

Abstract
The INK4a/ARF locus encodes upstream regulators of the retinoblastoma and p53 tumor suppressor gene products. To compare the impact of these loci on tumor development and treatment response, the Eμ–myc transgenic lymphoma model was used to generate genetically defined tumors with mutations in the INK4a/ARF, Rb, or p53 genes. Like p53 null lymphomas, INK4a/ARF null lymphomas formed rapidly, were highly invasive, displayed apoptotic defects, and were markedly resistant to chemotherapy in vitro and in vivo. Furthermore, INK4a/ARF−/− lymphomas displayed reduced p53 activity despite the presence of wild-type p53 genes. Consequently, INK4a/ARF and p53 mutations lead to aggressive tumors by disrupting overlapping tumor suppressor functions. These data have important implications for understanding the clinical behavior of human tumors.
Keywords: INK4a/ARF locus, lymphomagenesis, chemo resistance, p53
Mutations in the p53 tumor suppressor gene and at the INK4a/ARF locus are the two most frequent genetic lesions identified in human tumors (for reviews, see Haber 1997; Ruas and Peters 1998). p53 is a sequence-specific DNA-binding protein that can induce cell-cycle arrest or apoptosis in response to pathological insults such as DNA damage and expression of mitogenic oncogenes (Kastan et al. 1991, 1992; Lowe and Ruley 1993; Hermeking and Eick 1994; Serrano et al. 1997; for reviews, see Giaccia and Kastan 1998; Prives 1998). As a consequence, inactivation of p53 can promote oncogenic transformation and resistance to many anticancer agents (for reviews, see Giaccia and Kastan 1998; Prives 1998; Wallace-Brodeur and Lowe 1999). The INK4a/ARF locus encodes two tumor suppressors, designated p16INK4a and p19ARF. p16INK4a is a cyclin-dependent kinase inhibitor that acts upstream of the retinoblastoma (Rb) protein to promote cell-cycle arrest (Serrano et al. 1993; for reviews, see Haber 1997; Ruas and Peters 1998). p19ARF is translated in an alternative reading frame from p16INK4a and activates p53 by interfering with its negative regulator, Mdm2 (Kamijo et al. 1998; Pomerantz et al. 1998; Stott et al. 1998; Zhang et al. 1998; see also Tao and Levine 1999; Weber et al. 1999; Zhang and Xiong 1999). Consequently, INK4a/ARF mutations can disable both the Rb and p53 tumor suppressor pathways.
Recent studies indicate that p19ARF acts as an essential intermediate in oncogene signaling to p53 (Bates et al. 1998; de Stanchina et al. 1998; Palmero et al. 1998; Pomerantz et al. 1998; Zindy et al. 1998; for review, see Sherr 1998). For example, oncogenes such as E1A or c-myc induce ARF message and protein in normal mouse embryo fibroblasts, which correlates with their ability to activate p53 and promote apoptosis. In contrast, these oncogenes fail to activate p53 in ARF–null cells, and promote proliferation without substantial apoptosis (de Stanchina et al. 1998; Zindy et al. 1998). Together, these studies indicate that p19ARF acts as part of a p53-dependent fail-safe mechanism to counter hyperproliferative signals. Interestingly, p19ARF is not induced by DNA damage (Kamijo et al. 1997; Stott et al. 1998) but can cooperate with DNA damaging agents to induce apoptosis in oncogene expressing cells (de Stanchina et al. 1998). These studies predict that disruption of ARF, or the INK4a/ARF locus, should cooperate with mitogenic oncogenes during tumor development, in part, by disabling p53.
p53 mutations have been associated with aggressive cancers, poor prognosis, and drug resistance in human patients (Schmitt and Lowe 1999; Wallace-Brodeur and Lowe 1999). In principle, tumors with INK4a/ARF mutations might also display aggressive characteristics owing to extragenic defects in the p53 pathway. To test this, we examined the impact of INK4a/ARF mutations on tumor development and therapy using the Eμ–myc transgenic mouse. These mice constitutively express c-Myc in the B-cell lineage and develop B-cell lymphoma with associated leukemia (Adams et al. 1985; Adams and Cory 1991). This model was chosen for several reasons. First, because Myc induces p19ARF and activates p53 in cultured fibroblasts (Zindy et al. 1998), Eμ–myc transgenic mice provide a relevant setting for comparing the impact of INK4a/ARF and p53 mutations on tumor behavior. Second, Eμ–myc lymphomas/leukemias are easily monitored by lymph-node palpation or blood smears, a property that facilitates studies examining tumor responses to therapy. Finally, essentially pure tumor cells can be isolated from lymph nodes and studied ex vivo or expanded in genetically matched nontransgenic recipients. The tractable nature of this model is in stark contrast to human systems, which suffer from difficulties in obtaining well-characterized and comparable clinical material.
Results
Loss of the INK4a/ARF locus accelerates lymphomagenesis similarly to loss of p53
To generate lymphomas with defined alterations, we crossed the Eμ–myc transgenic to mice heterozygous for germ-line deletions in the Rb (Rb+/−), INK4a/ARF (INK4a/ARF+/−), or p53 (p53+/−) genes (Jacks et al. 1992, 1994; Serrano et al. 1996). Of note, the INK4a/ARF+/− animals harbor deletions that disrupt both p19ARF and p16INK4a, thereby recapitulating the common gross deletions seen in human tumors (Haber 1997; Ruas and Peters 1998). The onset of Eμ–myc lymphomas in Rb+/− animals was variable (Fig. 1A; green, b) and only slightly accelerated relative to that observed in the wild-type background (hereafter referred to as control) (Fig. 1A; black, a). In contrast, the onset of Eμ–myc lymphomas in INK4a/ARF+/− and p53+/− animals (Fig. 1A; blue, c, and red, d) was highly reproducible and greatly accelerated compared with controls (P < 0.0001 each); the timing of lymphoma development in INK4a/ARF+/− and p53+/− mice was virtually identical. Cell surface staining confirmed that all lymphomas were of the B-cell lineage (B220+; Thy1.2−), whereas the distribution of pre-B (IgM−) and B (IgM+) was similar between the genotypes.
Figure 1.
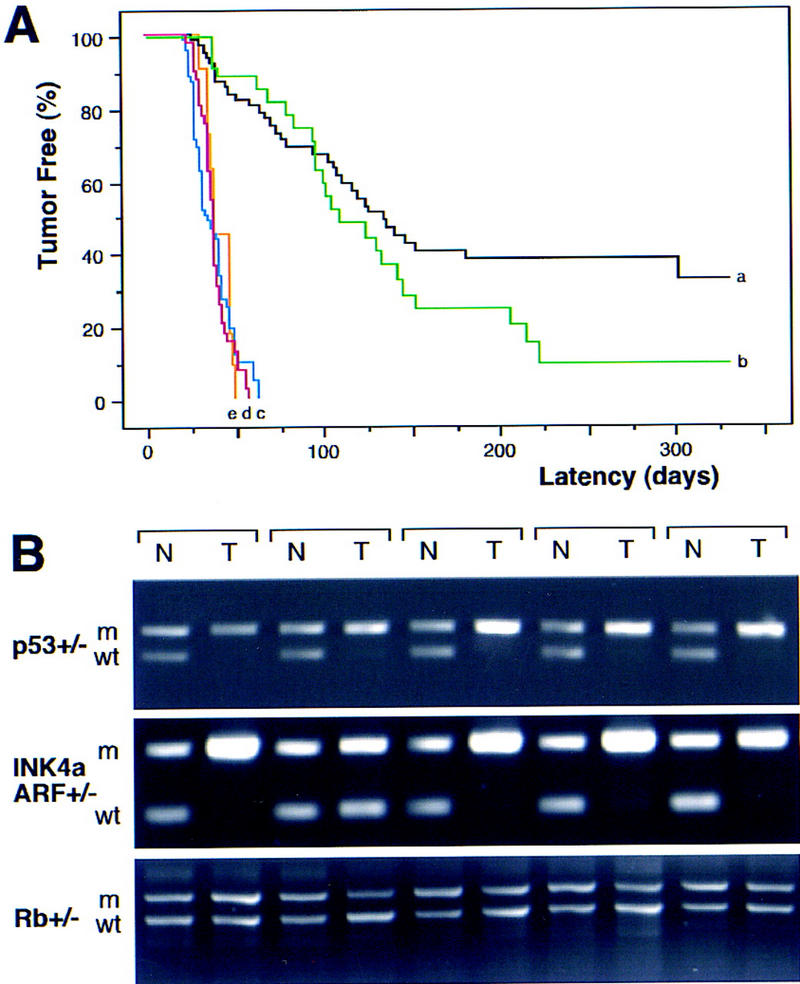
Tumor development in Eμ–myc transgenic mice. (A) Lymphoma incidence in Eμ–myc transgenic mice in the wild-type (control) background (n = 65; black, a) and in mice heterozygous for Rb (n = 39; green, b), INK4a/ARF (n = 41; blue, c), p53 (n = 73; red, d), and INK4a/ARF; p53 double heterozygotes (n = 11; orange, e). By day 70, all p53+/− and INK4a/ARF+/− mice developed lymphoma, whereas >75% of Rb+/− and control mice remained tumor free. (B) Matched normal (N) and tumor (T) DNA were isolated from tail and lymph nodes and analyzed by allele-specific PCR for the targeted gene [(m) mutated allele; (wt) wild-type allele]. Shown are results from five Eμ–myc transgenic mice in each genetic background. Note that tumors arising in the INK4a/ARF+/−; p53+/− double heterozygotes invariably lost the wild-type p53 allele but never the INK4a/ARF allele.
These data imply that p53 and products of the INK4a/ARF locus limit Myc-induced lymphomagenesis. Concordantly, Eμ–myc lymphomas arising in the p53+/− and INK4a/ARF+/− animals invariably lost the wild-type p53 or INK4a/ARF allele (93.8% and 88.2%, respectively) (Fig. 1B). Hence, these lymphomas were either p53-null (p53−/−) or INK4a/ARF-null (INK4a/ARF−/−). Virtually all control (6 out of 7), Rb+/− (4 out of 4), and INK4a/ARF−/− (9 out of 9) tumors retained wild-type p53 as indicated by RT–PCR and sequencing of p53 exons 4–8 (data not shown). The one p53 mutation identified (H190R in mouse; H193R in human) corresponds to a mutation observed in B-cell leukemias and a Burkitt's lymphoma (Beroud and Soussi 1998). Deletions of the INK4a/ARF locus were noted in 20% of control and Rb+/− tumors (4 out of 20) but never in p53−/− tumors (0 out of 10) (data not shown). In no instance did lymphomas arising in Rb+/− animals lose the wild-type Rb allele (Fig. 1B), confirming that Rb loss has a minimal impact on Myc-induced lymphomagenesis. Because Rb and p16INK4a loss should produce similar phenotypes (Haber 1997; Ruas and Peters 1998), these data imply that p19ARF is crucial for suppressing Myc-induced lymphomagenesis. The onset of Eμ–myc lymphomas is markedly accelerated in ARF-deficient mice (Eischen et al. 1999).
Loss of INK4a/ARF or p53 promotes lymphoma spreading into visceral organs
INK4a/ARF−/− and p53−/− lymphomas were highly invasive and infiltrated into various nonlymphoid organs. For example, in mice bearing INK4a/ARF−/− and p53−/− lymphomas, we observed extensive periportal invasion and spreading of lymphoma cell clusters throughout the liver parenchym and massive malignant pulmonary infiltration as consolidated aggregation of large mononuclear cells and within the distended interstitial capillaries (Fig. 2). Also, neoplastic cells accumulated in the submucosa of the urinary bladder, within the kidneys, in the serosal and mesenteric surfaces of the gasterointestinal tract, and along the meninges. In contrast, tumors in mice bearing control or Rb+/− lymphomas showed little systemic infiltration or remained localized to the lymph nodes and blood compartment despite a similarly large tumor burden. The invasive behavior of INK4a/ARF−/− and p53−/− tumors was reproduced following transplantation of the tumors into syngenic recipients (data not shown) and is indicative of a highly aggressive disease.
Figure 2.
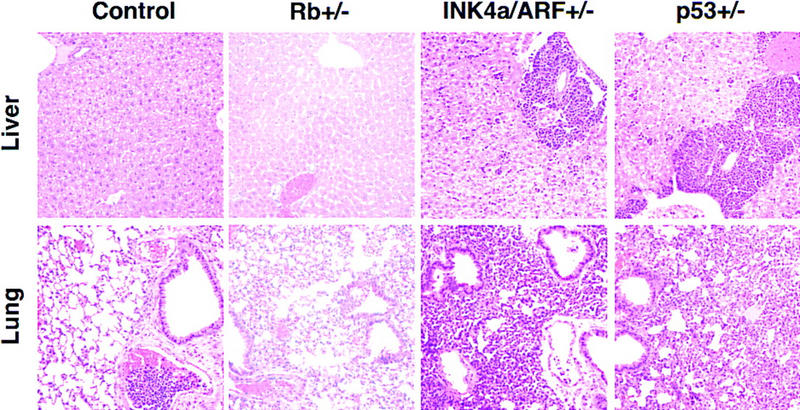
Invasiveness of Eμ–myc lymphomas in liver and lung (H.E. staining, 200×). Representative examples of control, Rb+/−, INK4a/ARF−/−, and p53−/− lymphomas are shown. Note the malignant embolus in the pulmonary vessel of the control—nonetheless, the lung itself remained tumor free. The relative congestion of the Rb+/− lung is a postmortem artifact.
INK4a/ARF−/− lymphomas show an apoptotic defect but no genomic instability
p53 mutations can decrease cell death, increase proliferation, and produce chromosomal instability depending on context (Schmitt and Lowe 1999; Wallace-Brodeur and Lowe 1999). To determine the impact of INK4a/ARF and p53 mutations on these characteristics, we examined apoptosis, mitotic index, and DNA content in control, INK4a/ARF−/− and p53−/− lymphomas. As revealed by histological staining and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling), control lymphomas contained large numbers of apoptotic cells that clustered (Fig. 3A). Apoptosis was much reduced in INK4a/ARF−/− or p53−/− lymphomas, and the apoptotic cells that appeared were isolated. Furthermore, primary INK4a/ARF−/− and p53−/− lymphoma cells explanted into culture survived much better than controls (Fig. 3B). The mitotic index (Fig. 3C) and S-phase fraction (Fig. 3D) of all lymphoma types analyzed were similar, implying that INK4a/ARF or p53 mutations did not affect the proliferation rate. DNA content analysis revealed one notable difference: Whereas most of the p53−/− tumors were aneuploid (10 out of 12), most control and INK4a/ARF−/− tumors remained diploid (13 out of 14 and 13 out of 14, respectively) (Fig. 3D). Together, these data demonstrate that highly aggressive lymphomas can occur in the absence of chromosomal instability and imply that the aggressive nature of INK4a/ARF−/− and p53−/− lymphomas is due to an apoptotic defect.
Figure 3.
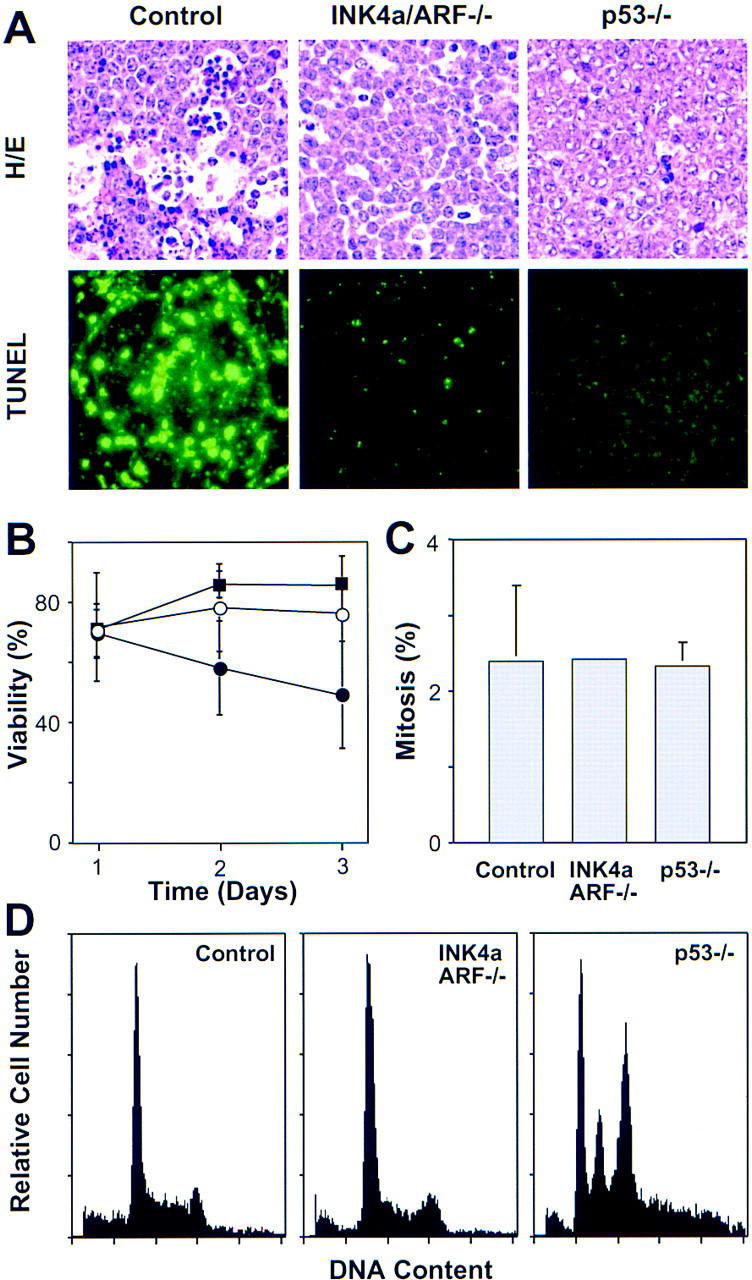
Analysis of apoptosis, proliferation, and chromosomal stability in Eμ–myc lymphomas. (A) Apoptosis in situ (lymph nodes) was visualized by HE staining and TUNEL. The reduced apoptotic rate observed in INK4a/ARF−/− tumors is consistent with a similar defect observed in the ocular lens of Rb−/−; INK4a/ARF−/− embryos (Pomerantz et al. 1998). (B) Viability of control (●), INK4a/ARF−/− (○), and p53−/− (█) lymphoma cells as measured by trypan blue exclusion after explanting onto feeder cells. (C) Proliferation as estimated by the percentage of mitotic figures in HE-stained lymphoma sections. (D) DNA content analysis of primary Eμ–myc lymphoma. The S-phase fractions of control (30.25% ± 8.31, n = 9) and INK4a/ARF−/− (29.98% ± 6.39, n = 10) were virtually identical, whereas subG1 fractions of control (3.26% ± 3.17) and INK4a/ARF−/− (0.30% ± 0.56) lymphomas were significantly different (P = 0.0097). Note that sub-G1 assessment recognizes only late apoptotic cells and gives lower estimates than TUNEL. Calculations of S-phase and sub-G1 fraction in p53−/− lymphomas were impossible due to aneuploidy. Representative profiles are shown. Note that low frequency of aneuploidy in control and INK4a/ARF−/− lymphomas (1 of 14 and 1 of 14, respectively) is consistent with the overall p53 mutation rate we observed in these tumors.
INK4a/ARF mutations compromise p53 function in vivo
The remarkable similarities between INK4a/ARF−/− and p53−/− lymphomas suggest that these mutations disrupt overlapping tumor suppressor functions. In agreement, Eμ–myc lymphomas arising in mice heterozygous for both genes (INK4a/ARF+/−; p53+/−) were detected at the same time as lymphomas in the INK4a/ARF+/− and p53+/− animals (50th percentile = 38 vs. 38 vs. 36 days to onset, respectively) (Fig. 1A; orange, e) and never displayed coincident loss of both wild-type INK4a/ARF and p53 alleles (data not shown). Therefore, inactivation of both loci produces no additional advantage to Eμ–myc lymphomas. In cultured fibroblasts, Myc activates p53 in an ARF-dependent manner (Zindy et al. 1998). Similarly, control Eμ–myc lymphomas displayed a variable but consistent increase in p53 levels and activity (as measured by expression of the p53 target p21) compared with normal splenocytes (Fig. 4A, cf. N with control). This increase appeared dependent on the INK4a/ARF locus, because INK4a/ARF−/− tumors displayed only a modest induction of p53 and virtually no increase in p21 (Fig. 4A). This implies that INK4a/ARF mutations can accelerate tumor progression and impair apoptosis by compromising p53 function.
Figure 4.


p53 levels and activity in untreated and CTX-treated Eμ–myc lymphomas. (A) Control (three independent tumors), INK4a/ARF−/− (four independent tumors), and p53−/− lymphoma lysates were probed against p53 and the p53 downstream target p21, reflecting p53's activity. Normal splenocytes (N) from nontransgenic mice were used for comparison. Tubulin (Tub) was used to verify protein loading. (B) Control, INK4a/ARF−/−, and p53−/− lymphoma cells were isolated from lymph nodes of untreated animals (NT) or 4 hr after CTX treatment (T1 and T2) and analyzed as above. For each tumor type, T1 and T2 were derived from separate primary tumors, whereas NT and T1 represent reconstituted lymphomas derived from the same primary tumor.
INK4a/ARF mutations reduce p53 activation following chemotherapy
The fact that p19ARF can cooperate with DNA-damaging agents to induce p53 and apoptosis raises the possibility that INK4a/ARF mutations might compromise cancer therapy (de Stanchina et al. 1998). To test this, we examined the impact of INK4a/ARF or p53 mutations on drug-induced responses in reconstituted lymphomas or following short-term culture. Reconstituted lymphomas were produced following intravenous injection of primary lymphoma cells into syngenic (nontransgenic) recipients, thereby eliminating the possibility that secondary malignancies might complicate scoring tumor responses. Importantly, these lymphomas were histopathologically identical to their respective primary tumors (data not shown). In control lymphomas, p53 and p21 levels were dramatically increased 4 hr after treatment with cyclophosphamide (CTX), an alkylating agent used to treat human leukemia and lymphoma (Fig. 4B). p53 and p21 levels also increased in INK4a/ARF−/− lymphomas, although this response was consistently reduced compared with controls. Therefore, in Eμ–myc lymphoma cells, INK4a/ARF mutations can reduce p53 activation by a DNA damaging agent.
INK4a/ARF mutations affect the short-term response to anticancer treatment
Loss of either INK4a/ARF or p53 had a profound effect on drug-induced cell death in vitro and in vivo. In short-term cultures, INK4a/ARF−/− or p53−/− lymphomas displayed a marked resistance to mafosphamide (a CTX analog active in vitro) (Fig. 5A). In peripheral blood, control animals harboring associated leukemias displayed a nearly 100-fold reduction in the white blood cell count (WBC) within 4 hr of CTX therapy, which coincided with a 6- to 10-fold accumulation of apoptotic cells (Fig. 5B,C). In contrast, INK4a/ARF−/− and p53−/− leukemias took 12–24 hr to achieve a similar reduction. Apoptosis was not detectable, perhaps because the slow death rate allowed clearance of apoptotic cells before they could accumulate. In lymph nodes, control lymphomas displayed massive apoptosis 5 hr after CTX therapy, whereas the INK4a/ARF−/− and p53−/− lymphomas displayed substantially fewer dying cells (Fig. 5D).
Figure 5.
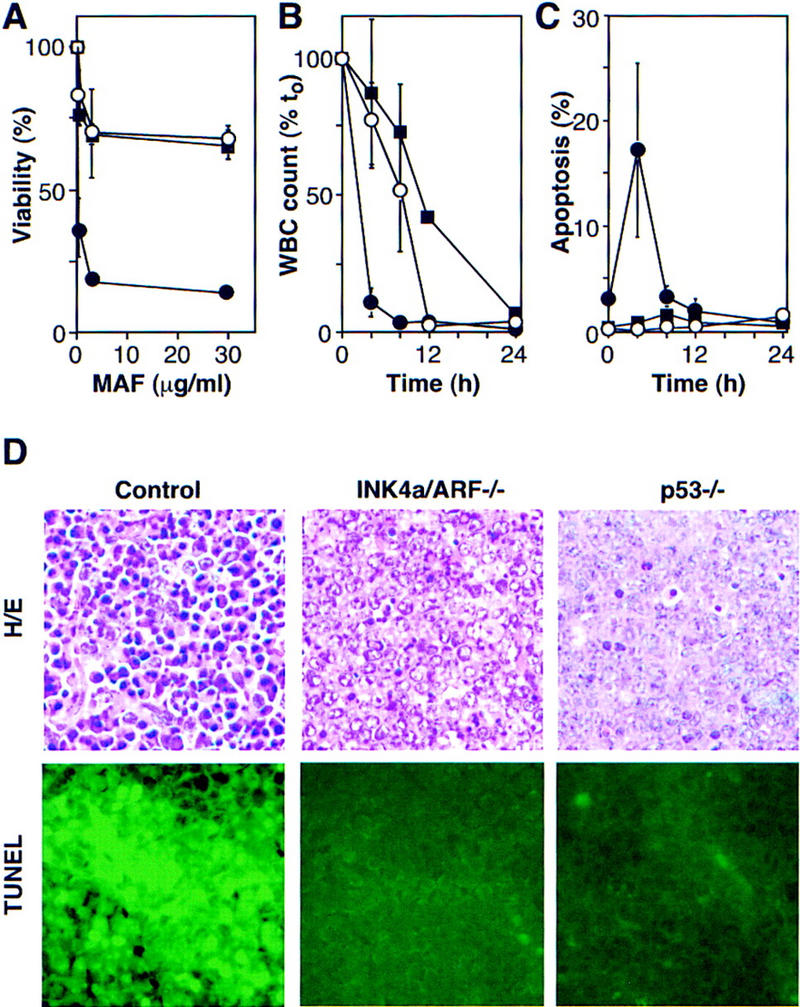
INK4a/ARF, p53, and short-term response to chemotherapy. (A) Explanted control (●), INK4a/ARF−/− (○), and p53−/− (█) lymphoma cells were treated with mafosphamide (MAF). Viability was analyzed after 24 hr by trypan blue exclusion; each value was normalized to untreated controls and represents the mean ± s.d. of two independently derived tumors reproduced in duplicate. (B) Leukemic mice were treated with CTX, and blood samples were taken at the indicated times. Each WBC is relative to its pretreatment value and represents the mean ± s.d. of three independent leukemias. Symbols are as in A. (C) Same as in B, except that blood samples were ethanol-fixed and stained with the DNA fluorochrome DAPI to visualize the chromatin condensation characteristic of apoptotic cells. Each value reflects the percentage of cells with apoptotic morphology (of 200 cells counted) and represents the mean ± s.d. of three independent leukemias. Symbols are as in A. (D) HE staining and TUNEL of lymph nodes harboring control, INK4a/ARF−/−, and p53−/− lymphomas 5 hr after CTX treatment.
INK4a/ARF mutations impair the long-term response to anticancer treatment
The ultimate determinant of drug-induced cell kill is tumor regression and the duration of remission. To assess long-term responses, animals harboring control, INK4a/ARF−/−, or p53−/− lymphomas were treated with CTX and monitored for remission and relapse by lymph node palpation and WBC. Control lymphomas responded extremely well to CTX treatment, and >70% remained in remission during the 100-day observation period (Fig. 6; black, a). In stark contrast, INK4a/ARF−/−- and p53−/−null tumors displayed an extremely poor response to CTX therapy: Despite initial responses, only 1 out of 14 p53−/− and 4 out of 35 INK4a/ARF−/− lymphomas remained in remission. p53−/− tumors (Fig. 6; red, c) were the most relapse prone (50th percentile = 20 days in remission, P < 0.0001 compared with control), although the defect in the INK4a/ARF−/− response (Fig. 6; blue, b) was also highly significant (50th percentile = 28 days in remission, P = 0.0053 compared with control). The response of INK4a/ARF+/−;p53−/− double-mutant lymphomas (Fig. 6; orange, d) was virtually identical to the p53−/− tumors (50th percentile = 20 days in remission), and the relapsed tumors never displayed loss of the wild-type INK4a/ARF allele (data not shown). Therefore, although INK4a/ARF mutations promote chemoresistance in the presence of wild-type p53 genes, they confer no additional survival advantage once p53 is mutated. These data demonstrate that INK4a/ARF mutations can compromise therapy, at least in part, by disabling p53.
Figure 6.
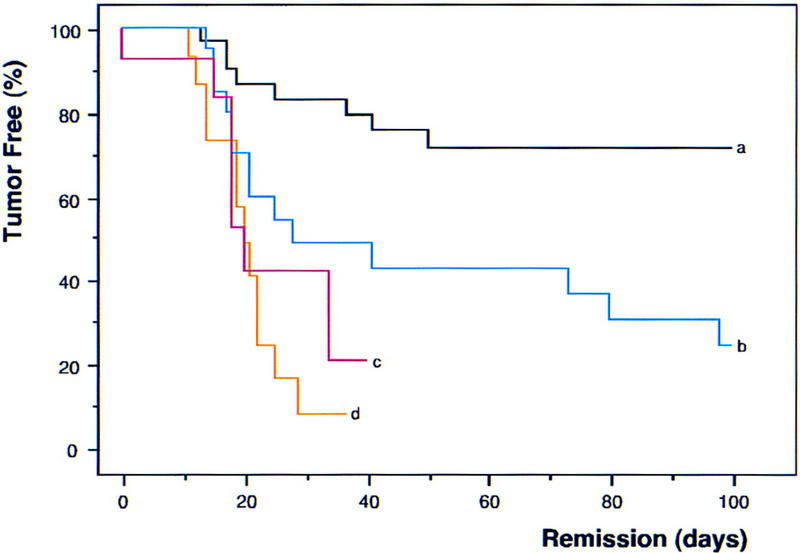
INK4a/ARF, p53, and long-term response to chemotherapy. Nontransgenic mice harboring reconstituted control (n = 60; black, a), INK4a/ARF−/− (n = 35; blue, b), p53−/− (n = 14; red, c), and INK4a/ARF+/−; p53−/− (n = 15; orange, d) lymphomas were treated with CTX and monitored for tumor regression and relapse. Importantly, CTX is not affected by classic multidrug resistance mechanisms that might complicate drug delivery. Tumor shrinkage to nonpalpability within 6 days after treatment is considered ‘remission’ and creates the tumor-free population at time 0. Relapse was defined by recurrent palpable lymph node enlargement. Values were plotted in Kaplan-Meier survival curve format and presented as percentage of mice in remission over the time post-therapy. Note that the overall rate of treatment failure in control lymphomas (∼25%–30%) is consistent with the combined frequency of INK4a/ARF and p53 mutations we observe in these tumors.
Discussion
By comparing the properties of Myc-induced lymphomas in several genetic backgrounds, we provide compelling evidence that INK4a/ARF deletions can impact tumor development and anticancer therapy by compromising p53 function. Like p53−/− tumors, INK4a/ARF−/− lymphomas formed rapidly, were highly invasive, displayed apoptotic defects, and were markedly resistant to chemotherapy. Furthermore, INK4a/ARF−/− lymphomas displayed attenuated p53 activity despite the presence of wild-type p53 genes. The profound impact of INK4a/ARF and p53 mutations on Myc-induced lymphomagenesis indicates that the ARF–p53 pathway contributes to oncogene-induced cell death in developing tumors and underscores the importance of this fail-safe mechanism in tumor suppression (also see Eischen et al. 1999; Jacobs et al. 1999). Furthermore, that INK4a/ARF mutations can compromise drug-induced cell death in Eμ–myc lymphomas implies that cooperative effects between oncogenes (in part via ARF) and DNA damage contribute to the remarkable drug sensitivity of some tumors.
The only substantial difference between INK4a/ARF−/− and p53−/− lymphomas was that the INK4a/ARF−/− lymphomas remained diploid, whereas the p53−/− lymphomas were aneuploid. This pattern is reminiscent of ARF−/− and p53−/− fibroblasts (Kamijo et al. 1997) and implies that p19ARF does not control the p53 functions involved in maintaining chromosome stability. Although we have not analyzed the secondary changes arising in INK4a/ARF−/− tumors in detail, these data argue that invasive, chemoresistant lymphomas can arise in the absence of substantial chromosomal instability. In turn, because INK4a/ARF mutations disable p53, the chromosomal instability observed in p53−/− lymphomas appears dispensable for the aggressive behavior of these tumors.
More likely, the increased invasiveness and drug resistance of INK4a/ARF−/− and p53−/− lymphomas arises from an apoptotic defect. INK4a/ARF−/− and p53−/− lymphomas displayed decreased apoptosis in situ and ex vivo (see Figs. 3 and 5), whereas there was no obvious relationship between tumor-cell genotype and proliferation, as measured by mitotic index in vivo, DNA content analysis ex vivo, and proliferation properties in vitro (see Fig. 3; data not shown). These results stand in contrast to an earlier report indicating that p53-null Eμ–myc lymphomas do not display an apoptotic defect (Hsu et al. 1995). Although we cannot explain this discrepancy, it is worth noting that disruption of apoptosis using a bcl-2 transgene is sufficient to accelerate Myc-induced tumorigenesis (Strasser et al. 1990). Moreover, ectopic expression of bcl-2 in the control lymphoma cells used in this study has no effect on proliferation but renders these cells highly invasive and chemoresistant following transplantation into syngenic mice (C.A. Schmitt and S.W. Lowe, unpubl.).
Although Eμ–myc lymphomas harboring INK4a/ARF or p53 mutations are defective in CTX-induced cell death, CTX therapy induces complete remissions irrespective of p53 status. We suggest that the p53-independent death is due to apoptosis, because Eμ–myc lymphomas expressing Bcl-2 are completely nonresponsive to CTX therapy at the maximally tolerated dose (C.A. Schmitt and S.W. Lowe, unpubl.). In contrast to CTX, doxorubicin fails to induce remissions in p53−/− lymphomas, although high doses induce p53-independent apoptosis in vitro (R.R. Wallace-Brodeur, M.E. McCurrach, and S.W. Lowe, unpubl.). Thus, the ability to achieve p53-independent killing depends on agent and dose. These data are consistent with the view that p53 is not an essential component of the apoptotic machinery but, rather, increases the probability that these agents trigger cell death (Lowe et al. 1993). In Eμ–myc lymphomas, this increased propensity for apoptosis can determine tumor cure or relapse.
Our data have important implications for the understanding of the clinical behavior of human tumors. First, they provide compelling evidence that disruption of apoptosis during tumor development can simultaneously select for chemoresistant cells. This pattern of coselection may explain why some tumors are de novo ‘resistant’ despite having no prior exposure to drug and why it is difficult to separate the impact of p53 mutations on treatment sensitivity from its contribution to overall patient prognosis. Second, our results demonstrate that tumors with extragenic mutations in the p53 pathway can display properties of p53 mutant tumors. This fundamental point is crucial for interpreting studies relating p53 mutations to clinical parameters in human patients, which typically classify tumors strictly by p53 gene or protein status. Our data imply that a substantial number of p53 ‘normal’ tumors would be misclassified by this approach (e.g., those harboring ARF mutations) and may explain why some studies fail to correlate p53 mutations with adverse clinical features (for review, see Brown and Wouters 1999).
This study provides the first evidence that INK4a/ARF mutations can have a negative impact on the outcome of cancer therapy and suggests that this defect arises from the failure of drugs to appropriately activate p53. Consequently, these data predict that disruption of the INK4a/ARF locus will contribute to chemoresistance in human tumors. As for p53 mutations, it seems likely that the overall impact of INK4a/ARF disruption on chemoresistance will depend on additional factors, such as tissue type, agent, and the mutational background of the tumor (for review, see Wallace-Brodeur and Lowe 1999; also see Bunz et al. 1999). However, it is noteworthy that p53 mutations are strongly associated with highly aggressive tumors and chemoresistance in human hematologic malignancies (e.g., see Elrouby et al. 1993; Diccianni et al. 1994; Fan et al. 1994; Wattel et al. 1994; Wilson et al. 1997), indicating that Eμ–myc lymphomas can recapitulate the behavior of human tumors. Therefore, we anticipate that this model will be useful for testing strategies to counter p53 and INK4a/ARF mutations in hematologic malignancies and other cancers.
Materials and methods
Mouse strains and tumor monitoring
All animal protocols used in this study were approved by the Cold Spring Harbor Laboratory Animal Care and Use Committee. Eμ–myc transgenic mice (C57BL/6 inbred strain) and Rb−/−, INK4a/ARF−/−, and p53−/− mice (C57BL/6 × 129/sv) were crossed, and the offspring was genotyped by allele-specific PCR (Jacks et al. 1992, 1994; Serrano et al. 1996). Transgenic mice of the F1 generation (pooled from the different crosses) or transgenics being heterozygous for the named loci were monitored twice a week by palpation of the prescapular and cervical lymph nodes. Enlargements of at least 5 mm in the longest diameter were considered ‘well palpable’ and reflect malignant disease. For determining white blood cell status, blood smears and 20 μl of peripheral blood were obtained by tail artery bleeding. After ammoniumchloride hemolysis of the PBS-diluted and K3–EDTA-anticoagulized blood sample, white blood cells were counted in a hemocytometer. Blood smears were fixed and stained using the Leukostat kit (Fisher Diagnostics). Mice having WBC > 3 × 105/μl and being positive for lymphoblastic cells in the blood stream were considered ‘leukemic’.
Histopathology
Animals harboring control, Rb+/−, INK4a/ARF−/−, and p53−/− lymphomas were sacrificed when prescapular lymph nodes reached a well-palpable size. Paraffin-embedded (7 μm), 4% neutral-buffered formalin-fixed tissue sections derived from lymph nodes and lung and liver specimens were stained with hematoxyilin–eosin (HE) to evaluate apoptotic nuclear morphology and invasiveness of lymphoma cells into visceral organs.
Lymphoma characterization, LOH analysis, and RT–PCR sequencing
After CO2 euthanasia, lymph nodes were dissected, minced in PBS, and filtered through a 35-μm nylon mesh. Single cell suspensions of freshly harvested lymphomas were immunophenotyped by flow cytometry using antibodies directed against Thy-1.2, B220, and IgM (Pharmingen). Pre-B-cell lymphomas are Thy-1.2−, B220+, and IgM−, whereas mature B-cell lymphomas are Thy-1.2−, B220+, and IgM+. To determine the mutational status of various genes, primary lymphoma cells were subjected to short-term culturing to eliminate normal cell contamination. Loss of the remaining wild-type allele [loss of heterozygosity (LOH)] in tumors arising in mice being heterozygous for an indicated tumor suppressor locus was detected by allele-specific PCR (Jacks et al. 1992, 1994; Serrano et al. 1996). Exons 4–8 of the p53 gene were sequenced by dye termination in an automated sequencer (Perkin-Elmer) after reverse transcription (SuperScript, GIBCO BRL) and PCR amplification of lymphoma cell total RNA. Finally, the gross integrity of the INK4a/ARF locus was assessed using PCR of exons 1β and exon 2 in a multiplex PCR reaction harboring primers to a positive control.
Lymphoma cell culture and in vitro treatment
Single cell suspensions of freshly extracted lymphoma cells (see above) were plated on irradiated (30 Gy) feeder layer (106 NIH-3T3 cells/2.4-cm plate) in 45% Iscove's modified Eagle medium, 45% Dulbecco's minimal essential medium, 10% fetal bovine serum, 100 U/ml penicillin and streptomycin, 4 mm l-glutamine, and 25 μm 2-mercaptoethanol. For in vitro drug assays, mafosphamide (cyclohexylammonium salt, a CTX analog active in vitro; a generous gift from Asta Medica, Germany) was added at 0, 0.3, 3, and 30 μg/ml, and viability was measured (see below) 24 hr later.
Assessment of viability, cell-cycle parameters, and apoptosis
Viability of short-term cultured lymphoma cells was analyzed by trypan blue dye exclusion. For analysis of ploidy, apoptosis (as percentage of cells in sub-G1 peak), and proliferation (as percentage of viable cells in S phase), 106 ethanol-fixed lymphoma cells were incubated for 30 min at room temperature in 1 ml of DNA staining solution (200 μg of propidium iodide and 2 mg of RNase in 10 ml of PBS), and DNA content was measured at 488 nm in a flow cytometer (FACScalibur, Becton Dickinson). In situ proliferation was estimated by counting of mitotic figures (cells in anaphase or telophase) relative to cell number in HE-stained lymphoma sections (four samples each genotype, seven different fields, 200 cells each). In situ apoptosis was visualized in lymphoma sections by HE staining and a fluorescence-based TUNEL assay. TUNEL assays were performed in accordance to the manufacturer's protocol (Boehringer Mannheim). Leukemias were analyzed for apoptotic nuclear morphology by fluorescence microscopy after ethanol fixation and DAPI (4‘,6-Diamidino-2-phenylindole) staining of peripheral blood samples.
Western blotting analysis
Whole-cell lymphoma cell or normal splenocyte lysates were generated by lysing of extracted cells in SDS sample buffer (60 mm Tris-HCl at pH 6.8, 10% glycerol, 2% SDS, and 5% 2-mercaptoethanol). Samples corresponding to 60 μg of protein (Bio-Rad Bradford protein assay) were separated on a SDS–polyacrylamide gel and transferred to Immobilon-P membranes (Millipore). p53 was detected using the polyclonal antibody CM5 (Novocastra, 1:2000 dilution), p21 using the polyclonal antibody C-19 (Santa Cruz, 1:500 dilution), and α-tubulin using the monoclonal antibody B-5-1-2 (Sigma, 1:2000 dilution). Protein detection was visualized by ECL (Amersham) or Supersignal (Pierce).
Lymphoma reconstitution and in vivo treatment
Immediately after extraction, 106 lymphoma cells in 100 μl of PBS were reconstituted by tail vein injection into genetically matched, nontransgenic recipient mice (two mice per individual lymphoma sample) to monitor response to treatment. Tumors derived from the INK4a/ARF+/− and p53+/− backgrounds were reconstituted in C57BL/6 × 129/sv mice (Jackson Laboratories). CTX was applied as a single 300-mg/kg dose i.p. when arising tumors became well palpable.
Statistical evaluation
Tumor onset data reflect the time between birth and first-time palpability of enlarged lymph nodes; treatment response data reflect the time between remission and relapse as first-time palpability of a recurrent lymph node enlargement. Individual time values were plotted in the Kaplan-Meier population–event–time course format and compared using the log-rank (Mantel-Cox) test. Comparisons of means and standard deviations (s.d.) were performed using the unpaired t-test. Ploidy, cell cycle distribution, and sub-G1 content were analyzed using the ModFit LT 2.0 software.
Acknowledgments
We thank T. Jacks for the Rb+/− and p53−/− mice; M. Serrano and D. Beach for the INK4a/ARF−/− mice; A. Harris for the Eμ–myc transgenic mice; K. Sokol for histopathology; L. Bianco and the CSHL animal facility for technical assistance; M. Ockler and J. Duffy of the CSHL Graphic Arts facility for help with the artwork; G. Ferbeyre, A. Lin, M. Soengas, and A. Samuelson for editorial advice; and M. Roussel, C. Sherr, and J. Cleveland for discussion of unpublished data. This work was supported by a Dr. Mildred Scheel Cancer Foundation fellowship (C.A.S), a DOD Breast Cancer Research fellowship (E.d.S.), a Kimmel Scholar Award (S.W.L.), and a grant (CA13106) from the National Cancer Institute (S.W.L.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lowe@cshl.org; FAX (516) 367-8454.
References
- Adams JM, Cory S. Transgenic models for haemopoietic malignancies. Biochim Biophys Acta. 1991;1072:9–31. doi: 10.1016/0304-419x(91)90004-5. [DOI] [PubMed] [Google Scholar]
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH. p14ARF links the tumour suppressors RB and p53. Nature. 1998;395:124–125. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- Beroud C, Soussi T. p53 gene mutation: Software and database. Nucleic Acids Res. 1998;26:200–204. doi: 10.1093/nar/26.1.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 1999;59:1391–1399. [PubMed] [Google Scholar]
- Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, Williams J, Lengauer C, Kinzler KW, Vogelstein B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–269. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Stanchina E, McCurrach ME, Zindy F, Shieh SY, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19ARF tumor suppressor. Genes & Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diccianni MB, Yu J, Hsiao M, Mukherjee S, Shao LE, Yu AL. Clinical significance of p53 mutations in relapsed T-cell acute lymphoblastic leukemia. Blood. 1994;84:3105–3112. [PubMed] [Google Scholar]
- Eischen, C.M., J.D. Weber, M.F. Roussel, C.J. Sherr, and J.L. Cleveland. 1999. Disruption of the ARF–Mdm2–p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Elrouby S, Thomas A, Costin D, Rosenberg CR, Potmesil M, Silber R, Newcomb EW. p53 gene mutation in B-cell chronic lymphocytic leukemia is associated with drug resistance and is independent of MDR1/MDR3 gene expression. Blood. 1993;82:3452–3459. [PubMed] [Google Scholar]
- Fan SJ, Eldeiry WS, Bae I, Freeman J, Jondle D, Bhatia K, Fornace AJ, Magrath I, Kohn KW, Oconnor PM. p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging agents. Cancer Res. 1994;54:5824–5830. [PubMed] [Google Scholar]
- Giaccia AJ, Kastan MB. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes & Dev. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- Haber DA. Splicing into senescence: The curious case of p16 and p19ARF. Cell. 1997;91:555–558. doi: 10.1016/s0092-8674(00)80441-9. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Eick D. Mediation of c-myc induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- Hsu B, Marin MC, Elnaggar AK, Stephens LC, Brisbay S, Mcdonnell TJ. Evidence that c-myc mediated apoptosis does not require wild-type p53 during lymphomagenesis. Oncogene. 1995;11:175–179. [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Jacobs, J.J.L., B. Scheijen, J.-W. Voncken, K. Kieboom, A. Berns, and M. van Lohuizen. (1999). Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc induced apoptosis via Ink4a/ARF. Genes & Dev. (This issue). [DOI] [PMC free article] [PubMed]
- Jawhs, J.J.L., B. Scheijen, J.-W. Voncken, K. Kreboom, A. Berns, M. van Lohuizen. 1999. Bmi-1 collaborates with cMyc in tumorigenesis by inhibiting c-Myc induced apoptosis via INK4a/ARF. Genes & Dev. (This issue). [DOI] [PMC free article] [PubMed]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci. 1998;95:8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace A., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE. Stabilization of the p53 tumor suppressor is induced by adenovirus E1A and accompanies apoptosis. Genes & Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:954–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- Palmero I, Pantoja C, Serrano M. p19ARF links the tumour suppressor p53 to Ras. Nature. 1998;395:125–126. doi: 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, Cordon-Cardo C, DePinho RA. The INK4a tumor suppressor gene product, p19ARF, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Prives C. Signaling to p53: Breaking the MDM2-p53 circuit. Cell. 1998;95:5–8. doi: 10.1016/s0092-8674(00)81774-2. [DOI] [PubMed] [Google Scholar]
- Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–F177. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, Lowe SW. Apoptosis and therapy. J Pathol. 1999;187:127–137. doi: 10.1002/(SICI)1096-9896(199901)187:1<127::AID-PATH251>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Tumor surveillance via the ARF–p53 pathway. Genes & Dev. 1998;12:2984–2991. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, Palmero I, Ryan K, Hara E, Vousden KH, Peters G. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17:5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- Tao W, Levine AJ. P19ARF stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci. 1999;96:6937–6941. doi: 10.1073/pnas.96.12.6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace-Brodeur RR, Lowe SW. Clinical implications of p53 mutations. Cell Mol Life Sci. 1999;55:64–75. doi: 10.1007/s000180050270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, Morel P, Fenaux P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994;84:3148–3157. [PubMed] [Google Scholar]
- Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nature Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- Wilson WH, Teruya-Feldstein J, Fest T, Harris C, Steinberg SM, Jaffe ES, Raffeld M. Relationship of p53, bcl-2, and tumor proliferation to clinical drug resistance in non-Hodgkin's lymphomas. Blood. 1997;89:601–609. [PubMed] [Google Scholar]
- Zhang Y, Xiong Y. Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol Cell. 1999;3:579–591. doi: 10.1016/s1097-2765(00)80351-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]