

Abstract
Procarboxypeptidase U (TAFI) is a recently discovered plasma procarboxypeptidase that upon activation by thrombin or thrombin-thrombomodulin turns into a potent antifibrinolytic enzyme. Its prominent bridging function between coagulation and fibrinolysis raised the interest of many research groups and of the pharmaceutical industry. The development of CPU inhibitors as profibrinolytic agents is an attractive concept and possibilities for rational drug design will become more available in the near future due to the recently published crystal structure.
Numerous studies have been performed and many of them show beneficial effects of CPU inhibitors for the improvement of endogenous fibrinolysis in different animal sepsis and thrombosis models. CPU inhibitors combined with t-PA seem to increase the efficiency of pharmacological thrombolysis allowing lower dosing of t-PA and subsequently fewer bleeding complications.
This review will focus on recently obtained in vivo data and the benefits/risks of targeting CPU for the treatment of thrombotic disorders.
Introduction
The coagulation and fibrinolytic systems safeguard the patency of the vasculature and surrounding tissues. Both cascades have long been considered as separate entities but the discovery of procarboxypeptidase U (proCPU) or thrombin activatable fibrinolysis inhibitor (TAFI) greatly improved our understanding of cross regulation of both systems [1–4].
Procarboxypeptidase U is a 60 kDa metallocarboxypeptidase produced by the liver and present in plasma. By the action of thrombin, the key protease of the coagulation, this inactive zymogen is proteolytically converted to the active enzyme carboxypeptidase U (CPU). CPU potently attenuates fibrinolysis by cleaving C-terminal lysines on partially degraded fibrin, thereby interfering with efficient plasminogen activation [2–5]. A recent report claimed that the zymogen proCPU also has an intrinsic antifibrinolytic activity, however, this finding was opposed by two other groups [6–8]. Therefore activation of proCPU still provides the explicit molecular link between coagulation and fibrinolysis.
Venous and arterial thromboembolism is the largest cause of disease and death in the Western World. Therapy available today includes thrombolytics, anticoagulants and antiplatelet drugs. However, the need for parenteral application, the risk for severe bleeding complications and in the case of the oral anticoagulants the requirement for close lab monitoring urgently require the development of orally active antithrombotic or thrombolytic drugs that are clinically safe and need less monitoring.
Because of its prominent bridging function between coagulation and fibrinolysis, the development of CPU inhibitors as pro-fibrinolytic agents is an attractive concept. Furthermore, since the coagulation cascade is unaffected, CPU inhibition may result in fewer bleeding complications than conventional therapy.
In recent years numerous small synthetic and naturally occurring CPU inhibitors have been evaluated in animal thrombosis models and existing in vivo data are intriguing and call for further evaluation in humans.
CPU basic research has been extensively reviewed in the recent past [eg. 9–13]. This rather brief review will focus on recently obtained in vivo data and the benefits/risks of targeting CPU for the treatment of thrombotic disorders.
CPU a key modulator of the fibrinolytic threshold
Intravascular fibrinolysis is initiated when plasminogen and its activator t-PA bind to the internal lysines on the fibrin surface. Plasmin formed by the action of t-PA on plasminogen cleaves fibrin after arginine or lysine residues, generating partially degraded fibrin containing C-terminal arginine and lysine residues (initial phase of fibrinolysis). These C-terminal lysine residues participate in a multifaceted positive feedback loop. First, plasmin generation is up-regulated by the increased affinity of plasminogen for plasmin-degraded fibrin and FDPs [5,14–17]. Second, plasmin converts C-terminal lysine bound Glu1-plasminogen to Lys78-plasminogen, a much better substrate for t-PA [18]. Finally, C-terminal lysine residues decrease the rate of plasmin inhibition, as plasmin bound to degraded fibrin and FDPs is protected from inactivation by α2-antiplasmin. As a result, the fibrinolytic efficiency increases dramatically (acceleration phase of fibrinolysis) [19,20].
Given the central role that C-terminal basic amino acids play in the regulation of fibrinolysis, it is not surprising that their removal from the degraded fibrin surface is also enzymatically controlled. Whereas plasmin up-regulates fibrinolysis via C-terminal lysine formation, the basic carboxypeptidase CPU downregulates fibrinolysis by removing C-terminal lysine residues from plasmin-degraded fibrin and FDPs [5,14,19,20]. The dependence of fibrinolysis on opposing processes that share components confers a threshold upon the system.
It was discovered independently by two research groups that CPU attenuates the fibrinolytic rate through a threshold dependent mechanism [21,22]. As long as CPU is present at or above a key threshold value, fibrinolysis stays in its initial phase, only to accelerate once the CPU activity decays to a level below this threshold value [21–24].
In general, thresholds are intrinsic properties of systems as a whole rather than a specific property of a single component in the system. Indeed, the critical threshold CPU concentration, defined as the concentration of CPU at which the accumulation of plasmin-catalyzed C-terminal lysine and arginine residues on fibrin is prevented, is determined by the basal steady state concentration of plasmin which on its turn is dependent on the rate of plasminogen activation (t-PA concentration) and plasmin inhibition (concentration of irreversible plasmin inhibitors) [21–24].
Leurs et al. [21] and Walker et al. [22] showed that the CPU threshold increases as the concentration of t-PA increases. With regard to the plasmin inhibitors, Walker and co-workers [24] provided evidence that the fibrinolytic process is not simply attenuated or prolonged but can be stopped by sufficient (above the threshold) and sustained basic carboxypeptidase activity. However, this is only possible when the aggregate concentration of irreversible inhibitors is in excess of the plasminogen in the system, as is the case in plasma. Indeed, the primary plasmin inhibitor in plasma, α2-antiplasmin, is present at only half the concentration of plasminogen (2µM) but when necessary it can act in concert with the secondary inhibitors α2-macroglobulin (3µM) and antithrombin (5µM). In the presence of this excess of plasmin inhibitors, a sustained carboxypeptidase activity above the threshold will completely stop the fibrinolytic process. This is most probably as a result of consumption of plasminogen prior to lysis [24].
The time interval over which the CPU level will stay above the threshold is determined by the plasma proCPU concentration, the extent of proCPU activation by the coagulation cascade and most importantly by the stability of CPU [21,23]. The latter can be influenced by a naturally occurring polymorphism in the proCPU gene : the Ile-325 variant has twice the half-life of the Thr-325 variant i.e. 15 minutes versus 7 minutes at 37°C [25,26].
It remains to be determined that at the site of a thrombus, steady state concentrations of CPU are achieved that are sufficient to completely shutdown fibrinolysis. Moreover, it is still not clear whether proCPU levels and proCPU genotype influence the process [24]. This hypothesis is plausible knowing that upon activation of platelets, proCPU can be secreted from α-granules boosting local proCPU concentrations at the site of the clot [27] and that (pro)CPU can be crosslinked to fibrin by factor XIIIa [28,29]. The latter can have an important clinical relevance because proCPU and IIa come in close proximity to each other at the surface of the thrombus, leading to efficient CPU generation [30]. Moreover, an interesting hypothesis is that the interaction of CPU with fibrin can lead to a stabilisation of the carboxypeptidase, increasing its antifibrinolytic potential dramatically [21–23]. The observation that “old” thrombi are plasminogen deficient and resistant to thrombolysis further suggests that this mechanism may occur in vivo [24].
CPU: a new drug target?
Drug design: the need for a crystal structure
Having a crystal structure at your disposal could be of great benefit for efficient rational drug design. However, due to the very low solubility of the enzyme, the glycosylation extent of the zymogen, and the pronounced thermal instability of the active enzyme, attempts to crystallize (pro)CPU remained unsuccessful. Mutagenesis studies of Knecht et al. [23] and Ceresa & coworkers [31,32] showed that limited mutagenesis in the protein-covering β-sheet 9 and α-helix 11 (residues 297–335) can dramatically increase the stability of CPU mostly with conserved wild-type characteristics. These mutants were a first step to understand the mechanism of inactivation of CPU and are potent tools to facilitate structural characterization. Most recently the crystal structure of proCPU was solved by Marx et al. [33]. ProCPU was expressed in a cell line lacking N-acetylglucosaminoyltransferase I yielding a proCPU recombinant form with homogenous N-linked glycans. This trick made it possible to grow properly diffracting crystals and to solve the proCPU structure [33]. The crystal structure explains the proCPU auto-regulation mechanism. Briefly, in the zymogen the activation peptide not only covers the active site but also stabilizes a dynamic segment of the enzyme moiety (residues 296–350). Proteolytic activation results in release of the activation peptide and concomitant increase in dynamic segment motility. The increased dynamics lead to conformational changes that disrupt the catalytic site [33]. Using this model we understand now that binding of reversible CPU inhibitors (see below) can stabilize CPU activity by reducing the mobility of the dynamic segment. The structural data of Marx were confirmed by two publications of another group [34,35].
CPU inhibitors: an overview
A specific inhibition of metallocarboxypeptidase activity can be achieved by agents such as o-phenantroline and EDTA that chelate the essential Zn ion in its active site [1,36,37]. CPU is also sensitive to dithiothreitol and 2-mercaptoethanol because of the presence of disulphide bridges in the active moiety [1,36,37]. The organic inhibitors 2-mercaptomethyl-3-guanidinoethylthiopropanoic acid (MERGETPA) and guanidinoethylmercaptosuccinic acid (GEMSA) are the most widely used inhibitors for both in vitro and in vivo studies with Ki values in the order of mM to µM [12]. However, both compounds have two major drawbacks from a drug discovery point of view. Firstly, both compounds are potent inhibitors of carboxypeptidase N (CPN). CPN is believed to have an important function in plasma as an inactivator of anaphylatoxins and in the processing of peptide hormones [38,39]. Consequently, selectivity towards CPN is an important parameter to consider during optimisation. Secondly, both compounds are extremely polar resulting in a low oral bioavailability [12]. Major efforts have been made to obtain more potent and selective drugs with favourable pharmacokinetic properties. An overview of synthetic CPU inhibitors is presented in Table 1. Although a high selectivity towards CPN is obtained by those inhibitors, obtaining selectivity towards the digestive basic carboxypeptidase, CPB, seems less straightforward [12 and references of Table 1]. However, little risk is anticipated for transient inhibition of pancreatic CPB since inhibition of this carboxypeptidase should not lead to intestinal malabsorption due to the presence of alternate active proteases available for digestion.
Table 1.
Small synthetic inhibitors and naturally occurring metallocarboxypeptidase inhibitors
| Compound | Structure | Inhibition constants |
Specificity vs. human CPN |
Specificity vs. human CPB |
Model | Ref |
|---|---|---|---|---|---|---|
| guanidinoethyl-mercaptosuccinic acid (GEMSA) | 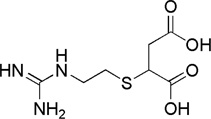 |
Ki = 10−6 – 10−4 | 0,025-fold | 0,011-fold | not available | 37,43, 44,45 |
| ε-amino caproic acid (ε-ACA) | 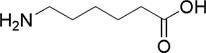 |
Ki = 10−3 – 10−2 | 0,8-fold | 0,9-fold | not available | 43,45, 46 |
| DL-2-mercapto methyl-3-guanidinoethyl-thiopropanoic acid (MERGETPA) | 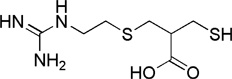 |
Ki = 10−7 – 10−6 | 0,009-fold | not available |
|
37,45, 47,48 |
| potato tuber carboxypeptidase inhibitor (PTCI) | 39 amino acid polypeptide | Ki= 10−10 – 10−9 | > 25 000-fold | 0,5-fold |
|
40,47, 49,50 |
| leech carboxypeptidase inhibitor (LCI) | 66 amino acid polypeptide | Ki= 10−9 | not available | not available | not available | 41 |
| tick carboxypeptidase inhibitor (TCI) | 75 amino acid cysteine-rich polypeptide | Ki= 10−9 | > 100 000-fold | 1,1-fold | not available | 42 |
| SAR-104772 | not available | not available | not available | not available | not available | 51 |
| Compound (−)-8 | 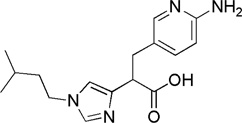 |
IC50 = 2 nM | > 25 000-fold | 1,35-fold | African green monkey model of vascular injury | 51–53 |
| UK-396,082 | 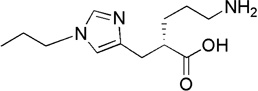 |
Ki = 10−8 – 10−9 | > 1000-fold | not available | rabbit model of venous thrombosis | 51,54 |
| Compound (−)-14 | 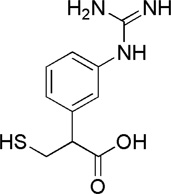 |
IC50 = 3 nM | 600-fold | 2-fold | rabbit model of venous thrombosis | 51,55 |
| AZD-9684 | not available | not available | not available | not available | phase II, single-blind, multicentre study on acute pulmonary embolism patients | 51, 56 |
| BX 528 | 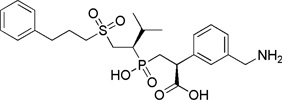 |
Ki = 10−9 | > 35 000-fold | 12-fold | four different animal models (rat, dog, rabbit) of thrombosis employing different thrombogenis stimuli (FeCl3, laser, ex vivo and batroxobin) in different tissues (artery, vein and lung microcirculation) | 51,57 |
| EF6265 | 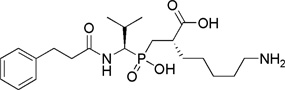 |
Ki = 10−8 | > 750-fold | not available | sepsis-induced organ dysfunction in rats | 51,60, 61 |
Apart from the synthetic inhibitors, protein inhibitors also have been described. The most familiar one is potato tuber carboxypeptidase inhibitor (PTCI), a 39 amino acid protein that competitively inhibits CPU with a Ki in the nM range [40]; others are the 66 residue leech carboxypeptidase inhibitor (LCI) found in Hirudo medicinalis [41] and the recently isolated tick carboxypeptidase inhibitor (TCI) from the soft tick Rhipicephalus bursa [42]. A major advantage of this kind of inhibitors is the high selectivity for CPU.
The promising results of CPU inhibitors have been somewhat overshadowed by the observation that in in vitro experiments several competitive inhibitors such as GEMSA and PTCI show a biphasic effect, prolonging clot lysis at low concentrations and enhancing lysis at high concentrations [44,62]. This can be explained by the notion that free and (inhibitor) bound CPU stay in equilibrium. While the free CPU inactivates irreversibly, the bound form, protected against spontaneous inactivation (due to decreased dynamic segment mobility), is released to replenish the free pool, so as to maintain the equilibrium [44,62]. As long as the free CPU concentration stays above the t-PA dependent threshold value, fibrinolysis will stay in its initial phase. Understandably this stabilising effect of CPU inhibitors has raised concerns, but one should keep in mind that in vitro observations are not always mirrored in the in vivo situations. Indeed, thus far in vivo data do not substantiate the in vitro observed paradoxical behaviour of competitive CPU inhibitors. However, this phenomenon needs to be further investigated, especially in in vivo animal models and in the clinic. Of note a minor or absent antifibrinolytic effect was observed at low concentrations of TCI and LCI [41,42]. These inhibitors show a 10-fold higher potency than PTCI. Moreover, their larger size (nearly twice of PTCI) and additional or more efficient contacts with the enzyme result in a higher stability of the enzyme inhibitor complex. This most probably results in the less apparent antifibrinolytic effects at low inhibitor concentrations [41,42]. With the available crystal structure, more efficient drug design will reveal potent non stabilising small synthetic CPU inhibitors in the near future.
A final pharmacological approach to target CPU is by hampering its generation from proCPU. Potential physiologic activators include thrombin, thrombin-thrombomodulin (T-TM) and plasmin. Several monoclonal antibodies have been described that inhibit the conversion of proCPU into CPU by T-Tm and plasmin [63]. Although activation of proCPU by thrombin or plasmin involves the same cleavage site, Arg92, Hillmayer et al. recently demonstrated that some of these antibodies can discriminate between plasmin or T/TM mediated activation, providing strong evidence that (i) those MA do not bind to the cleavage site and (ii) that different binding residues in proCPU exist for thrombin/plasmin [64]. Indeed, MA that inhibit exclusively the activation of proCPU by thrombin-thrombomodulin bind to Gly66 and MA that inhibits the activation by both T/TM and plasmin bind to Val41 [64]. Those MA are interesting tools to investigate physiological activation of proCPU in vivo. Recently mAbTAFI/TM#16, a MA antibody that interferes with the T/TM activation has been tested in vivo in a baboon model of E.coli induced sepsis [65]. In this model T-TM appeared to be the predominant activator of proCPU in vivo [65]. Of interest, those MA do not cross react with CPN or CPB theoretically favouring their therapeutic use, although one should consider the need for parenteral administration and their expensive nature.
Clinical utility of CPU inhibition or a role for CPU in vivo?
Data from knockout mice
Targeted gene disruption of the proCPU gene did not result in an overt phenotype [66]. ProCPU knockout mice are indistinguishable from their WT littermates concerning survival, development and fertility. Additionally, no major differences were revealed between KO and WT mice in hemostatic analyses including bleeding times following tail transaction [66].
Unexpectedly, Nagashima et al. did not observe significant differences between KO and WT mice in several thrombosis models including thrombin-induced thromboembolism, factor X coagulant protein-induced thromboembolism, endotoxin-induced thromboembolism, or time to vascular occlusion in phytochemical-induced vascular injury [66]. This raised questions about the in vivo significance of the proCPU/CPU system. However, by backcrossing proCPU deficient mice to a heterozygous plasminogen deficient background, a role for proCPU was demonstrated in models of pulmonary embolism and peritoneal inflammation indicating that proCPU can modulate the in vivo functions of plasmin(ogen) in fibrinolysis and cell migration [67]. Recently, new data on proCPU knock out mice with a normal plasminogen status have been published showing a significant in vivo role of CPU [68]. Wang et al. demonstrated that proCPU deficiency protects mice from 3.5% ferric chloride-induced vena cava thrombosis: a 34% reduction in thrombus weight occurred in proCPU−/− but not in proCPU+/− mice (3% reduction) compared to wild type littermates. However, in a more severe injury model (5% FeCl3) the difference in thrombus weight between proCPU−/− and proCPU+/+ mice was not significant anymore [68]. Mao et al. [69] demonstrated that, in vivo, in a batroxobin-induced pulmonary embolism model, proCPU knock out mice (with normal plasminogen status) displayed a lower retention of fibrin in the lungs than did WT littermates, thus linking proCPU deficiency with enhanced endogenous fibrinolysis
Improvement of endogenous fibrinolysis
Several studies have been conducted to evaluate whether a CPU inhibitor alone improves endogenous thrombolysis, albeit with contradictory results. The in vivo profibrinolytic efficiency of a CPU inhibitor alone depends on the type of thrombosis model and the studied animal species, the type of inhibitor and whether this inhibitor is administered before or after thrombus induction. Minnema et al. [70] showed that incorporation of anti-factor XI antibodies or PTCI in jugular vein thrombi resulted in both cases in an almost twofold increase in endogenous thrombolysis compared with controls, whereas inhibition of both factor XI and CPU activity had no additional effect. However, in two other studies, infusion of PTCI after thrombus formation in the isolated segment of the jugular vein [71], and in the abdominal aorta in rabbits [49], did not enhance endogenous thrombolysis. The latter studies suggest that administration of a CPU inhibitor alone is not efficient after thrombus formation. However, Muto et al. [47] and Suzuki et al. [61] demonstrated that tissue factor-induced microthrombi can be efficiently lysed by systemic injection of MERGETPA, PTCI or EF6265 alone even after formation of thrombi. Also Hashimoto & coworkers demonstrated enhancement of endogenous fibrinolysis on addition of PTCI or argatroban (a direct IIa inhibitor) after thrombus formation in an arterial thrombolysis model using rat mesenteric arterioles [72]. Interestingly, Wang et al. [50] elaborated on the importance of the magnitude of the thrombotic stimulus to see an overt effect of a CPU inhibitor alone. Indeed, PTCI, administered after thrombus formation, significantly inhibits murine thrombosis in the absence of exogenously administered t-PA using a model of ferric chloride (3.5 %)-induced vena cava thrombosis. However, in a more severe injury model (5% FeCl3) the effect of PTCI was not significant anymore. PTCI alone had also no effect on 3.5% FeCl3 –induced carotid artery thrombosis [50]. In a rat DIC model it has been demonstrated that inhibition of CPU by BX528 attenuated LPS induced resistance to endogenous fibrinolysis [58,59]. Most recently Muto et al. [60] showed in a rat endotoxemia and sepsis model that administration of EF6265 1 hour after the intravenous injection of lipopolysaccharide (LPS) and Pseudomonas aeruginosa, respectively, results in decreased deposition of fibrin in the kidney and liver without significant changes in platelet and fibrinogen concentrations. Moreover, this compound also significantly decreased levels of plasma lactate dehydrogenase and aspartate aminotransferase, markers of organ dysfunction [60].
Recently, a phase II, single-blind, multicentre study was presented investigating the effect of the novel CPU inhibitor AZD9684 in pulmonary embolism [56]. Fifty-eight patients with confirmed PE were randomized to receive AZD9684 or placebo, on top of once-daily dalteparin for 5–7 days. In the patient group receiving AZD9684, fibrinolysis biomarkers in plasma were higher and sustained for a longer period of time, implying that inhibition of CPU by AZD9684 stimulates endogenous fibrinolysis. Moreover, lung deficiency scintigraphy scores improved over the treatment period. In addition, no difference in the occurrence of adverse effects was seen between both treatment groups.
In conclusion, to date there is still a high need for more in vivo data to draw more definite conclusions about the efficiency of the administration of a CPU inhibitor alone in venous and arterial thrombosis models.
Adjuvants for thrombolytic therapy
Thrombolysis consists of the pharmacological dissolution of a blood clot by intravenous infusion of plasminogen activators that activate the fibrinolytic system. The clinical benefits of thrombolytic therapy in patients with acute myocardial infarction and ischemic stroke are well documented. However, all available thrombolytic agents still have significant shortcomings, including the need for large therapeutic doses, limited fibrin specificity and, most importantly, significant associated bleeding tendency and reocclusion [73]. For example, treatment of acute myocardial infarction with thrombolytic therapy is associated with a failure to lyse the clot in 15 to 50% of coronary thrombi, 10–25% of successfully recanalised vessels re-occlude and up to 2% of treated patients suffer from major bleeding complications [74,75].
Tremendous benefit could be expected from adjunctive therapy that potentiates the t-PA mediated thrombolytic effect and that consequently reduces the dose of plasminogen activators, hereby potentially reducing unfavourable side effects.
It is well known that thrombolytic therapy by t-PA or other thrombolytic agents induces a local procoagulant state. The observation of increased plasma fibrinopeptide A following such therapy is consistent with release of active IIa in the vasculature, and could help explain the observations of rethrombosis following successful thrombolysis [30,76]. Moreover, based on studies in both experimental animals and patients, it appears that IIa inhibitors accelerate thrombolysis and improve clinical outcome [76,77].
Mattsson et al. [78] demonstrated local in vivo proCPU activation in the coronaries during thrombolytic treatment in a dog model of coronary artery thrombosis. This CPU generation could be inhibited with a direct, small molecule IIa inhibitor (melagatran). This suggests that the profibrinolytic effects of direct IIa inhibitors may, at least partly, be due to an inhibition of IIa mediated activation of proCPU [78]. Indeed, the shortening in lysis time when rt-PA was combined with melagatran is comparable to the reduced lysis time seen on addition of MERGETPA to rt-PA in the same model [48]. We recently showed a similar CPU generation in patients with acute ischemic stroke being treated with intra-venous rt-PA or local intra-arterial urokinase administration [79] and showed that this CPU generation decreases the efficacy of thrombolytic therapy in these patients [80]. Indeed, the peak CPU activity during thrombolysis was associated with the evolution of the clinical deficit and the achieved recanalisation and the amount of proCPU consumption during thrombolysis was related to the risk of intracranial hemorrhage, mortality and final infarct volume [80]. Although these data need confirmation in larger studies it is the first step towards investigating the utility of CPU inhibitors as adjuvant treatment with tPA or urokinase in acute ischemic stroke in humans.
In recent years consistent data have been obtained in animal studies on the utility of CPU inhibitors as adjuvant therapy with thrombolytics. Klement et al. investigated the impact of a CPU inhibitor (PTCI) on t-PA induced clot lysis in a rabbit model of arterial thrombolysis [49]. They found that when the inhibitor was administered along with t-PA, numerous parameters associated with the efficiency of thrombolysis were enhanced. The time to reperfusion was reduced by an approximate factor of three. Vessel patency was similarly improved [49]. Similar studies were carried out by Nagashima et al. [71] in a rabbit model of venous thrombolysis, where the impact of PTCI on t-PA mediated clot lysis was determined. When PTCI was administered along with t-PA, the clot weight was reduced to approximately half of the control weight. t-PA alone at the same dose reduced clot weight only 26%. This combined effect of t-PA and PTCI could be obtained with three times the dose of t-PA in the absence of PTCI. Recently Wang et al. [58,59] demonstrated that administration of BX 528 enhances the thrombolytic effects of low dose t-PA in a dog model of femoral artery thrombosis induced by 10 % FeCl3 injury, a rat model of femoral artery thrombosis induced by photochemical injury and a rabbit ex vivo model of jugular vein thrombolysis.
Safety of CPU inhibition
One of the key concerns in antithrombotic therapy is the bleeding risk. Will the pharmacological inhibition of CPU result in an increased bleeding risk? It has been demonstrated that proCPU deficiency did not cause spontaneous bleeding or alterations in the bleeding time [66]. Moreover, the use of a CPU inhibitor in many in vivo animal models was not associated with an increased bleeding risk either when the inhibitor was used alone or in association with a low dose t-PA. There is only one study that shows a bleeding liability for PTCI [50]. The average bleeding time prolongation (mouse tail bleeding model) achieved at the top dose of PTCI was similar to that of aspirin (30 mg/kg); however none of the mice dosed with PTCI showed off-scale bleeding. In contrast, the 30 mg/kg dose of aspirin resulted in 36% of the mice-bleeding off-scale [50].
Important to realise is that besides its antifibrinolytic function, carboxypeptidase U may also play a role in inflammation [81,82]. Indeed, bradykinin and the complement factors C3a and C5a are well known CPU substrates [83–85] and recent studies show that proCPU plays a role in regulating complement-mediated vascular inflammation. It remains to be investigated whether CPU inhibitors can be used as profibrinolytic drugs in pathological conditions like atherosclerosis and arterial thrombosis where inflammation plays a pivotal role. A recent case control study reported that low plasma levels of proCPU are associated with increased risk of a first myocardial infarction [86]. These data, however, should be confirmed in a prospective study design where it should be investigated whether the low proCPU levels are causally linked to an increased risk of myocardial infarction or whether they are provoked by an increased activation to CPU due to a hypercoagulative state in these patients.
In the past years interesting animal studies have been published about the role of CPU in inflammation. It has been demonstrated that proCPU is a positive acute phase protein in mice as administration of bacterial lipopolysaccharide to these animals resulted in marked increase in hepatic CPB2 mRNA abundance and plasma proCPU protein concentrations [87]. However when interpreting these data one should keep in mind the important differences in the sequences of the proCPU promoters from human and mouse. Indeed unlike the mouse promoter, the human promoter is not induced by inflammatory cytokines [10]. In line with this observation, proCPU levels decreased significantly upon administration of low dose LPS to human volunteers whereas CRP increased more than 100-fold [88]. Also low proCPU plasma levels have been reported in patients with sepsis [89].
ProCPU knockout mice were protected from liver necrosis after intra peritoneal injection with Escherichia coli [90] and in proCPU/plasminogen double knock-out mice, the migration of leucocytes towards the peritoneum was increased in the proCPU-deficient animal models compared to the wild-types showing the importance of proCPU in plasminogen-dependent cell migration in vivo [83]. Moreover, it was shown that proCPU deficient mice have a wound-healing problem which may be related to this cell migration process [91]. These in vivo data suggest a role for CPU in inflammation, but at the same time call for further investigation.
Conclusions
Twenty years after its discovery, CPU has turned into a hot topic in the field of thrombosis and haemostasis. This carboxypeptidase is currently recognized as an important molecular link between coagulation and fibrinolysis, and selective CPU inhibitors have been designed and tested in several thrombosis models. Animal studies show improved endogenous fibrinolysis and an increased efficiency of t-PA mediated thrombolysis upon inhibition of the (pro)CPU system. Recently, a phase II, single-blinded, multicentre study in patients with pulmonary embolism also showed enhancement of endogenous fibrinolysis upon administration of a selective CPU inhibitor. These intriguing in vivo data call for further evaluation in humans.
References
- 1.Hendriks D, Scharpé S, van Sande M, Lommaert MP. Characterisation of a carboxypeptidase in human serum distinct from carboxypeptidase N. J Clin Chem Clin Biochem. 1989;27:277–285. doi: 10.1515/cclm.1989.27.5.277. [DOI] [PubMed] [Google Scholar]
- 2.Bajzar L, Manuel R, Nesheim ME. Purification and characterization of TAFI, a thrombin-activable fibrinolysis inhibitor. J Biol Chem. 1995;270:14477–14484. doi: 10.1074/jbc.270.24.14477. [DOI] [PubMed] [Google Scholar]
- 3.Nesheim M, Bajzar L. The discovery of TAFI. J Thromb Haemost. 2005;3:2139–2146. doi: 10.1111/j.1538-7836.2005.01280.x. [DOI] [PubMed] [Google Scholar]
- 4.Bertina RM, van Tilburg NH, Haverkate F, Bouma BN, von dem Borne PA, Meijers JC, Campbell W, Eaton D, Hendriks DF, Willemse JL. The discovery of thrombin activatable fibrinolysis inhibitor. J Thromb Haemost. 2006;4:256–257. doi: 10.1111/j.1538-7836.2005.01666.x. [DOI] [PubMed] [Google Scholar]
- 5.Sakharov DV, Plow EF, Rijken DC. On the mechanism of the antifibrinolytic activity of plasma carboxypeptidase B. J Biol Chem. 1997;272:14477–14482. doi: 10.1074/jbc.272.22.14477. [DOI] [PubMed] [Google Scholar]
- 6.Valnickova Z, Thøgersen IB, Potempa J, Enghild JJ. The intrinsic enzymatic activity of procarboxypeptidase U (TAFI) does not significantly influence the fibrinolytic rate: reply to a rebuttal. J Thromb Haemost. 2007;5:1336–1337. doi: 10.1111/j.1538-7836.2007.02593.x. [DOI] [PubMed] [Google Scholar]
- 7.Willemse JL, Heylen E, Hendriks DF. The intrinsic enzymatic activity of procarboxypeptidase U (TAFI) does not significantly influence the fibrinolysis rate: a rebuttal. J Thromb Haemost. 2007;5:1334–1336. doi: 10.1111/j.1538-7836.2007.02539.x. [DOI] [PubMed] [Google Scholar]
- 8.Foley JH, Kim P, Nesheim ME. Thrombin-activable fibrinolysis inhibitor zymogen does not play a significant role in the attenuation of fibrinolysis. J Biol Chem. 2008;283:8863–8867. doi: 10.1074/jbc.M800127200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willemse JL, Hendriks DF. A role for procarboxypepidase U (TAFI) in thrombosis. Front Biosci. 2007;12:1973–1987. doi: 10.2741/2203. [DOI] [PubMed] [Google Scholar]
- 10.Boffa MB, Koschinsky ML. Curiouser and curiouser: recent advances in measurement of thrombin-activatable fibrinolysis inhibitor (TAFI) and in understanding its molecular genetics, gene regulation, and biological roles. Clin Biochem. 2007;40:431–442. doi: 10.1016/j.clinbiochem.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 11.Mosnier LO, Bouma BN. Regulation of fibrinolysis by thrombin activatable fibrinolysis inhibitor, an unstable carboxypeptidase B that unites the pathways of coagulation and fibrinolysis. Arterioscler Thromb Vasc Biol. 2006;26:2445–2453. doi: 10.1161/01.ATV.0000244680.14653.9a. [DOI] [PubMed] [Google Scholar]
- 12.Leurs J, Hendriks D. Carboxypeptidase U (TAFIa): a metallocarboxypeptidase with a distinct role in haemostasis and a possible risk factor for thrombotic disease. Thromb Haemost. 2005;94:471–487. doi: 10.1160/TH04-07-0454. [DOI] [PubMed] [Google Scholar]
- 13.Willemse JL, Hendriks DF. Measurement of procarboxypeptidase U (TAFI) in human plasma: a laboratory challenge. Clin Chem. 2006;52:30–36. doi: 10.1373/clinchem.2005.055814. [DOI] [PubMed] [Google Scholar]
- 14.Wang W, Boffa MB, Bajzar L, Walker JB, Nesheim ME. A study of the mechanism of inhibition of fibrinolysis by activated thrombin-activable fibrinolysis inhibitor. J Biol Chem. 1998;273:27176–27181. doi: 10.1074/jbc.273.42.27176. [DOI] [PubMed] [Google Scholar]
- 15.Medved L, Nieuwenhuizen W. Molecular mechanisms of initiation of fibrinolysis by fibrin. Thromb Haemost. 2003;89:409–419. [PubMed] [Google Scholar]
- 16.Fleury V, Anglés-Cano E. Characterization of the binding of plasminogen to fibrin surfaces: the role of carboxy-terminal lysines. Biochemistry. 1991;30:7630–7638. doi: 10.1021/bi00244a035. [DOI] [PubMed] [Google Scholar]
- 17.Pannell R, Black J, Gurewich V. Complementary modes of action of tissue-type plasminogen activator and pro-urokinase by which their synergistic effect on clot lysis may be explained. J Clin Invest. 1988;81:853–859. doi: 10.1172/JCI113394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J Biol Chem. 1982;257:2912–2919. [PubMed] [Google Scholar]
- 19.Schneider M, Nesheim M. A study of the protection of plasmin from antiplasmin inhibition within an intact fibrin clot during the course of clot lysis. J Biol Chem. 2004;279:13333–13339. doi: 10.1074/jbc.M313164200. [DOI] [PubMed] [Google Scholar]
- 20.Schneider M, Brufatto N, Neill E, Nesheim M. Activated thrombin-activatable fibrinolysis inhibitor reduces the ability of high molecular weight fibrin degradation products to protect plasmin from antiplasmin. J Biol Chem. 2004;279:13340–13345. doi: 10.1074/jbc.M313211200. [DOI] [PubMed] [Google Scholar]
- 21.Leurs J, Nerme V, Sim Y, Hendriks D. Carboxypeptidase U (TAFIa) prevents lysis from proceeding into the propagation phase through a threshold-dependent mechanism. J Thromb Haemost. 2004;2:416–423. doi: 10.1111/j.1538-7836.2004.00605.x. [DOI] [PubMed] [Google Scholar]
- 22.Walker JB, Bajzar L. The intrinsic threshold of the fibrinolytic system is modulated by basic carboxypeptidases, but the magnitude of the antifibrinolytic effect of activated thrombin-activable fibrinolysis inhibitor is masked by its instability. J Biol Chem. 2004;279:27896–27904. doi: 10.1074/jbc.M401027200. [DOI] [PubMed] [Google Scholar]
- 23.Knecht W, Willemse J, Stenhamre H, Andersson M, Berntsson P, Furebring C, Harrysson A, Hager AC, Wissing BM, Hendriks D, Cronet P. Limited mutagenesis increases the stability of human carboxypeptidase U (TAFIa) and demonstrates the importance of CPU stability over proCPU concentration in down-regulating fibrinolysis. FEBS J. 2006;273:778–792. doi: 10.1111/j.1742-4658.2006.05110.x. [DOI] [PubMed] [Google Scholar]
- 24.Walker JB, Bajzar L. Complete inhibition of fibrinolysis by sustained carboxypeptidase B activity: the role and requirement of plasmin inhibitors. J Thromb Haemost. 2007;5:1257–1264. doi: 10.1111/j.1538-7836.2007.02541.x. [DOI] [PubMed] [Google Scholar]
- 25.Brouwers GJ, Vos HL, Leebeek FW, Bulk S, Schneider M, Boffa M, Koschinsky M, van Tilburg NH, Nesheim ME, Bertina RM, Gómez Garcia EB. A novel, possibly functional, single nucleotide polymorphism in the coding region of the thrombin-activatable fibrinolysis inhibitor (TAFI) gene is also associated with TAFI levels. Blood. 2001;98:1992–1993. doi: 10.1182/blood.v98.6.1992. [DOI] [PubMed] [Google Scholar]
- 26.Schneider M, Boffa M, Stewart R, Rahman M, Koschinsky M, Nesheim M. Two naturally occurring variants of TAFI (Thr-325 and Ile-325) differ substantially with respect to thermal stability and antifibrinolytic activity of the enzyme. J Biol Chem. 2002;277:1021–1030. doi: 10.1074/jbc.M104444200. [DOI] [PubMed] [Google Scholar]
- 27.Mosnier LO, Buijtenhuijs P, Marx PF, Meijers JC, Bouma BN. Identification of thrombin activatable fibrinolysis inhibitor (TAFI) in human platelets. Blood. 2003;101:4844–4846. doi: 10.1182/blood-2002-09-2944. [DOI] [PubMed] [Google Scholar]
- 28.Valnickova Z, Enghild JJ. Human procarboxypeptidase U, or thrombin-activable fibrinolysis inhibitor, is a substrate for transglutaminases. Evidence for transglutaminase-catalyzed cross-linking to fibrin. J Biol Chem. 1998;273:27220–27224. doi: 10.1074/jbc.273.42.27220. [DOI] [PubMed] [Google Scholar]
- 29.Björquist P, Andréasson AC, Boberg BM, Johansson T, Sarich TC, Strömqvist M, Jönsson AC. Immunihistochemical localization of carboxypeptidase U in human thrombi. Fibrinolysis & Proteolysis. 2008;14 supplement 1:O–88. [Google Scholar]
- 30.Mutch NJ, Robbie LA, Booth NA. Human thrombi contain an abundance of active thrombin. Thromb Haemost. 200;86:1028–1034. [PubMed] [Google Scholar]
- 31.Ceresa E, Van de Borne K, Peeters M, Lijnen HR, Declerck PJ, Gils A. Generation of a stable activated thrombin activable fibrinolysis inhibitor variant. J Biol Chem. 2006;281:15878–15883. doi: 10.1074/jbc.M509839200. [DOI] [PubMed] [Google Scholar]
- 32.Ceresa E, Peeters M, Declerck PJ, Gils A. Announcing a TAFIa mutant with a 180-fold increased half-life and concomitantly a strongly increased antifibrinolytic potential. J Thromb Haemost. 2007;5:418–420. doi: 10.1111/j.1538-7836.2007.02322.x. [DOI] [PubMed] [Google Scholar]
- 33.Marx PF, Brondijk TH, Plug T, Romijn RA, Hemrika W, Meijers JC, Huizinga EG. Crystal structures of TAFI elucidate the inactivation mechanism of activated TAFI: a novel mechanism for enzyme autoregulation. Blood. 2008;112:2803–2809. doi: 10.1182/blood-2008-03-146001. [DOI] [PubMed] [Google Scholar]
- 34.Sanglas L, Valnickova Z, Arolas JL, Pallarés I, Guevara T, Solà M, Kristensen T, Enghild JJ, Aviles FX, Gomis-Rüth FX. Structure of activated thrombin-activatable fibrinolysis inhibitor, a molecular link between coagulation and fibrinolysis. Mol Cell. 2008;31:598–606. doi: 10.1016/j.molcel.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 35.Anand K, Pallares I, Valnickova Z, Christensen T, Vendrell J, Wendt KU, Schreuder HA, Enghild JJ, Avilés FX. The crystal structure of thrombin-activable fibrinolysis inhibitor (TAFI) provides the structural basis for its intrinsic activity and the short half-life of TAFIa. J Biol Chem. 2008;283:29416–29423. doi: 10.1074/jbc.M804003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eaton DL, Malloy BE, Tsai SP, Henzel W, Drayna D. Isolation, molecular cloning, and partial characterization of a novel carboxypeptidase B from human plasma. J Biol Chem. 1991;266:21833–21838. [PubMed] [Google Scholar]
- 37.Wang W, Hendriks D, Scharpé S. Carboxypeptidase U, a plasma carboxypeptidase with high affinity for plasminogen. J Biol Chem. 1994;269:15937–15944. [PubMed] [Google Scholar]
- 38.Matthews KW, Mueller-Ortiz SL, Wetsel RA. Carboxypeptidase N: a pleiotropic regulator of inflammation. Mol Immunol. 2004;40:785–793. doi: 10.1016/j.molimm.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Willemse JL, Chen D, Hendriks DF. Major carboxypeptidase N deficiency. Clin Chim Acta. 2008;389:181–182. doi: 10.1016/j.cca.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 40.Redlitz A, Tan AK, Eaton DL, Plow EF. Plasma carboxypeptidases as regulators of the plasminogen system. J Clin Invest. 1995;96:2534–2538. doi: 10.1172/JCI118315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reverter D, Vendrell J, Canals F, Horstmann J, Avilés FX, Fritz H, Sommerhoff CP. A carboxypeptidase inhibitor from the medical leech Hirudo medicinalis. Isolation, sequence analysis, cDNA cloning, recombinant expression, and characterization. J Biol Chem. 1998;273:32927–32933. doi: 10.1074/jbc.273.49.32927. [DOI] [PubMed] [Google Scholar]
- 42.Arolas JL, Lorenzo J, Rovira A, Castellà J, Aviles FX, Sommerhoff CP. A carboxypeptidase inhibitor from the tick Rhipicephalus bursa: isolation, cDNA cloning, recombinant expression, and characterization. J Biol Chem. 2005;280:3441–3448. doi: 10.1074/jbc.M411086200. [DOI] [PubMed] [Google Scholar]
- 43.Boffa MB, Wang W, Bajzar L, Nesheim ME. Plasma and recombinant thrombin-activable fibrinolysis inhibitor (TAFI) and activated TAFI compared with respect to glycosylation, thrombin/thrombomodulin-dependent activation, thermal stability, and enzymatic properties. J Biol Chem. 1998;273:2127–2135. doi: 10.1074/jbc.273.4.2127. [DOI] [PubMed] [Google Scholar]
- 44.Walker JB, Hughes B, James I, Haddock P, Kluft C, Bajzar L. Stabilization versus inhibition of TAFIa by competitive inhibitors in vitro. J Biol Chem. 2003;278:8913–8921. doi: 10.1074/jbc.m205006200. [DOI] [PubMed] [Google Scholar]
- 45.Mao SS, Colussi D, Bailey CM, Bosserman M, Burlein C, Gardell SJ, Carroll SS. Electrochemiluminescence assay for basic carboxypeptidases: inhibition of basic carboxypeptidases and activation of thrombin-activatable fibrinolysis inhibitor. Anal Biochem. 2003;319:159-70µ. doi: 10.1016/s0003-2697(03)00252-5. [DOI] [PubMed] [Google Scholar]
- 46.Tan AK, Eaton DL. Activation and characterization of procarboxypeptidase B from human plasma. Biochemistry. 1995;34:5811–5816. doi: 10.1021/bi00017a012. [DOI] [PubMed] [Google Scholar]
- 47.Muto Y, Suzuki K, Sato E, Ishii H. Carboxypeptidase B inhibitors reduce tissue factor-induced renal microthrombi in rats. Eur J Pharmacol. 2003;461:181–189. doi: 10.1016/s0014-2999(03)01297-4. [DOI] [PubMed] [Google Scholar]
- 48.Björkman JA, Abrahamsson TI, Nerme VK, Mattsson CJ. Inhibition of carboxypeptidase U (TAFIa) activity improves rt-PA induced thrombolysis in a dog model of coronary artery thrombosis. Thromb Res. 2005;116:519–524. doi: 10.1016/j.thromres.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 49.Klement P, Liao P, Bajzar L. A novel approach to arterial thrombolysis. Blood. 1999;94:2735–2743. [PubMed] [Google Scholar]
- 50.Wang X, Smith PL, Hsu MY, Ogletree ML, Schumacher WA. Murine model of ferric chloride-induced vena cava thrombosis: evidence for effect of potato carboxypeptidase inhibitor. J Thromb Haemost. 2006;4:403–410. doi: 10.1111/j.1538-7836.2006.01703.x. [DOI] [PubMed] [Google Scholar]
- 51.Bunnage ME, Owen DR. TAFIa inhibitors in the treatment of thrombosis. Curr Opin Drug Discov Devel. 2008;11:480–486. [PubMed] [Google Scholar]
- 52.Barrow JC, Nantermet PG, Stauffer SR, Ngo PL, Steinbeiser MA, Mao SS, Carroll SS, Bailey C, Colussi D, Bosserman M, Burlein C, Cook JJ, Sitko G, Tiller PR, Miller-Stein CM, Rose M, McMasters DR, Vacca JP, Selnick HG. Synthesis and evaluation of imidazole acetic acid inhibitors of activated thrombin-activatable fibrinolysis inhibitor as novel antithrombotics. J Med Chem. 2003;46:5294–5297. doi: 10.1021/jm034141y. [DOI] [PubMed] [Google Scholar]
- 53.Nantermet PG, Barrow JC, Lindsley SR, Young M, Mao SS, Carroll S, Bailey C, Bosserman M, Colussi D, McMasters DR, Vacca JP, Selnick HG. Imidazole acetic acid TAFIa inhibitors: SAR studies centered around the basic P(1)(') group. Bioorg Med Chem Lett. 2004;14:2141–2145. doi: 10.1016/j.bmcl.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 54.Bunnage ME, Blagg J, Steele J, Owen DR, Allerton C, McElroy AB, Miller D, Ringer T, Butcher K, Beaumont K, Evans K, Gray AJ, Holland SJ, Feeder N, Moore RS, Brown DG. Discovery of potent & selective inhibitors of activated thrombin-activatable fibrinolysis inhibitor for the treatment of thrombosis. J Med Chem. 2007;50:6095–6103. doi: 10.1021/jm0702433. [DOI] [PubMed] [Google Scholar]
- 55.Islam I, Bryant J, May K, Mohan R, Yuan S, Kent L, Morser J, Zhao L, Vergona R, White K, Adler M, Whitlow M, Buckman BO. 3-Mercaptopropionic acids as efficacious inhibitors of activated thrombin activatable fibrinolysis inhibitor (TAFIa) Bioorg Med Chem Lett. 2007;17:1349–1354. doi: 10.1016/j.bmcl.2006.11.078. [DOI] [PubMed] [Google Scholar]
- 56.Eriksson H, Jensen E, Sandset PM, Wall U, Andersson M, Nerme V, Eriksson-Lepkowska M, Wåhlander K. CPU inhibition with AZD9684: profibrinolytic effects in acute patients. J Thromb Haemost. 2007;5 Supplement 2:P-S-367. [Google Scholar]
- 57.Adler M, Buckman B, Bryant J, Chang Z, Chu K, Emayan K, Hrvatin P, Islam I, Morser J, Sukovich D, West C, Yuan S, Whitlow M. Structures of potent selective peptide mimetics bound to carboxypeptidase B. Acta Crystallogr D Biol Crystallogr. 2008;64:149–157. doi: 10.1107/S0907444907057228. [DOI] [PubMed] [Google Scholar]
- 58.Wang YX, Zhao L, Nagashima M, Vincelette J, Sukovich D, Li W, Subramanyam B, Yuan S, Emayan K, Islam I, Hrvatin P, Bryant J, Light DR, Vergona R, Morser J, Buckman BO. A novel inhibitor of activated thrombin-activatable fibrinolysis inhibitor (TAFIa) - part I: pharmacological characterization. Thromb Haemost. 2007;97:45–53. [PubMed] [Google Scholar]
- 59.Wang YX, da Cunha V, Vincelette J, Zhao L, Nagashima M, Kawai K, Yuan S, Emayan K, Islam I, Hosoya J, Sullivan ME, Dole WP, Morser J, Buckman BO, Vergona R. A novel inhibitor of activated thrombin activatable fibrinolysis inhibitor (TAFIa) - part II: enhancement of both exogenous and endogenous fibrinolysis in animal models of thrombosis. Thromb Haemost. 2007;97:54–61. [PubMed] [Google Scholar]
- 60.Muto Y, Suzuki K, Iida H, Sakakibara S, Kato E, Itoh F, Kakui N, Ishii H. EF6265, a novel inhibitor of activated thrombin-activatable fibrinolysis inhibitor, protects against sepsis-induced organ dysfunction in rats. Crit Care Med. 2009;37:1744–1749. doi: 10.1097/CCM.0b013e31819ffc14. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki K, Muto Y, Fushihara K, Kanemoto K, Iida H, Sato E, Kikuchi C, Matsushima T, Kato E, Nomoto M, Yoshioka S, Ishii H. Enhancement of fibrinolysis by EF6265 [(S)-7-amino-2-[[[(R)-2-methyl-1-(3-phenylpropanoylamino)propyl]hydroxyphosphinoyl] methyl]heptanoic acid], a specific inhibitor of plasma carboxypeptidase B. J Pharmacol Exp Ther. 2004;309:607–615. doi: 10.1124/jpet.103.062729. [DOI] [PubMed] [Google Scholar]
- 62.Schneider M, Nesheim M. Reversible inhibitors of TAFIa can both promote and inhibit fibrinolysis. J Thromb Haemost. 2003;1:147–154. doi: 10.1046/j.1538-7836.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 63.Gils A, Ceresa E, Macovei AM, Marx PF, Peeters M, Compernolle G, Declerck PJ. Modulation of TAFI function through different pathways--implications for the development of TAFI inhibitors. J Thromb Haemost. 2005;3:2745–2753. doi: 10.1111/j.1538-7836.2005.01629.x. [DOI] [PubMed] [Google Scholar]
- 64.Hillmayer K, Vancraenenbroeck R, De Maeyer M, Compernolle G, Declerck PJ, Gils A. Discovery of novel mechanisms and molecular targets for the inhibition of activated thrombin activatable fibrinolysis inhibitor. J Thromb Haemost. 2008;6:1892–1899. doi: 10.1111/j.1538-7836.2008.03130.x. [DOI] [PubMed] [Google Scholar]
- 65.Binette TM, Taylor FB, Jr, Peer G, Bajzar L. Thrombin-thrombomodulin connects coagulation and fibrinolysis: more than an in vitro phenomenon. Blood. 2007;110:3168–3175. doi: 10.1182/blood-2007-03-078824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagashima M, Yin ZF, Zhao L, White K, Zhu Y, Lasky N, Halks-Miller M, Broze GJ, Jr, Fay WP, Morser J. Thrombin-activatable fibrinolysis inhibitor (TAFI) deficiency is compatible with murine life. J Clin Invest. 2002;109:101–110. doi: 10.1172/JCI12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swaisgood CM, Schmitt D, Eaton D, Plow EF. In vivo regulation of plasminogen function by plasma carboxypeptidase B. J Clin Invest. 2002;110:1275–1282. doi: 10.1172/JCI15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X, Smith PL, Hsu MY, Tamasi JA, Bird E, Schumacher WA. Deficiency in thrombin-activatable fibrinolysis inhibitor (TAFI) protected mice from ferric chloride-induced vena cava thrombosis. J Thromb Thrombolysis. 2007;23:41–49. doi: 10.1007/s11239-006-9009-4. [DOI] [PubMed] [Google Scholar]
- 69.Mao SS, Holahan MA, Bailey C, Wu G, Colussi D, Carroll SS, Cook JJ. Demonstration of enhanced endogenous fibrinolysis in thrombin activatable fibrinolysis inhibitor-deficient mice. Blood Coagul Fibrinolysis. 2005;16:407–415. doi: 10.1097/01.mbc.0000181175.62437.2a. [DOI] [PubMed] [Google Scholar]
- 70.Minnema MC, Friederich PW, Levi M, von dem Borne PA, Mosnier LO, Meijers JC, Biemond BJ, Hack CE, Bouma BN, ten Cate H. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J Clin Invest. 1998;101:10–14. doi: 10.1172/JCI781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nagashima M, Werner M, Wang M, Zhao L, Light DR, Pagila R, Morser J, Verhallen P. An inhibitor of activated thrombin-activatable fibrinolysis inhibitor potentiates tissue-type plasminogen activator-induced thrombolysis in a rabbit jugular vein thrombolysis model. Thromb Res. 200;98:333–342. doi: 10.1016/s0049-3848(00)00184-5. [DOI] [PubMed] [Google Scholar]
- 72.Hashimoto M, Yamashita T, Oiwa K, Watanabe S, Giddings JC, Yamamoto J. Enhancement of endogenous plasminogen activator-induced thrombolysis by argatroban and APC and its control by TAFI, measured in an arterial thrombolysis model in vivo using rat mesenteric arterioles. Thromb Haemost. 2002;87:110–113. [PubMed] [Google Scholar]
- 73.Collen D, Lijnen HR. Thrombosis fundamental and clinical aspects chapter 30. Leuven University Press; 2003. Novel thrombolytic agents for treatment of acute myocardial infarction; pp. 585–596. [Google Scholar]
- 74.Rapold HJ. Aspirin, heparin and new antithrombotic drugs as adjuncts to thrombolytic therapy for acute myocardial infarction. In: Beckers RC, editor. Textbook of coronary thrombosis and thrombolysis. Massachusetts: Kluwer academic Publishers; 1997. pp. 357–374. [Google Scholar]
- 75.Gurwitz JH, Gore JM, Goldberg RJ, Barron HV, Breen T, Rundle AC, Sloan MA, French W, Rogers WJ. Risk for intracranial hemorrhage after tissue plasminogen activator treatment for acute myocardial infarction. Participants in the National Registry of Myocardial Infarction 2. Ann Intern Med. 1998;129:597–604. doi: 10.7326/0003-4819-129-8-199810150-00002. [DOI] [PubMed] [Google Scholar]
- 76.Latacha MP, Schaiff WT, Eisenberg PR, Abendschein DR. Factor XII-dependent increases in thrombin activity induce carboxypeptidase-mediated attenuation of pharmacological fibrinolysis. J Thromb Haemost. 2004;2:128–134. doi: 10.1111/j.1538-7836.2004.00538.x. [DOI] [PubMed] [Google Scholar]
- 77.Mattsson C, Björkman JA, Ulvinge JC. Melagatran, hirudin and heparin as adjuncts to tissue-type plasminogen activator in a canine model of coronary artery thrombolysis. Fibrinolysis and Proteolysis. 1997;11:121–128. [Google Scholar]
- 78.Mattsson C, Björkman JA, Abrahamsson T, Nerme V, Schatteman K, Leurs J, Scharpé S, Hendriks D. Local proCPU (TAFI) activation during thrombolytic treatment in a dog model of coronary artery thrombosis can be inhibited with a direct, small molecule thrombin inhibitor (melagatran) Thromb Haemost. 2002;87:557–562. [PubMed] [Google Scholar]
- 79.Willemse JL, Brouns R, Heylen E, De Deyn PP, Hendriks DF. Carboxypeptidase U (TAFIa) activity is induced in vivo in ischemic stroke patients receiving thrombolytic therapy. J Thromb Haemost. 2008;6:200–202. doi: 10.1111/j.1538-7836.2007.02798.x. [DOI] [PubMed] [Google Scholar]
- 80.Brouns R, Heylen E, Sheorajpanday R, Willemse JL, Kunnen J, De Surgeloose D, Hendriks DF, De Deyn PP. Carboxypeptidase U (TAFIa) decreases the efficacy of thrombolytic therapy in ischemic stroke patients. Clin Neurol Neurosurg. 2009;111:165–170. doi: 10.1016/j.clineuro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 81.Bajzar L, Jain N, Wang P, Walker JB. Thrombin activatable fibrinolysis inhibitor: not just an inhibitor of fibrinolysis. Crit Care Med. 2004;32:S320–S324. doi: 10.1097/01.ccm.0000126361.00450.b1. [DOI] [PubMed] [Google Scholar]
- 82.Leung LL, Myles T, Nishimura T, Song JJ, Robinson WH. Regulation of tissue inflammation by thrombin-activatable carboxypeptidase B (or TAFI) Mol Immunol. 2008;45:4080–4083. doi: 10.1016/j.molimm.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shinohara T, Sakurada C, Suzuki T, Takeuchi O, Campbell W, Ikeda S, Okada N, Okada H. Pro-carboxypeptidase R cleaves bradykinin following activation. Int Arch Allergy Immunol. 1994;103:400–404. doi: 10.1159/000236661. [DOI] [PubMed] [Google Scholar]
- 84.Campbell WD, Lazoura E, Okada N, Okada H. Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol Immunol. 2002;46:131–134. doi: 10.1111/j.1348-0421.2002.tb02669.x. [DOI] [PubMed] [Google Scholar]
- 85.Nishimura T, Myles T, Piliponsky AM, Kao PN, Berry GJ, Leung LL. Thrombin activatable procarboxypeptidase B regulates activated complement C5a in vivo. Blood. 2007;109:1992–1997. doi: 10.1182/blood-2006-03-012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meltzer ME, Doggen CJ, de Groot PG, Meijers JC, Rosendaal FR, Lisman T. Low thrombin-activatable fibrinolysis inhibitor activity levels are associated with an increased risk of a first myocardial infarction in men. Haematologica. 2009;94:811–818. doi: 10.3324/haematol.2008.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sato T, Miwa T, Akatsu H, Matsukawa N, Obata K, Okada N, Campbell W, Okada H. Procarboxypeptidase R is an acute phase protein in the mouse whereas carboxypeptidase N is not. J Immunol. 2000;165:1053–1058. doi: 10.4049/jimmunol.165.2.1053. [DOI] [PubMed] [Google Scholar]
- 88.Verbon A, Meijers JC, Spek CA, Hack CE, Pribble JP, Turner T, Dekkers PE, Axtelle T, Levi M, van Deventer SJ, Reitsma PH, van der Poll T. Effects of IC14, an anti-CD14 antibody, on coagulation and fibrinolysis during low-grade endotoxemia in humans. J Infect Dis. 2003;187:55–61. doi: 10.1086/346043. [DOI] [PubMed] [Google Scholar]
- 89.Zeerleder S, Schroeder V, Hack CE, Kohler HP, Wuillemin WA. TAFI and PAI-1 levels in human sepsis. Thromb Res. 2006;118:205–212. doi: 10.1016/j.thromres.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 90.Renckens R, Roelofs JJ, ter Horst SA, van 't Veer C, Havik SR, Florquin S, Wagenaar GT, Meijers JC, van der Poll T. Absence of thrombin-activatable fibrinolysis inhibitor protects against sepsis-induced liver injury in mice. J Immunol. 2005;175:6764–6771. doi: 10.4049/jimmunol.175.10.6764. [DOI] [PubMed] [Google Scholar]
- 91.te Velde EA, Wagenaar GT, Reijerkerk A, Roose-Girma M, Borel Rinkes IH, Voest EE, Bouma BN, Gebbink MF, Meijers JC. Impaired healing of cutaneous wounds and colonic anastomoses in mice lacking thrombin-activatable fibrinolysis inhibitor. J Thromb Haemost. 2003;1:2087–2096. doi: 10.1046/j.1538-7836.2003.00404.x. [DOI] [PubMed] [Google Scholar]