

Abstract
Inositol hexakisphosphate kinase 2 (IP6K2), a member of the inositol hexakisphosphate kinase family, functions as a growth suppressive and apoptosis-enhancing kinase during cell stress. We created mice with a targeted deletion of IP6K2; these mice display normal embryogenesis, development, growth and fertility. Chronic exposure to the carcinogen 4-nitroquinoline 1-oxide (4-NQO, a UV-mimetic compound) in drinking water resulted in fourfold increased incidence of invasive squamous cell carcinoma (SCC) formation in the oral cavity and esophagus of the knockout (KO) mice compared to the wild-type (WT) littermates. Paradoxically, KO mice displayed relative resistance to ionizing radiation and exhibit enhanced survival following 8–10Gy total body irradiation. Primary KO fibroblasts displayed resistance to antiproliferative effects of interferon-β and increased colony forming units following ionizing radiation. Radioresistance of KO fibroblasts was associated with accelerated DNA repair measured by comet assay. Direct microinjection of 5-PP-Ins(1,2,3,4,6)P5 (the enzymatic product of IP6K2), but not InsP6 (the substrate of IP6K2) induced cell death in SCC22A squamous carcinoma cells.
Keywords: inositol hexakisphosphate kinase 2, chemical carcinogenesis, ionizing radiation, proliferation, inositol polyphosphates, apoptosis
Introduction
Inositol hexakisphosphate kinases (IP6Ks) catalyse the synthesis of inositol polyphosphates, high energy compounds that play diverse biologic roles, including regulation of nonhomologous end joining (NHEJ; Hanakahi et al., 2000; Ma and Lieber, 2002; Byrum et al., 2004), regulation of endocytic trafficking (Saiardi et al., 2002), protein phosphorylation (Saiardi et al., 2004), chemotaxis (Luo et al., 2003), regulation of telomere length (Saiardi et al., 2005; York et al., 2005) and apoptosis (Morrison et al., 2001; Nagata et al., 2005). Using inositol hexakisphosphate (InsP6) as a substrate, the enzyme inositol hexakisphosphate kinase 2 (IP6K2), encoded by the gene IHPK2, catalyses the synthesis of diphosphoinositol pentakisphosphate (5-PP-Ins(1,2,3,4,6)P5, also known as PP-InsP5 or InsP7; Figure 1). Overexpression of IP6K2 sensitizes ovarian carcinoma cell lines to the growth suppressive and apoptotic effects of interferon (IFN)-β, IFN-α2 treatment and ionizing radiation (Morrison et al., 2002). Snyder and colleagues demonstrated that IP6K2 enhanced the cytotoxic actions of several different cell stressors, including cisplatin, staurosporine, hydrogen peroxide and hypoxia (Nagata et al., 2005). Importantly, IP6K enzymatic activity was enhanced during death induction. York and colleagues recently discovered two gene products, VIP1 and VIP2, that function as both InsP6 and PP-InsP5 kinases (Fridy et al., 2007; Mulugu et al., 2007). Hence, IP6K2 catalyses formation of InsP7, whereas VIP1 directs the synthesis of both InsP7 and InsP8.
Figure 1.
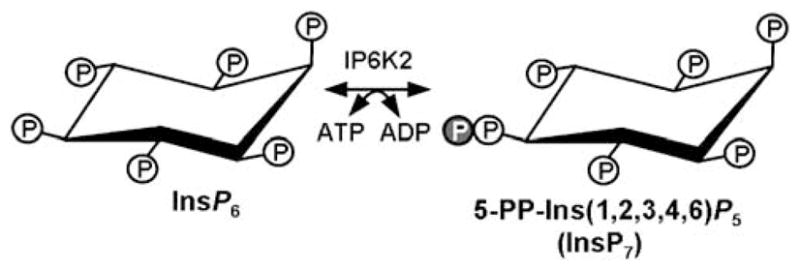
Inositol hexakisphosphate kinase 2 (IP6K2) catalyses the conversion of inositol hexakisphosphate (InsP6) plus ATP into 5-PP-Ins(1,2,3,4,6)P5, also known as InsP7. Circled P indicates phosphate group.
Recently, targeted deletion of the IHPK1 gene has been performed (Bhandari et al., 2008), resulting in mice that display growth retardation, decreased insulin release from pancreatic β-cells and sterility in males secondary to defective spermatogenesis. Because of the proapoptotic function of IP6K2, we hypothesized that loss of this gene product would confer relative resistance to cell death in certain tissues or predispose animals to neoplastic transformation. To determine the physiologic role of IP6K2, we created mice with a targeted deletion of exon 2. Unlike animals lacking IP6K1, IP6K2 knockout (KO) mice displayed normal embryogenesis, development, growth, blood chemistries, serum insulin levels and fertility. However, when exposed to chronic administration of a carcinogen (4-nitroquinoline 1-oxide; 4-NQO) in drinking water IP6K2 KO mice displayed accelerated development of invasive aerodigestive tract epithelial carcinoma, including squamous cell carcinoma (SCC) of the tongue, oral cavity and esophagus.
Results
Nucleotides 1–202 of exon 2 of the IHPK2 locus, encoding amino acids 1–67 of the protein, were targeted for deletion (Figure 2a) in the embryonic stem cell line. Male and female heterozygous mice (IHPK2+/−) were mated to produce KO (IHPK2−/−), heterozygous and wild-type (WT; IHPK2+/+) littermates. PCR performed on genomic DNA from tail clips identified genotypes of F1 and F2 mice (Figure 2b). To prove that site-specific, rather than random genomic, recombination had occurred, PCR using vector-specific (neo) primers were paired with primers specific for flanking regions immediately adjacent to (and outside of) the insertion site; only products of the predicted size were observed (Figure 2c). Immunoblot analysis of tail clip lysates indicated reduced levels of IP6K2 protein in tissues of heterozygous mice, and protein was undetectable in KO mice (Figure 2d).
Figure 2.
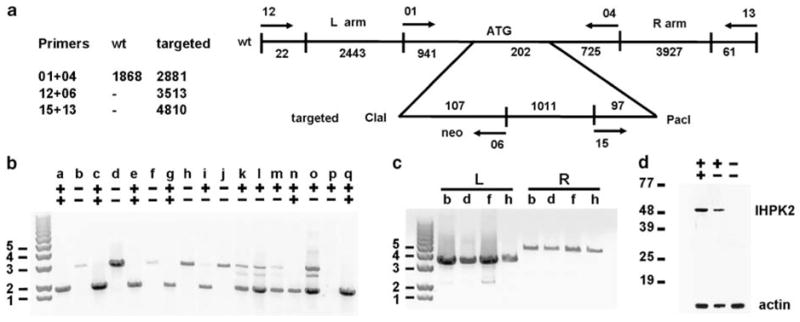
(a) Targeting scheme for generation of IHPK2−/− mice. The targeting construct was created from a mouse strain 129 SvEv bacterial artificial chromosome (BAC) clone that was used to generate the homologous targeting arms. The first 202 bp of exon 2 of IHPK2 were replaced with 1215bp derived from the targeting vector that contained a neo resistance marker. Utilizing primers 01 and 04, the wild-type allele generated a 1868 bp PCR product and the targeted allele gave a 2881 bp product. Primers specific for genomic flanking DNA outside the homology arms were designed to rule out random insertion of the targeting construct into host chromosomes. Pairing of flanking primers with neo-specific primers gave left-arm and right-arm products of 3515 and 4810 bp, respectively. (b) Genotype of knockout mice. Genomic DNA was isolated from tail clips of F1 and F2 mice and subject to PCR using the Roche Long Template PCR System. Primers 01 and 04 yielded a PCR product of 1868 bp from wild-type (IHPK2+/+) mice (lanes a, c, e, g, n and q); homozygous knockout (IHPK2−/−) mice gave a 2881 bp product (lanes b, d, f, h and j); heterozygous (IHPK2+/−) mice displayed both products (lanes i, k, l, m and o). (c) Neo-specific primers (06 or 15) were paired with a primer specific for the left-flanking region (primer 12) or the right-flanking region (primer 13), both outside of the homology region, to ensure that site-specific recombination had occurred. IHPK2−/− mice b, d, f and h yielded 3513 bp and 4810 bp PCR products when analysed using left flanking (L) and right flanking (R) primer pairs, respectively. (d) Immunoblot performed on tail snip lysates from WT, heterozygous IHPK2+/−, and IHPK2 knockout mice using polyclonal anti-IP6K2 antibody.
Heterozygous and KO mice displayed normal behavior, growth, development, fertility and life span. Extensive phenotyping of male and female 9-week-old KO and WT littermates, including examination of 56 different organs, complete blood count, chemistry-20 panel and radiographic studies revealed no abnormality associated with the IHPK2−/− genotype. Blood glucose and pancreatic histology were also normal. One distinguishing characteristic of IHPK1−/− mice, decreased serum insulin (Bhandari et al., 2008), was not observed in IHPK2−/− mice (4 h fasting serum insulin 0.95±0.071 and 1.08±0.065, 16 h fasting serum insulin 0.34±0.055 and 0.28±0.044 ng/ml in WT and KO mice, respectively, n=8), suggesting that even though IP6K1 and IP6K2 share similar enzymatic activity, they play fundamentally different physiologic roles. RNA silencing of IP6K1, but not IP6K2, resulted in reduced insulin exocytosis from pancreatic β-cells (Illies et al., 2007). IP6K1 phosphorylates InsP5 and InsP6 equally well, whereas IP6K2 displays a strong preference for InsP6 over InsP5 (Shears, 2004). It is predicted, therefore, that knocking out IP6K1 would effect both InsP7 and PP-InsP4 levels, whereas knocking out IP6K would chiefly effect InsP7.
To obtain an index of enzymatic activity, primary fibroblast cultures from WT and KO mice were incubated with [3H]-inositol. Whole cell lysates were subject to high performance liquid chromatography and levels of 5-PP-Ins(1,2,3,4,6)P5 were determined (Figure 3). 5-PP-Ins(1,2,3,4,6)P5 production in KO fibroblasts was 62% that of WT fibroblasts (2.1 vs 3.4% incorporation of [3H]-inositol, KO vs WT, respectively, P<0.003, Student’s t-test, n=4), suggesting that less than half the IP6K activity in primary cultured fibroblasts was attributed to IP6K2. The ratio of cellular PP-Ins(1,2,3,4,6)P5 to InsP6, expressed as 100−(PP-Ins(1,2,3,4,6)P5/InsP6), was 17.4±0.78% and 10.7±0.71% in WT and KO fibroblasts, respectively (P=0.0007, n=4). There was no difference in cellular [3H]-InsP6 levels between KO and WT fibroblasts (6.31±0.85and 5.44±0.79 pmol/mg cell pellet, respectively, n=4).
Figure 3.
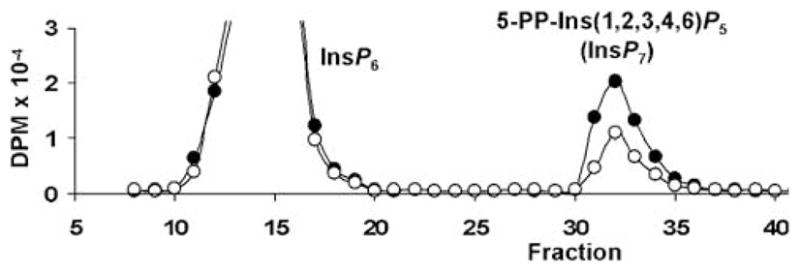
Primary fibroblasts (2−105 in 48-well plates) were grown in the presence of [3H]-inositol for 5days, washed and placed in Krebs solution buffered with HEPES (25mM). Cells were lysed by aspiration of Krebs solution, addition of cold 0.6M perchloric acid and 0.2mM InsP6, then neutralized with K2CO3 and Na2EDTA. Samples were centrifuged and supernatants loaded onto a PartiSphere 5mM SAX high performance liquid chromatography (HPLC) column. Mobile phase (1 ml/min) was a gradient of buffer A (1mM Na2EDTA) and buffer B (buffer A +1.3M (NH4)2HPO4 pH 3.85: 0–10 min 0% B, 10–25 min 0–35% B, 25–105 min 35–100% B, 105–115 min 100% B. Fractions were counted after addition of scintillant. Representative elution profiles for wild–type (WT; black) and knockout (KO; white) fibroblasts are depicted. Experiment was performed three times with similar results.
We sought to determine whether IHPK2−/− mice had an altered response to stressors. IHPK2−/− and IHPK2+/+ mice were subject to lethal total body irradiation (TBI) via a cesium source, and survival was determined. After 8Gy TBI, median survival of WT and KO mice was 10.5 days and 17 days, respectively (Figure 4a). There were 12.5% long-term KO survivors (>40 days) in the 8-Gy group. After 10 Gy TBI, survival of WT and IHPK2−/− mice was 8 days and 13 days, respectively (Figure 4b). Compared to WT primary fibroblasts, KO fibroblasts isolated from tail clips were relatively resistant to the growth-suppressive effects of IFN-β (Figure 4c). Similarly, KO fibroblasts exhibited enhanced survival and increased colony-forming units when compared to WT fibroblasts (Figure 4d). To determine whether the KO phenotype was primarily due to diminished InsP6K enzymatic activity or due to decreased IP6K2 protein, KO fibroblasts were rescued by transfection of constructs encoding WT or kinase-dead (Morrison et al., 2002) IHPK2 (Figure 4f). WT IHPK2, but not the kinase-dead variant, was able to reverse the KO phenotype, suggesting that InsP6K enzymatic activity contributes to radiosensitivity.
Figure 4.
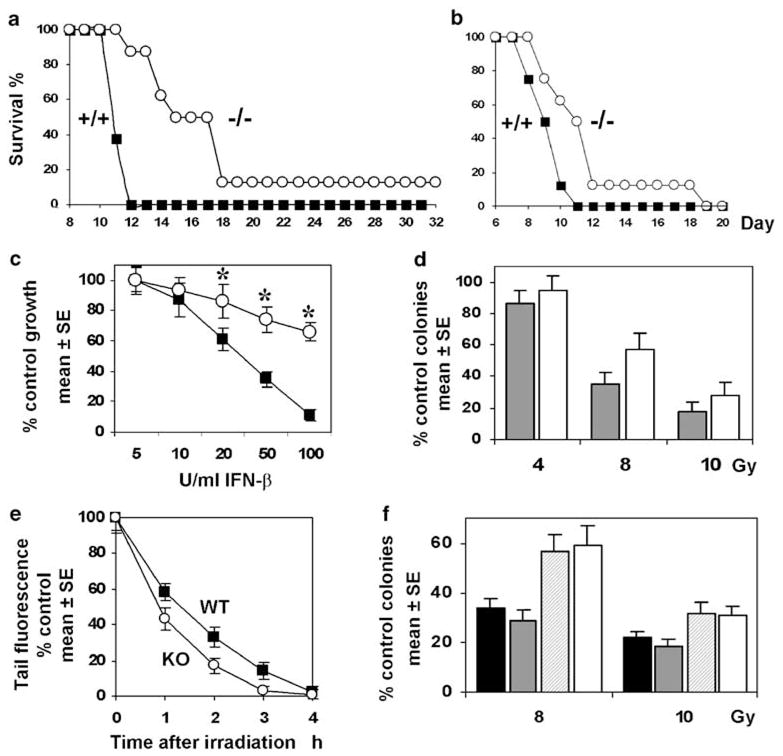
Kaplan–Meier survival curves for wild–type (WT) mice (squares) and homozygous IHPK2−/− mice (circles) following (a) 8Gy (P<0.01) or (b) 10 Gy total body irradiation (P<0.05). (c) In vitro antiproliferative effect of murine interferon (IFN)-β against WT fibroblasts (squares) or IHPK2−/− fibroblasts (circles) Primary WT and knockout (KO) fibroblasts were grown in the presence of 5–100 U/ml IFN-β. After 4 days, cells were fixed and stained with sulforhodamine B. Absorbance of bound dye was expressed as percent of untreated controls (n=8, each datapoint). Primary KO fibroblasts displayed relative resistance to the antiproliferative effects of IFN-β; Asterisk indicates P<0.001; n=8. (d) Fibroblast survival: Colony counts produced by WT (gray) and KO (white) fibroblasts expressed as % control (P<0.05, all points, n=6). (e) The neutral comet assay was used to quantitate repair of DNA double strand breaks (DSB). WT and KO fibroblasts (5−105) were plated in 60-mm dishes, exposed to 8Gy γ-irradiation at 25 °C, and incubated at 37 °C for various times. Cells were then detached using trypsin, suspended in 0.5ml phosphate-buffered saline (PBS), and mixed with 4.5ml low melting-point agarose that was dissolved in PBS at 90 °C and cooled to 37 °C. The mixture was placed onto microscope slides, incubated at 4 °C for 30 min, and cells were lysed at 4 °C for1 h in 2.5M NaCl, 100mM EDTA, 10mM Tris-HCl, 1% N-lauroylsarcosine (pH 7) and subjected to neutral electrophoresis at 4 °C. For image analysis, cells were treated with 50 μl CYBR Green and covered with a coverslip. Fluorescent comet images were captured using Image-Pro Plus 6.2 and DSB frequencies were determined from the percentage of total fluorescence (100% at 0 h) in the comet tail (P<0.05at 1, 2, 3 h; Student’s t-test, n=6 each datapoint). (f) KO fibroblasts were transfected with empty vector (white bars), kinase-dead IHPK2 (striped bars) or IHPK2 (gray bars), and compared to WT fibroblasts (black bars) in colony forming assays as in (d) above; P<0.01: vector and kinase-dead-expressing KO fibroblasts vs IHPK2-expressing KO fibroblasts and WT fibroblasts (both 8 and 10 Gy exposure).
As these data suggested that the IHPK2−/− genotype may confer some degree of radioresistance, we studied the DNA repair response to γ-irradiation-induced double-stranded breaks (DSB) using neutral comet assays (Wojewodzka et al., 2002; Figure 4e). In the 3-h time interval following 8Gy irradiation, KO fibroblasts demonstrated accelerated DSB rejoining compared to WT cells, associated with a more rapid disappearance of tail fluorescence. Enhanced DSB repair in KO animals may, therefore, explain their relative resistance to TBI.
As loss of IP6K2 expression has been associated with enhanced tumor cell proliferation (Morrison et al., 2001; Nagata et al., 2005), we hypothesized that tissues of KO mice might display enhanced proliferative activity when subjected to stress. We utilized 4-NQO, a UV-mimetic carcinogen that induces hyperproliferation, hyperkeratinization and tumorigenesis in the mucosa of the gastrointestinal tract following chronic administration. This water-soluble compound causes DNA adduct formation (Panigrahi and Walker, 1990), and induces SCC in the oral cavity of mice following a progression from dysplasia, preneoplasia to invasive carcinoma (Hawkins et al., 1994). These sequential changes closely mimic the temporal series of events that occur in cellular transformation and development of carcinoma in the human aerodigestive tract. After 23 weeks of continuous exposure to 4-NQO in drinking water, 100% of KO mice developed carcinoma in the esophagus, oral cavity or stomach, whereas 25% of WT mice developed neoplastic lesions at these sites (P=0.007; Table 1). Mucosal hyperplasia and hyperkeratosis were especially common in the esophagi of KO mice, and when advanced, caused distortion and partial obstruction of the lumen (Figure 5). Dysplastic lesions were common and scattered in location (from the mouth to the duodenum) in both WT and KO mice. Invasion of epithelial cells into the muscularis was fourfold more frequent in KO mice (Table 1, PFigures 6a–c). However, vascular invasion was observed only in KO mice (Figure 6d). Survival of KO and WT mice after 30 weeks of 4-NQO exposure was 0 and 75%, respectively, <0.01 (Figure 6e).
Table 1.
Carcinogenesis in IP6K2 knockout mice
| Mouse % | Survival at 23 weeks | Incidence hyperplasia | Incidence dysplasia | Incidence invasive ca | Incidence vascular invasion |
|---|---|---|---|---|---|
| +/+ | 87.5 | 87.5% | 62.5% | 25.0% | 0.0% |
| −/− | 37.5 | 100.0% | 100.0% | 100.0% | 37.5% |
| P | 0.12 | 1.00 | 0.20 | 0.007 | 0.20 |
Eight animals per group (four male, four female); P values determined by Fisher’s exact test.
Figure 5.
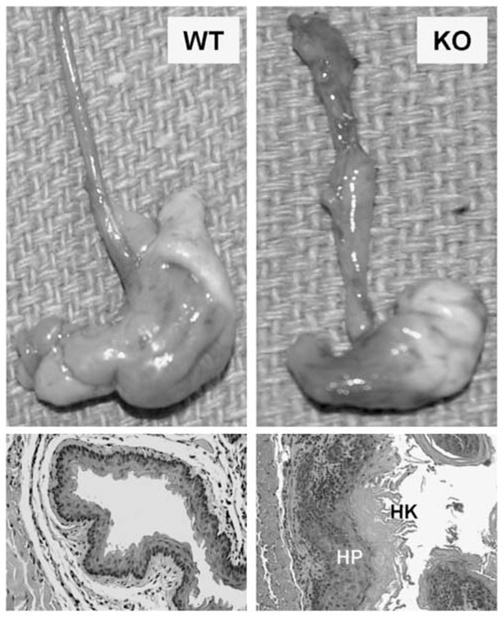
Chronic carcinogen exposure: Wild-type (WT) and IHPK2 knockout (KO) mice received the UV-mimetic drug 4 nitroquinoline 1-oxide (4-NQO) in drinking water continuously for 6 months. Upper panels: Gross appearance of stomach and esophagus from representative mice. Lower panels: Hematoxylin and eosin stained (H&E) sections—hyperproliferation (HP) and hyperkeratosis (HK) of the esophageal mucosa were more pronounced in KO mice.
Figure 6.
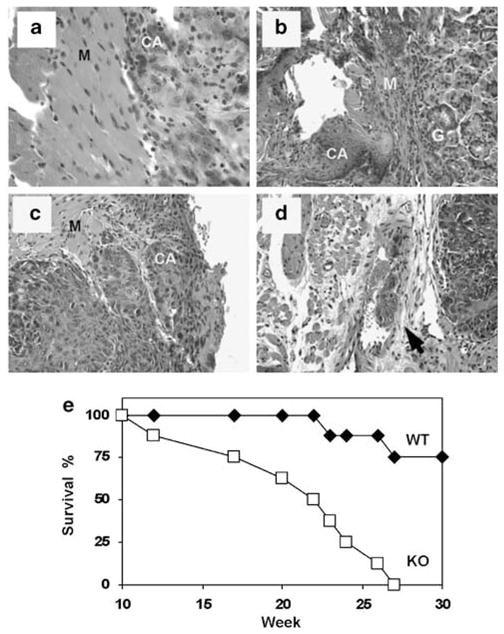
Induction of invasive carcinoma in the aerodigestive tract of IHPK2 KO mice following 4 nitroquinoline 1-oxide (4-NQO) exposure—hematoxylin and eosin stained (H&E) sections: (a) Muscularis (M) layer of esophagus demonstrating microinvasive carcinoma (CA). (b) Lesion at gastroesophageal junction with microinvasive carcinoma (CA) infiltrating the muscularis (M) near the gastric mucosa (g). (c) Dorsal surface of the tongue with carcinoma (CA) invading into muscular (M) layer. (d) Same tongue lesion as above with mass of tumor cells invading blood vessel (arrow). (e) Kaplan–Meier survival curve for wild-type mice (diamonds) and homozygous IHPK2−/− mice (squares) following 30 week exposure to 100 μM 4-NQO in drinking water ad libitum, n=8.
The enhanced propensity for development of aerodigestive tract carcinoma in KO mice that received 4-NQO led us to examine the role of IP6K2 expression in human head and neck squamous cell carcinomas (HNSCC). The IHPK2 locus in humans is 3p21, a region frequently deleted in HNSCC (Maestro et al., 1993; Li et al., 1994; Chakraborty et al., 2003) and lung carcinoma (Kok et al., 1987). We assessed IP6K2 protein expression in different cell lines by immunohistochemistry. The HNSCC cell line RW strongly expressed IP6K2 that localized to the cytoplasm and perinuclear regions; in contrast, SCC22A cells lacked expression (Figure 7, upper panel). Reverse transcription PCR (RT–PCR) performed on OVCAR-3, HeLa and five HNSCC cell lines demonstrated that IHPK2 mRNA was detectable in all cell lines. Western blot analysis indicated that IP6K2 protein was present in OVCAR-3, HeLa, RW and SCC22B cells, but was undetectable in SCC17A, SCC22A and UMSCC cells (Figure 7, lower panel). Hence, IP6K2 protein appears to be unstable in these latter three cell lines. The index of enzymatic activity (5-PP-Ins(1,2,3,4,6)P5, as performed in Figure 3) in SCC22A cells was 17% that of RW cells (0.8 vs 4.7% total cellular incorporation of [3H]-inositol into 5-PP-Ins(1,2,3,4,6)P5, respectively, P<0.002). There was no difference in total cellular [3H]-InsP6 levels between SCC22A and RW cells (3.29±0.60 and 4.18±0.69 pmol/mg cell pellet, respectively, n=4), indicating there was no impediment to cellular uptake of the inositol substrate in either cell line.
Figure 7.
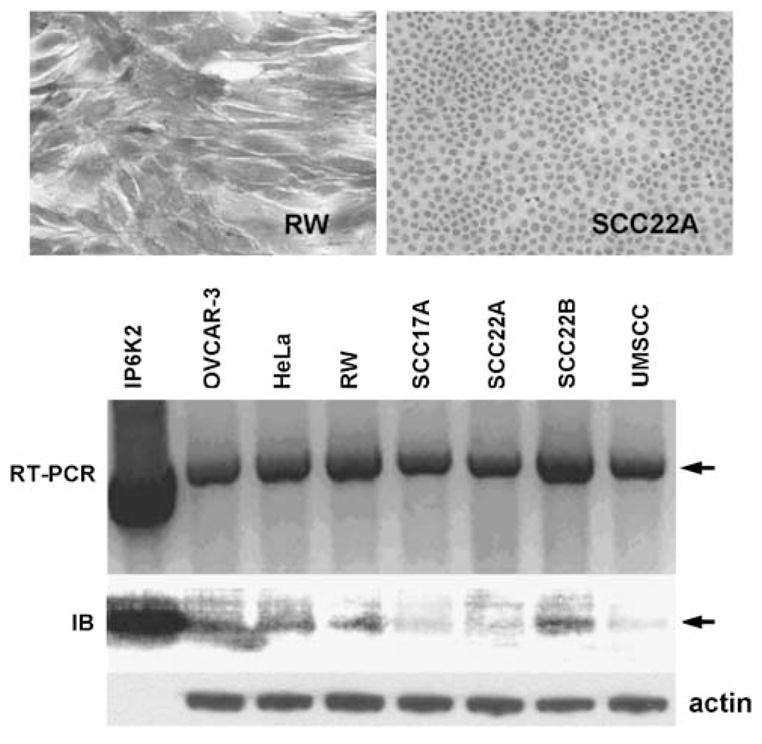
Human head and neck squamous cell carcinoma (HNSCC) cell lines in culture. Immunohistochemistry with mouse anti-human inositol hexakisphosphate kinase 2 (IP6K2) and peroxidase conjugated goat anti-mouse secondary Ab: RW cell line displayed intense staining cytoplasmic staining for hIP6K2, whereas SCC22A did not express IP6K2. Reverse transcription PCR (RT–PCR), utilizing primers to detect full length IHPK2 cDNA, demonstrated the presence of IHPK2 mRNA (upper arrow) in OVCAR-3, HeLa, and five HNSCC cell lines; plasmid encoding IHPK2 served as positive control. Immunoblot of same panel of cell lines demonstrated variable expression of IP6K2 (lower arrow); recombinant IP6K2 protein (20 ng) served as positive control.
We performed whole-genome expression profiling (Illumina, San Diego, CA, USA) analysis on IHPK2 KO mice and their WT littermates to determine if there was differential mRNA expression in various tissues. RNA was isolated from skin, brain, kidney, bladder, ovary and tongue of WT and KO animals (n=3), and prepared for hybridization according to the manufacturers instructions. We selected tongue as a representative tissue of the aerodigestive tract, one that gives rise to HNSCC and is easily biopsied. We selected skin as an epithelial tissue closely related to tongue, and brain, kidney, bladder and ovary because IHPK2 is expressed at relatively high levels in these tissues.
Compared to WT mice, in all six tissues of the KO mice there was relative overexpression of two genes, TTF1 and TWISTNB, both suggested to function as oncogenes (Figure 8). TTF1 is a transcription factor that is active during lung development (Kendall et al., 2007); it promotes proliferation of immortalized lung epithelial cells, and is overexpressed in lung carcinoma cell lines. TWISTNB may be overexpressed in colorectal carcinoma (Lu et al., 2006). Conversely, in all six tissues of IHPK2−/− mice there was consistent downregulation of two genes, DUSP16 and EXT2, both purported to function as tumor suppressors. DUSP16 dysregulation leads to abnormal keratinocyte function and skin formation (Magnusdottir et al., 2007). DUSP16 was suppressed in tumor biopsies from prostate cancer patients (Kibel et al., 2004). DUSP16 may exert tumor suppressor activity via dephosphorylation of c-Jun NH2-terminal kinase (Hoornaert et al., 2003; Teng et al., 2007). EXT2 (and EXT1) are often mutated in hereditary multiple exostoses, an autosomal dominant condition that causes protuberances at the ends of the long bones that can progress to become osteosarcomas (Hecht et al., 1995; Raskind et al., 1995). Hence, IHPK2−/− mice have decreased expression of these tumor suppressors.
Figure 8.
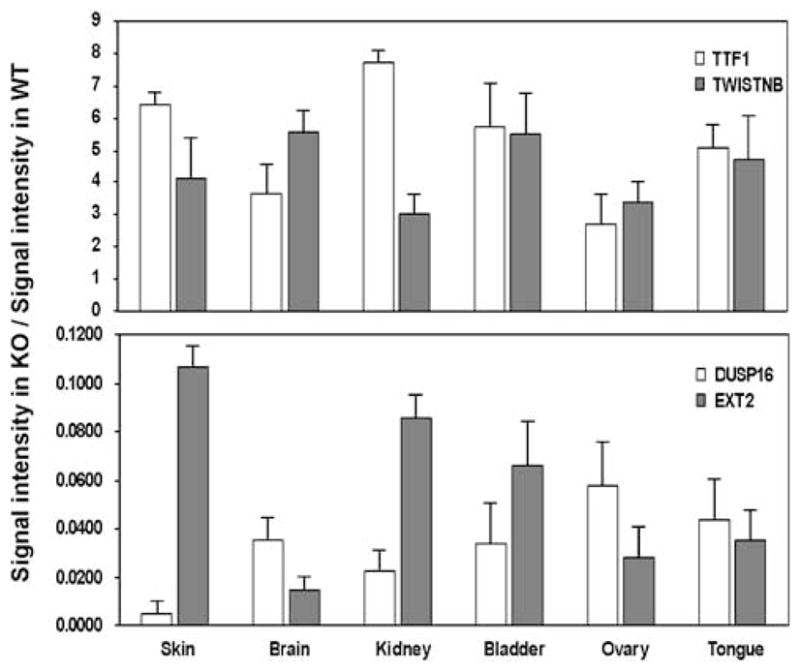
Whole-genome expression profiling in wild-type (WT) and inositol hexakisphosphate kinase 2 (IP6K2) knockout (KO) mice. Total RNA was prepared from six tissues (skin, brain, kidney, bladder, ovary, tongue) from WT and KO mice, which was then reverse transcribed into cDNA (n=3). Gene expression levels were determined using the Illumina platform and BeadStudio3 software (Illumina). Only hybridization signals with a minimum value of 25(arbitrary units, after background subtraction) and P<0.05(compared to background signal intensity) and a fold difference >5between WT and KO were considered for analysis. Data were expressed as fold change (mean KO signal/mean WT signal). Therefore, in the upper panel, genes are expressed at higher levels in KO mice; in the lower panel, genes are expressed at higher levels in WT mice. Four detailed datasets (spreadsheets containing all genes that were upregulated fivefold or greater in WT tongue, WT skin, KO tongue, KO skin) are included as Supplementary data.
Heat maps generated from the whole-genome expression analysis indicated there were eight tumor suppressor genes that were expressed in WT tongue at levels that were fivefold or greater compared to expression in KO tongue (Figure 9, left panel). Three putative oncogenes were differentially overexpressed in KO tongue compared to WT tongue (Figure 9, right panel). It is likely that loss of tumor suppressor function, coupled with a relative increase in oncogene expression, contribute to tumorigenesis in KO mice. Judging by the signal intensity and number of genes involved, loss of tumor-suppressor function may play a larger role than enhancement of oncogene expression during tumorigenesis. Notably, when differential gene expression was detected (a fivefold difference in KO vs WT) it frequently occurred in all six tissues tested, not exclusively in the tongue. These data suggest that IHPK2 loss may predispose to neoplasms other than those that originate in the aerodigestive tract.
Figure 9.
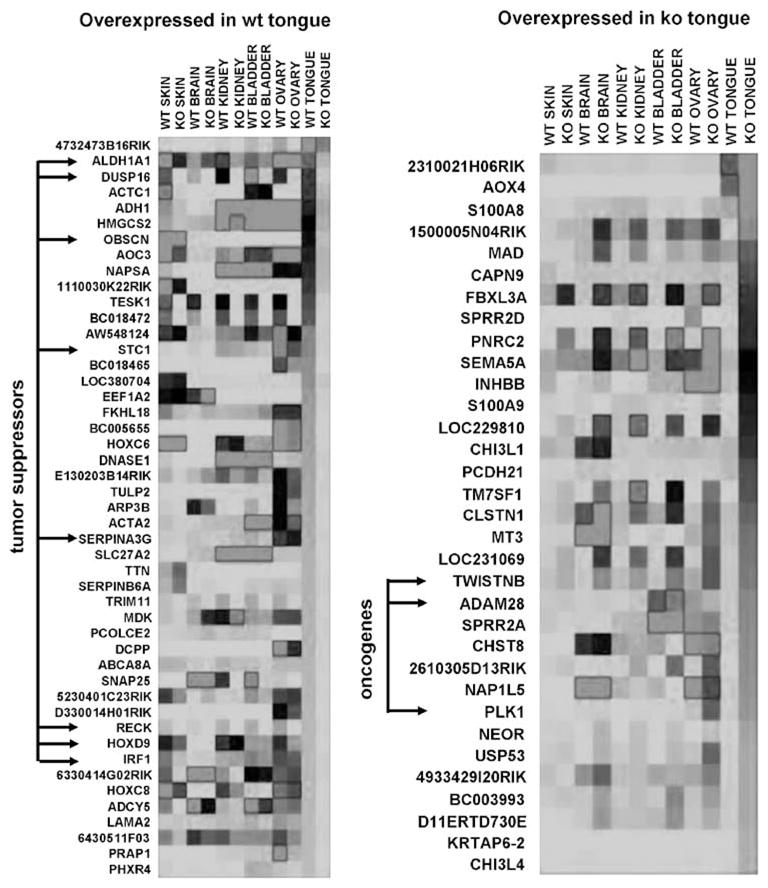
Heat maps: Whole-genome expression analysis. Illumina gene expression data in six murine tissues was selected for display using the same criteria as in Figure 8 (fold difference in gene expression >5(wild-type (WT) vs knockout (KO)); hybridization signal >25, P<0.05). Red indicates high-, black indicates moderate- and green indicates low-signal intensity. Left panel: genes ranked according to highest expression in WT tongue. Tumor suppressor genes indicated by arrows. Right panel: genes ranked according to highest expression in KO tongue. Putative oncogenes indicated by arrows. Four spreadsheets containing signal intensity data (WT, KO, tongue, skin) are included as Supplementary data. A full colour version of this figure is available at the Oncogene journal online.
Thus, whole-genome expression profiling data provide additional clues as to the mechanism of tumor suppression by mediators that are downstream of IHPK2. The molecular mechanism by which IP6K2 (or InsP6) modulates gene expression is not yet clear. DUSP16 and EXT2 gene expressions may be dependent on IHPK2 enzymatic activity. We are actively testing this hypothesis by expression of dominant negative IHPK2 constructs in HNSCC cell lines. Suppression of DUSP16 and EXT2 by DN-IHPK2 would support this theory. IHPK2 may function to normally suppress TTF1 and TWISTNB. In this case DN-IHPK2 will lead to enhanced TTF1 and TWISTNB expression. To establish the clinical relevance of these gene products, protein levels will be examined by immunohistochemistry performed on HNSCC specimens. We believe that tumors with low-level IHPK2 or mutant IHPK2 expression may have altered expression of these four markers.
To test the hypothesis that 5-PP-Ins(1,2,3,4,6)P5 might directly promote cell death, microinjection of SCC22A cells was performed. Time-lapse photomicroscopy suggested that introduction of 5-PP-Ins( 1,2,3,4,6)P5 into the cytoplasm of SCC22A cells inhibited the mitotic rate, slowed cellular migration, induced plasma membrane blebbing and promoted apoptotic body formation in the 24-h period following injection (animation WMV2). Microinjection with the same concentration of InsP6 (animation WMV1) did not induce these morphologic changes, and cells that received InsP6 behaved no differently than uninjected cells (data not shown). As the injection volume was estimated to be 10% of the cell volume, cytoplasmic concentration of 5-PP-Ins(1,2,3,4,6)P5 following injection was approximately 25 μM, fivefold higher than the concentration range normally observed in mammalian cells (1–5 μM) (Shears, 2004). Microinjection of 1-thiono-InsP7 a 1-pyrothiophosphate analog of InsP7, in which the nonhydrolysable pyrothiophosphate group was located at the 1-position rather than the 5-position, did not induce apoptosis (animation WMV3). These data suggest that pyrophosphorylation at the 5-position of the myo-inositol ring is critical for induction of HNSCC cell death.
Discussion
IP6K2 KO mice may display accelerated tumorigenesis following exposure to the carcinogen 4-NQO for several reasons. 5-PP-Ins(1,2,3,4,6)P5, the enzymatic product of IP6Ks, when microinjected into the cytoplasm, induced apoptosis in HNSCC cells that lacked it. Thus, changes in the substrate/product ratio (InsP6/5-PPIns( 1,2,3,4,6)P5) have been shown to regulate diverse cellular processes such as vesicular endocytosis (Saiardi et al., 2002), exocytosis (Illies et al., 2007) and DNA repair systems that utilize NHEJ (Hanakahi et al., 2000; Ma and Lieber, 2002; Byrum et al., 2004). Time-lapse video microscopy suggested that mitoses often appeared arrested, possibly secondary to inhibition of DNA repair. Commercially available lipid-based transfection agents were ineffective at delivery of 5-PP-Ins( 1,2,3,4,6)P5 into HNSCC cells and did not induce apoptosis (data not shown). Future efforts will focus on the use of synthetic compounds that function as shuttle vectors to enhance inositol polyphosphate delivery to the cytoplasm. This strategy may lead to development of a new class of chemotherapeutic agents.
A paradox: Why should IP6K2 KO animals show resistance to ionizing radiation, yet display increased susceptibility to a carcinogen, both stressors that drive neoplastic transformation? InsP6 promotes NHEJ, a process that repairs DSBs (Hanakahi et al., 2000; Cheung et al., 2008). The fidelity of NHEJ may depend on the cellular ratio of InsP7 to InsP6; NHEJ fidelity can be affected by small molecules such as caffeine (Kawata et al., 2005). TTF1, an oncogene upregulated in KO mice (Figure 8), is also upregulated during repair following lung injury (Pogach et al., 2007) and may modulate NHEJ. Effects of InsP7 on Ku70/80, a key component of the NHEJ complex known to bind InsP6 (Hanakahi et al., 2000), remain to be elucidated.
Materials and methods
Generation of IHPK2 knockout mice
The gene encoding IP6K2, IHPK2, is located on murine chromosome 9 (Ensembl gene model: ENSMUS00000032599), and has six exons. The OSDD2 vector (containing a Neo cassette flanked by LoxP sites) was designed to remove 201 bp of exon 2 (encoding amino acids 1–67) and throw the coding sequence in exon 3 out of frame (starting with translation from the next antithymocyte globulin (ATG)), when exon 1 (which is noncoding) was spliced to exon 3. The left homology arm, representing the sequence −3385 to −1 (with ATG at position 1), was engineered to contain XhoI and ClaI restriction sites. The right homology arm, 4671 bp in size, containing PacI and HindIII restriction sites, was amplified from bacterial artificial chromosomes using PCR. Both homology arms were digested and ligated into OSDD2. Final vector (14494 bp) was linearized with NotI before electroporation into 129SV/J ES cells. Neomycin-resistant ES cells were injected into C57Bl/6 blastocysts and implanted into pseudopregnant females. The resultant chimeric mice were crossed with outbred BL Swiss mice. Offspring of this breeding were interbred to generate KO (IHPK2−/−), heterozygous (IHPK2+/−) and WT (IHPK2+/+) mice; genotyping by PCR analysis of tail biopsies (Gentra; Qiagen, Valencia, CA, USA) confirmed the expected 1:2:1 KO/heterozygous/WT ratio. These mice, maintained on the 129SV-C57Bl/6 background, housed in microisolator cages under 12 h light/12 h dark cycles, were used for subsequent studies. All studies were approved by the Cleveland Clinic Institutional Animal Care and Use Committee.
Materials
Murine PEGylated IFN-β (Biogen-Idec, Cambridge, MA, USA) specific activity 3.4−108 U/mg was used in these studies.
Antiproliferative assays
Cells were treated with IFN-β during growth in RPMI-1640 (Mediatech Inc., Herndon, VA, USA) and 5% fetal bovine serum (HyClone, Logan, UT, USA). Cells were confirmed Mycoplasma free by PCR. Growth was monitored using a colorimetric assay (Skehan et al., 1990). Each treatment group contained eight replicates. Cells were fixed and stained with sulforhodamine B after 4 days. Bound dye was eluted from cells and absorbance (Aexp) was measured at 570 nm. One plate was fixed 8 h after plating to determine the absorbance representing starting cell number (Aini). Absorbance with this plate and that obtained with untreated cells at the end of the growth period (Afin) were taken as 0 and 100% growth, respectively. Thus,
Expressed as a percent of untreated controls, a decrease in cell number (relative to starting cell number) is a negative number on the y axis.
Carcinogen exposure
The carcinogen 4-NQO (Sigma Aldrich, St Louis, MO, USA) stock was prepared fresh weekly (5mg/ml) in propylene glycol and stored at 4 °C. Eight KO (four male, four female) and eight WT (four male, four female) mice were allowed free access to drinking water containing 4-NQO (100 μg/ml) that was changed weekly. After 24 weeks (or earlier if mice exhibited weight loss >15% body mass, dehydration, or lethargy), mice were killed, and gross lesions (>1mm diameter) of the tongue, esophagus and stomach were counted. Lesions were excised, fixed in formalin, embedded in paraffin and 4 μM sections were cut, deparaffinized and stained with hematoxylin and eosin.
Pathological examination
Lesions were classified as normal, epithelial hyperplasia, dysplasia and invasive carcinoma. Hyperplasia was defined as thickened epithelium with hyperkeratinization. Dysplasia was defined as nuclear pleomorphism, loss of epithelial cell polarity and increased/abnormal mitotic figures, confined to the epithelium. Invasive carcinoma was defined as migration of dysplastic cells into the subepithelial tissues.
RT–PCR
Total RNA was isolated from cell lines using the AllPrep DNA/RNA Mini Kit, first-strand cDNA synthesis (reverse transcription) was performed using the Omniscript RT Kit (both from Qiagen). PCR was performed using Roche Expand High Fidelity DNA Polymerase (Roche Applied Science, Mannheim, Germany), and the primers 5′-pATG AGC CCA GCC TTC AGG GCC ATG and 5′-p CTC CCC ACT CTC CTC ACT TAT CTC, an annealing temperature of 38 °C −45s, extension at 72 °C−60 s and 38 cycles of amplification.
Immunoblot analysis
Total cell protein (50 μg) was separated on 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membrane. Membranes were incubated with monoclonal antibody raised against IP6K2 (made in our laboratory) or GAPDH (Trevigen Inc., Gaithersburg, MD, USA). After washing, membranes were incubated with appropriate secondary antibody conjugated to horseradish peroxidase and developed using ECL reagents (Pierce, Rockford, IL, USA).
Synthesis of 5-PP-Ins(1,2,3,4,6)P5
Synthesis of 5-PP-Ins(1,2,3,4,6)P5 was performed as previously described; see Supplementary information (Bhandari et al., 2007). Description of a scalable synthetic route to 5-PP-Ins( 1,2,3,4,6)P5, its spectroscopic characterization and its validation in the ADP+InsP7↔ATP+InsP6 reaction catalysed by IP6K has recently been published (Zhang et al., 2009).
Microinjection
Cells were plated on coverslips in tissue culture dishes and microinjected 24 h later at 60–75% confluence. InsP6 or InsP7 or 1-thiono-InsP7 (250 μg/ml, dissolved in 200mM KPO4 pH 7.4) was microinjected into cells utilizing an Eppendorf Transinjector 5246, a Eppendorf Micromanipulator 5171, Eppendorf Femtotip injection pipettes and microloaders (used to fill microinjection needles) and a Nikon Diaphot 200 inverted microscope. Cytoplasmic injections were performed using the Z depth limit option (injection time of 0.5s, injection pressure of 65hPa, compensation pressure of 30 hPa). SCC22A cells normally grow as clusters, rather than dispersed single cells. Four clusters of 30–50 cells were injected with each compound. After microinjection, cells were washed once with phosphate-buffered saline (PBS) and fresh complete medium was applied.
Time-lapse photography
Time-lapse photography was performed using a Leica DMIRB Inverted microscope with a 20−/0.4 N.A. N Plan Phase lens (Leica Microsystems GmbH, Wetzlar, Germany), a PeCon Incubator (PeCon GmbH, Erbach, Germany), a Leica temperature controller and CO2 Incubation Chamber (Leica Microsystems GmbH), a Uniblitz shutter (Vincent Associates, Rochester, NY, USA), a Prior motorized stage and a linearly encoded controller with x/y/z drive for time-lapse imaging of multiple fields (Prior Scientific Inc., Rockland, MA, USA). Images were acquired using a Roper Scientific CoolSNAP HQ Cooled CCD camera (Roper Scientific, Tucson, AZ, USA) and MetaMorph Software (Molecular Devices, Downingtown, PA, USA). Phase contrast images of microinjected cells growing in a six-well plate were collected every 5min for 24 h. Time-lapse movies were created using ImagePro Plus software (Media Cybernetics, Bethesda, MD, USA).
Acknowledgments
We thank Philip Sanford, University of Cincinnati, for design of targeting vector and creation of IHPK2 knockout mice. Donna Kusewitt, Ohio State University, Director, Mouse Phenotyping Shared Resource, kusewitt.1@osu.edu, conducted the phenotyping studies. Judy Drazba, Cleveland Clinic Imaging Core, performed the time-lapse photography of microinjected cells. Dr Yong Xu, Dr Honglu Zhang and Dr Jianxing Zhang performed the synthesis of 5-PP-Ins(1,2,3,4,6)P5. These studies were supported by grants from NIH/NCI (CA095020) to DJL and from NIH (NS29632) to GDP.
Abbreviations
- IP6K2
inositol hexakisphosphate kinase 2
- 4-NQO
4-nitroquinoline 1-oxide
- IFN
interferon
- HNSCC
head and neck squamous cell carcinoma
- NHEJ
nonhomologous end joining
- PBS
phosphate-buffered saline
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
Conflict of interest
The authors declare no conflict of interest.
References
- Bhandari R, Juluri KR, Resnick AC, Snyder SH. Gene deletion of inositol hexakisphosphate kinase 1 reveals inositol pyrophosphate regulation of insulin secretion, growth, and spermiogenesis. Proc Natl Acad Sci USA. 2008;105:2349–2353. doi: 10.1073/pnas.0712227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari R, Saiardi A, Ahmadibeni Y, Snowman AM, Resnick AC, Kristiansen TZ, et al. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc Natl Acad Sci USA. 2007;104:15305–15310. doi: 10.1073/pnas.0707338104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrum J, Jordan S, Safrany ST, Rodgers W. Visualization of inositol phosphate-dependent mobility of Ku: depletion of the DNA-PK cofactor InsP6 inhibits Ku mobility. Nucleic Acids Res. 2004;32:2776–2784. doi: 10.1093/nar/gkh592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty SB, Dasgupta S, Roy A, Sengupta A, Ray B, Roychoudhury S, et al. Differential deletions in 3p are associated with the development of head and neck squamous cell carcinoma in Indian patients. Cancer Genet Cytogenet. 2003;146:130–138. doi: 10.1016/s0165-4608(03)00127-4. [DOI] [PubMed] [Google Scholar]
- Cheung JC, Salerno B, Hanakahi LA. Evidence for an inositol hexakisphosphate-dependent role for Ku in mammalian nonhomologous end joining that is independent of its role in the DNA-dependent protein kinase. Nucleic Acids Res. 2008;36:5713–5726. doi: 10.1093/nar/gkn572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridy PC, Otto JC, Dollins DE, York JD. Cloning and characterization of two human VIP1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. J Biol Chem. 2007;282:30754–30762. doi: 10.1074/jbc.M704656200. [DOI] [PubMed] [Google Scholar]
- Hanakahi LA, Bartlet-Jones M, Chappell C, Pappin D, West SC. Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair. Cell. 2000;102:721–729. doi: 10.1016/s0092-8674(00)00061-1. [DOI] [PubMed] [Google Scholar]
- Hawkins BL, Heniford BW, Ackermann DM, Leonberger M, Martinez SA, Hendler FJ. 4NQO carcinogenesis: a mouse model of oral cavity squamous cell carcinoma. Head Neck. 1994;16:424–432. doi: 10.1002/hed.2880160506. [DOI] [PubMed] [Google Scholar]
- Hecht JT, Hogue D, Strong LC, Hansen MF, Blanton SH, Wagner M. Hereditary multiple exostosis and chondrosarcoma: linkage to chromosome II and loss of heterozygosity for EXT-linked markers on chromosomes II and 8. Am J Hum Genet. 1995;56:1125–1131. [PMC free article] [PubMed] [Google Scholar]
- Hoornaert I, Marynen P, Goris J, Sciot R, Baens M. MAPK phosphatase DUSP16/MKP-7, a candidate tumor suppressor for chromosome region 12p12-13, reduces BCR-ABL-induced transformation. Oncogene. 2003;22:7728–7736. doi: 10.1038/sj.onc.1207089. [DOI] [PubMed] [Google Scholar]
- Illies C, Gromada J, Fiume R, Leibiger B, Yu J, Juhl K, et al. Requirement of inositol pyrophosphates for full exocytotic capacity in pancreatic beta cells. Science. 2007;318:1299–1302. doi: 10.1126/science.1146824. [DOI] [PubMed] [Google Scholar]
- Kawata T, Ito H, Saito M, Uno T, Okayasu R, Liu C, et al. Caffeine sensitizes nondividing human fibroblasts to x rays by inducing a high frequency of misrepair. Radiat Res. 2005;164:509–513. doi: 10.1667/rr3382.1. [DOI] [PubMed] [Google Scholar]
- Kendall J, Liu Q, Bakleh A, Krasnitz A, Nguyen KC, Lakshmi B, et al. Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc Natl Acad Sci USA. 2007;104:16663–16668. doi: 10.1073/pnas.0708286104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibel AS, Huagen J, Guo C, Isaacs WB, Yan Y, Pienta KJ, et al. Expression mapping at 12p12-13 in advanced prostate carcinoma. Int J Cancer. 2004;109:668–672. doi: 10.1002/ijc.20060. [DOI] [PubMed] [Google Scholar]
- Kok K, Osinga J, Carritt B, Davis MB, van der Hout AH, van der Veen AY, et al. Deletion of a DNA sequence at the chromosomal region 3p21 in all major types of lung cancer. Nature. 1987;330:578–581. doi: 10.1038/330578a0. [DOI] [PubMed] [Google Scholar]
- Li X, Lee NK, Ye YW, Waber PG, Schweitzer C, Cheng QC, et al. Allelic loss at chromosomes 3p, 8p, 13q, and 17p associated with poor prognosis in head and neck cancer. J Natl Cancer Inst. 1994;86:1524–1529. doi: 10.1093/jnci/86.20.1524. [DOI] [PubMed] [Google Scholar]
- Lu B, Xu J, Lai M, Zhang H, Chen J. A transcriptome anatomy of human colorectal cancers. BMC Cancer. 2006;6:40. doi: 10.1186/1471-2407-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo HR, Huang YE, Chen JC, Saiardi A, Iijima M, Ye K, et al. Inositol pyrophosphates mediate chemotaxis in dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell. 2003;114:559–572. doi: 10.1016/s0092-8674(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Ma Y, Lieber MR. Binding of inositol hexakisphosphate (IP6) to Ku but not to DNA-PKcs. J Biol Chem. 2002;30:30. doi: 10.1074/jbc.C200030200. [DOI] [PubMed] [Google Scholar]
- Maestro R, Gasparotto D, Vukosavljevic T, Barzan L, Sulfaro S, Boiocchi M. Three discrete regions of deletion at 3p in head and neck cancers. Cancer Res. 1993;53:5775–5779. [PubMed] [Google Scholar]
- Magnusdottir E, Kalachikov S, Mizukoshi K, Savitsky D, Ishida-Yamamoto A, Panteleyev AA, et al. Epidermal terminal differentiation depends on B lymphocyte-induced maturation protein-1. Proc Natl Acad Sci USA. 2007;104:14988–14993. doi: 10.1073/pnas.0707323104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BH, Bauer JA, Hu J, Grane RW, Ozdemir A, Chawla-Sarkar M, et al. Inositol hexakisphosphate kinase 2 sensitizes ovarian carcinoma cells to multiple cancer therapeutics. Oncogene. 2002;21:1882–1889. doi: 10.1038/sj/onc/1205265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BH, Bauer JA, Kalvakolanu DV, Lindner DJ. Inositol hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-beta in ovarian carcinoma cells. J Biol Chem. 2001;276:24965–24970. doi: 10.1074/jbc.M101161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulugu S, Bai W, Fridy PC, Bastidas RJ, Otto JC, Dollins DE, et al. A conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science. 2007;316:106–109. doi: 10.1126/science.1139099. [DOI] [PubMed] [Google Scholar]
- Nagata E, Luo HR, Saiardi A, Bae BI, Suzuki N, Snyder SH. Inositol hexakisphosphate kinase-2, a physiologic mediator of cell death. J Biol Chem. 2005;280:1634–1640. doi: 10.1074/jbc.M409416200. [DOI] [PubMed] [Google Scholar]
- Panigrahi GB, Walker IG. The N2-guanine adduct but not the C8-guanine or N6-adenine adducts formed by 4-nitroquinoline 1-oxide blocks the 3′-5′ exonuclease action of T4 DNA polymerase. Biochemistry. 1990;29:2122–2126. doi: 10.1021/bi00460a023. [DOI] [PubMed] [Google Scholar]
- Pogach MS, Cao Y, Millien G, Ramirez MI, Williams MC. Key developmental regulators change during hyperoxia-induced injury and recovery in adult mouse lung. J Cell Biochem. 2007;100:1415–1429. doi: 10.1002/jcb.21142. [DOI] [PubMed] [Google Scholar]
- Raskind WH, Conrad EU, Chansky H, Matsushita M. Loss of heterozygosity in chondrosarcomas for markers linked to hereditary multiple exostoses loci on chromosomes 8 and 11. Am J Hum Genet. 1995;56:1132–1139. [PMC free article] [PubMed] [Google Scholar]
- Saiardi A, Bhandari R, Resnick AC, Snowman AM, Snyder SH. Phosphorylation of proteins by inositol pyrophosphates. Science. 2004;306:2101–2105. doi: 10.1126/science.1103344. [DOI] [PubMed] [Google Scholar]
- Saiardi A, Resnick AC, Snowman AM, Wendland B, Snyder SH. Inositol pyrophosphates regulate cell death and telomere length through phosphoinositide 3-kinase-related protein kinases. Proc Natl Acad Sci USA. 2005;102:1911–1914. doi: 10.1073/pnas.0409322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiardi A, Sciambi C, McCaffery JM, Wendland B, Snyder SH. Inositol pyrophosphates regulate endocytic trafficking. Proc Natl Acad Sci USA. 2002;99:14206–14211. doi: 10.1073/pnas.212527899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shears SB. How versatile are inositol phosphate kinases? Biochem J. 2004;377:265–280. doi: 10.1042/BJ20031428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Teng CH, Huang WN, Meng TC. Several dual specificity phosphatases coordinate to control the magnitude and duration of JNK activation in signaling response to oxidative stress. J Biol Chem. 2007;282:28395–28407. doi: 10.1074/jbc.M705142200. [DOI] [PubMed] [Google Scholar]
- Wojewodzka M, Buraczewska I, Kruszewski M. A modified neutral comet assay: elimination of lysis at high temperature and validation of the assay with anti-single-stranded DNA antibody. Mutat Res. 2002;518:9–20. doi: 10.1016/s1383-5718(02)00070-0. [DOI] [PubMed] [Google Scholar]
- York SJ, Armbruster BN, Greenwell P, Petes TD, York JD. Inositol diphosphate signaling regulates telomere length. J Biol Chem. 2005;280:4264–4269. doi: 10.1074/jbc.M412070200. [DOI] [PubMed] [Google Scholar]
- Zhang H, Thompson J, Prestwich GD. A scalable synthesis of the IP(7) Isomer, 5-PP-Ins(1,2,3,4,6)P(5) Org Lett. 2009;11:1551–1554. doi: 10.1021/ol900149x. [DOI] [PMC free article] [PubMed] [Google Scholar]