

Abstract
The bmi-1 and myc oncogenes collaborate strongly in murine lymphomagenesis, but the basis for this collaboration was not understood. We recently identified the ink4a–ARF tumor suppressor locus as a critical downstream target of the Polycomb-group transcriptional repressor Bmi-1. Others have shown that part of Myc's ability to induce apoptosis depends on induction of p19arf. Here we demonstrate that down-regulation of ink4a–ARF by Bmi-1 underlies its ability to cooperate with Myc in tumorigenesis. Heterozygosity for bmi-1 inhibits lymphomagenesis in Eμ–myc mice by enhancing c-Myc-induced apoptosis. We observe increased apoptosis in bmi-1−/− lymphoid organs, which can be rescued by deletion of ink4a–ARF or overexpression of bcl2. Furthermore, Bmi-1 collaborates with Myc in enhancing proliferation and transformation of primary embryo fibroblasts (MEFs) in an ink4a–ARF dependent manner, by prohibiting Myc-mediated induction of p19arf and apoptosis. We observe strong collaboration between the Eμ–myc transgene and heterozygosity for ink4a–ARF, which is accompanied by loss of the wild-type ink4a–ARF allele and formation of highly aggressive B-cell lymphomas. Together, these results reinforce the critical role of Bmi-1 as a dose-dependent regulator of ink4a–ARF, which on its turn acts to prevent tumorigenesis on activation of oncogenes such as c-myc.
Keywords: Apoptosis, tumorigenesis, bmi-1, c-myc, ink4a–ARF
c-myc is a member of the myc family of bHLH/LZ transcription factors, which also includes the N-myc and L-myc genes. Myc is a crucial regulator of many cellular processes, such as cell proliferation and differentiation and its importance during development is underscribed by the death of c-myc−/− mice at embryonic days 9.5–10.5 (Davis et al. 1993). Myc expression is found to be deregulated in many human neoplasias (Nesbit et al. 1999) and transgenic animal models have demonstrated convincingly that Myc overexpression induces tumorigenesis (Langdon et al. 1986; Henriksson and Lusher 1996; Facchini and Penn 1998). However, beside being a growth-promoting oncogene, Myc is also a potent inducer of apoptosis via mechanisms that still remain to be clarified (Evan et al. 1992; Prendergast 1999). Part of the difficulty in unraveling these mechanisms stems from the observation that Myc can activate both p53-dependent and independent apoptosis pathways; the relative contribution of each of these pathways depends on cell type and context (Hermeking and Eick 1994; Wagner et al. 1994; Hsu et al. 1995; Sakamuro et al. 1995). Furthermore, Myc-induced apoptosis in primary mouse embryo fibroblasts (MEFs) was shown to require CD95 (Fas/APO-1) signaling and to be suppressed by IGF-1 signaling and Bcl-2 (Hueber et al. 1997). In vivo, suppression of apoptosis enables oncogenes such as Myc and E1A to acquire full oncogenic activity and allows for efficient neoplastic outgrowth. This is clearly illustrated by the acceleration of myc-transgene-induced tumorigenesis by overexpression of bcl-2 (Strasser et al. 1990a) or by deletion of p53 (Blyth et al. 1995; Elson et al. 1995).
In the past, MoMLV insertional mutagenesis with Eμ–myc transgenic mice has led to the identification of a number of genes that collaborate with c-myc in the onset of B-cell lymphomas. Among the collaborators identified by such screens is the bmi-1 oncogene, a member of the mammalian Polycomb-group of transcriptional repressors (Haupt et al. 1991; van Lohuizen et al. 1991; van Lohuizen 1998; Jacobs and van Lohuizen 1999). The synergism in tumorigenesis has been confirmed by the generation of bmi-1/myc double transgenic mice that die from massive leukemia as newborns (Haupt et al. 1993; Alkema et al. 1997). Whereas this clearly established the powerful in vivo cooperation of myc and bmi-1, the molecular basis for this remained unclear because of insufficient knowledge about the precise function and critical downstream targets of Bmi-1 and Myc. Recently, we found that Bmi-1 acts as a negative regulator of the ink4a–ARF locus, which encodes the two tumor suppressors p16 and p19arf (Jacobs et al. 1999). p16 inhibits cell cycle progression by inhibiting cyclin D-dependent kinases and thereby prevents the phosphorylation of the tumor suppressor Rb (Serrano et al. 1993), whereas p19arf prevents the degradation and inactivation of the tumor suppressor p53 by binding to Mdm2 (Pomerantz et al. 1998; Weber et al. 1999). bmi-1−/− mice suffer from severe proliferation defects in both the hematopoietic system and brain. Furthermore, in vitro, bmi-1−/− MEFs proliferate poorly and prematurely senesce. On the other hand, overexpression of Bmi-1 in MEFs was found to delay senescence and facilitate immortalization (Jacobs et al. 1999). Absence of Bmi-1 expression is accompanied by increased levels of p16 and p19arf, whereas Bmi-1 overexpression results in down-regulation of p16 and p19arf. The full rescue of the proliferation defects in bmi-1−/−;ink4a–ARF−/− MEFs and the dramatic rescue of the lymphoid and neurological defects in bmi-1−/−;ink4a–ARF−/− mice indicated that ink4a–ARF is the critical downstream target of Bmi-1 in regulation of cell proliferation (Jacobs et al. 1999). In addition, we observed that Bmi-1 acts in a dose-dependent manner in regulating ink4a–ARF. This parallels our observations in vivo, in which doubling of the Eμ–bmi-1 transgene dose in homozygous Eμ–bmi-1 transgenic mice lead to a significantly increased rate of tumorigenesis (Alkema et al. 1997). Others have shown that part of the p53-dependent apoptosis induced by Myc depends on the presence of p19arf, and that Myc up-regulates p19arf but not p16 protein levels in MEFs (Zindy et al. 1998). Recent reports have shown that the up-regulation of ink4a–ARF is not specific for Myc, but rather represents a more general and important fail-safe that is activated on aberrant mitogenic signaling, and prevents primary cells from immortalization and transformation (for review, see Evan and Littlewood 1998; Ruas and Peters 1998; Sherr 1998; Sharpless and DePinho 1999). On the basis of these recent observations, we investigated whether regulation of the ink4a–ARF locus by Bmi-1 is at the basis for the dramatic collaboration between Bmi-1 and Myc in tumorigenesis, and tested the hypothesis that the relative levels of p16 and p19arf are critical for their tumor suppressive role.
Results
Heterozygosity for Bmi-1 reduces lymphomagenesis in Eμ–myc mice by enhancing c-Myc-induced apoptosis
With the original aim to identify genes, other than bmi-1, that are able to accelerate the onset of B cell lymphomas in Eμ–myc transgenic mice, we crossed Eμ–myc transgenic mice into the bmi-1 mutant background and used these mice in a MoMLV insertional mutagenesis screen. We found that the Eμ–myc transgene was not able to rescue the proliferative defects in the hematopoeitic system of bmi-1−/− mice, which is in line with our previous findings in MEFs (Jacobs et al. 1999). In fact, due to the poor growth and severe neurological defects of bmi-1−/− and Eμ–myc;bmi-1−/− animals, these mice needed to be sacrificed before MoMLV infection of newborns had resulted in the formation of lymphomas in either genotype. Interestingly however, we observed a clear gene dosage effect of Bmi-1 on the onset of both MoMLV-induced and spontaneous lymphomas in Eμ–myc;bmi-1+/− mice. The most pronounced difference in tumor susceptibility was observed when comparing the onset and frequency of spontaneous (pre-) B cell lymphomas in Eμ–myc;bmi-1+/− mice with that in Eμ–myc;bmi-1+/+ mice (Fig.1 A,B).
To reveal the basis for the delayed onset of lymphomas in bmi-1+/− mice, we studied the B-cell composition within bone marrow and spleen of the Eμ–myc and Eμ–myc;bmi-1+/− in more detail by flow cytometry. Eμ–myc transgenic mice show a characteristic twofold increase in the amount of B220+ pre-B cells present in bone-marrow, which is the consequence of a higher proliferation rate (Fig. 1C; Langdon et al. 1986; Harris et al. 1988). Strikingly, in Eμ–myc;bmi-1+/− mice, this expansion of pre-B cells in bone marrow is almost completely absent and the number of B220+ cells is similar to that seen in wild-type mice (Fig. 1C). The absence of pre-B cell expansion in Eμ–myc;bmi-1+/− mice may be explained by the reduction of the proliferation rate of pre-B cells. Alternatively, bmi-1+/− mice might be partially blocked in B-cell differentiation, which cannot be overruled by c-Myc overexpression. Thirdly, c-Myc-induced apoptosis might be increased in bmi-1+/− mice. The first possibility is unlikely, because we found an equally increased cycling activity of pre-B cells in Eμ–myc;bmi-1+/− mice compared with Eμ–myc mice as indicated by the higher forward scatter signal (FSC-H; Fig. 1C). Furthermore, we have no indication for a block in differentiation, because bmi-1+/− mice have similar B-cell compositions in spleen and bone marrow as wild-type littermates (van der Lugt et al. 1994). We determined whether the absence of pre-B cell expansion in Eμ–myc;bmi-1+/− mice could be caused by an increase in apoptosis. Bone marrow cell suspensions of wild-type, Eμ–myc, and Eμ–myc;bmi-1+/− mice were cultured for 24 hr in the absence of specific growth factors and subsequently stained for both Annexin-V and cell surface B220. Flow-cytometric analysis shows that the apoptotic ratio (Annexin-V+/Annexin-V−) of Eμ–myc mice (6/2) is significantly increased compared with wild-type mice (3/4), confirming the notion that c-Myc overexpression induces apoptosis in B lymphocytes (Prasad et al. 1997). Interestingly, the apoptotic ratio is even increased further in the Eμ– myc;bmi-1+/− mice (11/2) (Fig. 1D). Viable B220+ lymphocytes (B220+/PI−; Fig. 1E) of Eμ–myc;bmi-1+/− mice show a 10-fold increase in commitment to apoptosis (Annexin-V+), as compared with Eμ–myc mice. These results indicate that the bone marrow compartment of bmi-1+/− mice is more susceptible to c-Myc-induced apoptosis, which results in a dramatic reduction in the expansion of pre-B cells in Eμ–myc;bmi-1+/− mice.
Figure 1.

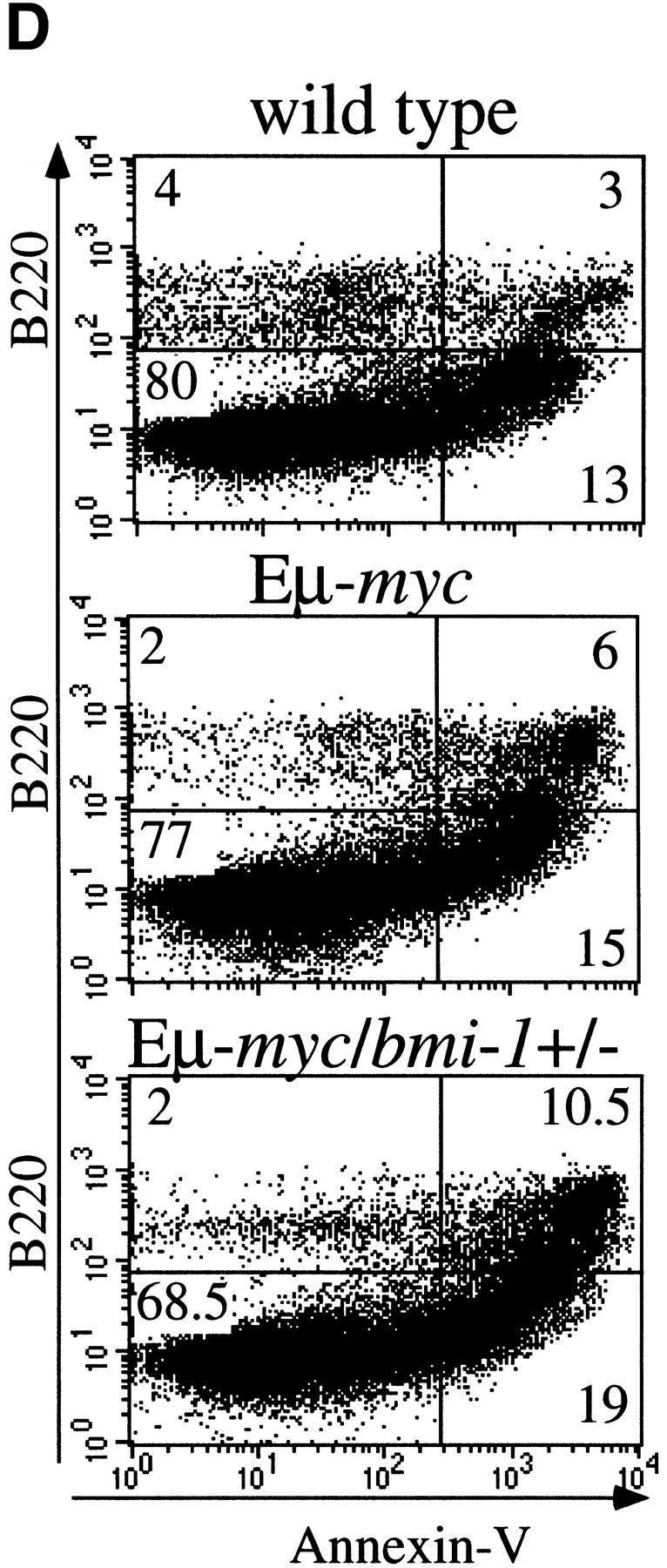
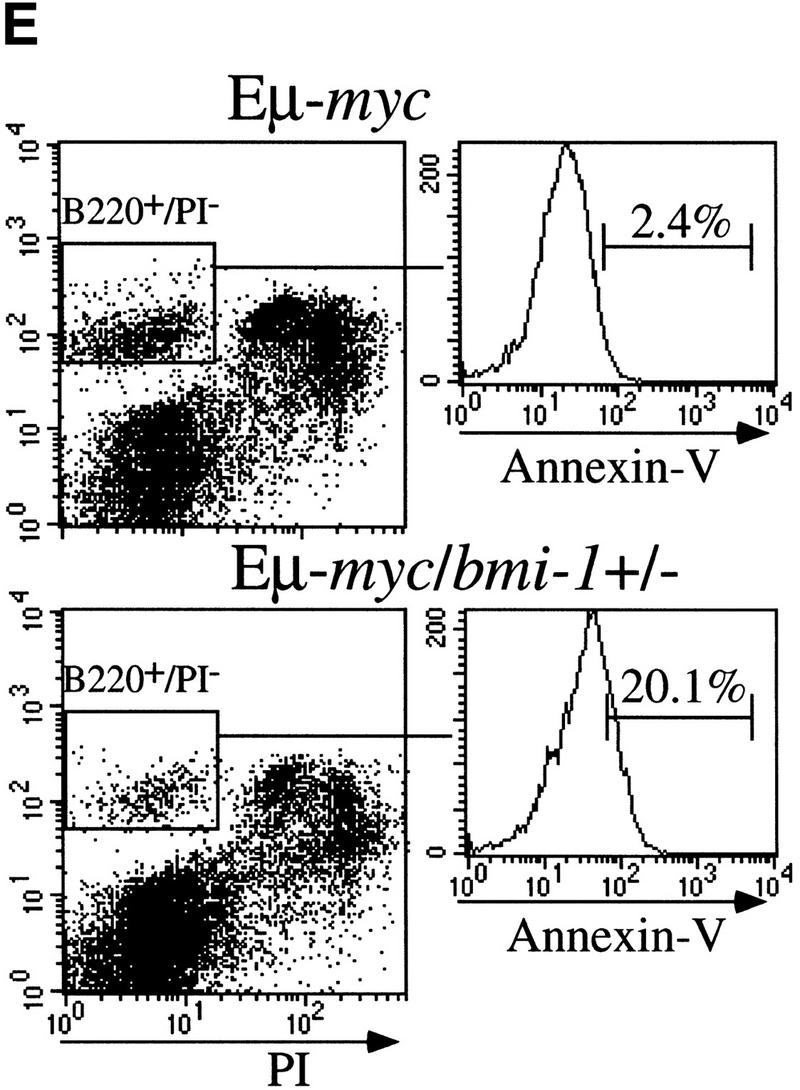

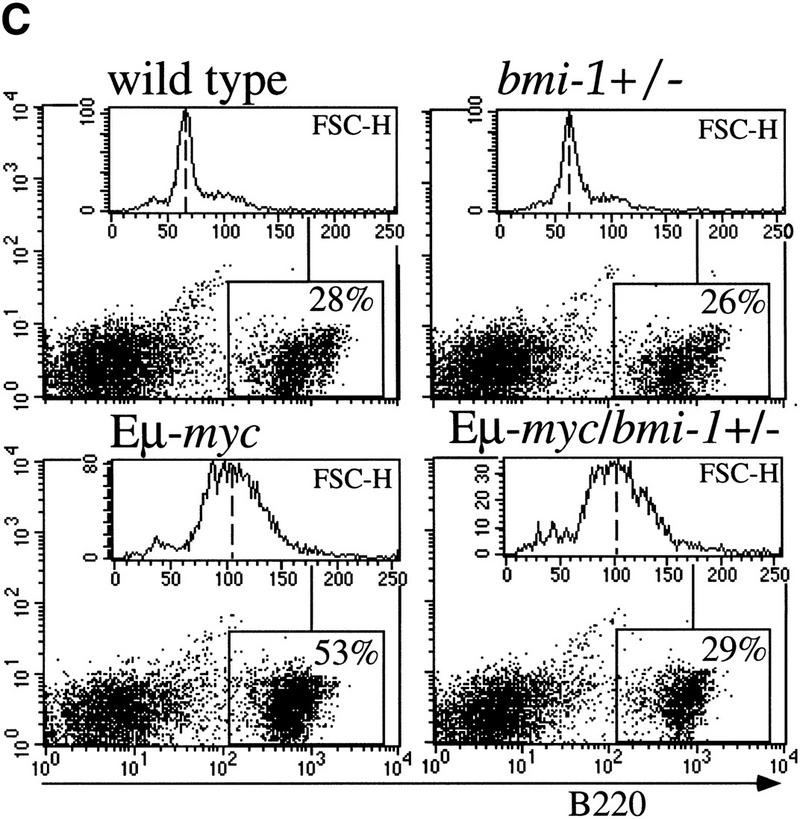
Heterozygosity for bmi-1 causes reduced susceptibility to lymphomagenesis, abrogation of c-Myc-induced pre-B cell expansion and increased c-Myc-induced apoptosis. (A) Kaplan-Meier survival plots of MoMLV-induced tumors in Eμ–myc, Eμ–myc;bmi-1+/−, bmi-1+/−, and wild-type control mice and (B) of spontaneous (pre-) B-cell lymphomas in Eμ–myc and Eμ–myc;bmi-1+/− mice. (C) Flow-cytometric analysis of bone marrow cell suspensions of wild-type, bmi-1+/−, Eμ–myc and Eμ–myc;bmi-1+/− mice at the age of 5–7 weeks. B220 cell-surface staining shows an increased cell size (FSC-H), due to higher cycling activity of pre-B cells (B220 positive lymphocytes) in Eμ–myc and Eμ–myc;bmi-1+/− mice and an expansion of the pre-B cell compartment in Eμ–myc but not in Eμ–myc;bmi-1+/− mice. (D) Flow-cytometric analysis of bone marrow cell suspensions of wild-type, Eμ–myc, and Eμ–myc;bmi-1+/− mice, cultured for 24 hr in 10% FBS/RPMI medium in the absence of specific growth factors and subsequently stained for B220 and Annexin-V. The apoptotic ratio Annexin-V+/Annexin-V− of B220+ lymphocytes indicates the apoptosis susceptibility of bone marrow-derived pre-B cells. (E) Analysis of the percentage of Annexin-V+ cells within the pool of viable B220+/PI−lymphocytes indicates a 10-fold increase in Eμ–myc;bmi-1+/− mice vs. Eμ–myc mice.
Bmi-1 inhibits Myc-induced apoptosis in MEFs by down-regulating ink4a–ARF
The increased c-Myc-induced apoptosis in Eμ–myc;bmi-1+/− mice and the resulting decrease in pre-B cell expansion and reduced susceptibility to lymphomas in these mice, suggested that Bmi-1 inhibits c-Myc-induced apoptosis and that this is the basis for their collaboration in tumorigenesis. To investigate this hypothesis under more defined conditions, we analyzed the ability of c-Myc to induce apoptotic cell death in primary MEFs overexpressing Bmi-1. Wild-type MEFs overexpressing different levels of Myc (see Materials and Methods) rapidly underwent apoptosis under low and high serum conditions (Fig. 2A). However, Myc-induced cell death was significantly reduced in MEFs infected with a bmi-1-encoding retrovirus, compared with control-infected MEFs, indicating that overexpression of Bmi-1 inhibits c-Myc-induced apoptosis (Fig. 2A).
Figure 2.



Bmi-1 inhibits c-Myc-induced apoptosis and strongly enhances proliferation in collaboration with myc in an ink4a–ARF-dependent manner. (A) Wild-type MEFs were infected at passage 1 with control (C) or bmi-1 (B) encoding retroviruses, at passage 2 with either control, mycER or mycHA-encoding retroviruses and analyzed for cell viability by trypan blue exclusion. mycER overexpressing cell populations were analyzed for cell death 0, 24, and 48 hr after transfer to 0.1% serum in the presence (circles) or absence (squares) of 125 nm 4-OHT (left). mycHA overexpressing cells were analyzed for cell death 0, 24, and 48 hr after transfer to 0.1% (circles) or 10% (squares) serum (right). Control-infected cultures remained viable for >95% during the entire experiment (not shown). Apoptotic cell death was confirmed by flow-cytrometric analysis of cells with a subdiploid DNA content. (B) Wild-type or ink4a–ARF−/− MEFs were infected at passage 1 with control (C, black bars) or bmi-1 (B, gray bars)-encoding retroviruses and subsequently at passage 2 with control or mycER retroviruses. After infection, cells were analyzed for subdiploid DNA content 24 hr after transfer to 0.1% serum (left), or for cell viability by trypan blue exclusion 0, 16, and 26 hr after transfer to 0.1% serum in the presence of 125 nm 4-OHT (right). (□ +/+C; █ +/+B; ○ −/−C; ● −/−B.) (C) Growth curves of wild-type (left) or ink4a–ARF−/− MEFs (right) infected at passage 1 with control (C) or bmi-1 (B) encoding retroviruses and at passage 2 with control or mycHA-encoding retroviruses. Experiments were performed at least three times, yielding highly reproducible results (all standard deviations were within 10% of the means shown) and similar data were obtained with lower levels of Myc by use of the mycER retrovirus in the absence of 4-OHT. (□ Control C; █ Control B; ○ MycHA C; ● MycHA B.)
Part of Myc-induced apoptosis is mediated via p19arf, and was shown to depend on p53 (Zindy et al. 1998). Because Bmi-1 acts as a negative regulator of p16 and p19arf expression, we analyzed to what extent inhibition of Myc-induced apoptosis by Bmi-1 is mediated via down-regulation of ink4a–ARF. To that end, the level of Myc-induced apoptosis in early-passage ink4a–ARF−/− primary MEFs and Bmi-1 overexpressing ink4a–ARF−/− MEFs was compared. As expected, ink4a–ARF−/− MEFs, which are both deficient for p19arf and p16 (Serrano et al. 1996), show an attenuated apoptotic response to Myc (Figs. 2B and 4A, below). In contrast, overexpression of Bmi-1 in ink4a–ARF−/− MEFs did not result in a significant further decrease in sensitivity to Myc-induced apoptosis when compared with ink4a–ARF−/− MEFs containing endogenous levels of Bmi-1 (Fig. 2B). This indicates that most of Bmi-1's ability to inhibit c-Myc-induced apoptosis depends on functional p19arf/p16.
Figure 4.



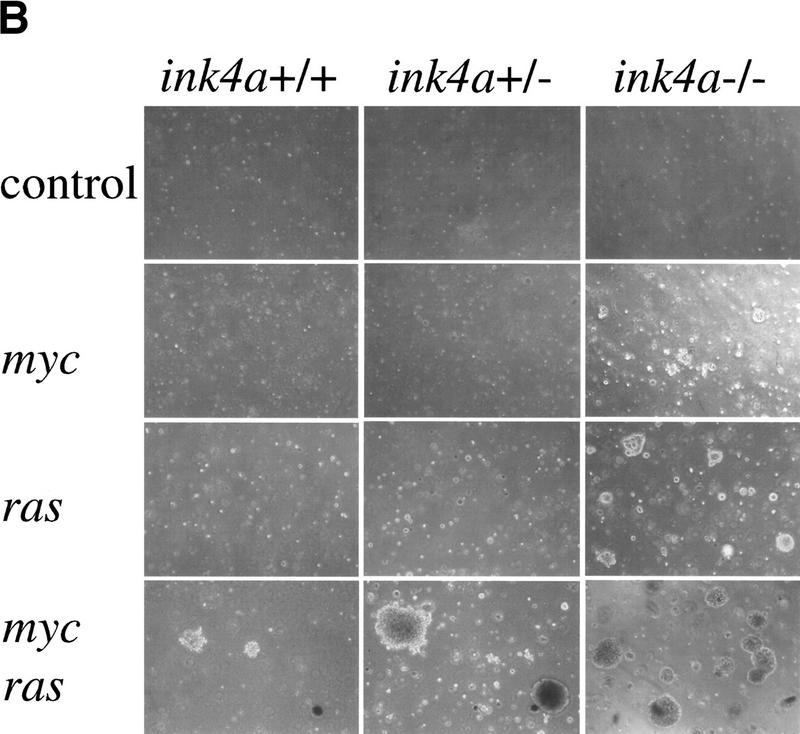

Dosage effects in ink4a–ARF+/− MEFs. (A) ink4a–ARF+/− MEFs proliferate faster than wild-type MEFs on overexpression of Myc. Growth curves of wild-type (+/+), ink4a–ARF+/− (right), and ink4a–ARF−/− (left) MEFs infected with mycER virus, in the presence (filled symbols) or absence (open symbols) of 250 nm 4-OHT. Analysis of GFP expression and Western analysis confirmed 100% infection and equal Myc protein levels for both wild-type and ink4a–ARF+/− MEFs. (B) ink4a–ARF+/− MEFs are more easily transformed by myc and ras oncogenes. First passage wild-type, ink4a–ARF+/−, and ink4a–ARF−/− MEFs were infected with control or rasV12-encoding retroviruses and subsequently infected with control or mycHA retroviruses, after which cells were analyzed for growth in soft agar. Photographs were taken 2 weeks after plating in soft agar. Similar data were obtained with the mycER virus in the absence of 4-OHT except that colonies were smaller. (C,D) ink4a–ARF+/−;ras/myc transformed colonies had retained the wild-type ink4a–ARF allele and wild-type p53. (C) PCR analysis of the wild-type (WT) and mutated (KO) ink4a–ARF allele for 9 of 16 tested ink4a–ARF+/−;ras/myc soft agar colonies, picked out of the agar 2 weeks after plating and directly subjected to DNA isolation and PCR. LOH was found for only one case. DNA isolated from wild-type, ink4a–ARF+/−, and ink4a–ARF−/− MEFs served as controls (D) Western blot analysis of mutant p53 in ink4a–ARF+/−;ras/myc soft agar colonies, picked out of the agar at 1.5 weeks after plating and expanded for 1.5 weeks prior to lysis. MEFs established as an immortal cell line according to a standard 3T3 protocol (lane 1) contained mutant p53, however primary wild-type MEFs (lane 2) and 6 of 12 tested ink4a–ARF+/−;ras/myc colonies (lanes 3–8) did not. Analysis of tubulin levels served as loading control. (E) Induction of p19arf in wild-type and ink4a–ARF+/− MEFs infected with mycER virus and cultured in the presence (+) or absence (−) of 250 nm 4-OHT.
In a population of fibroblasts overexpressing Myc and kept under 10% serum conditions, a balance exists between enhanced proliferation and apoptosis (Evan et al. 1992; Zindy et al. 1998). The resulting net proliferation rate can be increased by counteracting Myc-induced apoptosis. In our hands, the net proliferation rate under high serum conditions of MEFs overexpressing Myc was similar to that of control-infected MEFs (Fig. 2C). Bmi-1 overexpressing MEFs proliferate faster than control cells (Jacobs et al. 1999). Strikingly, when Bmi-1 and Myc were coexpressed, a synergistic increase in the proliferation rate was seen in wild-type MEFs (Fig. 2C). This effect requires functional ink4a–ARF, because such an increase was not observed in ink4a–ARF−/− MEFs overexpressing Myc and Bmi-1 (Fig. 2C). Western blot analysis of Bmi-1 and Myc expression levels showed equal Bmi-1 or Myc expression levels in the infected wild-type and ink4a–ARF−/− MEFs, respectively (Fig. 3B; data not shown). Taken together, this indicates that the synergistic effect of Bmi-1 and Myc overexpression on the proliferative capacity of MEFs is mostly mediated via ink4a–ARF.
Figure 3.
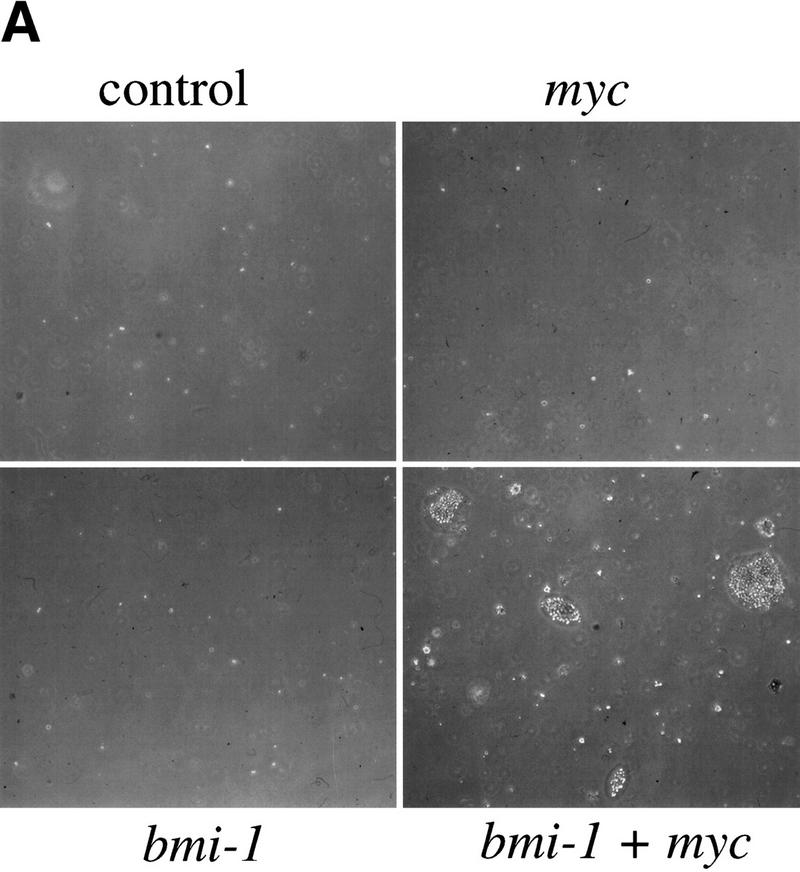

(A) Myc and Bmi-1 induce transformation of MEFs. Soft agar assay of MEFs infected at the first passage with control or bmi-1-encoding retroviruses and subsequently with control or myc-encoding retroviruses. (B) Bmi-1 inhibits induction of p19arf by Myc. Western blots showing p16, p19arf, MycHA, MycER, and Bmi-1 protein levels in wild-type MEFs infected first with control (C) or bmi-1 (B) retroviruses and subsequently with control, mycHA, or mycER retroviruses. Tubulin levels served as loading control. Bmi-1 overexpression leads to a down-regulation of p16 and p19arf levels, whereas overexpression of MycHA or MycER (in the absence of 4-OHT) induces p19arf but not p16. Combined overexpression of Bmi-1 and Myc completely abrogates the induction of p19arf by Myc.
Bmi-1 collaborates with Myc in transformation by inhibiting Myc-mediated p19arf up-regulation
To investigate whether combined overexpression of Bmi-1 and Myc, beside increasing cell proliferation, can also transform primary MEFs, we analyzed their ability to grow in semi-solid medium. Infection of wild-type, ink4a–ARF+/−, and ink4a–ARF−/− MEFs with control or bmi-1-encoding retroviruses did not cause transformation (Fig. 3A). Overexpression of Myc alone transformed ink4a–ARF−/−, but not ink4a–ARF+/− or wild-type MEFs (Figs. 3A and 4B). In contrast, these wild-type and ink4a–ARF+/− MEFs rapidly produced colonies in soft agar when overexpressing both Bmi-1 and Myc (Fig. 3A), indicating that Bmi-1 and Myc not only cooperate in transformation in vivo, but also in vitro in primary MEFs. However, the anchorage-independent growth observed here is less efficient, (yielding smaller colonies) than what is achieved by coexpression of Ras and Myc (Fig. 4B).
The experiments described above clearly indicate that the collaboration between myc and bmi-1 is mostly mediated via ink4a–ARF. Because Bmi-1 and Myc have opposite effects on p19arf expression, we were interested to see what happens to p19arf protein levels when both Bmi-1 and Myc are overexpressed in MEFs. Overexpression of Myc in MEFs results in the predicted increase of p19arf levels. However, in MEFs first infected with bmi-1 encoding retrovirus and subsequently with myc encoding virus, no induction of p19arf was found, whereas Myc levels were equally high in all the myc virus-infected MEFs (Fig. 3B, shown for different myc-retroviruses). This clearly indicates that Bmi-1 prohibits the up-regulation of p19arf by Myc and, in combination with the data above, strongly suggests that this is the underlying basis for their efficient collaboration in transformation.
Overexpression of Myc and Ras in ink4a-ARF+/− MEFs reveals dosage effects
Bmi-1 overexpression leads to a strong down-regulation of p16 and p19arf, however, it does not completely abrogate their expression (Fig. 3B). Nevertheless, we observe clear dose-dependent collaborative effects of Bmi-1 and Myc, which are largely mediated via the counteractive effect of Bmi-1 on the ink4a–ARF locus. This would imply that both Myc-induced apoptosis and transformation are sensitive to cellular ink4a–ARF dosage. Myc overexpressing ink4a–ARF+/− MEFs appeared to be more resistant to Myc-induced apoptosis and proliferated faster under high serum conditions than Myc overexpressing wild-type MEFs (Fig. 4A). To assess whether ink4a–ARF gene dosage correlates with increased efficiency in transformation, we infected wild-type, ink4a–ARF+/−, and ink4a–ARF−/− MEFs in two consecutive rounds with control or ras-encoding retroviruses and subsequently with control or myc-encoding retroviruses and analyzed their ability to proliferate in semi-solid medium (Fig. 4B). As has been reported by others (Serrano et al. 1996), wild-type and ink4a–ARF+/− MEFs were not easily transformed by Myc or Ras alone. In contrast, ink4a–ARF−/− MEFs could be transformed by either Ras or Myc. Transformation by a combination of Ras and Myc was achieved for all three genotypes and was highest for the ink4a–ARF−/− MEFs, with a much higher percentage of cells that was able to produce colonies in soft agar than in case of the wild-type or ink4a–ARF+/− MEFs (Fig. 4B). Whereas initially no large differences were observed in the percentage of dividing cells in ras+ myc-infected wild-type or ink4a–ARF+/− MEFs, from ∼1.5 week onward, many of the wild-type colonies stopped growing and died (Fig. 4B). In contrast, the ras+ myc-infected ink4a–ARF+/− MEFs resulted in much more and larger colonies that continued growing (Fig. 4B, bottom). Wild-type MEFs, and with higher efficiency, ink4a–ARF+/− MEFs, can be selected for resistance to Myc-induced apoptosis, through loss of the remaining wild-type ink4a–ARF allele or mutation of p53. (Zindy et al. 1998; J. Jacobs, unpubl.). However, analysis of the soft agar colonies for ink4a–ARF genotype by PCR and for p53 status by Western blotting with an antibody against mutant and wild-type p53 revealed that only 1 of 16 colonies showed loss of heterozygosity (LOH) for ink4a–ARF (Fig. 4C), and none of 12 colonies tested showed p53 mutation (Fig. 4D). This clearly illustrates that only a 50% reduction in ink4a–ARF dose already gives a selective growth advantage to MEFs overexpressing Ras and Myc. This is further reflected in the relative induction of p16 and p19arf by Ras and Myc, which is significantly lower in ink4a–ARF+/− MEFs when compared with wild-type MEFs (Fig. 4E, shown for Myc).
Strong collaboration in vivo between Myc overexpression and ink4a–ARF loss
Overexpression of Bmi-1 and Myc in mice strongly accelerates tumorigenesis, with the double transgenic mice already developing massive leukemia as newborns (van Lohuizen et al. 1991; Alkema et al. 1997). Here we show that in vitro most of the collaboration between Bmi-1 and Myc can be attributed to down-regulation of the ink4a–ARF locus by Bmi-1. Following these observations, we wished to asses whether ink4a–ARF−/− mice also show efficient acceleration of lymphomagenesis when crossed to Eμ–myc transgenic mice. However, we were unable to generate these mice because Eμ–myc;ink4a–ARF+/− mice rapidly became severely ill and all died before giving offspring, between the ages of 5.5 and 7.5 weeks (Fig. 5A). Histopathological analysis revealed that these mice all died with an unusually aggressive lymphoblastic leukemia. In most animals, the tumor cells invaded different organs, such as thymus and adjacent lymph nodes, liver, ovaria, uterus, fatpads of the mammary glands, and sometimes in lungs, kidneys, and meningae (illustrated in Fig. 5B, panels A and B, for liver and lungs). Without exception, the blood was extremely leukemic, reminiscent of our observations in Eμ–bmi-1; Eμ–myc double transgenics (Fig. 5B, cf. panel D with E). Flow cytometric analysis of tumor cell suspensions from thymus revealed that these tumors were of B-cell origin and were either highly or intermediately positive for sIgM (Fig. 5C). These results were confirmed by the presence of clonal or oligoclonal B-cell receptor heavy-chain rearrangements, and in cases of mature sIgM+ tumors, also clonal light-chain rearrangements, whereas no TCRβ-rearrangements were observed (data not shown). The tumor cells appear to retain the capacity to differentiate to some extent, because one animal that was sacrificed before overt tumor occurrence already showed a significant pre-B (tumor) cell population in the thymus (K1911, Fig. 5C). These results are remarkable, because no strong dosage effects have been reported for ink4a–ARF+/− mice in tumorigenesis in vivo before (Serrano et al. 1996). In vitro, Myc overexpression in p19arf+/− MEFs was shown to select for cells that have lost the remaining ink4a–ARF allele (Kamijo et al. 1997). Therefore, we examined the Eμ–myc;ink4a–ARF+/− B-cell tumors for LOH by Southern analysis. All 15 Eμ–myc;ink4a–ARF+/− tumors and in one CD2–myc;ink4a–ARF+/− B-cell tumor examined displayed LOH of the ink4a–ARF locus, whereas control Eμ–myc, CD2-myc, or Eμ–bmi-1;ink4a–ARF+/− tumors did not (Fig. 5D). In contrast to significant acceleration of tumor development in Eμ–myc;ink4a–ARF+/− mice, this was not observed in Eμ–bmi-1;ink4a–ARF+/− tumors, in line with the notion that Bmi-1 overexpression also acts via ink4a–ARF in tumorigenesis (Fig. 5A; data not shown). The presence of many pyknotic tumor cells as well as preliminary evidence from TUNEL staining of Eμ–myc tumors showed that a relatively high percentage of tumor cells underwent apoptosis, however, in the Eμ–myc;ink4a–ARF+/− B-cell tumors (showing LOH), the percentage of apoptotic tumor cells was lower (Fig. 5B,G, and H; data not shown). This suggests that as in vitro, in vivo loss of ink4a–ARF may result in decreased Myc-induced apoptosis, although effects on apoptotic rates caused by the different homing/surroundings or differentiation stage (thymus, mature B-cell lymphoma vs. lymph nodes and spleen in Eμ–myc pre-B cell lymphomas) can not be excluded.
Figure 5.

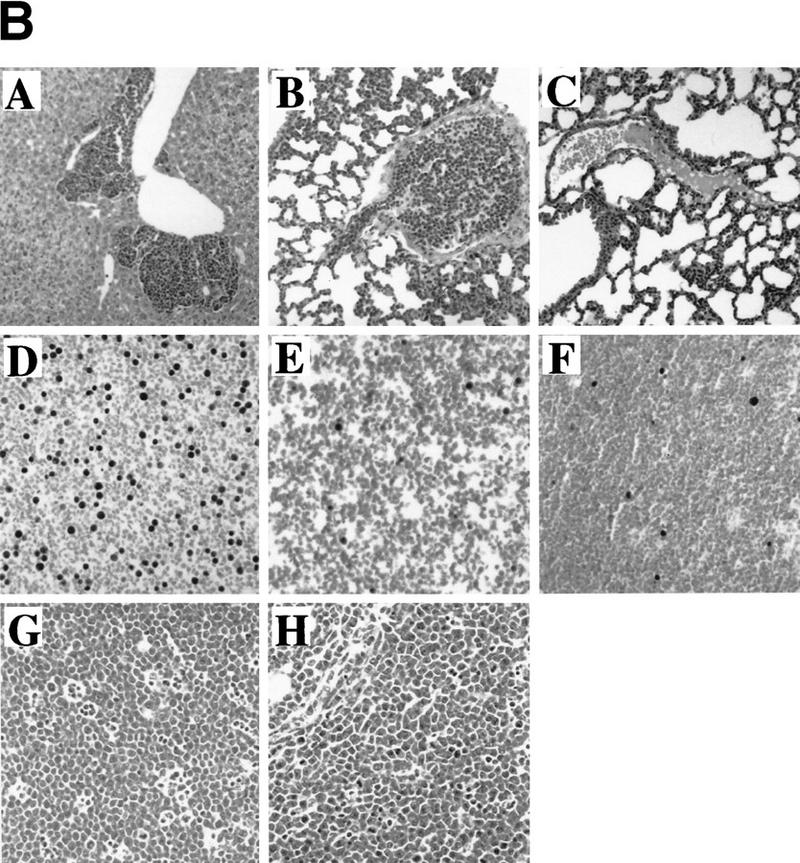
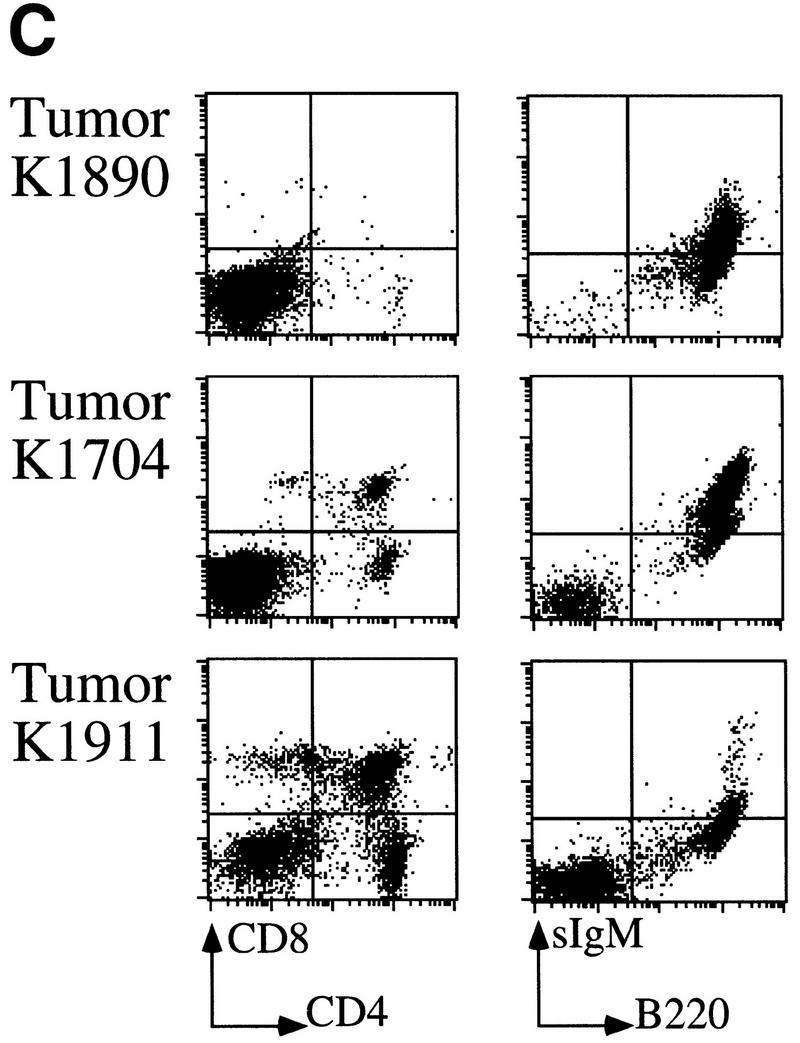

Severe acceleration of lymphomagenesis in Eμ–myc;ink4a–ARF+/− mice. (A) Eμ–myc;ink4a–ARF+/− mice quickly die of aggressive B-cell tumors. Kaplan-Meier survival plot of Eμ–myc; ink4a–ARF+/− mice and Eμ–myc mice. (B) Haematoxylin-eosin-stained sections of tumors that arose in Eμ–myc;ink4a–ARF+/− and Eμ–myc mice and of blood from these and ink4a–ARF−/− mice. (A) Representative example of an Eμ–myc;ink4a–ARF+/− tumor invading the liver; (B) a blood vessel in the lung of an Eμ–myc;ink4a–ARF+/− mouse filled with tumor cells. (C) A representative example of a blood vessel in the lung of an Eμ–myc mouse, which is free of tumor cells; (D-F) blood of Eμ–myc;ink4a–ARF+/− (D), Eμ–myc (E), and ink4a–ARF−/− (F) mice. Note that in contrast to the Eμ–myc and ink4a–ARF−/− mice, the blood of Eμ–myc;ink4a–ARF+/− mice is highly leukemic. (G, H) A higher magnification of a representative example of tumors that arose in Eμ–myc;ink4a–ARF+/− (G) and Eμ–myc (H) mice. Note the presence of more pyknotic tumor cells, which are indicative of apoptosis, in Eμ–myc tumors compared to Eμ–myc;ink4a–ARF+/− tumors. Photographs were taken at 10-fold (A-C) and 20-fold (D-H) magnification. (C) Flow-cytrometric analysis of cell suspensions of three Eμ–myc;ink4a–ARF+/− tumors after staining for cell surface CD8, CD4, sIgM, and B220. (D) Southern blot analysis of ink4a–ARF status of genomic DNA isolated from normal liver (L) or tumor (T) tissue showing LOH of the ink4a–ARF locus in tumors arising in Eμ–myc;ink4a–ARF+/− (lanes 1) and CD2–myc;ink4a–ARF+/− (lanes 3) mice but not in CD2–myc (lanes 2) Eμ–myc (lanes 5) and Eμ–bmi-1;ink4a–ARF+/− (lanes 4) mice.
Decreased cellularity of bmi-1−/− lymphoid compartment is caused by increased apoptosis and is rescued by ink4a–ARF deletion and by Bcl2 overexpression
bmi-1−/− mice show a severe reduction in the number of B and T lymphocytes in the spleen and thymus (van der Lugt et al. 1994). We have shown previously that this is accompanied by highly increased transcript levels of p16 and p19arf, and that this defect in cellularity is largely absent in bmi-1−/−;ink4a–ARF−/− mice (Jacobs et al. 1999). We investigated whether the severe reduction in cell numbers in bmi-1−/− is due to increased apoptosis rates. This is the case not only for splenocytes and bone marrow of bmi-1+/− mice challenged with Myc overexpression, but also in freshly isolated CD4+ bmi-1−/− thymocytes, as is evident from the increase in Annexin-V/PI-positive-staining T cells (Fig. 6, middle). CD4+ cells were selected because more mature T cells are most dramatically affected as opposed to immature CD4−/CD8− T cells (van der Lugt et al. 1994). This increase is dependent on ink4a–ARF, because in ink4a–ARF;bmi-1−/−, double knockout thymocytes, apoptosis is restored to wild-type levels (Fig. 6, bottom). As an independent means of assessing the in vivo contribution of apoptosis to the reduced cell numbers in bmi-1−/− spleen and thymus, we analyzed whether overexpression of the anti-apoptotic bcl2 oncogene could restore cellularity. Hereto, we crossed in the Eμ–bcl2-36SV transgene (hereafter abbreviated as SVbcl2) into the bmi-1−/− background. This transgene has been shown to effectively reverse potent apoptotic effects in hematopoietic cells, such as occurs in T cells at different developmental stages of IL-7 receptor-deficient mice (Akashi et al. 1997; Maraskovsky et al. 1997). We observed reproducibly a partial rescue of cellularity in both thymus and spleen in SVbcl2;bmi-1−/− mice when compared with bmi-1−/− mice (Fig. 7A, left). This effect was independently confirmed by introducing a different Eμ–bcl2 transgene, leading to a similar partial rescue (Fig. 7A, right). Analysis of the composition of T- and B-cell subsets by FACS analysis with standard B- and T-cell differentiation markers revealed that the rescue is most prominent in more mature CD4+/CD8+ and CD4+ T cells as well as in mature B220/sIgM-positive B cells (Fig. 7B; compare two independent SVbcl2;bmi-1−/− panels with the bmi-1−/− panel). This is of interest, because these more mature populations are among the most severely affected in bmi-1−/− mice. Overexpression of the SVbcl2 transgene alone did cause an increase in cellularity in the spleen, as has been observed previously (Strasser et al. 1990b), but did not lead to major changes in composition or cell proliferation (Fig. 7B, panel SVbcl2; data not shown). Together, these results clearly show that increased apoptosis contributes to the reduced cellularity in bmi-1−/− mice, which is mediated through up-regulation of ink4a–ARF.
Figure 6.
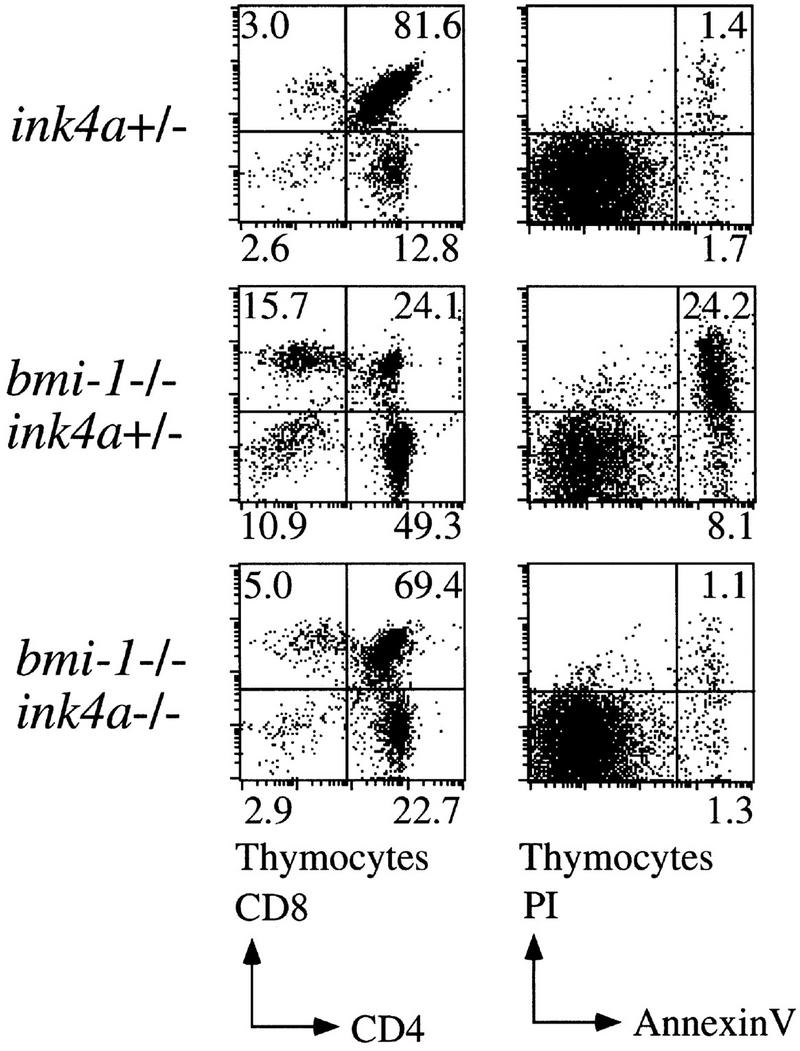
Increased apoptosis in the thymus of bmi-1−/− mice is rescued by deletion of ink4a–ARF. Flow-cytometric analysis of freshly isolated thymocytes of ∼6-week-old ink4a–ARF+/−, bmi-1−/−;ink4a–ARF+/−, and bmi-1−/−;ink4a–ARF−/− mice after staining for cell-surface CD4 and CD8 (left) and of CD4-positive thymocytes after staining for Annexin-V (right).
Figure 7.

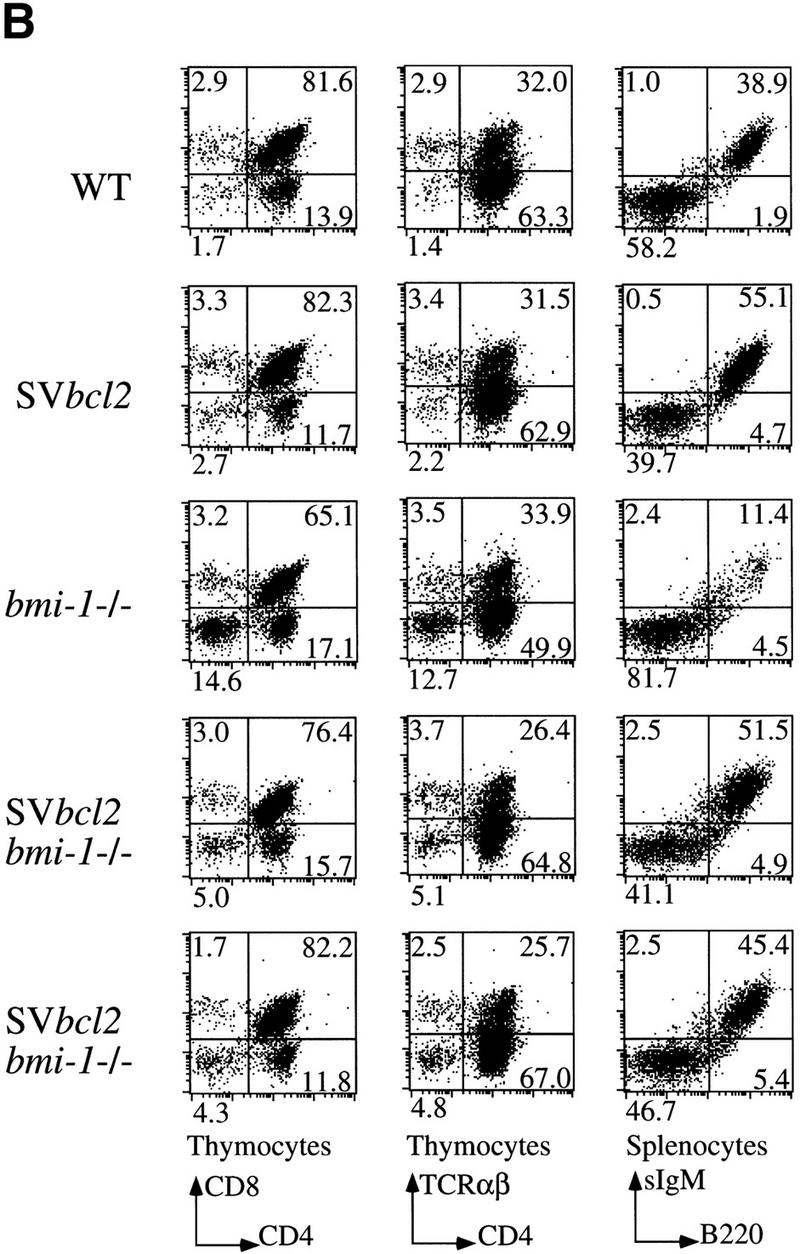
Bcl2 overexpression partially rescues cellularity in bmi-1−/− spleen (black bars) and thymus (gray bars). (A) Percent nucleated cells in thymus and spleen of ∼6-week-old wild-type, SVbcl2, bmi-1−/−, SVbcl2;bmi-1−/− mice (left) and of wild-type, Eμ–bcl2, bmi-1−/−, Eμ–bcl2;bmi-1−/− mice (right). (B) Flow-cytometric analysis of thymocytes and splenocytes of wild-type, SVbcl2, bmi-1−/−, and SVbcl2;bmi-1−/− mice.
Discussion
Bmi-1 affects apoptosis in vitro and in vivo, by regulating ink4a–ARF
Increased ink4a–ARF-dependent apoptosis clearly contributes to the dramatically reduced cellularity in thymus and spleen of bmi-1−/− mice. However, the total reduction is likely due to a combination of increased apoptosis and blocked proliferation caused by up-regulation of p16 and p19arf in bmi-1−/− cells (Jacobs et al. 1999). This is supported by the partial rescue by bcl2-transgene overexpression, as opposed to the almost complete rescue observed in bmi-1−/−;ink4a–ARF−/− mice (Fig. 7; Jacobs et al. 1999). This fits well with the notion that Bcl2 is known to prevent apoptosis but does not accelerate cell proliferation (Bissonette et al. 1992; Fanidi et al. 1992), whereas ink4a–ARF-loss both accelerates proliferation of primary cells (Serrano et al. 1996), and prevents apoptosis via the p19arf/p53 pathway (de Stanchina et al. 1998; Zindy et al. 1998). Increased apoptosis due to reduced levels of Bmi-1 has profound consequences for tumorigenesis; remarkably, only a twofold reduction in bmi-1 gene dose already results in significantly reduced lymphomagenesis on MoMLV-infection or Eμ–myc transgene overexpression, which could be largely ascribed to increased apoptosis. Conversely, overexpression of Bmi-1 in wild-type MEFs results in a significant reduction of Myc-induced ink4a–ARF-dependent apoptosis, which is evident at different levels of Myc overexpression. Significant apoptosis reduction by Bmi-1 is not affected by high serum conditions suggesting that bmi-1/ink4a–ARF signaling to the apoptotic machinery may bypass concomitant survival signals.
The powerful in vivo cooperation between Myc and Bmi-1 overexpression is mimicked well in vitro in MEFs. Bmi-1 and Myc not only cause a synergistic and dose-dependent increase in proliferation and decrease in apoptosis, but also can lead to transformation of wild-type MEFs, as assayed by anchorage-independent growth. In accordance with Bmi-1 acting in transformation by down-regulating ink4a–ARF, Myc overexpression is able to transform ink4a–ARF−/− MEFs, but not wild-type MEFs. The anchorage-independent growth observed here is, however, clearly less efficient than transformation by Ras + Myc, which yields more and larger colonies in soft agar. Importantly, Western blot analysis revealed that Bmi-1 overexpression not only results in reduced p16 and p19arf protein levels, but also prohibits the induction of p19arf by overexpression of Myc. This is in line with the role of Bmi-1 in Polycomb-complexes that act on chromatin to stably repress target genes (for review, see Jacobs and van Lohuizen 1999; van Lohuizen 1999). Taken together, the in vitro and in vivo data presented here strongly suggest that the Bmi-1-mediated prevention of p19arf induction by Myc forms the basis for the efficient cooperation in oncogenic transformation between bmi-1 and myc.
ink4a–ARF dosage effects: Implications for tumorigenesis
Beside to the Bmi-1 dose-dependent effects on ink4a–ARF, MEF transformation assays with myc and ras also revealed clear ink4a–ARF dose dependence in efficiency of anchorage-independent growth capacity. As in the classical REF cotransformation assay (Land et al. 1983), coexpression of Myc and Ras in wild-type MEFs cause colony growth in soft agar; however, this occurs with very low efficiency, given the high efficiency of infection of the viruses we used. Interestingly, the efficiency of transformation by Ras + Myc increases dramatically in an ink4a–ARF gene dose-dependent manner in ink4a–ARF+/− and ink4a–ARF−/− MEFs. Notably, the initial mitogenic effects of Myc + Ras in unattached wild-type REFs, followed by rapid cell death of most cells, has been shown to occur because of deprivation of matrix adhesion (McGill et al. 1997). Our results clearly suggest that ink4a–ARF is required in a dose-dependent manner for the apoptosis that occurs on disruption of matrix attachment. Because efficient Cyclin D1 up-regulation requires matrix attachment (for review, see Assoian 1997), this may suggest a role for p16 in this respect. We suggest that initial proliferation followed by apoptosis reflects the gradual increase in p16 and p19arf levels with each cell division; this is observed when embryos are disaggregated and MEFs are put in culture (Zindy et al. 1997, 1998). As a critical level of p16 and/or p19arf is reached, such primary cells normally enter a quiescent state called cellular senescence, whereas in the case of Myc (and perhaps Ras) overexpression, the fate of such cells now becomes ink4a–ARF-dependent apoptosis. In agreement with this hypothesis, we find efficient overgrowth of whole monolayers, when such Ras + Myc-transduced primary MEFs are plated on coated dishes in focus-formation assays, suggesting efficient proliferation of most cells under these conditions (not shown).
We further observe that MEFs coexpressing Ras and Myc retain hyperinduction of p16 and p19arf protein levels (our unpublished data). Importantly, analysis of Myc + Ras overexpressing soft agar colonies obtained in ink4a–ARF+/− MEFs revealed, at the (relatively early) time of analysis, no significant levels of p53-mutations or LOH for ink4a–ARF. If no other (and as yet unprecedented) mutations have occurred rapidly in other effectors of p16/p19arf, this could suggest that the twofold reduction in ink4a–ARF in these rapidly proliferating clones prevents the wild-type senescence threshold level to be reached. We consider this unlikely because of the significant induction of p16 and p19Arf levels by Ras + Myc. Rather, we favor the coexpression of Ras + Myc to render the MEFs relatively more insensitive to p16/p19arf arrest. We speculate that one way of achieving insensitivity could be by the known ability of Ras to prevent Myc-induced apoptosis via the PI3-kinase–AKT/PKB pathway (Kauffman-Zeh et al. 1997). Notably, the soft agar assays were performed under high-serum conditions, which may assist in tipping the balance toward survival. Alternatively, it is possible that Ras + Myc overexpression affects localization or activity of other modulators or effectors of ink4a–ARF, such as Mdm2, pRB, or p53. In this respect, it is of potential relevance that we fail to detect significant up-regulation of p53 in the Myc + Ras-infected ink4a–ARF+/− soft agar clones (not shown), whereas in wild-type MEFs, Myc has been shown to induce p53 and, concomitantly, the p53 target gene Mdm2, both of which are involved in mediating the p19arf inhibitory response (Zindy et al. 1998).
Perhaps the most dramatic illustration of ink4a–ARF tumor suppressive effects is the significant increase in onset and progression of lymphomagenesis in Eμ–myc;ink4a–ARF+/− mice, which is clearly reminiscent of the potent in vivo collaboration in Eμ–myc; Eμ–bmi-1 double-transgenic mice (Alkema et al. 1997). These mostly clonal tumors invariably showed loss of the remaining wild-type ink4a–ARF allele, conforming to the notion that loss of ink4a–ARF allows for full growth-promoting and oncogenic activity of Myc. This implicates that LOH must have occurred rapidly, perhaps reflecting the recently described role of Myc in eliciting genomic instability (McCormack et al. 1998; Felsher and Bishop 1999). The leukemias obtained show several unusual characteristics, in that they are highly aggressive mature B-cell leukemias that invade the thymus and/or adjacent lymphnodes as well as subcutaneous and mammary fatpads and several organs, such as liver, lungs, and pancreas. This is unlike the predominant pre-B-cell lymphomas in Eμ–myc mice that are largely confined to peripheral lymph nodes and spleen (Langdon et al. 1986), or the pre-B-cell lymphomas and predominant fibrosarcomas seen in ink4a–ARF−/− mice (Serrano et al. 1996). Given the unusual aggressive nature and early onset of the Eμ–myc;ink4a–ARF+/− B-cell tumors and the above-noted implication of ink4a–ARF in apoptosis on disruption of matrix-attachment, it is tempting to speculate that these tumor cells are perhaps more tolerant to loss of adhesion-mediated survival signals, due to ink4a–ARF-loss.
Implication of p16 and/or p19Arf?
Whereas the odds may seem in favor of p19arf as the culprit, on the basis of the results of C.J. Sherr and colleagues with p19arf−/− MEFs and mice (Kamijo et al. 1997), it may be that in cell types other than MEFs, p16 loss could contribute. Of notice in this regard is the observation that p19arf−/− mice develop fibrosarcomas and T-cell lymphomas rather than B-cell lymphomas, although possibly strain background differences could account for this. Interestingly, the slower rate of tumor formation in Eμ–myc;p19arf+/− mice (mean survival 11 weeks; Eischen et al. 1999) when compared with the mean survival of 7 weeks observed in Eμ–myc;ink4a–ARF+/− mice could point to a subtle additional effect of p16 loss in lymphomagenesis. Clearly, a definitive assignment of the relative contributions of p16, p19arf, or both awaits comparison to the effects on proliferation, apoptosis, and tumorigenesis in p16-specific knockout mice and MEFs.
In conclusion, we have shown that Bmi-1 cooperates efficiently with c-Myc in transformation and tumorigenesis, by preventing Myc-induced p19arf up-regulation and apoptosis. These studies reinforce the notion that Myc overexpression is not equivalent to ink4a–ARF loss and immortalization, but rather has severe additional transforming capacity, which becomes apparent when p19arf loss prevents Myc-induced apoptosis. Furthermore, our studies uncovered clear dosage effects of ink4a–ARF, in controlling proliferation and apoptosis, and showed that ink4a–ARF loss is required to prevent apoptosis by disruption of matrix attachment in Ras + Myc-transformed MEFs. Finally, the potent tumor-suppressor role of ink4a–ARF is uncovered in Eμ–myc;ink4a–ARF+/− mice, which shows an unsuspected potent predisposition to malignant B-cell lymphomas. If also applicable to human cancer, the results presented here suggest that ink4a–ARF levels, rather than full inactivation, need to be assayed. Furthermore, the potent collaboration between ink4a–ARF heterozygosity and Myc overexpression is of potential prognostic relevance for human multipe myeloma and Burkitts lymphoma, in which the hallmark myc-immunoglobulin translocations account for Myc overexpression.
Materials and methods
Generation of compound mutant mice and MoMLV infection
bmi-1+/− (van der Lugt et al. 1994) and ink4a–ARF+/− mice (Serrano et al. 1996) were crossed with Eμ–myc transgenic mice of founder line 186 (Verbeek et al. 1991). Eμ–myc;bmi-1+/− mice were subsequently intercrossed to generate bmi-1 mutant mice with and without the c-myc transgene. bmi-1+/− mice were crossed with Eμ–bcl2–36SV (Strasser et al. 1990b) and with Eμ–bcl2 (McDonnell 1990) transgenic mice to generate SV–bcl2;bmi-1+/− and Eμ–bcl2;bmi-1+/− mice, which were subsequently crossed with bmi-1+/− mice to generate SV–bcl2;bmi-1−/− and Eμ–bcl2;bmi-1−/− mice and control littermates. The generation of bmi-1−/−;ink4a–ARF−/− mice has been described elsewhere (Jacobs et al. 1999). All mice have been maintained on a FVB background and genotyped routinely by PCR or Southern blot analysis.
For the proviral-tagging experiment, newborn mice were injected with 50 μl of 104–105 infectious units of MoMLV clone 1A (Jaenisch et al. 1975), and sacrificed when they became terminally ill.
Flow cytometry and Annexin-V staining
Flow-cytometric analysis was performed on single-cell suspensions from thymus, spleen, and bone marrow, after staining the cells under standard conditions with directly fluorochrome-conjugated monoclonal antibodies (Pharmigen) against CD3ε (145-2C11), CD4 (RM4-5), CD8a (53-6.7), CD45R/B220 (RA3-6B2), TCRβ (H57-597), and sIgM (Biosource; LO-MM-9-F). For apoptosis analysis, thymocytes were stained immediately after isolation with CD4 and Annexin-V antibodies, whereas bone marrow cells were cultured for 24 hr in 10% FBS/RPMI medium in the presence of 50 μm β-mercaptoethanol before staining with B220 and Annexin-V antibodies according to the instructions of the supplier (Boehringer).
Cell culture and retroviral infection
MEFs were isolated as described previously (Jacobs et al. 1999), with the modification that fetal tissue was incubated for 45 min at 37°C in 400 μl of trypsin/EDTA prior to dissociation. MEFs were split 1:4 and maintained in DMEM (GIBCO) supplemented with 10% FBS (PAA). Retroviral infections with LZRS-iresGFP viruses and growth curves with crystal violet staining were performed as described (Jacobs et al. 1999). The use of high titer LZRS–iresGFP viruses ensures 100% infection efficiency without the need for drug selection of cells that can easily be checked for by analysis of GFP expression. For Myc, two versions were used, LZRS–mycHA–iresGFP and LZRS–mycER–iresGFP. The latter results in expression of inactive MycER fusion protein that can be induced into the active conformation by addition of 4-hydroxy tamoxifen (4-OHT) (Littlewood et al. 1995). However, this system is a bit leaky, resulting in considerable Myc activity in the absence of 4-OHT, which can be further increased by adding 4-OHT.
Growth curves, apoptosis, and soft agar assay
For growth curves, cells were plated into 12-well dishes and the number of cells was determined each day by crystal violet staining as described (Jacobs et al. 1999). For analysis of Myc-induced cell death, first passage MEFs were infected with control empty LZRS–iresGFP virus or LZRS–bmi-1PY–iresGFP virus for 48 hr; then the cells were split and infected for 48 hr with control-empty LZRS–iresGFP, LZRS–mycHA–iresGFP, or LZRS–mycER–iresGFP virus. Retrovirus-infected cells were subsequently seeded onto 12-well plates in 10% serum containing medium with or without 4-OHT. The next day, the medium was replaced with medium containing 0.1% or 10% serum (with or without 4-OHT), and 24 or 48 hr later adherent and nonadherent cells were pooled and analyzed for cell death by trypan blue exclusion. For measurement of subdiploid DNA content, cells were seeded onto 60-mm dishes, treated as above, and harvested, fixed, stained with propidium iodide, and analyzed by flow cytometry as described (Rowan et al. 1996).
For analysis of growth in semisolid medium, ∼5 × 104 cells were plated per well of a six-well dish in DMEM containing 10% serum and 0.4% low gelling temperature agarose (Sigma).
Western blotting
Cells and tissues were lysed (Jacobs et al. 1999), protein concentration was determined, equal amounts of protein were separated by SDS-PAGE, and blotted onto nitrocellulose or, for detection of p19arf, onto immobilon-P (Amersham) membranes. Western blot analysis was done according to standard methods with enhanced chemiluminescence (Amersham ). A list of antibodies used is available on request.
Acknowledgments
We thank M. van der Valk for histological analysis and L. Rijswijk, N. Bosnie, and C. Friedrich for animal care. Thanks to G.I. Evan and H. Hermeking for providing Myc–MER and HA–MYC fusion constructs, and to A. Strasser and S.J. Korsmeyer for providing Eμ–bcl-2-36 and Eμ–bcl-2 transgenic mice, respectively. LZRS-viral constructs and PHOENIX packaging cell lines were kindly provided by Dr. G.I. Nolan. We thank C.J. Sherr for communicating results prior to publication.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Lohuizen@nki.nl; FAX 31-20-512 1954.
References
- Akashi K, Kondo M, von Freeden-Jeffry U, Murray R, Weissman I. Bcl-2 rescues T lymphopoieses in Interleukin-7 receptor-deficient mice. Cell. 1997;89:1033–1041. doi: 10.1016/s0092-8674(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Alkema MJ, Jacobs H, van Lohuizen M, Berns A. Perturbation of B and T cell development and predisposition to lymphomagenesis in Eμ-Bmi1 transgenic mice require the Bmi1 RING finger. Oncogene. 1997;15:899–910. doi: 10.1038/sj.onc.1201262. [DOI] [PubMed] [Google Scholar]
- Assoian RK. Anchorage-dependent cell cycle progression. J Cell Biol. 1997;136:1–4. doi: 10.1083/jcb.136.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonette RP, Echeverri F, Mahboubi A, Green DR. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature. 1992;359:522–554. doi: 10.1038/359552a0. [DOI] [PubMed] [Google Scholar]
- Blyth K, Terry A, O'Hara M, Baxter EW, Campbell M, Stewart M, Donehower LA, Onions DE, Neil JC, Cameron ER. Synergy between a human c-myc transgene and p53 null genotype in murine thymic lymphomas; contrasting effects of homozygous and heterozygous p53 loss. Oncogene. 1995;10:1717–1723. [PubMed] [Google Scholar]
- Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes & Dev. 1993;7:671–682. doi: 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- de Stanchina E, McCurrach ME, Zindy F, Shieh S-Y, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19arf tumor suppressor. Genes & Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen, C.M., J.D. Weber, M.F. Roussel, C.J. Sherr, and J.L. Cleveland. 1999. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes & Dev. (This paper) [DOI] [PMC free article] [PubMed]
- Elson A, Deng C, Campos-Torres J, Donehower LA, Leder P. The MMTV/c-myc transgene and p53 null alleles collaborate to induce T-cell lymphomas, but not mammary carcinomas in transgenic mice. Oncogene. 1995;11:181–190. [PubMed] [Google Scholar]
- Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Water CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Facchini LM, Penn LZ. The molecular role of Myc in growth and transformation: Recent discoveries lead to new insights. FASEB J. 1998;12:633–651. [PubMed] [Google Scholar]
- Fanidi A, Harrington EA, Evan GI. Cooperative interaction between c-myc and bcl-2 proto-oncogenes. Nature. 1992;359:554–556. doi: 10.1038/359554a0. [DOI] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci. 1999;96:3940–3944. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The Eμ-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167:353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in Eμ-myc transgenic mice. Cell. 1991;65:753–763. doi: 10.1016/0092-8674(91)90383-a. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Bath ML, Harris AW, Adams JM. bmi-1 transgene induces lymphomas and collaborates with myc in tumorigenesis. Oncogene. 1993;8:3161–3164. [PubMed] [Google Scholar]
- Henriksson M, Luscher B. Proteins of the Myc network: Essential regulators of cell growth and differentiation. Adv Cancer Res. 1996;68:109–182. doi: 10.1016/s0065-230x(08)60353-x. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Eick D. Mediation of c-myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- Hsu B, Marin MC, El-Naggar AK, Stephens LC, Brisbay S, McDonnel TJ. Evidence that c-myc mediated apoptosis does not require wild-type p53 during lymphomagenesis. Oncogene. 1995;11:175–179. [PubMed] [Google Scholar]
- Hueber A-O, Zörnig M, Lyon D, Suda T, Nagata S, Evan GI. Requirement for the CD95 receptor-ligand pathway in c-Myc induced apoptosis. Science. 1997;278:1305–1309. doi: 10.1126/science.278.5341.1305. [DOI] [PubMed] [Google Scholar]
- Jacobs JJL, van Lohuizen M. Cellular memory of transcriptional states by Polycomb-group proteins. Sem Cell Dev Biol. 1999;10:227–235. doi: 10.1006/scdb.1999.0304. [DOI] [PubMed] [Google Scholar]
- Jacobs JJL, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Fan H, Croker B. Infection of preimplantation mouse embryos and of newborn mice with leukemia virus: Tissue distribution of viral DNA and RNA and leukemogenesis in the adult animal. Proc Natl Acad Sci. 1975;72:4008–4012. doi: 10.1073/pnas.72.10.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19arf. Cell. 1997;91:641–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kauffman-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncigenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- Langdon WY, Harris AW, Cory S, Adams J. The c-myc oncogene perturbs B lymphocyte development in Eμ-myc transgenic mice. Cell. 1986;47:11–18. doi: 10.1016/0092-8674(86)90361-2. [DOI] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraskovsky E, O'Reilly LA, Teepe M, Corcoran LM, Peschon JJ, Strasser A. Bcl-2 can rescue T lymphocyte development in Interleukin-7 receptor-deficient mice but not in rag-1−/− mice. Cell. 1997;89:1011–1019. doi: 10.1016/s0092-8674(00)80289-5. [DOI] [PubMed] [Google Scholar]
- McCormack SJ, Weaver Z, Deming S, Natarajan G, Torri J, Johnson MD, Livanage M, Ried T, Dickson RB. Myc/p53 interactions in transgenic mouse mammary development, tumorigenesis and chromosomal instability. Oncogene. 1998;16:2755–2766. doi: 10.1038/sj.onc.1201804. [DOI] [PubMed] [Google Scholar]
- McDonnell TJ, Nunez G, Platt FM, Hockenberry D, London L, McKearn JP, Korsmeyer SJ. Deregulated Bcl-2-immunoglobulin transgene expands a resting but responsive immunoglobulin M and D-expressing B-cell population. Mol Cell Biol. 1990;10:1901–1907. doi: 10.1128/mcb.10.5.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill G, Shimamura A, Bates RC, Savage RE, Fisher DE. Loss of matrix adhesion triggers rapid transformation-selective apoptosis in fibroblasts. J Cell Biol. 1997;138:901–911. doi: 10.1083/jcb.138.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit CE, Tersak JM, Prochownik EV. Myc oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee H-W, et al. The Ink4a Tumor suppressor gene product p19arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Prasad VS, LaFond RE, Zhou M, Jacobsen KA, Osmond DG, Sidman CL. Upregulation of endogenous p53 and induction of in vivo apoptosis in B-lineage lymphomas of Eμ-myc transgenic mice by deregulated c-myc transgene. Mol Carcinogen. 1997;18:66–77. doi: 10.1002/(sici)1098-2744(199702)18:2<66::aid-mc2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Prendergast GC. Mechanisms of apoptosis by c-Myc. Oncogene. 1999;18:2967–2987. doi: 10.1038/sj.onc.1202727. [DOI] [PubMed] [Google Scholar]
- Rowan S, Ludwig RL, Haupt Y, Bates S, Lu X, Oren M, Vousden KH. Specific loss of apoptotic but not cell-cycle arrest function in a human tumor derived p53 mutant. EMBO J. 1996;15:827–838. [PMC free article] [PubMed] [Google Scholar]
- Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–F177. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- Sakamuro D, Eviner V, Elliott K, Showe L, White W, Prendergast GC. c-Myc induces apoptosis in epithelial cells by both p53-dependent and p53-independent mechanisms. Oncogene. 1995;11:2411–2418. [PubMed] [Google Scholar]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H-W, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. 1999;9:22–30. doi: 10.1016/s0959-437x(99)80004-5. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Tumor surveillance via the ARF-p53 pathway. Genes & Dev. 1998;12:2984–2991. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumors induced in transgenic mice by cooperation between myc and bcl2. Nature. 1990a;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Vaux DL, Webb E, Bath ML, Adams JM, Cory S. Abnormalities of the immune system induced by dysregulated bcl-2 expression in transgenic mice. Curr Topics Microbiol Immun. 1990b;166:175–181. doi: 10.1007/978-3-642-75889-8_22. [DOI] [PubMed] [Google Scholar]
- van der Lugt NMT, Domen J, Linders K, van Roon M, Robanus-Maandag E, te Riele H, van der Valk M, Deschamps J, Sofroniew M, van Lohuizen M, et al. Posterior transformation, neurological abnormalities, and severe hematopoeitic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes & Dev. 1994;8:757–769. doi: 10.1101/gad.8.7.757. [DOI] [PubMed] [Google Scholar]
- van lohuizen M. Functional analysis of mouse Polycomb group genes. Cell Mol Life Sci. 1998;54:71–79. doi: 10.1007/s000180050126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— The trithorax-group and Polycomb-group chromatin modifiers: Implications for disease. Curr Opin Genet Dev. 1999;9:355–361. doi: 10.1016/s0959-437x(99)80053-7. [DOI] [PubMed] [Google Scholar]
- van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A. Identification of cooperating oncogenes in Eμ-myc transgenic mice by provirus tagging. Cell. 1991;65:737–752. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- Verbeek S, van Lohuizen M, van der Valk M, Domen J, Kraal G, Berns A. Mice bearing the Eμ-myc and Eμ-pim-1 transgenes develop pre-B-cell leukemia prenatally. Mol Cell Biol. 1991;11:1176–1179. doi: 10.1128/mcb.11.2.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AJ, Kokonitis JM, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21 waf1/cip1. Genes & Dev. 1994;8:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nature Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16ink4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signalling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]