > A) MVK mutation, associated with hyper-IgD and periodic fever syndrome, in the Netherlands. Eur J Hum Genet. 2003;11:196–200. doi: 10.1038/sj.ejhg.5200933. [DOI] [PubMed] [Google Scholar]
- 37.Drenth JP, Haagsma CJ, van der Meer JW. Hyperimmunoglobulinemia D and periodic fever syndrome. The clinical spectrum in a series of 50 patients. International Hyper-IgD Study Group. Medicine (Balt) 1994;73:133–44. [PubMed] [Google Scholar]
- 38.Houten SM, Frenkel J, Rijkers GT, Wanders RJ, Kuis W, Waterham HR. Temperature dependence of mutant mevalonate kinase activity as a pathogenic factor in hyper-IgD and periodic fever syndrome. Hum Mol Genet. 2002;11:3115–24. doi: 10.1093/hmg/11.25.3115. [DOI] [PubMed] [Google Scholar]
- 39.Ammouri W, Cuisset L, Rouaghe S, et al. Diagnostic value of serum immunoglobulinaemia D level in patients with a clinical suspicion of hyper IgD syndrome. Rheumatology. 2007;46:1597–600. doi: 10.1093/rheumatology/kem200. [DOI] [PubMed] [Google Scholar]
- 40.Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL. Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum. 2003;48:2645–51. doi: 10.1002/art.11218. [DOI] [PubMed] [Google Scholar]
- 41.Lachmann HJ, Goodman HJ, Andrews PA, et al. AA amyloidosis complicating hyperimmunoglobulinemia D with periodic fever syndrome: a report of two cases. Arthritis Rheum. 2006;54:2010–14. doi: 10.1002/art.21901. [DOI] [PubMed] [Google Scholar]
- 42.Hoffman HM, Wright FA, Broide DH, Wanderer AA, Kolodner RD. Identification of a locus on chromosome 1q44 for familial cold urticaria. Am J Hum Genet. 2000;66:1693–8. doi: 10.1086/302874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol. 2003;4:95–104. doi: 10.1038/nrm1019. [DOI] [PubMed] [Google Scholar]
- 44.Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–34. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 45.Hawkins PN, Lachmann HJ, McDermott MF. Interleukin-1-receptor antagonist in the Muckle–Wells syndrome. N Engl J Med. 2003;348:2583–4. doi: 10.1056/NEJM200306193482523. [DOI] [PubMed] [Google Scholar]
- 46.Leslie KS, Lachmann HJ, Bruning E, et al. Phenotype, genotype, and sustained response to anakinra in 22 patients with autoinflammatory disease associated with CIAS-1/NALP3 mutations. Arch Dermatol. 2006;142:1591–7. doi: 10.1001/archderm.142.12.1591. [DOI] [PubMed] [Google Scholar]
- 47.Muckle TJ, Wells MV. Urticaria, deafness and amyloidosis: a new heredo-familial syndrome. QJ Med. 1962;31:235–48. [PubMed] [Google Scholar]
- 48.Prieur AM, Griscelli C, Lampert F, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl. 1987;66:57–68. doi: 10.3109/03009748709102523. [DOI] [PubMed] [Google Scholar]
- 49.Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58:2443–52. doi: 10.1002/art.23687. [DOI] [PubMed] [Google Scholar]
- 50.Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360:2426–37. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet. 2002;11:961–9. doi: 10.1093/hmg/11.8.961. [DOI] [PubMed] [Google Scholar]
- 52.Blau EB. Familial granulomatous arthritis, iritis, and rash. J Pediatr. 1985;107:689–93. doi: 10.1016/s0022-3476(85)80394-2. [DOI] [PubMed] [Google Scholar]
- 53.Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- 54.Arostegui JI, Arnal C, Merino R, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56:3805–13. doi: 10.1002/art.22966. [DOI] [PubMed] [Google Scholar]
- 55.Marshall GS, Edwards KM, Butler J, Lawton AR. Syndrome of periodic fever, pharyngitis, and aphthous stomatitis. J Pediatr. 1987;110:43–6. doi: 10.1016/s0022-3476(87)80285-8. [DOI] [PubMed] [Google Scholar]
- 56.Feder HM, Salazar JC. A clinical review of 105 patients with PFAPA (a periodic fever syndrome) Acta Paediatr. 2010;99:178–84. doi: 10.1111/j.1651-2227.2009.01554.x. [DOI] [PubMed] [Google Scholar]
- 57.Padeh S, Brezniak N, Zemer D, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome: clinical characteristics and outcome. J Pediatr. 1999;135:98–101. doi: 10.1016/s0022-3476(99)70335-5. [DOI] [PubMed] [Google Scholar]
- 58.Tasher D, Somekh E, Dalal I. PFAPA syndrome: new clinical aspects disclosed. Arch Dis Child. 2006;91:981–4. doi: 10.1136/adc.2005.084731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gattorno M, Caorsi R, Meini A, et al. Differentiating PFAPA syndrome from monogenic periodic fevers. Pediatrics. 2009;124:e721–8. doi: 10.1542/peds.2009-0088. [DOI] [PubMed] [Google Scholar]
- 60.Garavello W, Romagnoli M, Gaini RM. Effectiveness of adenotonsillectomy in PFAPA syndrome: a randomized study. J Pediatr. 2009;155:250–3. doi: 10.1016/j.jpeds.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 61.Peridis S, Koudoumnakis E, Theodoridis A, Stefanaki K, Helmis G, Houlakis M. Surgical outcomes and histology findings after tonsillectomy in children with periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis syndrome. Am J Otolaryngol. 2010;31:472–5. doi: 10.1016/j.amjoto.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 62.Schnitzler L, Schubert B, Boasson M, Gardais J, Tourmen A. Chronic urticaria, bone lesions, IgM macroglobulinemia – Waldenstroem disease. Bull Soc Française Dermatol Syphiligr. 1974;81:363. [Google Scholar]
- 63.de Koning HD, Bodar EJ, van der Meer JW, Simon A. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum. 2007;37:137–48. doi: 10.1016/j.semarthrit.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 64.Lipsker D. The Schnitzler syndrome. Orphanet J Rare Dis. 2010;5:38. doi: 10.1186/1750-1172-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pizzirani C, Falzoni S, Govoni M, et al. Dysfunctional inflammasome in Schnitzler's syndrome. Rheumatology (Oxf) 2009;48:1304–8. doi: 10.1093/rheumatology/kep222. [DOI] [PubMed] [Google Scholar]
- 66.Lachmann HJ, Goodman HJ, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356:2361–71. doi: 10.1056/NEJMoa070265. [DOI] [PubMed] [Google Scholar]
- 67.Lachmann HJ, Goodman HJB, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356:2361–71. doi: 10.1056/NEJMoa070265. [DOI] [PubMed] [Google Scholar]
- 68.Ledue TB, Weiner DL, Sipe JD, Poulin SE, Collins MF, Rifai N. Analytical evaluation of particle-enhanced immunonephelometric assays for C-reactive protein, serum amyloid A and mannose-binding protein in human serum. Ann Clin Biochem. 1998;35:745–53. doi: 10.1177/000456329803500607. [DOI] [PubMed] [Google Scholar]