

Abstract
Movement of transposable genetic elements requires the cleavage of each end of the element genome and the subsequent joining of these cleaved ends to a new target DNA site. During Mu transposition, these reactions are catalyzed by a tetramer of four identical transposase subunits bound to the paired Mu DNA ends. To elucidate the organization of active sites within this tetramer, the subunit providing the essential active site DDE residues for each cleavage and joining reaction was determined. We demonstrate that recombination of the two Mu DNA ends is catalyzed by two active sites, where one active site promotes both cleavage and joining of one Mu DNA end. This active site uses all three DDE residues from the subunit bound to the transposase binding site proximal to the cleavage site on the other Mu DNA end (catalysis in trans). In addition, we uncover evidence that the catalytic activity of these two active sites is coupled such that the coordinated joining of both Mu DNA ends is favored during recombination. On the basis of these results, we propose that the DNA joining stage requires a cooperative transition within the transposase–DNA complex. The cooperative utilization of active sites supplied in trans by Mu transposase provides an example of how mobile elements can ensure concomitant recombination of distant DNA sites.
Keywords: Transposase, DDE motif, phosphoryl transfer, active site, protein–DNA assembly, Mu transposition
Transposition and retroviral integration, like all forms of site-specific recombination, require a series of DNA cleavage and joining reactions to accomplish DNA movement. The propagation of mobile genetic elements requires the spatial and sequential coordination of several DNA regions and is orchestrated by one or two proteins, called transposases or integrases, in the context of multimeric, nucleoprotein complexes. These proteins form a family related by sequence within their catalytic domains (Kulkosky et al. 1992; Baker and Luo 1994; Polard and Chandler 1995; Rice et al. 1996). Mu transposase (MuA) is one well-studied member of this transposase/integrase family.
Multimers of transposase and retroviral integrase catalyze recombination using similar mechanisms that share two reaction steps: (1) cleavage of the element–host DNA junctions to yield a 3′ OH at each end of the element genome and (2) joining of these two 3′ OH ends, through single-step transesterification, to two phosphodiester bonds on opposite strands of a new DNA site (the target site) in a process called strand transfer (for review, see Mizuuchi 1992; Kleckner et al. 1995). Elements that transpose via a nonreplicative, or cut-and-paste, mechanism (e.g., Tn10, Tn7, and P elements) also cleave the 5′ strand at each element end to excise the element completely from its old location before joining the 3′ ends to a new DNA site (Bainton et al. 1991; Benjamin and Kleckner 1992; Kaufman and Rio 1992). Although these reactions appear to be catalyzed within nucleoprotein complexes, the active multimeric state of most transposases and integrases is not well understood. In contrast, it is well established that a stable tetramer of the Mu transposase catalyzes 3′-end cleavage and strand transfer during transposition (Lavoie et al. 1991; Mizuuchi et al. 1992; Baker et al. 1993).
Several stable nucleoprotein complexes (transpososomes) that are intermediates in Mu transposition have been characterized (for review, see Chaconas et al. 1996). Each transpososome minimally contains a tetramer of transposase subunits bound to the two Mu DNA ends. Transpososomes containing two paired but unreacted Mu DNA ends are called stable synaptic complexes (SSCs or Type 0 complexes) (Mizuuchi et al. 1992). In the presence of Mg2+, transposase catalyzes 3′-end cleavage to yield the cleaved donor complex (CDC or Type 1 complex); joining the Mu DNA ends to a new target DNA site generates the strand transfer complex (STC or type II complex) (Surette et al. 1987). Specific sequences at each end of the Mu genome (called the Mu DNA end binding sites) are bound by transposase within these transpososomes (Craigie et al. 1984). Transpososomes assembled on plasmid substrates containing the left and right Mu DNA ends protect three of these sites (L1, R1, and R2) as well as the DNA surrounding each cleavage site (located 5 bp away from the L1 or R1 sites) from chemical and enzymatic digestion (Lavoie et al. 1991; Mizuuchi et al. 1991; Zou et al. 1991). Active transpososomes can also be assembled on substrates containing two copies of the right end of the Mu genome (Savilahti et al. 1995). In these transpososomes, the two right ends are paired, and both pairs of R1 and R2 sites are similarly protected (Mizuuchi et al. 1991).
Each transposase subunit (75 kD) contains a catalytic core domain and several DNA-binding regions. In vitro, domains Iβγ, II, and IIIa comprise the minimal length of transposase necessary for assembly and catalysis (Aldaz et al. 1996), although domain IIIb is also required for stimulation of transposition by the phage-encoded MuB activator protein (Baker et al. 1991; Leung and Harshey 1991; Wu and Chaconas 1994). Transposase binds to the Mu DNA end sites through domain Iβγ (Nakayama et al. 1987; Leung et al. 1989; Schumacher et al. 1997), whereas regions from domains II and IIIa contain nonspecific DNA-binding activity (Nakayama et al. 1987; Wu and Chaconas 1995). The catalytic core is structurally related to the corresponding domain of retroviral integrases (Rice and Mizuuchi 1995; Rice et al. 1996). This domain (IIa) contains the three acidic amino acids [two aspartates (D) and one glutamate (E), (DDE)] of the conserved DDE motif that are specifically required for both cleavage and strand transfer reaction steps (Baker and Luo 1994; Kim et al. 1995; Krementsova et al. 1998). These DDE residues are proposed to form part of the active site by coordinating a Mg2+ ion(s) required for catalysis (for review, see Mizuuchi 1997). Crystal structures of the avian sarcoma virus and human immunodeficiency virus (HIV) integrase core domains show the two aspartates coordinating a single Mg2+ ion (Bujacz et al. 1995; Goldgur et al. 1998; Maignan et al. 1998).
To begin to understand how the ends of the Mu genome and the target DNA are coordinated within the transpososome, we have investigated the arrangement of subunits supplying the active site DDE residues for cleavage and strand transfer. Previous studies led to the proposal of a model in which complete transposition required DDE residues from each subunit of the tetramer (Baker et al. 1994). More recent studies have yielded conflicting results that were interpreted to suggest that complete transposition required DDE residues from either four or two subunits of the tetramer, depending on the type of Mu DNA substrate used (Namgoong and Harshey 1998). Here, we clarify the location and number of catalytic centers functioning in the Mu transpososome by identifying the particular subunits that supply DDE residues for each reaction step. Our results show that two active sites, each containing a set of DDE residues from a separate transposase subunit, catalyze complete transposition. The subunit donating DDE residues to the active site catalyzing recombination of one Mu DNA end is bound to the R1 site of the partner Mu DNA end (catalysis in trans). Furthermore, we discover that, in the absence of MuB, the presence of an uncleaved DNA end in one active site greatly inhibits strand transfer of a cleaved DNA end within the partner active site. Based on these results, we propose a model for Mu transposition in which strand transfer requires a cooperative transition between the two active sites. This transition is sensitive to the DNA immediately outside the genome ends and is stimulated by MuB.
Results
Strategy
To gain insight into the functional organization of the Mu transpososome, we designed a method to identify the subunit(s) supplying active site DDE residues for one specific cleavage reaction. The approach included three steps: (1) assembly of transpososomes containing theoretically random arrangements of DDE+ and DDE− transposase subunits, (2) isolation of a subset of these transpososomes based on their ability to cleave a specific Mu DNA end, and (3) identification of the type of subunit (DDE+ or DDE−) bound to each Mu DNA end binding site within the transpososomes by protein–DNA cross-linking. A description of this strategy follows.
Transpososomes containing mixtures of DDE+ and DDE− transposase were assembled on two types of Mu DNA end substrates. In reactions containing these two transposase forms and two DNA substrates, there are 16 possible configurations of DDE+ and DDE− subunits within the tetramer and three different possible combinations of substrate pairs to give a total of 48 different nucleoprotein complexes (Fig. 1A, not all combinations are shown). The two substrates were identical except for the presence or absence of four biotinylated thymidines at the 3′ end of the strand subject to cleavage by transposase (Fig. 1B).
Figure 1.

Schematic of experimental and substrate designs. (A) Transpososomes containing DDE+ (white circle) and DDE− (gray circle) transposase subunits and the nonbio and bio (denoted by B in figure) Mu DNA end substrates (vertical lines) were assembled in Ca2+. Transpososomes containing a bio substrate were then immobilized on an avidin matrix (⋄). The addition of Mg2+ allowed hydrolysis by transposase, and complexes that catalyzed cleavage of the bio substrate were released from the avidin matrix. Although mixed tetramer complexes that contained two bio substrates could be released from the matrix, the majority of released complexes contained one nonbio and one bio substrate in these experiments (data not shown). (B) Mu DNA end substrates. Molecules containing the R1 and R2 MuA-binding sites (shaded boxes) were made by annealing the indicated single-stranded oligonucleotides. The bio substrate is shown; the nonbio substrate lacks the five 3′ nucleotides (TBBBB) of TB407. Oligonucleotides TB237 and TB636 contained two cross-linking nucleotides (U) that replaced two thymidines in the natural DNA sequence; these oligonucleotides are individually 32P-phosphorylated at their 5′ ends to label subunits bound to the R1 and R2 sites, respectively, upon cross-linking. All unlabeled oligonucleotides within each substrate contained a 5′ OH. The adenosine directly 5′ to the phosphodiester bond that is cleaved by transposase is indicated by the arrow. (B) Biotin-dT; (U) 5-iodo-2′-deoxyuridine (IdU in text). (C) Denaturing PAGE analysis of the bio substrate in complexes released from the matrix; oligonucleotide TB407 was 32P-phosphorylated at its 5′ end. Numbers above lanes 3–5 indicate the molar ratio of DDE+ to DDE− transposase included in the initial reaction.
To isolate transpososomes that had cleaved a particular DNA end, complexes that contained a biotinylated substrate were immobilized on an avidin matrix (Fig. 1A). Transpososomes (mixed tetramer complexes) were initially assembled in the presence of Ca2+ (a condition that does not support hydrolysis; Mizuuchi et al. 1992); the addition of Mg2+ to the immobilized complexes allowed active transpososomes to catalyze cleavage. Although both nonbiotinylated (nonbio) and biotinylated (bio) substrates can be cleaved, only hydrolysis of the bio substrate released complexes from the avidin matrix. Thus, complexes that cleaved the bio Mu DNA end were isolated. Because mutations in the DDE residues inhibit catalysis (e.g., D269N, E392Q double mutant accumulated ∼50 times less product than DDE+ transposase; data not shown), nearly all of the released complexes will be those containing DDE+ subunits in the positions responsible for cleaving the bio substrate.
The ability of this method to isolate complexes that had cleaved the bio Mu DNA end was verified by labeling the 5′ end of the cleaved strand in this substrate. Denaturing PAGE analysis of the material released from the matrix upon Mg2+ addition showed that nearly all (90%–99%) of this substrate was cleaved (Fig. 1C). As expected, the amount of cleaved substrate recovered depended on the fraction of DDE+ transposase included in the reaction. Native agarose gel electrophoresis of the DNA released from the matrix indicated that most (≥93 ± 7%) was bound within stable transpososomes, regardless of the ratio of DDE+ to DDE− transposase included in the initial reaction. Thus, this method successfully purifies complexes that have cleaved the bio substrate.
To determine which subunits supplied the DDE residues essential for this cleavage reaction, protein–DNA cross-linking substrates were used to label subunits bound to each of the Mu DNA end binding sites. To create these cross-linking substrates, two thymidines in each R1 and R2 site were replaced by the cross-linking nucleotide 5-iodo-2′-deoxyuridine (IdU) (Fig. 1A). We define a subunit as bound to a site if it was cross-linked by this zero-length cross-linker. Each substrate consisted of a set of unligated oligonucleotides; by 32P-labeling the 5′ end of one R1 or R2 IdU-containing oligonucleotide (TB237 or TB636; Fig. 1A), the subunit cross-linked to that site was indirectly labeled. To label each of the four subunits one at a time, four separate reactions were performed that differed in the location of the 32P-labeled oligonucleotide. The R1 and R2 binding sites are distinguishable based on their location relative to the cleavage site of the bio substrate: those of the bio substrate are called the cis sites, whereas those of the nonbio substrate are called the trans sites (Fig. 2A). The DDE+ and DDE− transposase forms differed in the absence or presence of domain IIIb (amino acids 606–663, which are not required for catalysis). Thus, once labeled by cross-linking, the subunit bound to each DNA end site was easily identified as DDE+ or DDE− by the difference in size of these two proteins. In this way, the fraction of cross-linked subunits that were DDE+ at a given site could be quantitated.
Figure 2.



Cleavage requires D269 and E392 from the R1 trans subunit. (A) Schematic depicting the location of each subunit relative to the cleavage site (arrow) on the bio Mu DNA end. (B, C) SDS-PAGE of cross-linked (and therefore labeled) complexes from the total reaction (Total) and complexes released from the matrix upon cleavage of the bio substrate (Cleaved). The entire gel is shown only for R1 trans and R1 cis Total (wells at top). Percent DDE+ transposase cross-linked to each site is shown below each panel. The molar ratio of DDE+ to DDE− initially included in each reaction is indicated above each lane in B, Total; equivalent lanes in all other panels included the same ratio. Transpososomes were cross-linked before (C) or after (B) the cleavage step.
Cleavage of one Mu DNA end requires all three DDE residues of the R1 trans subunit
Using the cross-linking strategy described above, the position(s) of the subunit(s) carrying the DDE residues necessary for cleavage of the bio substrate was determined. This analysis compared the fraction of subunits cross-linked to each Mu DNA end site that were DDE+ between complexes that had cleaved the bio substrate and the total pool of complexes. A requirement for DDE residues was identified as an over-representation of DDE+ transposase cross-linked to a particular site in complexes released from the matrix as compared with the total pool. One DDE− transposase form used in these experiments contained mutations in two of the DDE motif residues (D269N and E392Q, referred to as DE/NQ). Each of these mutations individually inhibits the catalytic activity of transposase (Baker and Luo 1994). A requirement for more than one DDE+ transposase subunit in reactions containing DE/NQ would suggest that catalysis required at least one of these residues from one of the DDE+ subunits, whereas a unique DDE+ subunit requirement would indicate a necessity for both D269 and E392 from that subunit.
Analysis of complexes that cleaved the bio substrate revealed a preferential recovery of DDE+ transposase cross-linked to the R1 trans, but not the R1 cis, site. SDS-PAGE of the total pool of complexes showed that the DDE+ and DDE− transposase forms cross-linked to either R1 site at frequencies that largely reflected their input ratio (Fig. 2B, Total). At the R1 cis site, the fraction of cross-linked subunits that were DDE+ (% DDE+) did not change between complexes released from the matrix and the total pool of complexes (Fig. 2B, cf. lanes 8–10 between Total and Cleaved). In contrast, even in reactions containing the smallest fraction of DDE+ transposase, greater than 90% of the transposase cross-linked to the R1 trans site was DDE+ in released complexes (Fig. 2B, cf. lanes 3–5 between Total and Cleaved). Similar results were obtained whether complexes were cross-linked before (Fig. 3) or after (Fig. 2B) the cleavage step and when the DDE+ transposase form, rather than the DDE− form, contained domain IIIb (data not shown). Analysis of complexes immobilized on the matrix (before the addition of Mg2+) showed DDE+ and DDE− transposase cross-linked to the R1 trans site at the same frequency as observed in the total pool (data not shown). Thus, the biased recovery of DDE+ transposase cross-linked to the R1 trans site specifically reflected cleavage of the bio substrate. The less than 100% bias may be the result of (1) release of a small number of complexes that did not cleave the bio substrate at the cleavage site (see Fig. 1C), (2) residual cleavage activity by the DDE− transposase, and/or (3) incomplete correction for tailing of labeled protein peaks during quantitation (see Materials and Methods).
Figure 3.

Summary of the %DDE+ cross-linked to each of the four R1 and R2 sites in the total reaction (Total) and in complexes released from the matrix (Cleaved). Reactions were as described in Figs. 1 and 2; complexes were cross-linked before the cleavage step. The input molar ratio of 77-605,DDE+ to 77-663,DE/NQ MuA is indicated above each set. Each number within a cluster represents a separate experiment. (NA) Not available.
Preferential recovery of DDE+ transposase was not detected at either R2 site in released complexes, although the DDE+ transposase reproducibly cross-linked to both R2 sites in the total pool at a frequency higher than that observed for either R1 site (Figs. 2C and 3). This overabundance in the total pool may reflect either an increased affinity of this protein for the R2 sites or an increased ability to be cross-linked to these sites. No bias for the DDE+ transposase at the R2 sites in complexes that cleaved the bio substrate was observed whether complexes were cross-linked before or after cleavage (data not shown) or when the DDE+ transposase, instead of the DDE− transposase, contained domain IIIb (data not shown).
These experiments ascertained the relative location within the transpososome of the subunit providing both residues D269 and E392 for cleavage. To determine whether cleavage required DDE motif residue D336 from the same subunit, mixed tetramer complexes containing DDE+ and D336N transposase were similarly analyzed. Preferential recovery of DDE+ transposase was again observed exclusively at the R1 trans site in complexes that had cleaved the bio substrate (Table 1). Together, these results demonstrate that cleavage requires all three DDE motif residues from the subunit cross-linked to the R1 trans site.
Table 1.
Cleavage requires D336 from the R1 trans subunita
| Input ratio of DDE+ to DDE− transposaseb
|
Percent of total protein cross-linked to the Mu DNA end binding site that was DDE+ (%DDE+)
|
|||
|---|---|---|---|---|
| R1 trans
|
R2 trans
|
|||
| Total
|
Cleaved
|
Total
|
Cleaved
|
|
| 1:1 | 33 ± 4 | 91 ± 2 | 53 ± 2 | 56 ± 2 |
| 1:3 | 18 ± 2 | 84 ± 8 | 29 ± 2 | 32 ± 2 |
| 1:5 | 13 ± 2 | 81 ± 10 | 21 ± 4 | 24 ± 2 |
| R1 cis | R2 cis | |||
| Total | Cleaved | Total | Cleaved | |
| 1:1 | 31 ± 4 | 31 ± 2 | 47 ± 2 | 50 ± 1 |
| 1:3 | 18 ± 1 | 19 ± 1 | 25 ± 5 | 30 ± 1 |
| 1:5 | 13 ± 1 | 14 ± 1 | 17 ± 6 | 23 |
Cleavage and strand transfer require the DDE residues of the R1 trans subunit
Previous experiments that bypassed the cleavage step by using a precleaved substrate indicate that strand transfer of one Mu DNA end requires DDE residues from the R1 trans subunit (Aldaz et al. 1996). The cross-linking studies described here reveal the same DDE+ subunit requirement for the cleavage step. It is possible that the DDE residues of other subunits are required when these two steps are not isolated. To determine whether the DDE residues of the R1 trans subunit are the only set required for the sequential cleavage and strand transfer of one Mu DNA end, the strategy used above was slightly modified (Fig. 4A). Strand transfer of only one DNA end was evaluated by creating an unjoinable substrate that lacked the adenosine normally joined to the target DNA as well as the nucleotides 3′ to this adenosine. Mixed tetramer complexes containing the unjoinable and bio substrates were purified from the avidin matrix and cross-linked as described above. A circular target DNA (φX174 RFI) was then added, and complexes that catalyzed strand transfer into this DNA were purified by native agarose gel electrophoresis. SDS-PAGE analysis of these strand transfer complexes (STCs) indicated that, as expected, nearly all of the transposase cross-linked to the R1 trans site carried the wild-type DDE residues (Fig. 4B; Table 2). Examination of the other three subunits in these complexes showed no preferential recovery of DDE+ transposase cross-linked to any other site.
Figure 4.


The same R1 trans subunit provides DDE residues in trans for sequential cleavage and strand transfer of one Mu DNA end. (A) Reactions were performed as described in Materials and Methods and Fig. 1 except that the nonbio substrate was replaced with an unjoinable substrate (see text). Released complexes were incubated with target DNA to permit strand transfer. Complexes that catalyzed strand transfer were isolated by separating these complexes from inactive complexes on a native agarose gel. (B) SDS-PAGE of gel-purified STC. Numbers above and below each lane are as described in Fig. 2.
Table 2.
Sequential cleavage and strand transfer of one Mu DNA end requires DDE residues only from the R1 trans subunita
| Input ratio of DDE+ to DDE− transposaseb
|
Percent of total protein cross-linked to the Mu DNA end binding site that was DDE+ (%DDE+)
|
|||
|---|---|---|---|---|
| R1 trans
|
R2 trans
|
|||
| Total
|
STC
|
Total
|
STC
|
|
| 1:1 | 56,54 | 100,100 | 60,63 | 69,73 |
| 1:3 | 32,34 | 100,100 | 35,40 | N.A.,55 |
| 1:5 | 23,29 | 100,100 | 28,31 | 42,32 |
| R1 cis | R2 cis | |||
| Total | STC | Total | STC | |
| 1:1 | 57,68 | 74,54 | 60,62 | 69,79 |
| 1:3 | 32,37 | 51,26 | 34,38 | 37,51 |
| 1:5 | 22,24 | 35,18 | 24,25 | 31,36 |
Numbers from two experiments are shown. Experiments were as described in Fig. 4.
DDE+ = 77-605, DDE− = 77-663,DE/NQ.
These results suggest that only the R1 trans subunit must have functional DDE residues for the transpososome to catalyze recombination of one Mu DNA end. However, this conclusion depends on the assumption that complexes did not reassemble between the cleavage and strand transfer reaction steps. To detect possible complex reassembly, an excess (10×–50×) of full-length transposase (amino acids 1–663) was included with Mg2+ at the cleavage step in a set of reactions that were otherwise identical to those in Figure 4. SDS-PAGE analysis of the challenged complexes indicated that transpososomes did not reassemble at any point after the addition of Mg2+ (data not shown). Thus, the sequential cleavage and strand transfer of one Mu DNA end requires DDE residues from only the R1 trans subunit.
An uncleaved Mu DNA end inhibits strand transfer of the partner end
Extrapolation of the above cross-linking results to both Mu DNA ends suggests that the DDE residues of only two subunits of the tetramer should be sufficient to catalyze complete transposition. Two extreme models that could describe the activities of these transpososomes were considered (Fig. 5): In one model, recombination of the two Mu DNA ends occurs independently within the transpososome, whereas in the second model, strand transfer of one Mu DNA end is coupled to the cleaved state of the partner DNA end (strand transfer of one DNA end requires cleavage of both DNA ends). The spectrum of products predicted to be generated by mixed tetramer complexes differs between these two models. Both models predict that mixed tetramer complexes containing two DDE+ R1 subunits will generate products in which both Mu DNA ends are joined to the target. However, the independent model predicts that complexes containing one DDE+ and one DDE− R1 subunit will generate products in which one Mu DNA end is joined; cleaved but unjoined DNA ends are not predicted to accumulate. In contrast, the coupled model predicts that complexes containing one DDE+ and one DDE− R1 subunit will not be able to generate single-end strand transfer products (only one DNA end joined) but will generate cleaved but unjoined Mu DNA ends. Thus, the independent and coupled models differ in the predicted ability of complexes containing one DDE+ and one DDE− R1 subunit to catalyze single-end strand transfer.
Figure 5.

Relationship of partial products to subunit composition predicted by the independent and coupled two active site models. Coupled refers to a requirement for both Mu DNA ends to be cleaved for strand transfer to proceed. The DDE+ (white circles) and DDE− (gray circles) transposase subunits of mixed tetramer complexes are shown; the DNA has been omitted for clarity.
To determine whether the independent or coupled model could describe the Mu transpososome, we determined if strand transfer of one cleaved Mu DNA end depended on whether the partner DNA end was cleaved. For this analysis, complexes that contained a cleaved Mu DNA end (the bio substrate) were isolated from the avidin matrix, and the efficiency with which these complexes catalyzed strand transfer of this cleaved ‘biotinylated’ substrate was examined. (The cleaved bio substrate will be called bioc, even though it no longer contains the biotinylated nucleotides.) To determine whether strand transfer occurred more efficiently when the partner DNA end was cleaved, strand transfer of bioc was examined in mixed tetramer complexes that contained either the nonbio or precleaved partner substrates. Nearly all complexes released from the matrix contained a DDE+ subunit at the R1 trans position due to selection for cleavage of the bio substrate. If recombination occurred independently within the transpososome, the DDE residues of the R1 trans subunit would be sufficient to catalyze strand transfer of bioc. Thus, nearly all released mixed tetramer complexes would promote strand transfer of bioc, regardless of whether they contained the nonbio or precleaved partner DNA end. In contrast, if recombination of the two Mu DNA ends were coupled, the fraction of released mixed tetramer complexes catalyzing strand transfer of bioc would be limited to those complexes that contained a cleaved partner substrate.
The efficiency of strand transfer of bioc in mixed tetramer complexes did not support the independent model. Strand transfer of bioc was followed by labeling the 5′ end of the cleaved strand in this substrate, in reactions similar to those described in Figure 4A. As expected, when complexes contained the precleaved partner substrate, nearly all mixed tetramer complexes joined bioc to the target DNA (Fig. 6A, white bars), regardless of the fraction of DDE− transposase included in the initial reaction. Similar results were obtained when complexes contained the unjoinable partner substrate (data not shown). However, when released complexes contained the nonbio partner substrate, strand transfer of bioc decreased as the fraction of DDE+ transposase included in the reaction decreased (Fig. 6A, solid bars). These data suggest that although the DDE residues of the R1 trans subunit were sufficient to catalyze recombination of one Mu DNA end when the partner substrate was precleaved (or unjoinable), the DDE residues of at least one other subunit were required when the partner substrate was not already cleaved.
Figure 6.

An uncleaved Mu DNA end inhibits strand transfer of the cleaved partner Mu DNA end. (A) Percent of the bioc substrate that was joined. Reactions were performed as described in Fig. 4. The 5′ end of the cleaved strand of the bio substrate was 32P-labeled. Reactions contained either the nonbio partner substrate (solid bars) or the precleaved partner substrate (open bars). (B) Types of complexes released from the avidin matrix in reactions containing the nonbio partner substrate. (white circles) DDE+ transposase; (black circles) DDE− transposase; (striped circles) DDE+ or DDE− transposase. DNA 3′ of cleavage site is dashed and light gray to signify that it has been cleaved. (C) Agarose gel electrophoresis of strand transfer products generated by complexes released from the matrix in reactions containing the nonbio partner. (Lanes 1–5) The products of complexes that have cleaved both Mu DNA ends (DECL complexes). The products of DECL complexes were observed by labeling the 5′ end of the cleaved strand of the nonbio substrate. (Lanes 6–10) The products of both DECL complexes and SECL complexes; these products were observed by similarly labeling the bio substrate. Products in which both Mu DNA ends (DEST) or a single Mu DNA end (SEST) are joined to the target DNA comigrate with linear and nicked target DNA, respectively. Numbers (average of three to five experiments, ± 1 s.d.) below lanes 1, 3–5 were calculated by dividing the percent of nonbio substrate joined by the percent of nonbio substrate cleaved (before addition of the target DNA); numbers >1.0 could reflect a stimulation of cleavage by the target DNA. The percent of observed strand transfer products that were single ended (%SEST) (average of three to four experiments) was calculated by comparing the amount of radioactivity in the SEST and DEST bands. The predicted %SEST was calculated based on the probability of obtaining DDE+ transposase at the R1 cis position.
This additional DDE+ subunit requirement suggests that complexes that had cleaved both Mu DNA ends [double-end cleaved (DECL)] efficiently catalyzed strand transfer, whereas complexes containing an uncleaved DNA end [single-end cleaved (SECL)] catalyzed strand transfer poorly, as predicted by the coupled model. To verify the activity of DECL complexes, the fraction of DECL complexes released from the matrix that catalyzed strand transfer was ascertained. Because the fraction of released complexes that contained a cleaved nonbio partner substrate equaled the fraction of DECL complexes, the strand transfer activity of DECL complexes could be calculated by comparing the fraction of nonbio substrate that was cleaved in the released complexes to the fraction that was joined to the target DNA. The percent of nonbio substrate joined equaled or exceeded the percent cleaved (cleavage assayed before target addition), regardless of the fraction of DDE− transposase included in the reaction (Fig. 6C, ratio %nonbio joined to %nonbio cleaved shown under lanes 1, 3–5). Furthermore, all of the nonbio substrate was joined in conjunction with bioc to yield double-end strand transfer (DEST) products. Together, these data indicate that all complexes that had cleaved both Mu DNA ends (and thus contained two DDE+ R1 subunits) also catalyzed strand transfer of both DNA ends.
The coupled model predicts that complexes containing one DDE+ and one DDE− R1 subunit (SECL complexes) will not catalyze strand transfer. To verify this prediction, the activity of SECL complexes was determined by analyzing the ratio of single-end strand transfer (SEST) products to DEST products generated by complexes released from the matrix. By labeling the bioc substrate, the activities of both SECL and DECL complexes were observed. Because DECL complexes generated only DEST products, SEST products must come from SECL complexes. Therefore, the fraction of SEST products observed provides a measure of the activity of SECL complexes. Inspection of the reaction products observed by labeling bioc shows that very few SEST products were generated (Fig. 6C, lanes 6–10). The percent of observed strand transfer products that were single ended was much lower than the percent expected if all SECL complexes catalyzed strand transfer (cf. %SEST observed to %SEST predicted, shown under lanes 6, 8–10). Thus, as suggested from the difference in strand transfer activity between complexes containing the nonbio or precleaved partner substrates (see Fig. 6A), the presence of an uncleaved Mu DNA end inhibits strand transfer of the partner DNA end such that the cleaved DNA end remains unjoined. Consistent with these results, cross-linking studies of purified STCs from reactions containing the nonbio and bio substrates showed a biased recovery of DDE+ transposase at both of the R1 sites but neither of the R2 sites (data not shown). Although the coupled model predicts that no SECL complexes will promote strand transfer, a small number of SEST products were observed (Fig. 6C, lanes 6–10). These results indicate that an uncleaved Mu DNA end did not completely inhibit strand transfer of the partner DNA end. Thus, transpososomes catalyze strand transfer most efficiently when both Mu DNA ends are cleaved, although recombination of one Mu DNA end is not obligatorily coupled to cleavage of the partner DNA end.
MuB stimulates the formation of SEST products
Inhibition of strand transfer by an uncleaved partner DNA end causes most complexes containing one DDE+ and one DDE− R1 subunit to stall after cleavage; few of these complexes generate SEST products. However, previous experiments show a large accumulation of SEST products in mixed tetramer reactions as well as DEST products and cleaved but unjoined substrate molecules (Baker et al. 1994). These previous experiments were performed in the presence of the MuB activator protein, whereas the experiments described above were performed in the absence of MuB. MuB stimulates transposition by the wild-type transposase both in vivo and in vitro (Coelho et al. 1982; Chaconas et al. 1985; Baker et al. 1991). We were therefore interested in determining the effect of MuB on the catalytic activities of mixed tetramer complexes.
To determine whether MuB influenced the accumulation of SEST products, the ratio of SEST to DEST products was quantitated in reactions performed in the presence or absence of MuB. Transpososomes were assembled in the presence of target DNA and Mg2+, which allowed assembly, cleavage, and strand transfer to occur in an unstaged manner. When reactions contained Mu DNA end substrates that required cleavage before strand transfer, the majority of strand transfer products in the mixed tetramer reactions were double ended in the absence of MuB (Fig. 7, lanes 1–5), consistent with the poor strand transfer activity of SECL complexes. Inclusion of MuB in these reactions increased strand transfer overall and specifically increased the fraction of SEST products (Fig. 7, lanes 6–10). As expected, stimulation by MuB required transposase domain IIIb (data not shown). This stimulation of SEST was not accompanied by an increase in cleaved but unjoined Mu DNA ends in purified STCs (data not shown), suggesting that MuB did not promote SEST by stimulating cleavage of the partner Mu DNA end. Together, these results support a model for the activity of the Mu transpososome in which transposition is catalyzed through the coupled activity of two active sites. Coupling between active sites reflects a preference for two cleaved Mu DNA ends for efficient strand transfer; this preference is not absolute and is influenced by MuB.
Figure 7.

SEST products accumulate in the presence of MuB. Strand transfer reactions containing the indicated ratios of DDE+ to DDE− transposase were performed in Mg2+ as described in Materials and Methods. Reactions contained a 10 to 1 ratio of unlabeled to labeled substrate, so that most DEST products contained only one labeled substrate. Reaction products are labeled as in Fig. 6. The percent of strand transfer products that were single ended (%SEST) (average ± 1 s.d. of three to four experiments, calculated as in Fig. 6) is indicated below each lane. The higher level of SEST products observed in lanes 1,3–5 as compared with equivalent lanes in Fig. 6 was due to the unstaged protocol used in these experiments.
Discussion
Catalytic organization in the Mu transposase tetramer
A tetramer of Mu transposase binds specific sites at each end of the phage genome and catalyzes the two 3′-end-cleavage and two 3′-end-joining reactions that occur during transposition. Here, we demonstrate that cleavage of one Mu DNA end requires all three DDE motif residues from the same subunit. Of the two paired Mu DNA ends bound within the transpososome, the subunit supplying the DDE residues for cleavage of one DNA end cross-linked to the R1 site of the other DNA end (the DNA end trans to that being cleaved). Furthermore, the DDE residues from this same subunit are the only ones strictly required to catalyze cleavage and strand transfer of the same Mu DNA end, indicating that one set of DDE residues are used for these two reaction steps.
These results are consistent with previous work that investigated the location of the subunits supplying the DDE residues required for the strand transfer step (Aldaz et al. 1996; Savilahti and Mizuuchi 1996; Yang et al. 1996; Namgoong and Harshey 1998). The DDE+ requirement for cleavage from the R1 trans subunit also agrees with previous work showing that cleavage requires E392 from one or two subunits bound to the partner Mu DNA end (Savilahti and Mizuuchi 1996), although this result disagrees with the conclusion that cleavage required E392 from an R2 subunit (Yang et al. 1996). In a recent study, which used transposase variants with altered DNA end-binding specificity and substrates carrying mutant transposase binding sites, cleavage appeared to require E392 from an R2 subunit when reactions contained oligonucleotide substrates, although in reactions containing plasmid substrates, the R1 and L1 subunits donated E392 for cleavage in trans (Namgoong and Harshey 1998). By directly identifying the type of subunit at each position of the tetramer through protein–DNA cross-linking and by making a quantitative assessment of the catalytic activities of mixed tetramer complexes, we unambiguously demonstrate that recombination of one Mu DNA end requires the DDE residues of only the R1 trans subunit. These results confirm the conclusion (based on plasmid substrates) that the two sequential steps of transposition require E392 in trans from the R1 and L1 subunits (Namgoong and Harshey 1998).
Mu transposase is a member of a large family of transposases and retroviral integrases that cleave and join the 3′ ends of their genomes in a chemically related manner. These proteins are related by amino acid sequence surrounding the strictly conserved DDE motif residues and presumably share a similar catalytic domain structure. Individual mutations in the DDE residues of transposases and integrases inactivate cleavage and strand transfer activities to similar extents, suggesting that both reactions require these residues (Engelman and Craigie 1992; Kulkosky et al. 1992; van Gent et al. 1992; Baker and Luo 1994; Bolland and Kleckner 1996; Sarnovsky et al. 1996). Although the arrangement of subunits supplying active site DDE residues for specific cleavage and joining reactions has been elucidated only for Mu transposase, the activity of mixtures of DDE+ and DDE− Tn10 transposase indicates that recombination of one end of the Tn10 element uses DDE residues from only one transposase subunit (Bolland and Kleckner 1996). Furthermore, as seen for Mu transposase, the negligible activity of complexes containing mixtures of single mutant proteins suggests that all three DDE residues from the same subunit are used for catalysis in both Tn7 transposition and HIV integration (E. Krementsova and T.A. Baker, unpubl.; van Gent et al. 1993; Baker et al. 1994; Sarnovsky et al. 1996). This similarity in DDE requirements among these family members may suggest that the repeated utilization of the same set of DDE residues for 3′-end cleavage and strand transfer of one DNA end is conserved throughout this class of recombinase.
Our results allow us to propose a model for the arrangement of active sites within the Mu transpososome (Fig. 8). Within the tetramer, the two subunits bound to the L1 and R1 sites at each end of the Mu genome donate all three active site DDE residues for 3′ end cleavage and strand transfer of the partner Mu DNA end. Sharing of DDE residues for cleavage and strand transfer of one Mu DNA end implies a common active site for these two reactions; thus, two active sites catalyze recombination of the two Mu DNA ends. Within an active site, at least some of the amino acids of the DDE motif may interact (through a bound Mg2+ ion or ions) with water, the phosphodiester bonds undergoing attack, and/or the 3′ OH at the cleaved Mu DNA end (Mizuuchi 1997). Each active site probably contains additional protein determinants for binding the DNA surrounding the cleavage site and the target DNA; whether such determinants remain the same or change between the two catalytic steps is unknown.
Figure 8.
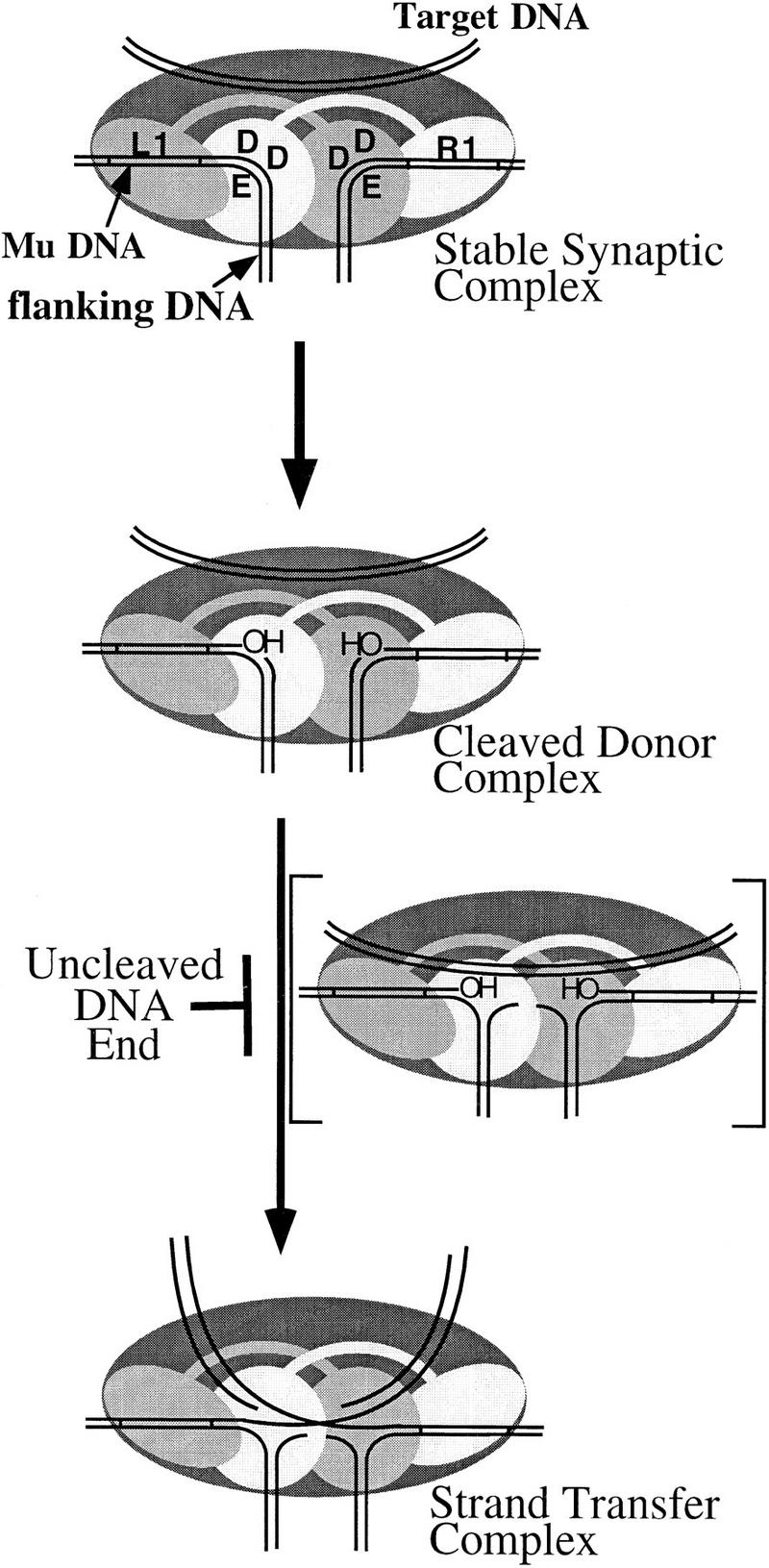
Organization and dynamics of the MuA tetramer. Transpososome complexes for each stage of the transposition reaction are shown. The complex in brackets depicts a hypothetical transition involving a movement of DNA components within the active sites that must occur prior to strand transfer. Because this transition must occur for the efficient progression from cleavage to strand transfer, it is placed between these two steps in the reaction pathway. However, the point along the reaction pathway at which this transition occurs is unknown. The end-type DNA-binding domain (oval) and catalytic core (circle) of the L1 and R1 bound subunits are indicated.
Although the other two subunits of the tetramer (not individually shown in Fig. 8) do not supply DDE residues to the active sites catalyzing 3′-end cleavage and strand transfer, they are likely to play other important roles. These subunits appear to be critical to the structural integrity of the transpososome (Lavoie et al. 1991) and may contribute transposase domains IIb and IIIa to the two active sites as additional DNA-binding components (Wu and Chaconas 1995; Aldaz et al. 1996; Naigamwalla et al. 1998). It is also possible that the DDE residues of the other two subunits could be required for 5′ strand cleavage during the relatively uncharacterized nonreplicative (cut-and-paste) pathway of Mu transposition, in a manner analogous to that proposed for the TnsA protein of the TnsA/TnsB transposase of Tn7 (Sarnovsky et al. 1996).
Cooperative utilization of active sites within the Mu transpososome
By quantitating the strand transfer efficiency of each Mu DNA end in transpososomes of known subunit composition, we find that Mu transposase catalyzes strand transfer most efficiently when both Mu DNA ends are cleaved. Thus, even when an active site contains all of the transposase components necessary for the catalytic steps, it rarely promotes strand transfer when the partner active site contains an uncleaved Mu DNA end. This situation arises in complexes containing one DDE+ and one DDE− active site. Because the wild-type active site efficiently catalyzes strand transfer when the partner DDE− active site contains a precleaved Mu DNA end, the DDE motif residues of the partner active site are not directly required for strand transfer by the wild-type active site. The poor strand transfer activity of complexes containing an uncleaved Mu DNA end can not be attributed to an instability of these complexes, because nearly all Mu DNA ends are bound within these transpososomes. Thus, we demonstrate communication between active sites of the cleaved state of the Mu DNA ends; this communication specifically affects the progression from cleavage to strand transfer. Communication between DNA ends during assembly and perhaps cleavage has also been inferred from the effects of terminal mutations of the Mu genome and retroviral elements (Surette et al. 1991; Murphy and Goff 1992; Kukolj and Skalka 1995).
The impaired ability of complexes containing an uncleaved DNA end to catalyze strand transfer can be easily explained if there is a cooperative transition within the Mu transpososome that must occur prior to strand transfer. This transition occurs most efficiently when both Mu DNA ends are cleaved. A working model for such a transition is depicted in Figure 8. Cleavage of each Mu DNA end yields a 3′ OH at each end of the Mu genome and a 5′-phosphate (not shown) on the DNA that was previously covalently attached to the 3′ end (the old flanking DNA). Strand transfer via direct transesterification requires that the 3′ OH come within close proximity of the target DNA phosphodiester bond. This requirement implies a shift in the relative positions of these DNA components within each active site as the complex proceeds from cleavage to strand transfer. Although this shift is represented (Fig. 8) by a movement of the old flanking DNA away from the 3′ OH ends and an accompanying introduction of target DNA within close proximity to each 3′ OH, the exact nature or sequence of any movement of these DNA elements is not well understood. Furthermore, the point along the transposition pathway at which the target DNA enters the active sites is unknown, although transpososomes can bind MuB and the target DNA during early stages of the reaction (Naigamwalla and Chaconas 1997). It is attractive to consider that the presence of an uncleaved Mu DNA end may block a cooperative conformational change required to position each 3′ OH adjacent to the target DNA phosphodiester bonds and/or may destabilize target DNA binding within the two active sites. Roles for transposase domains IIb and IIIa in contacting the DNA at or surrounding the cleavage sites during assembly and/or strand transfer have been proposed (Baker et al. 1993; Krementsova et al. 1998; Naigamwalla et al. 1998; Namgoong et al. 1998).
We show that MuB can stimulate strand transfer even in complexes containing an uncleaved Mu DNA end. In reactions containing mixtures of DDE+ and DDE− transposase, this stimulation leads to the accumulation of a large proportion of SEST products. These results are consistent with a role for MuB in stimulating the transition otherwise inhibited by the presence of an uncleaved Mu DNA end and provide an alternate interpretation for previous studies. Thus, although a transpososome containing four active sites was proposed to explain the yields of cleaved but unjoined, SEST, and DEST products generated in reactions containing mixtures of DDE+ and DDE− transposase (Baker et al. 1994), these products can be generated by transpososomes containing two active sites depending on the degree of inhibition an uncleaved DNA end imposes on the cooperative transition required for strand transfer and the level of MuB stimulation. Consistent with this explanation, reactions containing mixtures of DDE+ and DDE− transposase catalyze cleavage but complete strand transfer only poorly when one of the transposase proteins carries a deletion of the MuB interaction domain (domain IIIb; Mizuuchi et al. 1995). In a separate study, mixed tetramer complexes in which the DDE+ transposase lacks domain IIIb (and a portion of domain IIIa) catalyze cleavage but not strand transfer unless a precleaved substrate is used (Yang et al. 1995). To accommodate these results, the authors proposed a reciprocal domain sharing model between the DDE domain and domain IIIa for cleavage and strand transfer such that two separate subunits supplied DDE motif residues for recombination of one DNA end. Given our results, it is likely that the differential activities of these mixed tetramer complexes that contained precleaved or uncleaved substrates was due to a differential requirement for stimulation by MuB to efficiently complete strand transfer. Thus, by studying the inhibitory effect of an uncleaved DNA end and the compensatory effect of MuB, we provide evidence for a specific role for MuB in stimulating a cooperative transition necessary for strand transfer.
All transposases and integrases must coordinate the DNA surrounding the cleavage site at each end of the element as well as the target DNA within their active sites. Although little is known about how members of this transposase/integrase family may manipulate these DNA components to accomplish recombination, disruption of base pairs at or surrounding the cleavage site may be an important step during transposition (Savilahti et al. 1995; Scottoline et al. 1997). During Mu replicative transposition, strand transfer may be proceeded by the cooperative movement of at least some of these DNA components. Tn10 transposase follows an interesting parallel to Mu transposase in that only complexes that have completely excised both ends of the element from the old DNA location can be committed to strand transfer to a new target site (Bolland and Kleckner 1995; Sakai and Kleckner 1997). The investigators propose a model in which the old flanking DNA and the new target DNA occupy the same position within the active site (Sakai and Kleckner 1997); as a result, the presence of flanking DNA at an uncleaved DNA end physically blocks the introduction of target DNA into the active sites. However, within the Mu transpososome, the presence of an uncleaved DNA end is unlikely to physically block binding of the target DNA within the active sites, because some complexes containing an uncleaved DNA end are able to promote strand transfer and because MuB can stimulate SEST in these complexes without promoting cleavage of the partner DNA end. Furthermore, in contrast to the excised Tn10 genome, the 5′ strand at each DNA end remains covalently attached to the old flanking DNA even as each 3′ end is joined to the target DNA during Mu replicative transposition, suggesting that Mu transposase simultaneously binds and manipulates all of these DNA components within the active sites to catalyze strand transfer. Therefore, although both nonreplicative and replicative elements must manipulate flanking and target DNAs within their active sites, the nature of the events that occur during the transition from cleavage to strand transfer may differ. Understanding how these DNA regions are arranged and coordinated among the active sites of these transpososomes is essential to elucidating the mechanism by which this class of recombinase operates.
Materials and methods
DNA
Oligonucleotides were synthesized using standard phosphoramidite chemistry by the MIT Biopolymer Laboratory. The biotin–dT and 5-iodo–2′-deoxyuridine phosphoramidites were purchased from Glen Research. Purification of oligonucleotides, 5′ 32P-phosphorylation, and annealing of the donor substrates were essentially as described previously (Aldaz et al. 1996) except that 50 μl annealing reactions contained 60 pmoles of each oligonucleotide. The sequence of oligonucleotides used to make the bio donor substrates are shown in Figure 1. Identical oligonucleotides were used to make the nonbio substrate except that TB601 (5′-TTTTTCGTGCGCCGCTTCACTAGCAGCTTGGCGTAATCGGGCGTAATGC-3′) replaced TB407. For the unjoinable substrate, TB259 (5′-TTTTTCGTGCGCCGCTTC-3′) replaced TB407; TB259a (5′-TTTTTCGTGCGCCGCTTCA-3′) replaced TB407 in the precleaved substrate. None of the component oligonucleotides contained a 5′-phosphate except for the oligonucleotide that was labeled at the 5′ end with [γ-32P]ATP. The target DNA was φX174 RFI DNA (GIBCO BRL).
Proteins
MuA 77-663,D269N/E392Q; MuA 77-663,D336N; and MuA 77-605,D269N/E392Q were cloned by standard techniques and verified by sequencing. MuA 77-605; MuA 77-663,D269N/E392Q; MuA 77-663,D336N; and MuA 77-605,D269N/E392Q were purified essentially as described previously (Baker et al. 1993) through fraction III, except that the proteins were overexpressed in NovaBlue cells (Novagen) and KCl was replaced by NaCl. The conductivity of fraction III was adjusted to 300 mm NaCl and loaded onto a Mono-S HR 5/5 column (Pharmacia) equilibrated in HEDG + 300 mm NaCl (HEDG is 25 mm HEPES at pH 7.3, 0.1 mm EDTA, 1 mm DTT, 10% glycerol). The column was developed with a five-volume gradient from 0.3 m to 1 m NaCl in HEDG. Peak fractions (fraction IV) were pooled, frozen in liquid nitrogen, and stored at −80°C. MuA 77-663 (Baker et al. 1993) and MuB (Yamauchi and Baker 1998) were purified as described.
Transposition reactions
Cleavage reactions (100 μl in most cases) were assembled in buffer C (25 mm Tris-HCl at pH 8, 2 mm DTT, 100 mg/ml BSA, 15% glycerol, 100 mm NaCl, 12% DMSO, 0.1% Triton X-100) and contained 10 mm CaCl2, 2.4 pmoles each donor substrate, and a total of 10 pmoles of the MuA derivatives. The reaction mixture was incubated at 30°C for 1 hr, a 5-μl aliquot was set aside (‘total’), and 50-μl neutravidin beads (pre-equilibrated in buffer C + 10 mm CaCl2) (Pierce) was added. This slurry was incubated at room temperature for 30 min to allow transpososomes containing the bio substrate to bind to the beads. The supernatant was removed, and the beads were washed twice with 200 μl of buffer C + 10 mm CaCl2, twice with 200 μl buffer C + 10 mm CaCl2 + 400 mm NaCl (this wash step included a 30-min incubation at 30°C), and twice more with 200 μl of buffer C + 10 mm CaCl2. To allow cleavage to occur, the beads were resuspended in buffer C + 70 mm MgCl2. The supernatant (purified CDCs) was collected after incubating 30 min at 30°C. To assay strand transfer, 250 ng of φX174 RFI was added to 20 μl of the supernatant, and the mixture was incubated at 30°C for 30 min. STCs were purified by cutting the appropriate band from a preparative agarose/BSA/heparin gel essentially as described (Aldaz et al. 1996).
For DNA–protein cross-linking experiments, the reaction mixture was irradiated in the wells of a polystyrene microtiter plate (Corning Glassworks) with UV light (302 nm, UVP transilluminator) at a distance of 2.5 cm for 10 min. When cross-linking was done prior to cleavage, the mixture was irradiated before adding the neutravidin beads. For cross-linking experiments investigating strand transfer, the DNA and MuA proteins were present at a concentration five times the standard reaction. Cross-linked proteins were separated on a 7% polyacrylamide–SDS gel in 1× Tris-glycine running buffer. The percent of protein cross-linked to each transposase DNA-binding site that was DDE+ was quantitated by comparing the amount of radioactivity in peaks corresponding to DDE+ and DDE− transposase using the Molecular Dynamics ImageQuant software. The close spacing of peaks on the gel resulted in the overlap of peak tails, and peaks were defined without subtracting this overlap. This lead to a small artificial increase in the amount of radioactivity associated with the DDE− peak.
The percent of substrate bound within transpososomes was quantitated by mobility shift on a 2% metaphor agarose (FMC) gel in 0.5× TBE at 0°C, run at 6 V/cm for 2 hr. Gels were fixed in 10% acetic acid for 30 min. Strand transfer products were separated on a 1% HGT agarose (SeaKem) gel in 1× TBE, 7 V/cm, for 2 hr. To analyze DNA products of cleavage, SDS and EDTA were added to aliquots to a final concentration of 0.2% SDS, 20 or 80 mm EDTA. Samples were extracted once with 25:24:1 phenol/chloroform/isoamyl alcohol, the DNA was recovered by EtOH precipitation, and samples were analyzed by denaturing PAGE. All gels were quantitated using the Molecular Dynamics ImageQuant software after drying and exposing them to a Molecular Dynamics PhosphorImager screen.
The composition of transposition reactions testing MuB was essentially identical to that described above except that 10 mm CaCl2 was replaced with 10 mm MgCl2. Reactions (20 μl) were mixed on ice, and then 4 μl of the cold φX174/ATP premix ± MuB was added. The premix consisted of 15.5 mm HEPES, 62 μm EDTA, 12.4% glycerol, 0.62 m NaCl, 0.62 mm DTT, 62.5 ng/μl φX174, 12.6 mm ATP (pH 7), ± 100 ng/μl MuB. Reactions were incubated at 30°C for 1 hr.
Acknowledgments
We thank Steve Bell, Tony Schwaca, Graham Walker, Marjorie Oettinger, Sara Cherry, David Freed, and members of the Baker lab for helpful discussions and clarifying the manuscript. Early experiments performed by Hector Aldaz are also gratefully acknowledged. This work was supported by a U.S. Public Health Service grant (GM499224) from the National Institutes of Health. T.A.B., E.L.J., and A.C. are employees of the Howard Hughes Medical Institute. T.L.W. is the recipient of a National Science Foundation graduate fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tabaker@mit.edu; FAX (617) 252-1852.
References
- Aldaz H, Schuster E, Baker TA. The interwoven architecture of the Mu transposase couples DNA synapsis to catalysis. Cell. 1996;85:257–269. doi: 10.1016/s0092-8674(00)81102-2. [DOI] [PubMed] [Google Scholar]
- Bainton R, Gamas P, Craig NL. Tn7 transposition in vitro proceeds through an excised transposon intermediate generated by staggered breaks in DNA. Cell. 1991;65:805–816. doi: 10.1016/0092-8674(91)90388-f. [DOI] [PubMed] [Google Scholar]
- Baker TA, Luo L. Identification of residues in the Mu transposase essential for catalysis. Proc Natl Acad Sci. 1994;91:6654–6658. doi: 10.1073/pnas.91.14.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, Mizuuchi M, Mizuuchi K. MuB protein allosterically activates strand transfer by the transposase of phage Mu. Cell. 1991;65:1003–1013. doi: 10.1016/0092-8674(91)90552-a. [DOI] [PubMed] [Google Scholar]
- Baker TA, Mizuuchi M, Savilahti H, Mizuuchi K. Division of labor among monomers within the Mu transposase tetramer. Cell. 1993;74:723–733. doi: 10.1016/0092-8674(93)90519-v. [DOI] [PubMed] [Google Scholar]
- Baker TA, Krementsova E, Luo L. Complete transposition requires four active monomers in the Mu transposase tetramer. Genes & Dev. 1994;8:2416–2428. doi: 10.1101/gad.8.20.2416. [DOI] [PubMed] [Google Scholar]
- Benjamin HW, Kleckner N. Excision of Tn10 from the donor site during transposition occurs by flush double-strand cleavages at the transposon termini. Proc Natl Acad Sci. 1992;89:4648–4652. doi: 10.1073/pnas.89.10.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolland S, Kleckner N. The two single-strand cleavages at each end of Tn10 occur in a specific order during transposition. Proc Natl Acad Sci. 1995;92:7814–7818. doi: 10.1073/pnas.92.17.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— The three chemical steps of Tn10/IS10 transposition involve repeated utilization of a single active site. Cell. 1996;84:223–233. doi: 10.1016/s0092-8674(00)80977-0. [DOI] [PubMed] [Google Scholar]
- Bujacz G, Jaskolski M, Alexandratos J, Wlodawer A, Merkel G, Katz RA, Skalka AM. High-resolution structure of the catalytic domain of avian sarcoma virus integrase. J Mol Biol. 1995;253:333–346. doi: 10.1006/jmbi.1995.0556. [DOI] [PubMed] [Google Scholar]
- Chaconas G, Giddens EB, Miller JL, Gloor G. A truncated form of the bacteriophage Mu B protein promotes conservative integration, but not replicative transposition, of Mu DNA. Cell. 1985;41:857–865. doi: 10.1016/s0092-8674(85)80066-0. [DOI] [PubMed] [Google Scholar]
- Chaconas G, Lavoie BD, Watson MA. DNA transposition: Jumping gene machine, some assembly required. Curr Biol. 1996;6:817–820. doi: 10.1016/s0960-9822(02)00603-6. [DOI] [PubMed] [Google Scholar]
- Coelho A, Maynard-Smith S, Symonds N. Abnormal cointegrate structures mediated by gene B mutants of phage Mu: Their implications with regard to gene function. Mol & Gen Genet. 1982;185:356–362. doi: 10.1007/BF00330812. [DOI] [PubMed] [Google Scholar]
- Craigie R, Mizuuchi M, Mizuuchi K. Site-specific recognition of the bacteriophage Mu ends by the MuA protein. Cell. 1984;39:387–394. doi: 10.1016/0092-8674(84)90017-5. [DOI] [PubMed] [Google Scholar]
- Engelman A, Craigie R. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J Virol. 1992;66:6361–6369. doi: 10.1128/jvi.66.11.6361-6369.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R, Davies DR. Three new structures of the core domain of HIV-1 integrase: An active site that binds magnesium. Proc Natl Acad Sci. 1998;95:9150–9154. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman PD, Rio DC. P element transposition in vitro proceeds by a cut-and-paste mechanism and uses GTP as a cofactor. Cell. 1992;69:27–39. doi: 10.1016/0092-8674(92)90116-t. [DOI] [PubMed] [Google Scholar]
- Kim K, Namgoong SY, Jayaram M, Harshey RM. Step-arrest mutants of phage Mu transposase. Implications in DNA-protein assembly, Mu end cleavage, and strand transfer. J Biol Chem. 1995;270:1472–1479. doi: 10.1074/jbc.270.3.1472. [DOI] [PubMed] [Google Scholar]
- Kleckner N, Chalmers RM, Kwon D, Sakai J, Bolland S. Tn10 and IS10 transposition and chromosome rearrangements: Mechanism and regulation in vivo and in vitro. Curr Top Microbiol Immunol. 1995;204:49–82. doi: 10.1007/978-3-642-79795-8_3. [DOI] [PubMed] [Google Scholar]
- Krementsova E, Giffin MJ, Pincus D, Baker TA. Mutational analysis of the Mu transposase. J Biol Chem. 1998;273:31358–31365. doi: 10.1074/jbc.273.47.31358. [DOI] [PubMed] [Google Scholar]
- Kukolj G, Skalka AM. Enhanced and coordinated processing of synapsed viral DNA ends by retroviral integrases in vitro. Genes & Dev. 1995;9:2556–2567. doi: 10.1101/gad.9.20.2556. [DOI] [PubMed] [Google Scholar]
- Kulkosky J, Jones KS, Katz RA, Mack JP, Skalka AM. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol Cell Biol. 1992;12:2331–2338. doi: 10.1128/mcb.12.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie BD, Chan BS, Allison RG, Chaconas G. Structural aspects of a higher-order nucleoprotein complex: Induction of an altered DNA structure at the Mu-host junction of the Mu Type 1 transpososome. EMBO J. 1991;10:3051–3059. doi: 10.1002/j.1460-2075.1991.tb07856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung PC, Harshey RM. Two mutations of phage Mu transposase that affect strand transfer or interactions with B protein. J Mol Biol. 1991;219:189–199. doi: 10.1016/0022-2836(91)90561-j. [DOI] [PubMed] [Google Scholar]
- Leung PC, Teplow DB, Harshey RM. Interaction of distinct domains in Mu transposase with Mu DNA ends and an internal transpositional enhancer. Nature. 1989;338:656–658. doi: 10.1038/338656a0. [DOI] [PubMed] [Google Scholar]
- Maignan S, Guilloteau J-P, Zhou-Liu Q, Clement-Mella C, Mikol V. Crystal structures of the catalytic domain of HIV-1 integrase free and complexed with its metal cofactor: High level of similarity of the active site with other viral integrases. J Mol Biol. 1998;282:359–368. doi: 10.1006/jmbi.1998.2002. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K. Transpositional recombination: Mechanistic insights from studies of Mu and other elements. Annu Rev Biochem. 1992;61:1011–1051. doi: 10.1146/annurev.bi.61.070192.005051. [DOI] [PubMed] [Google Scholar]
- ————— Polynucleotidyl transfer reactions in site-specific DNA recombination. Genes Cells. 1997;2:1–12. doi: 10.1046/j.1365-2443.1997.970297.x. [DOI] [PubMed] [Google Scholar]
- Mizuuchi M, Baker TA, Mizuuchi K. DNase protection analysis of the stable synaptic complexes involved in Mu transposition. Proc Natl Acad Sci. 1991;88:9031–9035. doi: 10.1073/pnas.88.20.9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Assembly of the active form of the transposase-Mu DNA complex: A critical control point in Mu transposition. Cell. 1992;70:303–311. doi: 10.1016/0092-8674(92)90104-k. [DOI] [PubMed] [Google Scholar]
- ————— Assembly of phage Mu transpososomes: Cooperative transitions assisted by protein and DNA scaffolds. Cell. 1995;83:375–385. doi: 10.1016/0092-8674(95)90115-9. [DOI] [PubMed] [Google Scholar]
- Murphy JE, Goff SP. A mutation at one end of Moloney murine leukemia virus DNA blocks cleavage of both ends by the viral integrase in vivo. J Virol. 1992;66:5092–5095. doi: 10.1128/jvi.66.8.5092-5095.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naigamwalla DZ, Chaconas G. A new set of Mu DNA transposition intermediates: Alternate pathways of target capture preceding strand transfer. EMBO J. 1997;16:5227–5234. doi: 10.1093/emboj/16.17.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naigamwalla DZ, Coros CJ, Wu Z, Chaconas G. Mutations in domain IIIα of the Mu transposase: Evidence suggesting an active site component which interacts with the Mu-host junction. J Mol Biol. 1998;282:265–274. doi: 10.1006/jmbi.1998.2011. [DOI] [PubMed] [Google Scholar]
- Nakayama C, Teplow DB, Harshey RM. Structural domains in phage Mu transposase: Identification of the site-specific DNA-binding domain. Proc Natl Acad Sci. 1987;84:1809–1813. doi: 10.1073/pnas.84.7.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namgoong SY, Harshey RM. The same two monomers within a MuA tetramer provide the DDE domains for the strand cleavage and strand transfer steps of transposition. EMBO J. 1998;17:3775–3785. doi: 10.1093/emboj/17.13.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namgoong SY, Kim K, Saxena P, Yang JY, Jayaram M, Giedroc DP, Harshey RM. Mutational analysis of domain II beta of bacteriophage Mu transposase: Domains II alpha and II beta belong to different catalytic complementation groups. J Mol Biol. 1998;275:221–232. doi: 10.1006/jmbi.1997.1466. [DOI] [PubMed] [Google Scholar]
- Polard P, Chandler M. Bacterial transposases and retroviral integrases. Mol Microbiol. 1995;15:13–23. doi: 10.1111/j.1365-2958.1995.tb02217.x. [DOI] [PubMed] [Google Scholar]
- Rice P, Mizuuchi K. Structure of the bacteriophage Mu transposase core: A common structural motif for DNA transposition and retroviral integration. Cell. 1995;82:209–220. doi: 10.1016/0092-8674(95)90308-9. [DOI] [PubMed] [Google Scholar]
- Rice P, Craigie R, Davies DR. Retroviral integrases and their cousins. Curr Opin Struct Biol. 1996;6:76–83. doi: 10.1016/s0959-440x(96)80098-4. [DOI] [PubMed] [Google Scholar]
- Sakai J, Kleckner N. The Tn10 synaptic complex can capture a target DNA only after transposon excision. Cell. 1997;89:205–214. doi: 10.1016/s0092-8674(00)80200-7. [DOI] [PubMed] [Google Scholar]
- Sarnovsky RJ, May EW, Craig NL. The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. EMBO J. 1996;15:6348–6361. [PMC free article] [PubMed] [Google Scholar]
- Savilahti H, Mizuuchi K. Mu transpositional recombination: Donor DNA cleavage and strand transfer in trans by the Mu transposase. Cell. 1996;85:271–280. doi: 10.1016/s0092-8674(00)81103-4. [DOI] [PubMed] [Google Scholar]
- Savilahti H, Rice PA, Mizuuchi K. The phage Mu transpososome core: DNA requirements for assembly and function. EMBO J. 1995;14:4893–4903. doi: 10.1002/j.1460-2075.1995.tb00170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher S, Clubb RT, Cai M, Mizuuchi K, Clore GM, Gronenborn AM. Solution structure of the Mu end DNA-binding ibeta subdomain of phage Mu transposase: Modular DNA recognition by two tethered domains. EMBO J. 1997;16:7532–7541. doi: 10.1093/emboj/16.24.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scottoline BP, Chow S, Ellison V, Brown PO. Disruption of the terminal base pairs of retroviral DNA during integration. Genes & Dev. 1997;11:371–382. doi: 10.1101/gad.11.3.371. [DOI] [PubMed] [Google Scholar]
- Surette MG, Bush SM, Chaconas G. Transpososomes: Stable protein-DNA complexes involved in the in vitro transposition of bacteriophage Mu DNA. Cell. 1987;49:253–262. doi: 10.1016/0092-8674(87)90566-6. [DOI] [PubMed] [Google Scholar]
- Surette MG, Harkness T, Chaconas G. Stimulation of the Mu A protein-mediated strand cleavage reaction by the Mu B protein, and the requirement of DNA nicking for stable type 1 transpososome formation. J Biol Chem. 1991;266:3118–3124. [PubMed] [Google Scholar]
- van Gent DC, Oude Groeneger AAM, Plasterk RH. Mutational analysis of the integrase protein of human immunodeficiency virus type 2. Proc Natl Acad Sci. 1992;89:9598–9602. doi: 10.1073/pnas.89.20.9598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gent DC, Vink C, Oude Groeneger AAM, Plasterk RH. Complementation between HIV integrase proteins mutated in different domains. EMBO J. 1993;12:3261–3267. doi: 10.1002/j.1460-2075.1993.tb05995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Chaconas G. Characterization of a region in phage Mu transposase that is involved in interaction with the Mu B protein. J Biol Chem. 1994;269:28829–28833. [PubMed] [Google Scholar]
- ————— A novel DNA binding and nuclease activity in domain III of Mu transposase: Evidence for a catalytic region involved in donor cleavage. EMBO J. 1995;14:3835–3843. doi: 10.1002/j.1460-2075.1995.tb00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi M, Baker TA. An ATP-ADP switch controls progression of the Mu transposition pathway. EMBO J. 1998;17:5509–5518. doi: 10.1093/emboj/17.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Kim K, Jayaram M, Harshey RM. A domain sharing model for active site assembly within the Mu A tetramer during transposition: The enhancer may specify domain contributions [published erratum appears in EMBO J. 1995 Jul 17;14: 3596] EMBO J. 1995;14:2374–2384. doi: 10.1002/j.1460-2075.1995.tb07232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Jayaram M, Harshey RM. Positional information within the Mu transposase tetramer: Catalytic contributions of individual monomers. Cell. 1996;85:447–455. doi: 10.1016/s0092-8674(00)81122-8. [DOI] [PubMed] [Google Scholar]
- Zou A, Leung PC, Harshey RM. Transposase contacts with Mu DNA ends. J Biol Chem. 1991;266:20476–20482. [PubMed] [Google Scholar]