

Abstract
Tumour necrosis factor (TNF)-α plays a critical role in the pathogenesis of T helper type 1-mediated colitis such as Crohn's disease. However, the roles of its two receptors in mediating pathology remain largely unknown. In this study, trinitrobenzene sulphonic acid (TNBS) was used to induce colitis in TNF-receptor single or double knock-out (DKO) BALB/c mice and in wild-type counterparts. TNF-R1−/− mice had significantly less weight loss, reduced mortality, colon shortening and oedema, colon histological damage and lower levels of colon myeloperoxidase compared with wild-type (WT) BALB/c mice. A similar manifestation was also observed in TNF-R2−/− and TNF-R1−/−TNF-R2−/− (TNF-R DKO) mice. Strikingly, systemic inflammatory response (including splenomegaly and monocyte expansion) was found in WT and TNF-R1−/− mice after TNBS, instead of TNF-R2−/− and TNF-R DKO mice. Attenuated pathology of colitis in TNF-R1−/− or TNF-R2−/−mice correlated with lower amounts of interleukin (IL)-6, IL-1β, monocyte chemotactic protein (MCP)-1, IL-12p70 and interferon (IFN)-γ production in the colons. Importantly, ablation of TNF-R1 or TNF-R2 reduced the number of terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labelling (TUNEL)-positive apoptotic epithelial cells in the affected colons compared with WT TNBS-instilled controls, which might be due to the heightened ratio of Bcl-2/Bax and reduced activity of nuclear factor (NF)-κB. These findings suggest that either TNF-R1 or TNF-R2 plays a pathogenic role in the pathology of colitis and TNF signalling via TNF-R1 or TNF-R2 alone is not sufficient for inducing mucosal damage.
Keywords: apoptosis, proinflammatory mediator, TNBS colitis, TNF receptor
Introduction
Tumour necrosis factor (TNF) plays a critical role in immunity and inflammation based on its pleiotropic function on differentiation, growth and apoptotic cell death of both immune and non-immune cell subsets [1,2]. It is well established that TNF is intimately involved in the pathogenesis of inflammatory and autoimmune diseases such as rheumatoid arthritis, multiple sclerosis and inflammatory bowel diseases (IBD) [3]. TNF mediates its functions by binding as a trimer to either TNF receptor (TNF-R) 1 or 2. TNF-R1, which is expressed on most cell types, has a death domain that mediates apoptosis through caspase activation and activates nuclear factor (NF)-κB, leading to transcription of inflammatory cytokines, chemokines and anti-apoptotic proteins [4]. TNF-R2, which is expressed dominantly by haematopoietic cells, does not contain the death domain but can mediate apoptosis through the kinase receptor-interacting protein (RIP) [5].
There is increasing evidence to support the intimate correlation of TNF-R expression with the pathogenesis of IBD. During active stages of the disease, TNF-R2 expression by colonic epithelial cells is increased in patients with IBD and in mice with experimental colitis [6,7]. Furthermore, it has been reported that polymorphisms in the TNF-R2 gene are associated with a higher susceptibility to Crohn's disease (CD) [8]. These observations uniformly show a disease-promoting role of TNF signalling via TNF-R2 during IBD development. However, a recent study demonstrates an opposite effect of TNF-R expression by CD4+ T cells on the course of experimental colitis [9]. The transfer of colitogenic CD4+CD45hiTNF-R2−/− T cells into RAG−/− recipients leads to an accelerated onset of disease and to more severe signs of inflammation. In terms of TNF-R1, the conflicting data supporting or not supporting the protective effects of TNF-R1 during the course of colitis also exist. Ebath et al. [10] demonstrated that TNF-R1−/− C57Bl/6 mice lost more weight and had increased mortality after trinitrobenzene sulphonic acid (TNBS) instillation. In contrast, Nakai et al. [11] showed that TNF-R1 deficiency attenuated tissue damage induced by TNBS administration, which was related to reduced nuclear factor κB (NF-κB) activity. Overall, these studies underscore the complexity of TNF-mediated function through TNF-R1 or -R2 during the onset and perpetuation of colonic inflammation, which may be influenced by different TNF-R expression patterns and different colitis models used.
Conversely, although a chimeric monoclonal antibody against TNF-α (infliximab) has shown definite therapeutic effects in patients with refractory CD [12], blockade of TNF has been associated with the reactivation of tuberculosis and the possible development of other opportunistic infections such as histoplasmosis, listeriosis and pneumocystis [13]. Therefore, the beneficial outcome of complete blocking of TNF may be obtained from blocking the signalling from only one of the two receptors. This prompts us to deeply dissect the roles of TNF-R1 or 2 in the pathogenesis of experimental colitis, respectively.
In this study, we investigated the effect of TNF-R knock-out on TNBS-induced intestinal inflammation in BALB/c mice. As a result, TNF-R1 or TNF-R2 knock-out mice show reduced weight loss and attenuated inflammation in colonic tissues compared with wild-type (WT) controls after TNBS. Strikingly, lacking TNF-R2 led to dampened systemic inflammatory response after TNBS, while TNF-R1 deficiency did not.
Materials and methods
Animals
BALB/c WT mice were purchased from Jackson Laboratory (Bar Harbor, MI, USA). TNF-R1−/−, TNF-R2−/− and TNF-R1−/−TNF-R2−/− mice from Dr Zhihai Qin (Institute of Biophysics, Chinese Academy of Sciences, Beijing, China) were back-crossed for 12 generations onto the BALB/c background [14]. Animals were housed in specific pathogen-free conditions with an alternating light/dark cycle. All experiments were performed using 6–8-week-old male mice. The animal experiments were conducted in accordance with the institutional guidelines of the Institute of Basic Medical Sciences.
Colitis induction
TNBS was prepared using 5% TNBS (Sigma, St Louis, MO, USA) and diluted to a final concentration of 2% TNBS and 50% ethanol with phosphate-buffered saline (PBS). Mice were anaesthetized with ether. A 3·5F umbilical catheter was inserted 4 cm into the rectum, and 150 µl (3 mg) of TNBS was injected. The mouse was held vertically for 30 seconds. Control mice underwent identical procedures but were instilled with ethanol 50% dissolved in PBS. Weights were measured before induction of colitis and daily thereafter. At the end of the experiments mice were killed by cervical dislocation under anaesthesia.
Histology
Colons were removed, fixed then processed conventionally. Haematoxylin and eosin (H&E)-stained sections were examined for evidence of colitis using criteria described previously by Scheiffele and Fuss [15].
Detection and quantitation of apoptotic cells
For detection of apoptotic cells in tissues, labelling of degraded DNA specific to apoptotic cells was performed using a modification of the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labelling (TUNEL) technique by application of in-situ cell death detection kits (Roche Diagnostics, Indianapolis, IN, USA), according to the manufacturer's instructions. Levels of apoptosis were quantified by counting the numbers of TUNEL-positive cells per 100 nuclei. The apoptotic index was obtained from the ratio of apoptotic to total cells.
Colon homogenates
A 1-cm segment was divided from the distal 4 cm of the harvest colon. Wet weight was recorded separately for the whole distal 4 cm and the portion taken for homogenation. Colon tissue samples were homogenized in PBS containing a cocktail of protease inhibitors (1 µl to 20 mg of tissue according to the manufacturer's protocol; Roche Diagnostics) with a Polytron homogenizer and centrifuged at 12 000 g for 10 min. The supernatants were stored at −20°C until used for enzyme-linked immunosorbent assay (ELISA) analysis.
Determination of colon oedema
Colons were dissected at indicated time-points after TNBS. A piece of the affected colon was then collected, weighed, placed in an 80°C oven for 24 h, then reweighed, and the wet-to-dry weight ratio was determined as a measure of colon oedema [16].
Measurement of myeloperoxidase (MPO) activity
Tissue MPO activity was determined by a standard enzymatic procedure, as described previously [17]. Results for colon MPO content were converted to absorbance units per gram of tissue.
Immunoblotting
Colon tissues were homogenized and sonicated in RIPA lysis buffer (Santa Cruz Laboratories, Santa Cruz, CA, USA), supplemented with protease inhibitors. After centrifugation at 20 000 g for 15 min, 30 µg of the supernatants were separated onto 10% sodium dodecyl sulphide-polyacrylamide gel and transferred onto an Immunobilon-P Transfer membrane (Millipore, Billerica, MA, USA). After being blocked with 5% skimmed milk, the membrane was incubated with antibodies to Bcl-2 (1:1000), Bax (1:1000), or IκBα (1:1000). Rabbit anti-glyceraldehyde 3-phosphate dehydrogenase antibody (1:1000) was used as an internal control. ImmunoPure peroxidase-conjugated anti-rabbit immunoglobulin (Ig)G was used as secondary antibody. The blotted membrane was then treated with the Super Signal West Dura Extended Duration Substrate (Pierce, Rockford, IL, USA) and signals were detected by LAS-3000 mini CCD camera (Fuji Film, Tokyo, Japan). Primary antibodies used for immunoblotting were purchased from eBioscience (San Diego, CA, USA).
Cytokine analysis
Serum and colonic TNF-α, interleukin (IL)-6, IL-1β, monocyte chemotactic protein (MCP)-1, IL-12p70 and interferon (IFN)-γ were measured by ELISA according to the manufacturer's instructions. The IFN-γ ELISA kit was purchased from BD Pharmingen (San Diego, CA, USA). Other cytokine ELISA kits were purchased from eBioscience.
Flow cytometry
Single-cell suspensions were prepared from spleen by passing them through nylon mesh and stained with fluorescein isothiocyanate (FITC)-labelled anti-murine Gr-1 (RB6-8C5; Biolegend, San Diego, CA, USA) or F4/80 (CI : A3-1; Biolegend) antibody in PBS with 2% heat-inactivated fetal calf serum and 0·1% sodium azide on ice for 25 min. These cells were fixed using PBS with 1% paraformaldehyde. Data collection and analysis were performed on a fluorescence activated cell sorter (FACS)Calibur flow cytometer using CellQuest software (Becton Dickinson, San Diego, CA, USA).
Real-time reverse transcription–polymerase chain reaction (RT–PCR) for TNF expression
RNA extracted from colon tissues was reverse-transcribed into cDNA using the SuperScript III first-strand cDNA synthesis system (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized from 0·5 µg RNA using random hexamer primers and SuperScript III (Invitrogen). Real-time RT–PCR was performed on a Bio-Rad iCycler to quantify mRNA levels of TNF. The primers for real-time PCR were as follows: TNF forward primer 5′-ACC CTC ACA CTC AGA TCA TC-3′ and reverse primer 5′-GAG TAG ACA AGG TAC AAC CC-3′; glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward primer 5′-TGA AGG TCG GAG TCA ACG GAT TTG GT-3′ and reverse primer 5′-CAT GTG GGC CAT GAG GTC CAC CAC-3′. The data were analysed using Q-Gene software and expressed as fold change mean normalized expression (MNE) from control value.
Data analysis
Student's t-test and one-way analysis of variance (anova) was used to determine significance, with P < 0·05 considered significant. The Kaplan–Meier test was used for survival analysis with log rank P < 0·05 to determine significance.
Results
TNF-R ablation confers protection against weight loss and mortality
First, we evaluated the effects of TNF-R1 or TNF-R2 deficiency on the course of TNBS colitis in a BALB/c background. WT mice lost an average of 14·5 ± 2·7% of their baseline weight 2 days after TNBS. TNF-R1−/−, TNF-R2−/− and TNF-R double knock-out (DKO) mice lost 4·2 ± 2·5%, 2·2 ± 3·4% and 5·8 ± 3·3%, respectively (Fig. 1a). By day 7 after TNBS, although there was a recovery of weight to a certain extent in all groups, weight loss in WT mice was significantly higher than TNF-R-deficient counterparts. Furthermore, weight gain shown in Fig. 1a may be due to some of the mice with the greatest weight loss dying prior to the end of the experiment. There was initial weight loss in all control mice, but these mice were back at normal weight by day 2 after ethanol instillation. In accordance with this, at day 7, 80% (16 of 20) of TNF-R1−/−, 94% (16 of 17) of TNF-R2−/− and 89% (16 of 18) of TNF-R DKO mice had survived, whereas survival was 50% (six of 12) in WT mice (Fig. 1b). Ethanol-treated mice had 100% survival.
Fig. 1.
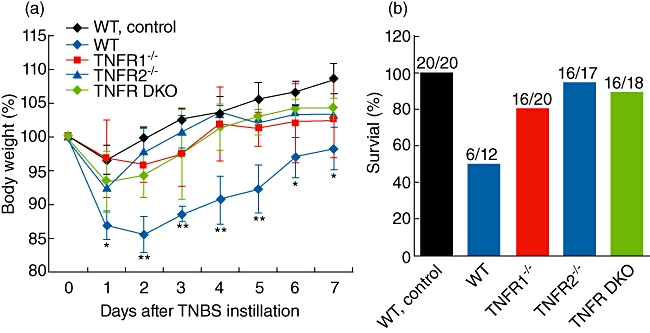
Tumour necrosis factor receptor 1 (TNF-R1)- or TNF-R2-deficient mice have less weight loss and increased survival after trinitrobenzene sulphonic acid (TNBS). TNBS colitis was established as described in Materials and methods. Weight (a) in each group was monitored daily at 7-day intervals. Weight was presented as percentage of the initial weight at day 0. Survival rate was calculated at the end of experiments (b). Data are pooled from four independent experiments (n = 15–32 mice per group). *P < 0·05; **P < 0·01 versus TNF-R knock-out mice after TNBS.
Absence of TNF-R1 or TNF-R2 attenuates intestinal damage after TNBS
Next, we examined the influence of TNF-R1 or TNF-R2 deficiency on macroscopic and microscopic injury of colons after TNBS. WT mice exhibited significant shortening and thickening of the colon 5 days after TNBS (Fig. 2a). In contrast, the colons from TNF-R1−/−, TNF-R2−/− and TNF-R DKO groups did not show considerable injury (Fig. 2a), resembling the colon of ethanol-treated control mice. Furthermore, wet-to-dry weight ratios of colons from all three TNBS-administrated TNF-R-deficient groups were remarkably decreased compared with TNBS-instilled WT mice (Fig. 2b). Consistent with macroscopic changes, WT mice after TNBS showed marked, transmural infiltration with inflammatory cells and injury with ulceration. TNF-R1 or TNF-R2 ablation rendered less severity of histological features of colitis (Fig. 2c). When quantified by histological system for evidence of inflammation and injury, these histological changes were highly significant (Fig. 2d).
Fig. 2.
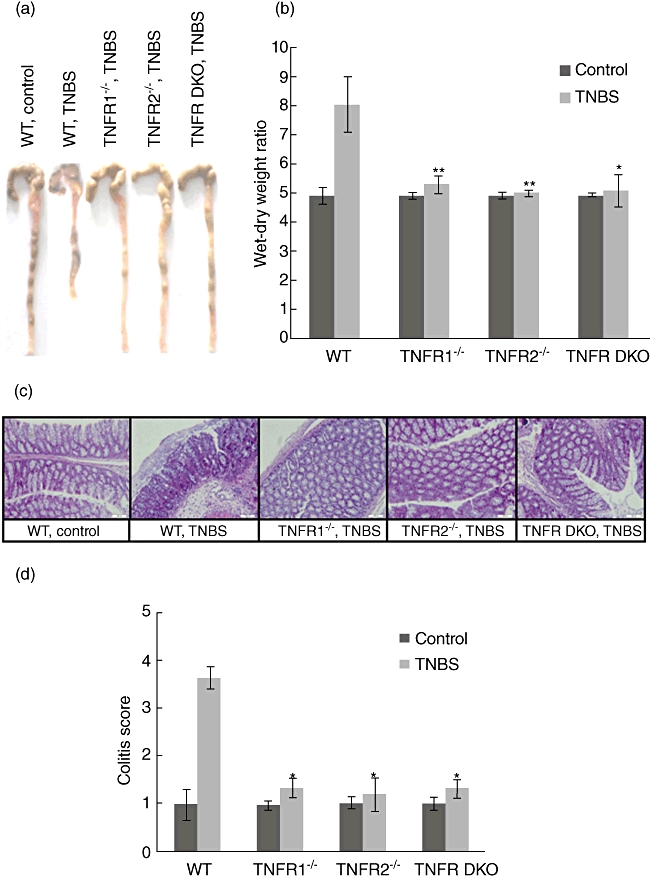
Tumour necrosis factor receptor 1 (TNF-R1) or TNF-R2 deficiency protects against mucosal damage after trinitrobenzene sulphonic acid (TNBS). Colons from each group were collected at day 5 after TNBS instillation. (a) Macroscopic images of the colons are shown. (b) Colon oedema was determined by wet-to-dry weight ratios. (c) Microscopic images of the colons are shown by haematoxylin and eosin staining. Magnification × 200. (d) Colitis score was determined as described in Materials and methods. Data represent the mean ± standard deviation of six to eight mice per group. *P < 0·05; **P < 0·01 versus wild-type mice after TNBS.
Mice lacking either TNF-R1 or TNF-R2 have lower levels of MPO in colons after TNBS
To determine whether decreased infiltrate of neutrophil contributed to mild signs of colitis in mice lacking either TNF-R1 or TNF-R2, we detected the levels of MPO, indices for neutrophil infiltration, in the colon 5 days after TNBS. Levels of MPO in the colons of WT colitis mice were significantly higher than in ethanol-treated control mice (Fig. 3). In mice lacking TNF-R1, TNF-R2 or two receptors, the levels of colonic MPO were reduced to similar levels to those of the ethanol-instilled animals (Fig. 3).
Fig. 3.
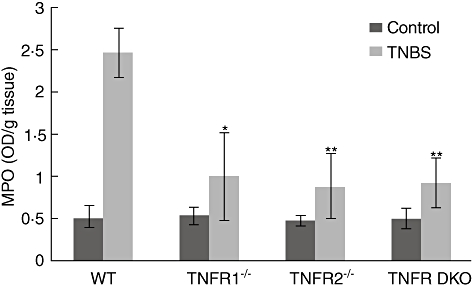
Tumour necrosis factor receptor 1 (TNF-R1)- or TNF-R2-deficient mice have reduced infiltrates of neutrophils into the colons after trinitrobenzene sulphonic acid (TNBS). Levels of myeloperoxidase (MPO) in the colons of mice in each group were measured at day 5 after colitis induction. Data represent the mean ± standard deviation of five to six mice per group. *P < 0·05; **P < 0·01 versus wild-type mice after TNBS.
TNF-α levels are higher in mice lacking TNF-R2
To determine whether differences in weight loss and colitis scores could be secondary to differences in TNF-α production, serum TNF-α concentrations were examined by ELISA. TNBS administration led to increased production of TNF-α in WT and TNF-R knock-out mice. TNF-α levels were elevated in serum of TNF-R1−/−TNF-R2−/− mice compared with TNF-R1−/− mice (Fig. 4a). This may be due to the fact that soluble TNF-R2 has been reported as a TNF-α sink [4].
Fig. 4.
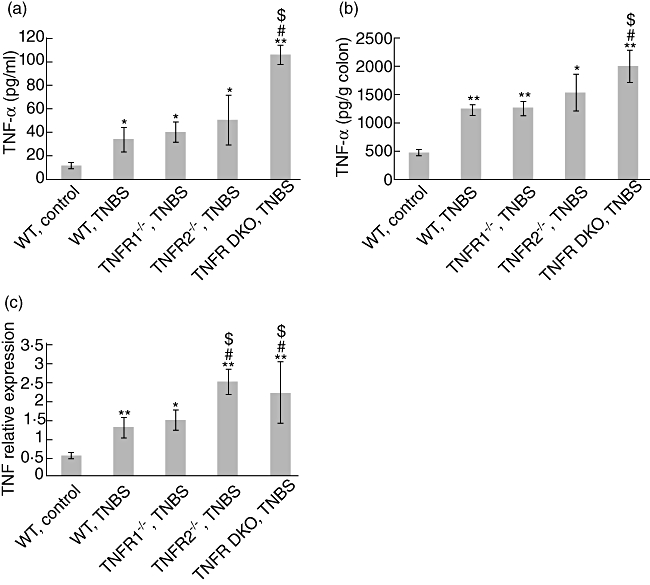
Tumour necrosis factor (TNF) levels in TNF-R−/− and wild-type (WT) mice after trinitrobenzene sulphonic acid (TNBS). (a) TNF concentrations in serum of mice were examined by enzyme-linked immunosorbent assay (ELISA) at day 5 after TNBS. (b) At the same time-points, colons were collected and homogenized. TNF levels in the supernatants of homogenates were examined by ELISA. (c) Simultaneously, colon RNA were extracted and gene expression of TNF was determined by quantitative reverse transcription–polymerase chain reaction (RT–PCR). Data represent the mean ± standard deviation of five to six mice per group. *P < 0·05; **P < 0·01 versus (WT) control mice; #P < 0·01 versus WT mice after TNBS; $P < 0·05 versus TNF-R1−/− mice after TNBS.
To further explore differences in TNF-α concentration in sites of inflammation, the levels of TNF-α proteins in the colons were examined. Administration of TNBS increased colonic production of TNF in all groups. TNF-R1−/−TNF-R2−/− colons contained greater amounts of TNF-α than TNF-R1−/− or WT colons (Fig. 4b). Furthermore, TNF-R2 ablation also resulted in enhanced expression of TNF mRNA after TNBS (Fig. 4c), although heightened TNF protein levels in the serum and colon tissues of TNF-R2−/− mice were not seen. Overall, these data suggest the presence of a compensatory regulation of TNF expression in the absence of TNF-R2.
Mice lacking TNF-R2 have ameliorated systemic inflammatory responses
As TNBS-administered mice also exhibit marked systemic inflammatory responses [10], we next examined the effect of TNF-R knock-out on these disease parameters. Five days after TNBS, WT mice showed visible splenomegaly in the term of spleen weights. Similar effects were observed in TNF-R1−/− TNBS-treated mice, but no significant difference was found between WT and TNF-R1−/− mice (Fig. 5a). Intriguingly, TNF-R2−/− groups (e.g. TNF-R2−/− and TNF-R DKO mice) did not exhibit splenic enlargement after TNBS (Fig. 5a). This observation was more pronounced in TNF-R DKO mice, indicating a key role of TNF signalling via TNF-R2 for TNBS-induced systemic inflammatory reaction. In addition, it is worthy of note that relative reduction of spleen weight in TNF-R2−/− and TNF-R DKO mice was seen compared to WT controls. This suggests that TNF-R2 ablation may influence the development of lymphoid organs, which needs further investigation.
Fig. 5.
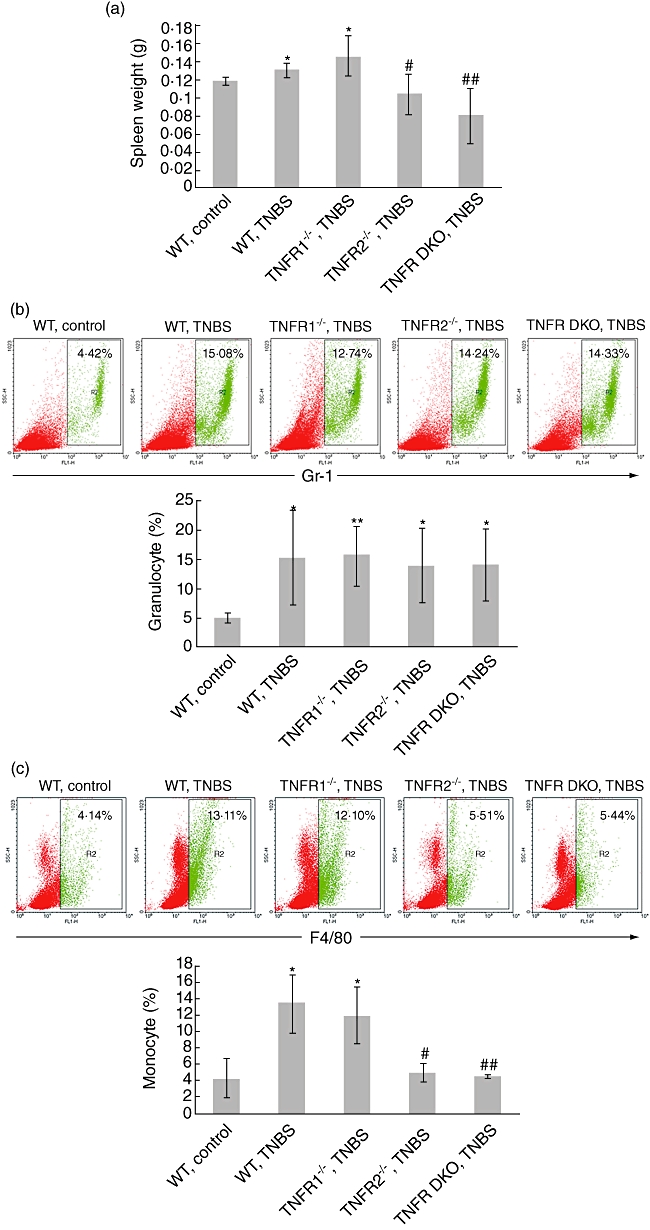
Systemic inflammatory response in tumour necrosis factor (TNF)-R−/− and wild-type (WT) mice after trinitrobenzene sulphonic acid (TNBS). (a) Spleen was isolated and weighed at day 5 after colitis induction. The percentages of granulocytes (Gr-1+) (b) or monocytes (F4/80+) (c) in the spleens were examined by flow cytometry. Representative plots were shown. Data represent the mean ± standard deviation of three to five mice per group from three independent experiments. *P < 0·05; **P < 0·01 versus WT control mice; #P < 0·05; ##P < 0·01 versus TNF-R1−/− mice after TNBS.
To define the characteristics of this systemic inflammatory response induced by TNBS administration, FACS analysis for quantity of granulocytes and monocytes in spleen was performed. The percentage of granulocytes (Gr-1+) was increased dramatically in all TNBS-instilled groups (Fig. 5b). Monocyte percentage was also increased in WT and TNF-R1−/− TNBS-instilled mice compared with ethanol-treated control mice. This effect, however, was not found in TNF-R2−/− and TNF-R DKO mice (Fig. 5c), indicating that the proliferation of monocytes is restricted in the absence of TNF signalling via TNF-R2 instead of TNF-R1.
TNF-R-deficient colons contain fewer amounts of proinflammatory mediators after TNBS
To determine whether ameliorated mucosal damage in TNF-R knock-out mice is related to restrained expression of colitis-associated proinflammatory cytokines and chemokines, the colons were dissected, homogenized and assayed at day 5 after TNBS. TNBS administration resulted in significant up-regulation of IL-6, IL-1β, MCP-1, IFN-γ and IL-12p70 levels in the colonic tissues of WT mice (Fig. 6). As expected, in agreement with reduced colitis scores, the colons of TNF-R1−/−, TNF-R2−/− and TNF-R DKO mice contained lower amounts of these cytokines and chemokines (Fig. 6).
Fig. 6.
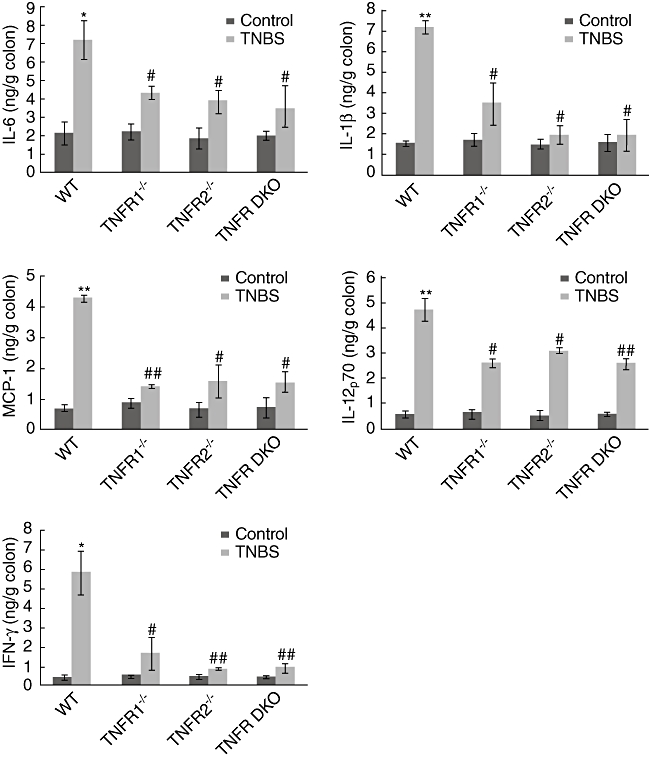
Tumour necrosis factor receptor (TNF-R) deficiency leads to reduction of proinflammatory mediator expression in the colons after trinitrobenzene sulphonic acid (TNBS). Colons were isolated and homogenized at day 5 after colitis induction. Interleukin (IL)-6, IL-1β, monocyte chemotactic protein (MCP)-1, IL-12p70 and interferon (IFN)-γ concentrations in the supernatants of homogenates were determined by enzyme-linked immunosorbent assay (ELISA). Data represent the mean ± standard deviation of seven to eight mice per group from two independent experiments. *P < 0·05; **P < 0·01 versus wild-type (WT) control mice; #P < 0·05; ##P < 0·01 versus WT mice after TNBS.
TNF-R deficiency reduces colonic pro-apoptotic Bax expression and NF-κB activity after TNBS
It is well established that TNF is a key mediator for epithelial cell death and mucosal injury during inflammation [18,19], so we hypothesized that lacking TNF signalling via TNF-R1 or TNF-R2 leads to weakened apoptosis of colonic epithelial cells, thereby to less severity of colitis. To address this, colon samples from WT and TNF-R knock-out groups were stained using the TUNEL method. In colonic mucosa of WT control mice, the apoptotic epithelial cells were scarcely visible. When TNBS colitis was established, numerous apoptotic cells in the epithelium were seen (Fig. 7a). In agreement with the grade of mucosal damage, TNF-R1 or TNF-R2 knock-out mice, which had lower colitis scores, exhibited significant reduction of apoptotic epithelial cells (Fig. 7a). Interestingly, when lamina propria mononuclear cells were detected, we did not find notable differences in the degree of apoptosis of these cells in all groups.
Fig. 7.
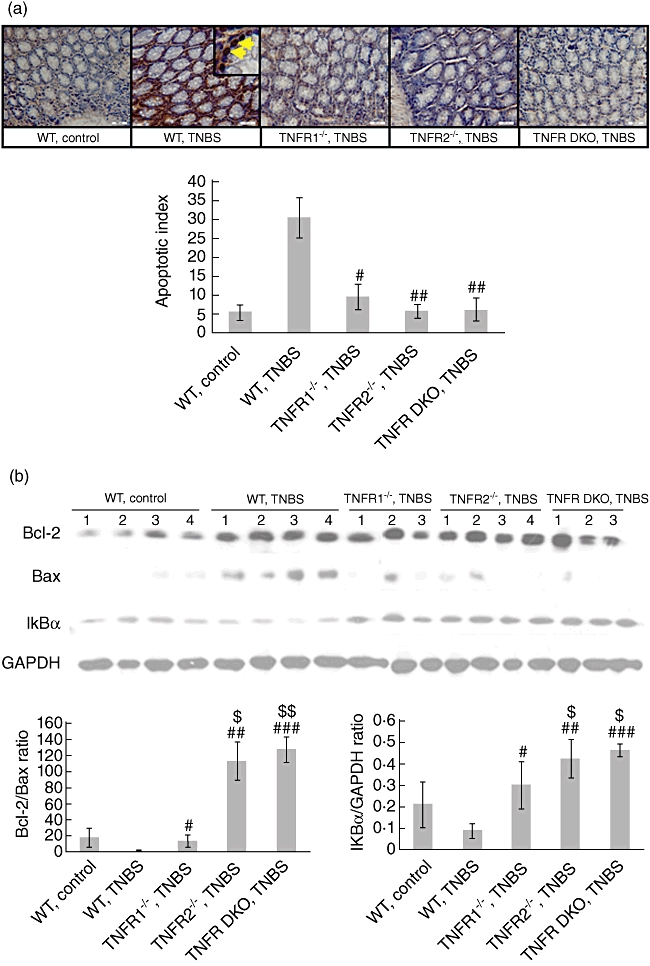
Tumour necrosis factor receptor 1 (TNF-R1) or TNF-R2 deficiency reduces apoptosis of colonic epithelial cells after trinitrobenzene sulphonic acid (TNBS). (a) Colons were dissected at day 5 after colitis induction. Apoptosis in colonic epithelial cells was determined by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labelling (TUNEL) staining (brown nuclei). Magnification × 400. Yellow arrows denote TUNEL-positive epithelial cells. Histology is representative for more than 15 mice per group. Quantification of apoptosis was shown as apoptotic index (see Materials and methods). Data represent the mean ± standard deviation of 10–15 mice per group. (b) At the same time-points, colonic protein was extracted. Bcl-2, Bax, and IκBα expression in the colons was examined by Western blotting. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as internal control. Changes in quantity of expression of these factors were determined by densitometric analysis. Data are representative of four independent experiments. #P < 0·05; ##P < 0·01; ###P < 0·001 versus wild-type mice after TNBS; $P < 0·05; $$P < 0·01 versus TNF-R1−/− mice after TNBS.
Next, we investigated whether reduction in apoptosis of colonic epithelial cells in TNF-R knock-out mice after TNBS is related to enhanced anti-apoptosis signals. The results showed that TNBS administration dramatically augmented the expression of pro-apoptotic factor Bax in the colon of WT mice (Fig. 7b). Ablation of TNF-R1 or TNF-R2 (e.g. TNF-R1−/−, TNF-R2−/− and TNF-R DKO mice) reduced Bax expression to the baseline, resembling those in control mice (Fig. 7b). This effect was most pronounced in TNF-R DKO groups. Although Bcl-2 expression, a member of Bcl-2 family with anti-apoptotic function, was enhanced in the colon of all TNBS-instilled groups, densitometry analysis showed that the ratio of Bcl-2/Bax in TNF-R1−/− mice was significantly higher than in WT mice after TNBS (Fig. 7b). The same effects were also found in TNF-R2−/− groups, which might account for reduced apoptosis of colonic epithelial cells in these groups after TNBS. Further, to clarify the effects of TNF-R deficiency on NF-κB activity, we detected IκBα expression in the colon after TNBS. The amounts of IκBα in WT TNBS-treated mice were much lower than those in control mice, suggesting NF-κB activation in the sites of inflammation after TNBS (Fig. 7b). TNF-R knock-out (e.g. TNF-R1−/−, TNF-R2−/− and TNF-R DKO mice) restrained NF-κB activity, as the colon of three groups contained more amounts of IκBα proteins (Fig. 7b). Furthermore, NF-κB activity was suppressed more significantly in TNF-R2−/− and TNF-R DKO mice compared to TNF-R1−/− counterparts (Fig. 7b). Thus, local reduced expression of pro-apoptotic proteins and restrained NF-κB activity may account for improved signs of colitis after TNBS.
Discussion
TNF plays a central role in the immunopathogenesis of IBD. However, little information has been available so far on the role of TNF-dependent signal transduction via TNF-R1 or TNF-R2 in chronic intestinal inflammation. In this study, we demonstrated clearly that TNF signalling via TNF-R1 or TNF-R2 was required for mucosal damage during inflammation in TNBS-induced colitis. Furthermore, co-existence of these two receptors in the intestinal epithelium seemed to be indispensible for TNF-mediated injury, as TNF signalling via TNF-R1 or TNF-R2 alone failed to elicit a colitogenic response (Fig. 1a). This observation might be important for designing therapeutic intervention targeting TNF/TNF-R signalling. Blocking TNF signalling via either TNF-R1 or TNF-R2 might be beneficial for treatment of patients suffered from inflammatory bowel disease, especially for CD, considering that colonic inflammation induced by TNBS in BALB/c strain displays human CD-like features [20].
It is worth noting, as described above, that the present data about the role of TNF receptors in the course of TNBS-induced colitis appear to be paradoxical [10,11]. The data presented in this paper showed a disease-promoting role of TNF receptors (either TNF-R1 or TNF-R2) in the pathogenesis of colonic inflammation. In the TNBS model in BALB/c mice, we found that both TNF-R1−/− and TNF-R2−/− mice had less loss of body weight and improved survival than WT mice. In accord with this, colon injury was attenuated and colitis score was lower in these mice. These data are consistent with a previous study reporting that mucosa damage scores were significantly lower in TNF-R1 knock-out and TNF-R DKO C57Bl/6 mice [11], but is not in agreement with Stenson's observations [10] which showed that TNF-R1−/− mice lost more weight and had increased mortality after TNBS. This may be explained by the fact that different mouse strains were used in these studies, as BALB/c strains used in our study are well known to be predisposed to a Th2 response, while C57Bl/6 strains did not. Strikingly, in terms of TNF-R2, our finding of a disease-promoting role of TNF/TNF-R2 signalling in TNBS-induced colitis coincides with Stenson's work [10], showing that TNF-R2-deficient C57Bl/6 mice lost less weight, had normal temperatures and had improved survival, which indicates that it is necessary to uncover further the roles of TNF receptors in the course of colitis. A pathogenic role of TNF signalling via TNF-R2 was evidenced further by the fact that TNF-R2 expression is increased in colonic epithelial cells in both ulcerative colitis and CD, and TNF-R2 is significantly up-regulated in the lamina propria and peripheral blood T cells in CD [6,7]. This concept is also confirmed by studies in severe combined immunodeficiency (SCID) mice reconstituted with CD4+CD62L+ T cells from transgenic mice over-expressing TNF-R2, which develop worse colonic inflammation than those receiving T cells expressing normal amounts of TNF-R2 [6]. Intriguingly, a recent study showed that, using a CD4+ T cell transfer model of colitis, mice injected intravenously with TNF-R2−/− CD4+CD45RBhi T cells exhibited an accelerated onset of disease and more severe signs of inflammation, indicating a protective role of TNF-R2 in T cell-mediated experimental colitis [9]. The difference in these studies may reflect distinct effects of TNF/TNF-R2 signalling on lymphoid and non-lymphoid populations, as transfer of TNF-R2−/− non-lymphoid cells into RAG−/− mice did not influence the course of colitis [9]. Overall, these data underscore the complexity of functions of TNF signalling via TNF-R1 or TNF-R2 on various cell types and in different models of colitis.
The examination of TNF concentration in serum and colons showed that TNBS-treated TNF-R1−/−TNF-R2−/− mice had increased TNF levels in blood and colon samples compared to WT and TNF-R1−/− counterparts (Fig. 4). This is consistent with the finding that TNF is increased in TNF-R2−/− mice challenged by lipopolysaccharide (LPS) [21] or TNBS [10] compared to WT mice. However, Peschon et al. [22] reported that TNF levels were also increased in mice lacking TNF-R1 and markedly increased in mice lacking both receptors. It is possible that TNF is elevated in TNF-R2-deficient mice because TNF-R2 down-regulates TNF responses by soluble TNF-R2 binding of TNF or by signalling for decreased transcription of TNF. Indeed, TNF-R2-deficient mice have increased levels of TNF gene expression after TNBS. Therefore, elevated levels of TNF in TNF-R2-deficient mice are secondary to increased synthesis and decreased uptake by soluble TNF-R2. This indicates that signalling via TNF-R2 down-regulates TNF transcription. However, the details in this process remain elusive.
One of the hallmarks of intestinal inflammation is the infiltration of neutrophils into colons and release of large amounts of oxygen radicals [23,24]. Our results showed significant reduction of neutrophil infiltrates in sites of inflammation in TNF-R1 or -R2-deficient mice when TNBS-induced colitis was established (Fig. 3). Thus, we believe that the absence of TNF signalling through TNF-R1 or TNF-R2 affects neutrophil recruitment into colonic tissues when colitis is established. This viewpoint is supported by previous studies showing that TNF is able to induce integrin CD11b/CD18 up-regulation on human neutrophils, enhance cell adhesion and recruit and retain leucocytes in inflammatory sites [25]. Furthermore, lacking TNF-R1 or TNF-R2 led to down-regulation of proinflammatory cytokine and chemokine expression in the sites of inflammation (Fig. 6). Although the cell types which secreted these mediators need to be uncovered, these data account in part for restriction of inflammation in TNF-R knock-out mice.
TNF is an important factor for survival of colonic epithelial cells in physiological conditions. However, in pathological settings, large amounts of TNF are released and it has deleterious effects on epithelial cells [26]. Our results showed that TNBS administration led to increased expression of TNF in serum and colons (Fig. 4). These TNF may induce apoptosis of colonic epithelial cells by enhancing expression of pro-apoptotic protein Bax and NF-κB activity, and finally resulted in mucosal damage (Fig. 7), which is also reported by previous studies [18,19]. Lacking TNF signalling via TNF-R1 or TNF-R2 could efficiently block this apoptosis-promoting process. Besides Bcl-2 family members, TNF signalling also affects other apoptosis-related factors, for example cytochrome C and caspases [27]. Although the pathways involved in TNF-mediated apoptosis of colonic epithelial cells need to be elucidated further, the present data indicate that decreased apoptosis of colonic epithelial cells is one of the reasons for improved signs of colitis in TNF-R−/− mice after TNBS.
The importance of TNF in the pathology of IBD renders it necessary to dissect the role of TNF signalling via TNF-R1 or TNF-R2 in experimental colitis, for example TNBS-induced colitis. In the current study, we demonstrated a disease-promoting role of TNF-R1 or TNF-R2 in this model of colitis, which is associated with reduced infiltration of granulocytes, restrained release of proinflammatory mediators and decreased apoptosis of epithelial cells. These data suggest that targeting one of the receptors specifically might be an alternative approach to therapy for patients with CD, considering that blockade of TNF has been associated with an increase risk of certain infections such as tuberculosis, histoplasmosis, Cryptococcus and Aspergillus[28]. Furthermore, we showed that lacking TNF-R1 did not influence the increased number of granulocytes and monocytes in spleen after TNBS, while lacking TNF-R2 failed to expand monocyte population specifically (Fig. 5). This result allows us to postulate that targeting TNF-R1 may be more beneficial than blockade of TNF-R2. Further studies in other models, for example dextran sulphate sodium-induced colitis, and in humans are warranted.
Acknowledgments
The authors would like to thank Professor Zhihai Qin for supplying TNFR knock-out mice. This work was supported by grants from the National Key Basic Research Program of China (2007CB512406; 2009CB522408) and the National Natural Science Foundation of China (30801029).
Disclosure
The authors have no financial conflicts of interest.
References
- 1.Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;182:1281–90. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med. 2000;51:289–98. doi: 10.1146/annurev.med.51.1.289. [DOI] [PubMed] [Google Scholar]
- 3.Kollias G, Douni E, Kassiotis G, Kontoyiannis D. On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immunol Rev. 1999;169:175–94. doi: 10.1111/j.1600-065x.1999.tb01315.x. [DOI] [PubMed] [Google Scholar]
- 4.MacEwan DJ. TNF receptor subtype signaling: differences and cellular consequences. Cell Signal. 2002;14:477–92. doi: 10.1016/s0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- 5.Pimental-Munios FX, Seed B. Regulated commitment of TNF signaling: a molecular switch for death or activation. Immunity. 1999;11:783–93. doi: 10.1016/s1074-7613(00)80152-1. [DOI] [PubMed] [Google Scholar]
- 6.Holtmann MH, Douni E, Schutz M, et al. Tumor necrosis factor-receptor 2 is up-regulated on lamina propria T cells in Crohn's disease and promotes experimental colitis in vivo. Eur J Immunol. 2002;32:3142–51. doi: 10.1002/1521-4141(200211)32:11<3142::AID-IMMU3142>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 7.Mizoguchi E, Mizoguchi A, Takedatsu H, et al. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology. 2002;122:134–44. doi: 10.1053/gast.2002.30347. [DOI] [PubMed] [Google Scholar]
- 8.Sashio H, Tamura K, Ito R, et al. Polymorphisms of the TNF gene and the TNF receptor superfamily member 1B gene are associated with susceptibility to ulcerative colitis and Crohn's disease, respectively. Immunogenetics. 2002;53:1020–7. doi: 10.1007/s00251-001-0423-7. [DOI] [PubMed] [Google Scholar]
- 9.Dayer Schneider J, Seibold I, Saxer-Sekulic N, Paredes BE, Saurer L, Mueller C. Lack of TNFR2 expression by CD4+ T cells exacerbates experimental colitis. Eur J Immunol. 2009;39:1743–53. doi: 10.1002/eji.200839132. [DOI] [PubMed] [Google Scholar]
- 10.Ebach DR, Newberry R, Stenson W. Differential role of tumor necrosis factor receptors in TNBS colitis. Inflamm Bowel Dis. 2005;11:533–40. doi: 10.1097/01.mib.0000163698.34592.30. [DOI] [PubMed] [Google Scholar]
- 11.Nakai M, Sudo K, Yamada Y, et al. The role of the tumor necrosis factor receptor in 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis in mice. Dig Dis Sci. 2005;50:1669–76. doi: 10.1007/s10620-005-2913-1. [DOI] [PubMed] [Google Scholar]
- 12.Blam ME, Stein RB, Lichtenstein GR. Integrating anti-tumor necrosis factor therapy in inflammatory bowel disease: current and future perspectives. Am J Gastroenterol. 2001;96:1977–97. doi: 10.1111/j.1572-0241.2001.03931.x. [DOI] [PubMed] [Google Scholar]
- 13.Weisman MH. What are the risks of biological therapy in rheumatoid arthritis? An update on safety. J Rheumatol Suppl. 2002;65:33–8. [PubMed] [Google Scholar]
- 14.Zhao X, Mohaupt M, Jiang J, Liu S, Li B, Qin Z. Tumor necrosis factor receptor 2-mediated tumor suppression is nitric oxide dependent and involves angiostasis. Cancer Res. 2007;67:4443–50. doi: 10.1158/0008-5472.CAN-07-0185. [DOI] [PubMed] [Google Scholar]
- 15.Scheiffele F, Fuss IJ. Induction of TNBS colitis in mice. Curr Protoc Immunol. 2002;Chapter 15:Unit 15.19.1–14. doi: 10.1002/0471142735.im1519s49. [DOI] [PubMed] [Google Scholar]
- 16.Rachmilewitz D, Simon PL, Schwartz LW, Griswold DE, Fondacaro JD, Wasserman MA. Inflammatory mediators of experimental colitis in rats. Gastroenterology. 1989;97:326–37. doi: 10.1016/0016-5085(89)90068-1. [DOI] [PubMed] [Google Scholar]
- 17.Krawisz JE, Sharon P, Stenson WF. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–50. [PubMed] [Google Scholar]
- 18.Yamaoka T, Yan F, Cao H, et al. Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc Natl Acad Sci USA. 2008;105:11772–7. doi: 10.1073/pnas.0801463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fish RJ, Kruithof EK. Evidence for serpinB2-independent protection from TNF-α-induced apoptosis. Exp Cell Res. 2006;312:350–61. doi: 10.1016/j.yexcr.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 21.Erickson SL, de Sauvage FJ, Kikly K, et al. Decreased sensitivity to tumor-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature. 1994;372:560–3. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- 22.Peschon JJ, Torrance DS, Stocking KL, et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–52. [PubMed] [Google Scholar]
- 23.Chin AC, Parkos CA. Neutrophil transepithelial migration and epithelial barrier function in IBD: potential targets for inhibiting neutrophil trafficking. Ann NY Acad Sci. 2006;1072:276–87. doi: 10.1196/annals.1326.018. [DOI] [PubMed] [Google Scholar]
- 24.Naito Y, Takagi T, Yoshikawa T. Molecular fingerprints of neutrophil-dependent oxidative stress in inflammatory bowel disease. J Gastroenterol. 2007;42:787–98. doi: 10.1007/s00535-007-2096-y. [DOI] [PubMed] [Google Scholar]
- 25.Montecucco F, Steffens S, Burger F, et al. Tumor necrosis factor-alpha (TNF-α) induces integrin CD11b/CD18 (Mac-1) up-regulation and migration to the CC chemokine CCL3 (MIP-1α) on human neutrophils through defined signaling pathways. Cell Signal. 2008;20:557–68. doi: 10.1016/j.cellsig.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 26.Hehlgans T, Pfeffer K. The intriguing biology of the tumor necrosis factor/tumor necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115:1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu X, Guo R, Chen P, Wang Q, Cunningham PN. TNF induces caspase-dependent inflammation in renal endothelial cells through a Rho- and myosin light chain kinase-dependent mechanism. Am J Physiol Renal Physiol. 2009;297:F316–26. doi: 10.1152/ajprenal.00089.2009. [DOI] [PubMed] [Google Scholar]
- 28.Ellerin T, Rubin RH, Weinblatt ME. Infections and anti-tumor necrosis factor α therapy. Arthritis Rheum. 2003;48:3013–22. doi: 10.1002/art.11301. [DOI] [PubMed] [Google Scholar]