

Abstract
Appendicitis followed by appendectomy (AA) at a young age protects against inflammatory bowel disease (IBD). Using a novel murine appendicitis model, we showed that AA protected against subsequent experimental colitis. To delineate genes/pathways involved in this protection, AA was performed and samples harvested from the most distal colon. RNA was extracted from four individual colonic samples per group (AA group and double-laparotomy control group) and each sample microarray analysed followed by gene-set enrichment analysis (GSEA). The gene-expression study was validated by quantitative reverse transcription–polymerase chain reaction (RT–PCR) of 14 selected genes across the immunological spectrum. Distal colonic expression of 266 gene-sets was up-regulated significantly in AA group samples (false discovery rates < 1%; P-value < 0·001). Time–course RT–PCR experiments involving the 14 genes displayed down-regulation over 28 days. The IBD-associated genes tnfsf10, SLC22A5, C3, ccr5, irgm, ptger4 and ccl20 were modulated in AA mice 3 days after surgery. Many key immunological and cellular function-associated gene-sets involved in the protective effect of AA in experimental colitis were identified. The down-regulation of 14 selected genes over 28 days after surgery indicates activation, repression or de-repression of these genes leading to downstream AA-conferred anti-colitis protection. Further analysis of these genes, profiles and biological pathways may assist in developing better therapeutic strategies in the management of intractable IBD.
Keywords: appendectomy, appendicitis, colitis, Th17 system
Introduction
Regarded as vestigial and useless despite the abundant lymphoid follicles it contains, the human vermiform appendix is constantly exposed to an array of intestinal flora. Unlike its human counterpart, the appendix of other mammals such as the rabbit has been recognized to play a pivotal role in systemic and mucosal immunity [1–3]. The lifetime risk of appendicitis has been estimated to be 8·7% in men and 6·7% in women [4], making it the most common human abdominal emergency requiring surgical intervention.
Uncertainty has persisted about the causality of acute appendicitis, although the most popular theory posits luminal obstruction and incarceration of secretions, leading to increased intraluminal pressure, culminating in mucosal ischaemia and bacterial overgrowth. Potential causes of appendiceal obstruction include lymphoid hyperplasia, faecoliths and malignancy [5]. Mortality due to acute appendicitis is around 0·3%, rising to 1·7% if perforation is present [6]. Although acute appendicitis can occur at any age, the peak age of incidence of appendicitis without perforation is in the second and third decades [7].
There has been a paucity of immunological data from appendicitis, in contrast to histopathological data. Similarly, the immunopathology and complex interactions between genetic predisposition, bacterial flora and the intestinal immune system in inflammatory bowel diseases (comprised of ulcerative colitis and Crohn's disease) have not been elucidated satisfactorily. The critical role of appendicitis followed by appendicectomy in ameliorating or preventing development of human ulcerative colitis [8–10] and Crohn's disease [9,11] has been demonstrated reproducibly, despite controversies surrounding that role in Crohn's disease [12]. However, the protective effect is limited to patients having surgery before 20 years of age [10]. Additionally, studies in three different murine models including the T cell receptor-α mutant colitis model [13], the dextran sulphate sodium-induced colitis model [14] and the adoptive T lymphocyte transfer colitis model [15] have demonstrated that removal of the caecum prevented the development of experimental colitis.
We recently developed a murine model of appendicitis by constructing a pouch and ligature – occluding the murine equivalent of the human appendix, the caecal patch, followed by appendicectomy (removal of the murine caecal patch) [16]. The appendiceal histopathology in this appendicitis model closely resembles human appendicitis and reveals an age-dependent protection against trinitrobenzene sulphonic acid (TNBS)-induced colitis offered by appendicitis and appendicectomy [16]. Appendicitis per se or appendectomy per se was not protective. This protection offered by appendicitis followed by appendicectomy was dependent upon appendiceal interleukin (IL)-10-producing CD4+ and CD8+ regulatory T lymphocytes which proliferated in the appendix and migrated to the distal colon (Ng et al., submitted).
Conventional gene expression analysis strategies have been used successfully to elucidate differences in individual gene expression between two groups or specific states. However, this approach excludes the biological reality of cellular processes concertedly effecting changes in series of genes as diverse as transcriptional mediators, stress-responses, metabolic processes, subcellular transport changes and cytokine fluxes, etc. These changes may be subtle or undetectable at the level of individual genes, but are evident at the level of gene-sets. For example, just one-fifth of an increase in the expression of genes which are components of a pathway may significantly change the flux via the pathway, increasing the contribution of one gene 20-fold [17].
Previous studies have elucidated the pathogenic gene pathways involved in human inflammatory bowel disease (IBD) [18–23] and experimental models of IBD [24,25], or the expression pathways involved in the therapy of human IBD [26] and intervention in experimental models of IBD [27–29]. In contrast, our novel study presented in this paper identifies several key gene expression profiles and biological pathways involved in the protective effect of appendicitis and appendectomy in experimental colitis and paves a way towards manipulating various aspects of these pathways to develop better therapeutic strategies in the management of intractable IBD.
Materials and methods
Animal experiments
Specific pathogen-free Balb/c mice (male, 5 weeks old), were purchased from the Animal Resource Centre, Perth, Western Australia and kept in the University of New South Wales holding and care facility in physical containment level 2 rooms. The mice were kept in filtered plastic cages and permitted to acclimatize for 1 week before the studies commenced. All experiments were approved and monitored by the University of New South Wales Animal Care and Ethics Committee. Mice were anaesthetized with xylazine (5 mg/kg) and ketamine (100 mg/kg) intraperitineally (i.p.) followed by allocation into two treatment groups, the appendicitis group or the sham surgery group [16]. Surgical manipulations were performed as described previously [16]. Briefly, mice were randomized to have either appendicitis or sham operation. Appendicitis was induced by constructing an appendiceal pouch from the caecal lymphoid patch. This murine appendix was obstructed by rubber band ligation using standardized negative aspiration. Sham surgery entailed a similar procedure, but without continuous obstruction by band ligation of the caecal patch and the placement of a sterile rubber band in the abdominal cavity as a control for foreign body reaction. Seven days following initial surgery, appendicitis mice underwent appendicectomy [appendicitis and appendectomy (AA) group] while sham mice underwent a second sham surgery [sham and sham (SS) group]. All mice were monitored daily for grooming, weight loss, mobility and evidence of bowel obstruction. Normal saline was administered subcutaneously daily to ensure adequate hydration.
Processing of colonic specimens for RNA extraction
Transmural colonic samples were cleaned of faecal contents with normal saline and transferred immediately to TRIzol® reagent (Invitrogen Australia Pty Limited, Mulgrave, Victoria, Australia; 50–75 mg of tissue in 600 µl of TRIzol® reagent), snap-frozen in liquid nitrogen and stored at −80°C until the microarray analysis. Further extraction entailed chloroform and isopropanol treatment and centrifugation followed by washing the resultant pellet with 75% ethanol, air-drying and final reconstitution in nuclease-free H2O. Concentration and purity of RNA were determined by automated optical density evaluation [optical density (OD) 260/OD 280 ≥ 1·8 and OD 260/OD 230 ≥ 1·8] using Nanodrop ND-1000 (Nanodrop Technologies, Wilmington, DE, USA). The degree of RNA degradation was analysed by the Agilent electrophoresis bioanalyzer 2100 (Agilent Technologies Inc., Santa Clara, CA, USA) with the RNA integrity number (RIN) values consistently above 7.
Experimental design of microarray studies
All experiments were designed to be compliant with minimum information about a microarray experiment (MIAME) standards [30,31]. To ensure adequate accountability for intrabatch and interbatch variability, colonic samples from two batches, each batch encompassing colonic samples from two AA mice and two SS mice. For Affymetrix array experiments, four individual test samples were used per group (AA group versus SS group; one colonic sample per mouse) with each sample hybridized to an individual slide (Table 1).
Table 1.
Overview of differentially regulated gene-set groups in appendicitis–appendectomy (AA) mice compared to sham–sham (SS) mice.
| Genes set group | Category | No. of gene-sets | Gene-sets up-regulated in AA mice | |
|---|---|---|---|---|
| 1 | Kegg pathways | KEGG | 150 | 9 |
| c2_kegg.GseaPreranked | ||||
| 2 | Micro RNAs | MIR | 200 | 0 |
| c3_mir.GseaPreranked | ||||
| 3 | Transcription factors | TFT | 579 | 41 |
| c3_tft.GseaPreranked | ||||
| 4 | Biological processes | BP | 536 | 7 |
| c5_bp.GseaPreranked | ||||
| 5 | Others | OTH | 1387 | 209 |
| c2_all.GseaPreranked | ||||
| Total | 2852 | 266 |
The gene-set groups chosen for further evaluation had stringent cut-off values (FDR < 1% and P < 0·001). Using these criteria, there were no gene-sets up-regulated in the SS group. However, 266 gene-sets were up-regulated in the AA group. SS group: sham and sham group; AA group: appendicitis and appendectomy group; FDR: false discovery rate.
Affymetrix array process – labelling, hybridization, scanning and normalization
For Affymetrix arrays, 100 ng of RNA from each sample was labelled using the Whole Transcript Sense Target Labelling Assay as described previously [32] (Affymetrix). Labelled cRNA samples were then hybridized to Affymetrix mouse gene 1·0 ST arrays (28 853 well-annotated genes) (Ramaciotti Centre for Gene Function Analysis, University of New South Wales, Australia) before being scanned using a Affymetrix GCS3000 7G four-colour gene array scanner with autoloader (Affymetrix). The Gene Expression Omnibus Accession number for microarray data reported here, inclusive of MIAME-compliant experimental details [30,31], is GSE23914, and the relevant link is http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE23914.
Microarray preprocessing and filtering
All non-control probesets from the eight arrays were imported into Partek (version 6·4; Partek Inc., St Louis, MO, USA), and then normalized using RMA [33]. Using principle components analysis, a batch effect was evident in principle component 1, which was removed using the batch removal tool in Partek, using default parameters. The probability of each probeset being expressed was determined using the detected above background procedure, using Affymetrix Power Tools (version 1·10·2), excluding 13 probes from probeset 10338063 which had very low GC, and thus did not have matched controls. Probesets were excluded if none of the samples were detected above background (P = 10−5). To assess the degree of differential expression between AA and SS groups, a two-way analysis of variance (anova) on treatment and batch was fitted to each probeset using Partek. To correct for multiple hypothesis testing, we used the qvalue/positive false discovery rate (FDR) [34].
Gene-set enrichment analysis (GSEA)
We compared gene expression profiles to the c2_all collection of curated gene-sets from the molecular signatures database (version 2·5) [35]. This collection contains gene-sets that are experimentally derived, as well as from expert curated pathway databases. A preranked file was created, containing the average difference between AA and SS for each probeset, sorted from most up-regulated in SS to most down-regulated. We used the na28 annotation csv file from http://www.affymetrix.com to determine the gene symbol for each probeset and collapsed probesets to unique genes using the default, max_probe option, resulting in 18 600 unique genes. GSEA (version 2·0) [35] was run in preranked mode, using default parameters (gene-set sizes between 15 and 500 leaving 1387 gene-sets, 1000 permutations, images on the top 50 gene-sets).
Validation and analysis of gene expression with reverse transcription–polymerase chain reaction (RT–PCR)
We used mRNA extracted from distal colons obtained from four SS and four AA mice for RT–PCR confirmation of our gene expression study. Reverse transcription to produce cDNA was performed using RT2 First Strand Kits (SA Biosciences, Frederick, MD, USA), according to the manufacturer's instructions. RT–PCR was performed utilizing the LightCycler 480 real-time PCR system (Roche Applied Science, Mannheim, Germany) with RT2 SYBR green PCR master mix according, to the manufacture's protocol (SA Biosciences). Predesigned primers for genes of interest (slpi, s100A8, lbp, CD68, IL18R1, IL33, ccl8, cxcl10, ccl12, pf4, ccl5, ccl7, fpr1 and ccr5) were obtained from SA Biosciences. For reference genes we evaluated three candidates, β-actin, β-glucuronidase and 18S rRNA. Beta-glucuronidase was selected based on similar expression patterns to most of our genes of interest and also because it was expressed invariantly between the groups. Hence, each sample was normalized on the basis of its β-glucuronidase content. Thermal cycling was performed as follows: initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Each assay was performed in duplicate. The quantification points generated from quantitative RT–PCR (qRT–PCR) were normalized against a reference gene using this formula: normalized value of gene of interest with β-glucuronidase = 2–(QPGOI–QPRG), where QP = quantitative point, GOI = gene of interest and RG = reference gene (i.e. β-glucuronidase).
Time–course expression study of selected genes over 4 weeks using RT–PCR
We used the same 14 genes that we used for RT–PCR confirmation of our microarray study. We collected distal colonic samples from 3 days, 14 days and 28 days after the last (second) surgery. For each of the time-points we used four SS and four AA mice. The colons were collected, stored and processed for RT–PCR as described earlier.
Statistics
Group comparisons were analysed using the Mann–Whitney U-test with GraphPad Prism (Graphpad Software, San Diego, CA, USA). The differences were considered to be significant if P < 0·05.
Results
Groups of gene-sets enriched
We utilized GSEA, developed by Mootha [17], to delineate related genes and biological pathways that were consistently altered in the distal colons of all AA mice when compared to the all the control SS mice. Distal colons were selected as this is the site of migration of protective appendiceal lymphocytes (Ng et al., submitted). Our approach merges data from groups of gene-sets described previously in the literature to detect significant expression differences. These gene-set groups were Kegg pathways (150 gene-sets), micro-RNAs (200 gene-sets), transcription factors (579 gene-sets), biological processes (536 gene-sets) and others (1387 gene-sets). We used stringent statistical cut-offs: false discovery rates (FDR) values < 1% and P value < 0·001. Expression of 266 gene-sets was up-regulated significantly in AA group samples; distributed across Kegg pathways (9 gene-sets), transcription factors (41 gene-sets), biological processes (seven gene-sets) and others (209 gene-sets) as depicted in Table 1. The 266 gene-sets up-regulated in the AA group (Table S1) included immunity-related and unrelated gene-sets. No gene-sets were up-regulated in the SS group when compared to the AA group.
Correlation of IBD-linked genes with up-regulated post-AA distal colonic genes
The tnfsf10 gene was up-regulated 1·46-fold, the SLC22A5 gene (OCTN2) 1·31-fold, the C3 gene 1·74-fold, the ccr5 gene 1·5-fold, the irgm gene 1·66-fold and the ptger4 gene 1·43-fold in the AA mice 3 days after surgery. Conversely, the ccl20 gene was decreased 0·6-fold in the AA mice 3 days after surgery.
Quantitative RT–PCR validation of our gene-expression study
We selected 14 genes for confirmation of our gene expression studies. These genes were immunological genes of interest which were up-regulated in the AA group in this study. They broadly belonged to four major groups: innate immunity (slpi, s100A8, lbp, CD68), immune mediators (IL18R1, IL33), cell migration-chemokines (ccl8, cxcl10, ccl12 or mcp5, pf4, ccl5, ccl7 or mcp3) and cell migration-receptors (fpr1, ccr5). The RT–PCR results (Fig. 1) indicate that eight of the total 14 genes tested were up-regulated significantly in the AA group; three of these genes just missed statistical significance, and three genes showed no difference between the SS and AA groups. These RT–PCR results validate our microarray data.
Fig. 1.
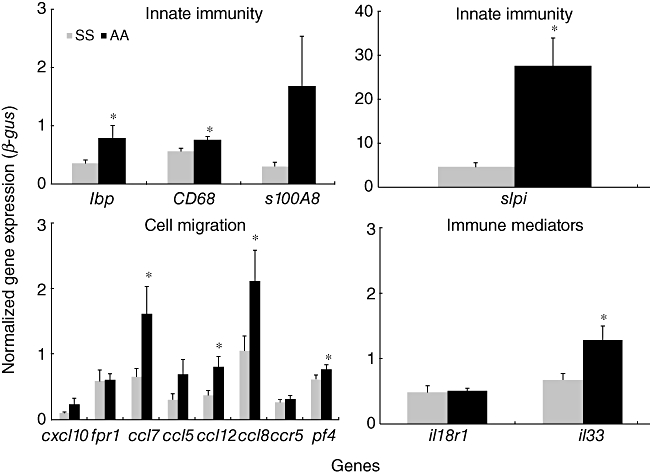
Validation of microarray differential gene expression. Validation of microarray differential gene expression in AA mice compared to SS mice using reverse transcription–polymerase chain reaction (RT–PCR) quantitative analysis. AA: appendicitis–appendectomy; SS: sham–sham. *P < 0·05. SS group, sham and sham group; AA group, appendicitis and appendectomy group.
Time–course expression study of selected genes over 4 weeks using RT–PCR
Distal colonic samples from 3 days, 14 days and 28 days after the last (second) surgery from SS and AA mice were assessed. SS and AA expression levels of all 14 genes analysed (except for the pf4 gene) either decreased or remained level. Pertaining to the four innate immunity genes that were quantified (slpi, s100A8, lbp, CD68), slpi was reduced significantly in the AA group when compared to the SS group at the 28 day post-surgery time-point, in contrast to the 3-day post-surgery time-point (Fig. 2). CD68 was relatively up-regulated in the SS group, although being expressed to a relatively lesser extent in the AA group (Fig. 2). Of the eight cell migration genes that were quantified (ccl8, cxcl10, ccl12, pf4, ccl5, ccl7, fpr1, ccr5), except for the pf4 gene, both SS and AA expression levels either decrease or remain steady for all the genes (Fig. 3).
Fig. 2.
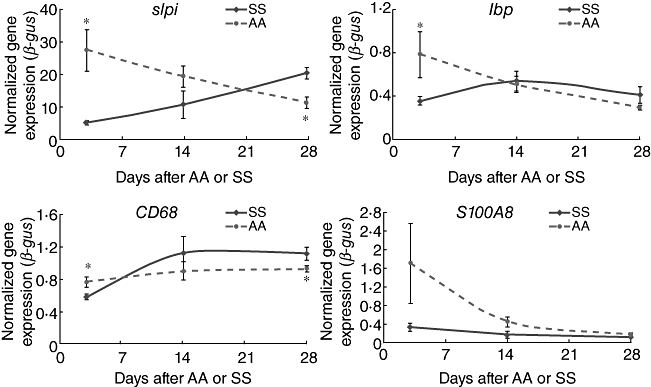
Time–course reverse transcription–polymerase chain reaction (RT–PCR): innate immunity. Longitudinal RT–PCR study comparing transcript levels in SS and AA mice at 7, 14 and 28 days after the second surgery. The four genes in this panel play roles in innate immunity. Both SS and AA expression levels either decrease or remain level. However, at the 28-day post-surgery time-point, slpi expression was relatively reduced in the AA group and CD68 was relatively up-regulated in the SS group. AA: appendicitis-appendectomy; SS: sham–sham. *P < 0·05. SS group, sham and sham group; AA group, appendicitis and appendectomy group.
Fig. 3.

Time–course reverse transcription–polymerase chain reaction (RT–PCR): cell migration. Longitudinal RT–PCR study comparing transcript levels in SS and AA mice at 7, 14 and 28 days after the second surgery. The six genes in this panel play roles in cell migration. Except for the pf4 gene, both SS and AA expression levels either decrease or remain steady for all the genes. AA: appendicitis–appendectomy; SS: sham–sham. *P < 0·05. SS group, sham and sham group; AA group, appendicitis and appendectomy group.
Of the two immune-mediator genes that were quantified (IL18R1, IL33), only IL18R1 expression was reduced significantly in the AA group when compared to the SS group at the 28-day post-surgery time-point (Fig. 4).
Fig. 4.
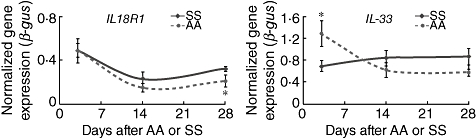
Time–course reverse transcription–polymerase chain reaction (RT–PCR): immune mediators. Longitudinal RT–PCR study comparing transcript levels in SS and AA mice at 7, 14 and 28 days after the second surgery. The two genes in this panel play roles in immune mediation. Both SS and AA expression levels either decrease or remain level. However, at the 28-day post-surgery time-point, il18R1 expression was reduced significantly in the AA group when compared to the SS group. AA: appendicitis–appendectomy; SS: sham–sham. *P < 0·05. SS group, sham and sham group; AA group, appendicitis and appendectomy group.
Discussion
Utilizing the first murine appendicitis model (developed by us), we have shown previously that although appendicitis alone or appendectomy alone or no intervention alone were not protective, appendicitis followed by appendectomy (AA) provided significant protection against subsequent experimental colitis [16]. We chose the distal-most colonic samples carefully, avoiding the caecum and the rest of the colon, not only for the obvious reason of pathological relevance, but also to minimize bacterial contamination and severely acute inflammatory changes in the acutely inflamed caecum. We have also avoided delving into minutiae regarding specific immunological systems, such as the markedly suppressed T helper type 2 (Th17) system in AA which will be expounded in another manuscript, for the sake of space, brevity and focus.
We used gene-set enrichment analysis (GSEA) to elucidate the pathways involved in this protective effect. Distal colonic expression of 266 gene-sets was significantly up-regulated in AA group samples and the study was validated by quantitative RT–PCR of 14 selected genes. However, time–course experiments involving these genes displayed down-regulation of these genes over a period of 28 days in both SS and AA groups.
Many key immunological, apoptosis-related and cellular function-associated gene-sets involved in the protective effect of AA in experimental colitis were identified. The up-regulated gene-sets not known to be involved directly in immunity include those participating in cellular cytoskeleton, apoptosis, cell cycle, growth and growth factors, non-immune development and differentiation, enzyme activity and regulation, protein metabolism, injury, healing and angiogenesis, reactive species stress-related, malignancy and intervention-related and transcription factors. Up-regulated gene-sets known to play well-established roles in immunity include those participating in antigen processing, cellular adhesion, extracellular matrix and receptor interactions, nuclear factor-kappaB (NF-κB)-related pathways, cellular signalling, immune system development and differentiation, injury, healing and angiogenesis, responses to pathogens, chemokine and cytokine-related pathways, interferon and other immune-factor-related or -induced pathways.
The IBD-associated genes tnfsf10, SLC22A5, C3, ccr5, irgm, and ptger4 were up-regulated and ccl20 gene (also IBD-associated) was decreased in AA mice 3 days after surgery. Of immunologically relevant IBD genes that were modulated, tnfsf10[36] encodes a cytokine belonging to the tumour necrosis factor (TNF) ligand family, which binds to several members of the TNF receptor superfamily and triggers activation of MAPK8/JNK and caspases. The C3 gene [37] product plays a central role in the activation of both classical and alternative complement pathways in ‘innate’ immunity. Ccr5[38] encodes a member of the beta chemokine receptor family, which is expressed by T cells and macrophages, and has ligands known to be important in the intestine [39]. The ptger4 gene [40] encodes a G-protein coupled receptor for prostaglandin E2 (PGE2), which activates T cell factor signalling, and ccl20 is a crucial intestinal chemotactic factor which aids formation and function of mucosal lymphoid tissues by attracting lymphocytes and dendritic cells towards epithelial cells, and in addition possesses anti-bacterial activity [41]. The SLC22A5 gene (OCTN2) gene [42] encodes an organic cation transporter critical for elimination of endogenous organic cations, drugs and environmental toxins. The irgm product [43] regulates autophagy in response to intracellular pathogens. All these identified genes are crucial to the immune features of the intestine relevant to bacterial and toxin handling, and they share fundamental importance in our current understanding of IBD pathogenesis.
By 28 days after AA (data not shown), only the tnfsf10 gene (1·6-fold) and the irgm gene (1·7-fold) remained up-regulated and the ccl20 gene (0·63-fold) was sustainedly down-regulated, buttressing suggested roles for these genes in IBD pathogenesis and appendicitis-related protection against IBD.
The genes chosen for RT–PCR validation were representative of immune functions pertaining to innate immunity (slpi, s100A8, lbp, CD68), cell migration (ccl8, cxcl10, ccl12, pf4, ccl5, ccl7, fpr1, ccr5) and immune-mediation (IL18R1, IL33). Additionally, these genes were represented well across many gene-sets up-regulated in the AA group (data not shown).
Although the RT–PCR data at the 3-day time-point validated our microarray data, the subsequent down-regulation of 13 of the 14 selected genes shown by RT–PCR over 28 days after surgery is indeed intriguing. This may indicate activation, repression or de-repression of these or related genes leading to downstream gene-products, culminating in the milieu responsible for the durable AA-conferred protection against colitis. Inexplicably, CD68 was up-regulated in the SS group, although being expressed to a relatively lesser extent in the AA group.
Preliminary microarray data at the 28-day post-surgery time-point indicate fundamentally different gene-sets may be implicated in the durable effect of appendicitis and appendectomy. These genes and gene-sets may indicate downstream gene expression changes owing to repression or de-repression of genes modulated at earlier (3-day) time-points (data not shown).
Further analysis of these profiles and biological pathways will assist in the utilization of these gene products and manipulating various aspects of these pathways to develop better therapeutic strategies in the management of intractable IBD.
Acknowledgments
National Health and Medical Research Council (NHMRC) for funding this study. We acknowledge Warren Kaplan and Mark J Cowley from Peter Wills Bioinformatics Centre, Garvan Institute of Medical Research, Sydney, Australia who conducted the Gene Set Enrichment Analysis for us.
Disclosure
None.
Supporting information
Additional supporting information may be found in the online version of this article.
Table S1. Differentially expressed gene-sets [from gene-set enrichment analysis (GSEA)] in the distal colon of appendicitis-appendectomy (AA) mice compared to the distal colon of sham-sham (SS) mice.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Becker RS, Knight KL. Somatic diversification of immunoglobulin heavy chain VDJ genes: evidence for somatic gene conversion in rabbits. Cell. 1990;63:987–97. doi: 10.1016/0092-8674(90)90502-6. [DOI] [PubMed] [Google Scholar]
- 2.Dasso JF, Howell MD. Neonatal appendectomy impairs mucosal immunity in rabbits. Cell Immunol. 1997;182:29–37. doi: 10.1006/cimm.1997.1216. [DOI] [PubMed] [Google Scholar]
- 3.Weinstein PD, Anderson AO, Mage RG. Rabbit IgH sequences in appendix germinal centers: VH diversification by gene conversion-like and hypermutation mechanisms. Immunity. 1994;1:647–59. doi: 10.1016/1074-7613(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 4.Addiss DG, Shaffer N, Fowler BS, Tauxe RV. The epidemiology of appendicitis and appendectomy in the United States. Am J Epidemiol. 1990;132:910–25. doi: 10.1093/oxfordjournals.aje.a115734. [DOI] [PubMed] [Google Scholar]
- 5.Larner AJ. The aetiology of appendicitis. Br J Hosp Med. 1988;39:540–2. [PubMed] [Google Scholar]
- 6.Velanovich V, Satava R. Balancing the normal appendectomy rate with the perforated appendicitis rate: implications for quality assurance. Am Surg. 1992;58:264–9. [PubMed] [Google Scholar]
- 7.Marudanayagam R, Williams GT, Rees BI. Review of the pathological results of 2660 appendicectomy specimens. J Gastroenterol. 2006;41:745–9. doi: 10.1007/s00535-006-1855-5. [DOI] [PubMed] [Google Scholar]
- 8.Koutroubakis IE, Vlachonikolis IG, Kouroumalis EA. Role of appendicitis and appendectomy in the pathogenesis of ulcerative colitis: a critical review. Inflamm Bowel Dis. 2002;8:277–86. doi: 10.1097/00054725-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Lopez Ramos D, Gabriel R, Cantero Perona J, Moreno Otero R, Fernandez Bermejo M, Mate Jimenez J. Association of MALTectomy (appendectomy and tonsillectomy) and inflammatory bowel disease: a familial case-control study. Rev Esp Enferm Dig. 2001;93:303–14. [PubMed] [Google Scholar]
- 10.Andersson RE, Olaison G, Tysk C, Ekbom A. Appendectomy and protection against ulcerative colitis. N Engl J Med. 2001;344:808–14. doi: 10.1056/NEJM200103153441104. [DOI] [PubMed] [Google Scholar]
- 11.Radford-Smith GL, Edwards JE, Purdie DM, et al. Protective role of appendicectomy on onset and severity of ulcerative colitis and Crohn's disease. Gut. 2002;51:808–13. doi: 10.1136/gut.51.6.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersson RE, Olaison G, Tysk C, Ekbom A. Appendectomy is followed by increased risk of Crohn's disease. Gastroenterology. 2003;124:40–6. doi: 10.1053/gast.2003.50021. [DOI] [PubMed] [Google Scholar]
- 13.Mizoguchi A, Mizoguchi E, Chiba C, et al. Cytokine imbalance and autoantibody production in T cell receptor-alpha mutant mice with inflammatory bowel disease. J Exp Med. 1996;183:847–56. doi: 10.1084/jem.183.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krieglstein CF, Cerwinka WH, Laroux FS, et al. Role of appendix and spleen in experimental colitis. J Surg Res. 2001;101:166–75. doi: 10.1006/jsre.2001.6223. [DOI] [PubMed] [Google Scholar]
- 15.Farkas SA, Hornung M, Sattler C, et al. Preferential migration of CD62L cells into the appendix in mice with experimental chronic colitis. Eur Surg Res. 2005;37:115–22. doi: 10.1159/000084543. [DOI] [PubMed] [Google Scholar]
- 16.Watson Ng WS, Hampartzoumian T, Lloyd AR, Grimm MC. A murine model of appendicitis and the impact of inflammation on appendiceal lymphocyte constituents. Clin Exp Immunol. 2007;150:169–78. doi: 10.1111/j.1365-2249.2007.03463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 18.Flach CF, Eriksson A, Jennische E, Lange S, Gunnerek C, Lonnroth I. Detection of elafin as a candidate biomarker for ulcerative colitis by whole-genome microarray screening. Inflamm Bowel Dis. 2006;12:837–42. doi: 10.1097/01.mib.0000232469.23574.11. [DOI] [PubMed] [Google Scholar]
- 19.Seidelin JB, Nielsen OH. Expression profiling of apoptosis-related genes in enterocytes isolated from patients with ulcerative colitis. APMIS. 2006;114:508–17. doi: 10.1111/j.1600-0463.2006.apm_116.x. [DOI] [PubMed] [Google Scholar]
- 20.Cao D, Wilentz RE, Abbruzzese JL, Ho L, Maitra A. Aberrant expression of maspin in idiopathic inflammatory bowel disease is associated with disease activity and neoplastic transformation. Int J Gastrointest Cancer. 2005;36:39–46. doi: 10.1385/IJGC:36:1:039. [DOI] [PubMed] [Google Scholar]
- 21.Costello CM, Mah N, Hasler R, et al. Dissection of the inflammatory bowel disease transcriptome using genome-wide cDNA microarrays. PLoS Med. 2005;2:e199. doi: 10.1371/journal.pmed.0020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uthoff SM, Eichenberger MR, Lewis RK, et al. Identification of candidate genes in ulcerative colitis and Crohn's disease using cDNA array technology. Int J Oncol. 2001;19:803–10. doi: 10.3892/ijo.19.4.803. [DOI] [PubMed] [Google Scholar]
- 23.Dieckgraefe BK, Stenson WF, Korzenik JR, Swanson PE, Harrington CA. Analysis of mucosal gene expression in inflammatory bowel disease by parallel oligonucleotide arrays. Physiol Genomics. 2000;4:1–11. doi: 10.1152/physiolgenomics.2000.4.1.1. [DOI] [PubMed] [Google Scholar]
- 24.Rivera E, Flores I, Appleyard CB. Molecular profiling of a rat model of colitis: validation of known inflammatory genes and identification of novel disease-associated targets. Inflamm Bowel Dis. 2006;12:950–66. doi: 10.1097/01.mib.0000231575.11678.8c. [DOI] [PubMed] [Google Scholar]
- 25.de Buhr MF, Mahler M, Geffers R, et al. Cd14, Gbp1, and Pla2g2a: three major candidate genes for experimental IBD identified by combining QTL and microarray analyses. Physiol Genomics. 2006;25:426–34. doi: 10.1152/physiolgenomics.00022.2005. [DOI] [PubMed] [Google Scholar]
- 26.Dooley TP, Curto EV, Reddy SP, et al. Regulation of gene expression in inflammatory bowel disease and correlation with IBD drugs: screening by DNA microarrays. Inflamm Bowel Dis. 2004;10:1–14. doi: 10.1097/00054725-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 27.Kristensen NN, Olsen J, Gad M, Claesson MH. Genome-wide expression profiling during protection from colitis by regulatory T cells. Inflamm Bowel Dis. 2008;14:75–87. doi: 10.1002/ibd.20277. [DOI] [PubMed] [Google Scholar]
- 28.Bernstein H, Holubec H, Bernstein C, et al. Deoxycholate-induced colitis is markedly attenuated in Nos2 knockout mice in association with modulation of gene expression profiles. Dig Dis Sci. 2007;52:628–42. doi: 10.1007/s10620-006-9608-0. [DOI] [PubMed] [Google Scholar]
- 29.Nakajima A, Wada K, Katayama K, et al. Gene expression profile after peroxisome proliferator activator receptor-gamma ligand administration in dextran sodium sulfate mice. J Gastroenterol. 2002;37(Suppl 14):62–6. doi: 10.1007/BF03326416. [DOI] [PubMed] [Google Scholar]
- 30.Brazma A. Minimum Information About a Microarray Experiment (MIAME)–successes, failures, challenges. Scientific World J. 2009;9:420–3. doi: 10.1100/tsw.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brazma A, Hingamp P, Quackenbush J, et al. Minimum information about a microarray experiment (MIAME) – toward standards for microarray data. Nat Genet. 2001;29:365–71. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- 32.Affymetrix Online User Manual – GeneChip® Whole Transcript (WT) Sense Target Labeling Assay. Available at: http://www.affymetrix.com/support/downloads/manuals/wt_sensetarget_label_manual.pdf.
- 33.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brost S, Koschny R, Sykora J, et al. Differential expression of the TRAIL/TRAIL-receptor system in patients with inflammatory bowel disease. Pathol Res Pract. 2010;206:43–50. doi: 10.1016/j.prp.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Lundgren BA, Rorsman F, Portela-Gomes GM, et al. Identification of complement C3 as an autoantigen in inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2010;22:429–36. doi: 10.1097/MEG.0b013e32833283b1. [DOI] [PubMed] [Google Scholar]
- 38.Oki M, Ohtani H, Kinouchi Y, et al. Accumulation of CCR5+ T cells around RANTES+ granulomas in Crohn's disease: a pivotal site of Th1-shifted immune response? Lab Invest. 2005;85:137–45. doi: 10.1038/labinvest.3700189. [DOI] [PubMed] [Google Scholar]
- 39.Grimm MC, Newman R, Hassim Z, et al. Cutting edge: vasoactive intestinal peptide acts as a potent suppressor of inflammation in vivo by trans-deactivating chemokine receptors. J Immunol. 2003;171:4990–4. doi: 10.4049/jimmunol.171.10.4990. [DOI] [PubMed] [Google Scholar]
- 40.Libioulle C, Louis E, Hansoul S, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaser A, Ludwiczek O, Holzmann S, et al. Increased expression of CCL20 in human inflammatory bowel disease. J Clin Immunol. 2004;24:74–85. doi: 10.1023/B:JOCI.0000018066.46279.6b. [DOI] [PubMed] [Google Scholar]
- 42.Rioux JD, Silverberg MS, Daly MJ, et al. Genomewide search in Canadian families with inflammatory bowel disease reveals two novel susceptibility loci. Am J Hum Genet. 2000;66:1863–70. doi: 10.1086/302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waterman M, Xu W, Stempak JM, et al. Distinct and overlapping genetic loci in Crohn's disease and ulcerative colitis: correlations with pathogenesis. Inflamm Bowel Dis. 2010 doi: 10.1002/ibd.21579. e-publication ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.