

Abstract
It is estimated that ∼2500 genes are essential for the normal development of a zebrafish embryo. A mutation in any one of these genes can result in a visible developmental defect, usually followed by the death of the embryo or larva by days 5–7 of age. We are performing a large-scale insertional mutagenesis screen in the zebrafish with the goal of isolating ∼1000 embryonic mutations. We plan to clone a significant fraction of the mutated genes, as these are the genes important for normal embryogenesis of a vertebrate. To achieve this goal, we prepared ∼36,000 founder fish by injecting blastula-stage embryos with one of two pseudotyped retroviruses. We estimate that together these fish harbor between 500,000–1,000,000 proviral insertions in their germ lines. The protocol we have devised and the size of our facility allow us to breed ∼80,000–150,000 of these insertions to homozygosity within 2 years. Because a pilot screen conducted earlier in our laboratory revealed that the frequency of mutations obtained with this type of insertional mutagen is 1 embryonic lethal mutation per 70–100 proviral insertions, screening 100,000 insertions should yield at least 1000 mutants. Here we describe the protocol for the screen and initial results with the first of the two retroviral vectors used, a virus designated F5. We screened an estimated 760 insertions among F3 progeny from 92 F2 families and obtained 9 recessive embryonic lethal mutations. Thus, the efficiency of mutagenesis with this viral vector is approximately one-ninth that observed with the chemical mutagen ENU in zebrafish. We have also obtained two dominant mutations, one of which is described here. As expected, mutated genes can be readily identified. So far, genes mutated in four of the nine recessive mutants and one of the two dominant mutants have been cloned. Further improvements to this technology could make large-scale insertional mutagenesis screening and rapid gene cloning accessible to relatively small zebrafish laboratories.
Keywords: Zebrafish, mutagenesis screen, embryogenesis, development
Introduction
Genetic screens have been the most successful approach for identifying genes required for developmental processes. Applied on a sufficiently large scale, a genetic screen can identify all of the genes, which when mutated one at a time impact the phenotype of interest. Genetic screens make no assumptions about the genes involved in the biological processes of interest and thus can reveal novel genetic pathways underlying important phenotypes.
Although it has long been the primary method for identifying the genetic basis of phenotypes in invertebrate organisms, genetic screening is rarely performed in vertebrate animals, and a saturation screen has never been achieved in any vertebrate. This is because the number of animals that must be raised, maintained, and screened is hundreds of thousands for a moderate-sized screen, and millions to achieve saturation. Nonetheless, many small-scale screens in zebrafish (Grunwald et al. 1988; Kimmel et al. 1989; Kimmel 1989) and mice (Shedlovsky et al. 1986; Rinchik 1991; Vitaterna et al. 1994), have been highly successful, and two large-scale screens have been carried out in the zebrafish (Driever et al. 1996; Haffter et al. 1996). The genetic screens that have already been performed in vertebrate animals hint at the great potential of this approach. In zebrafish, simple visual screens of embryos in the first 5 days after fertilization can reveal mutations in genes essential for the normal development of most of the major organ systems, including the nervous system, heart, blood, gut, liver, jaws, eyes, and ears. Future screens should reveal the genetic basis of many additional phenotypes important in vertebrate development, physiology, and behavior.
Simply identifying mutant phenotypes in a genetic screen can be informative by revealing both the kinds of phenotypes that can occur and the number of genes involved in the process of interest. However, to understand how genes specify a biological process, it is essential to identify the mutated genes. Because high mutation frequencies can be obtained with chemical mutagens, most large-scale genetic screens have used these agents (Grunwald et al. 1985; Mullins et al. 1994; Solnica-Krezel et al. 1994). Although the mutation frequency can vary widely for different loci, chemical mutagens can induce mutations in most genes. Despite these advantages, the chemical mutagens that are commonly used are disadvantageous because cloning mutant genes is difficult, as these chemicals usually cause point mutations. Decades of work have made positional cloning of chemically induced mutants feasible, but in vertebrate animals with large genomes, cloning remains expensive and laborious.
An alternative approach to chemical mutagenesis that greatly speeds the cloning of mutant genes is insertional mutagenesis (Kidwell 1986; Gridley et al. 1987). The integration of exogenous DNA sequences into a genome can be mutagenic, and the inserted DNA serves as a tag to clone mutated genes. Insertional mutagens are usually less efficient than chemicals, presumably because fewer lesions are obtained per genome. Furthermore, insertional mutagens seldom, if ever, integrate entirely randomly into host DNA (Spradling et al. 1995). Despite these limitations, the development of insertional mutagenesis by P elements in Drosophila revolutionized the cloning of mutated genes, and even today, P-element-induced alleles are preferred for cloning mutant fly genes (Cooley et al. 1988). Insertional mutagens have also been shown to work in mice. Both DNA and retroviruses have been used successfully (Jaenisch 1988; Meisler 1992; Moyer et al. 1994). Despite these successes and despite the extraordinary potential of the method, however, the cost of generating and breeding large numbers of transgenic mice makes the use of insertional mutagenesis even more daunting than chemical mutagenesis for large-scale screens in this organism.
Given the feasibility of performing large-scale genetic screens in the zebrafish but the difficulty of identifying genes mutated by chemicals, our laboratory has undertaken to develop a method of insertional mutagenesis for this vertebrate. This is a substantial undertaking because the zebrafish genome is estimated to be 1.6 × 109 bp; thus, to achieve saturation, one would have to screen on the order of 500,000 randomly placed integrations.
Several years ago, our laboratory found that mouse retroviral vectors pseudotyped with a VSV-G envelope could infect the fish germ line following injection of virus into blastula-stage embryos at the 1000–2000-cell stage (Zavada 1972; Emi et al. 1991; Burns et al. 1993; Lin et al. 1994). Retroviruses were attractive candidates for insertional mutagens, because they had been shown to integrate into many different sites in mammalian and avian chromosomes and to be effective mutagens in mice (Withers-Ward et al. 1994; Jaenisch 1988). Importantly, they integrate without rearrangement of their own sequences or significant alterations to host DNA sequences at the site of insertion, essential features for easily cloning genes disrupted by insertions (Varmus 1982). Although initially the number of proviral insertions we could generate in the fish germ line was too low to make a genetic screen feasible, further advances in our laboratory allowed us to achieve high transgenesis rates, making it possible to generate hundreds of thousands of insertions (Gaiano et al. 1996a). In a pilot screen, we demonstrated that proviral insertions can induce mutations in zebrafish and that mutated genes could be easily cloned (Allende et al. 1996; Gaiano et al. 1996b). These advances set the stage for a large-scale insertional mutagenesis screen in the zebrafish.
We devised a strategy for the most efficient breeding and screening of proviral insertions so that we could perform a large-scale screen. By breaking this multistep experiment into component projects and designing a protocol for each, we established a workable method. Here we describe the method and report on the first mutants obtained in an ongoing screen and the rapid cloning of the mutated genes.
Results
To generate founder fish, we injected virus among the cells of embryos between the 1000- and 2000-cell stages (Lin et al. 1994). Studies from our laboratory have suggested that at this time there are four primordial germ cells, which divide over the next several hours to produce 20–30 cells (Yoon et al. 1997). Analysis of proviral insertions in F1 progeny of founders has revealed that different germ cells are infected independently, and with high titer virus stocks they often have multiple integrations. Any given insertion is transmitted mosaically to between 1% and 40% of the F1 progeny. Individual F1 fish can inherit multiple insertions, and proviral insertions in F1 fish are transmitted in a Mendelian fashion.
In a pilot screen, we had outcrossed founder fish, identified F1 fish with single proviral insertions, generated an F2 family for each insertion, and then inbred transgenic F2 fish and examined F3 progeny to identify mutations (Gaiano et al. 1996b). This approach made it easy to determine the mutation frequency and to clone mutated genes, but it was far too inefficient for a large scale screen because each insertion was inbred individually, thus requiring its own F2 family and a separate tank. To screen many more insertions with a given number of tanks, we took advantage of the fact that founders can transmit multiple insertions to individual F1 fish (Gaiano et al. 1996a). By crossing founders to each other, we enrich for F1 fish with multiple insertions and use these to generate F2 families in which many insertions can be screened simultaneously. Figure 1 depicts the protocol we devised for a large, diploid (F3) insertional mutagenesis screen.
Figure 1.
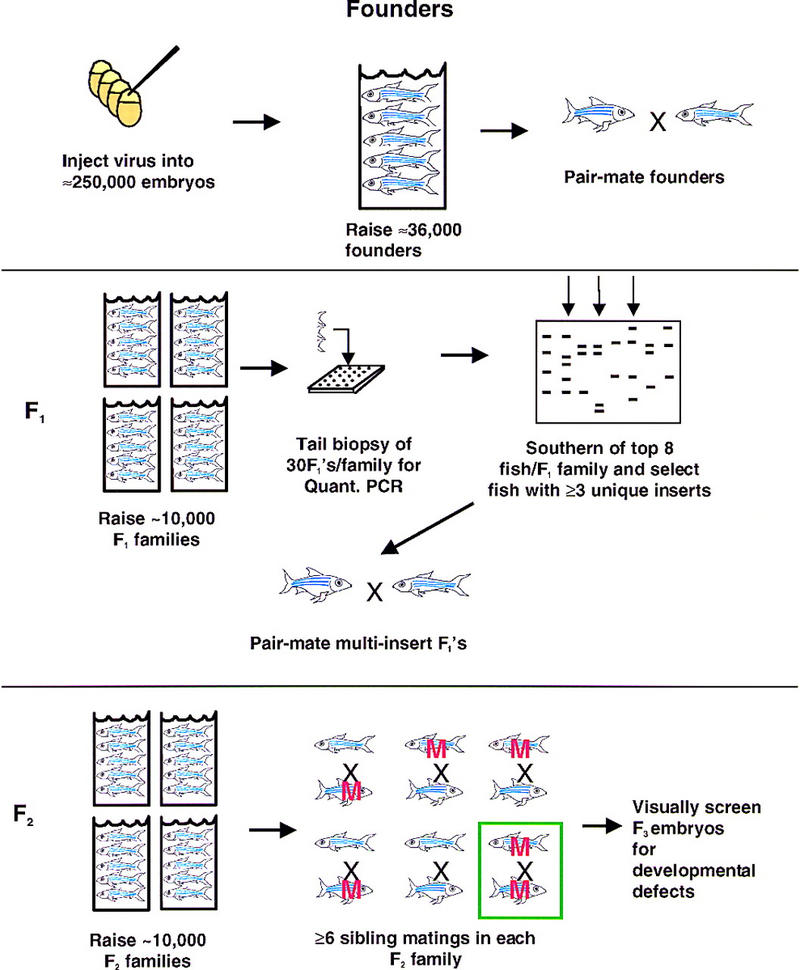
Schematic diagram of the protocol for the large-scale screen. We have finished generating founder fish. As of 8/99, 3800 F1 families have been born, 2500 have been genotyped by fin clips, and 950 F2 families have been born.
The size of the screen is based on the number of F1 and F2 families our facility could accommodate, taking into account the rate at which fish reach sexual maturity and thus can move through the facility. The protocol in Figure 1 is being carried out over 3.5 years to yield ∼1000 mutants. The critical determinant of the number of insertions screened for a given number of F2 families is the average number of unique inserts in F1 fish used to generate the F2 families. Thus, it was necessary to have excellent founder fish and methods for selecting F1 fish with the maximum number of unique inserts. Protocols were devised to achieve these tasks reliably in a high throughput manner.
Protocols for component tasks of a large-scale screen
1.
Preparation of high titer stocks of F5 virus We used two viruses to generate founder fish. Results with the first virus, F5, are described in this work. The preparation of the second virus, designated GT, was by a different and novel method and we have not yet begun to screen for mutants induced by this virus. Preparation of GT virus and results obtained with it in the screen will be published separately (W.-B. Chen, S. Burgess, and N. Hopkins, unpubl.).
To prepare a cell line producing high titer virus, we obtained a packaging cell line 293 gp/bsr (Miyoshi et al. 1997), infected it with a virus, SFGnlslacZ (Gaiano et al. 1996a), and selected a clone of cells designated F5 that yielded virus with high titer on both mouse 3T3 cells and a fish cell line PAC2 (Culp 1994), as determined by lacZ staining. Virus stocks were prepared by calcium phosphate transfection (Graham and van der Eb 1973). During the course of the work, we found that lacZ titering of viruses on PAC2 cells was unreproducible, so we developed an assay to titer viruses on injected embryos.
2.
Injecting virus: monitoring successful injections by the embryo assay To assess the efficiency with which injected embryos are infected, we used either quantitative Southern blotting or quantitative PCR. Two to five days after every injection session, several injected embryos were lysed and their DNA extracted for analysis. Two genomic sequences were probed, a single-locus gene RAG2 (Willett et al. 1997) and proviral sequences. The ratio of these signals was normalized to signals from DNA of a fish heterozygous for a single insertion. The result, designated the embryo assay value, was used as a measure of the average number of proviral integrations per cell. Injected eggs that were raised were assumed to have the same embryo assay value as those that were sampled from the same injected batch.
To determine that the embryo assay was a good predictor of efficient germ-line transmission of proviral insertions, founder fish from batches of injected embryos with a range of embryo assay values were tested to determine the amount of provirus they could transmit to their F1. We outcrossed the founders and used the quantitative assay for RAG2 versus proviral sequences on DNA extracted from pools of their F1 progeny. Although there was considerable variation between founders from injections that had yielded the same embryo assay value, there was a definite correlation between embryo assay and average provirus transmission rate. Most founders from injections with embryo assays below 2 did not transmit well enough for our purposes, about half the founders with embryo assays of 2–5 transmitted sufficiently well, and nearly all founders with embryo assays over 5 transmitted well with an average of greater than one insert per gamete. At this transmission rate, we found that a substantial proportion of F1 fish inherit multiple proviral insertions. With F5 virus, we kept batches of embryos from injections in which the embryo assay values ranged from 2 to 11.4. Injections to make 36,000 founder fish, of which ∼15,000 were made with F5 virus, were performed 5 days/week for 11 months by one to two injectors per day. We estimate that ∼250,000 embryos were injected, and hence the overall survival of injected embryos to adulthood was ∼15%.
3.
Generating F1 families, selecting multi-insert F1 fish, and identifying dominant visible mutations To generate F1 families, we mate founder fish to each other. There is considerable variation in the number of inserts between fish in a single F1 family, as well as between F1 families. To identify fish with the most nonoverlapping inserts, 30 F1 fish from each cross are raised for 8 to 12 weeks and then their tail fins are clipped (Westerfield 1995). The fish are held in individual cups while DNA is extracted from the fin clips. A small amount of DNA is analyzed by real time quantitative PCR (Heid et al. 1996) to identify the eight fish with the greatest number of inserts in each family, and the rest of the sample from these eight fish is used for Southern blot analysis. As shown in Figure 2, the F1 fish with the greatest number of inserts are often derived from the same germ cell(s) and hence share proviral insertions. We keep fish that have at least three unique insertions. For F1 families bred from F5-injected founders, we have kept an average of 2.4 F1 fish per family and these fish have an average of 4.8 unique proviral insertions per fish.
Figure 2.

Southern analysis of the top eight fish from two different F1 families. Note that in family A, the majority of the insertions are the same from fish to fish, implying that most of them came from the same germ cell from one of the founders. In this case, we would keep fish 3 (seven inserts), fish 6 (three new inserts), and no others, as none would give more than two new inserts. Family B has much greater diversity of inserts. In this case, we would keep fish 6 (nine inserts), fish 7 (seven new inserts), and fish 2 (five new inserts).
Whereas only these selected insertions can be inbred to screen for recessive mutations, the 30 fish originally raised in each F1 family represent 25–30 insertions that can be screened for dominant viable phenotypes. So far, we have limited such screening to simple visible phenotypes, such as pigmentation and fin shape, and have found two dominant mutations, which were confirmed by outcrossing and finding the phenotype in half of their progeny. In each case, a single proviral insertion (identified by Southern analysis) cosegregated with the phenotype (see Table 1).
Table 1.
Linkage analysis of insertional mutants
| Recessive mutants
|
Homozygotes observed in
|
|
|---|---|---|
| phenotypic embryos
|
wild-type embryos
|
|
| no knack | 31/31 | 0/110 |
| bubble brain | 31/31 | 0/105 |
| nearly normal | 142/145* | 8/132* |
| bleached blond | 68/68 | 0/299 |
| hi37 | 30/30 | 0/66 |
| hi43 | 63/63 | 0/73 |
| hi61 | 18/18 | 0/54 |
| hi63 | 28/28 | 0/59 |
| hi96 | 34/34 | 0/81 |
| Dominant mutants | Heterozygotes observed in | |
| phenotypic fish | wild-type fish | |
| HagoramoD2 | 54/54 | 0/61 |
| hiD862 | 33/33 | 0/36 |
For recessive mutants, heterozygotes were crossed, embryos were sorted as phenotypic or wild-type, and DNA was extracted from individual embryos and analyzed by Southern analysis for genotyping. For dominant mutants, heterozygotes were crossed to wild-type fish, juvenile fish were sorted by phenotype, and DNA was extracted from fin clips and analyzed by Southern analysis or PCR *In the case of nearly normal, because the sole embryonic phenotype is the lack of a swim bladder and sometimes embryos can fail to develop swim bladders for nongenetic reasons, we believe that the three “phenotypic” embryos that were not homozygous for the insertion are phenocopies, not recombinants. Additionally, the eight wild-type mutants analyzed that were homozygous appear to be due to incomplete penetrance of the embryonic phenotype; all observed surviving homozygotes (in other crosses) grew up to be about one-fourth the size of their siblings (see text and Fig. 4).
4.
Generating F2 families, screening F3 embryos, and demonstrating that mutants are caused by proviral insertions To generate F2 families, multi-insert F1 fish are mated and 50–70 embryos from each pair are raised. We perform sibling crosses of F2 fish at 3 months of age or older and examine their F3 embryos in a dissecting microscope to identify mutants. We examine embryos at 24 and 48 hr after fertilization and at 5 days of age (∼120 hr after fertilization). At day 5, embryos are screened for swimming behavior, then anesthetized, and visible structures are examined for defects.
To identify which of 10 or so insertions segregating in an F2 family is linked to an identified mutation, Southern analysis is performed on DNA extracted from fin clips of parents of all the crosses screened in the family. A specific insert (Southern band) must be shared by both parents of every cross that showed the phenotype, and must be in only one or neither of the parents of all crosses that did not show the phenotype. We also perform Southern analysis on DNA from individual mutant embryos to look for the presence of this band. An unlinked band would only be present in three-fourths of the embryos, whereas a linked band must be in all of them. Often one can tell from the relative intensity of the bands that the candidate band is homozygous in mutant embryos. However, to obtain stronger evidence of tight linkage, we use a probe to genomic DNA flanking the candidate band.
Once a candidate band is identified, a junction fragment from either or both sides of this insertion is (are) cloned by inverse PCR (Ochman et al. 1988). The strategy used to clone the correct junction fragment from families with many inserts is shown in Figure 3. The junction fragment is then used for two purposes: First, to distinguish chromosomes with and without the putative mutagenic insertion in Southern blots, and hence to determine whether mutant embryos are invariably homozygous for the mutagenic insert, whereas their wild-type siblings never are. Table 1 shows the linkage data for the nine recessive mutations obtained by this analysis. Second, the junction fragment, when sequenced, may have homology to a known gene or EST in the public database.
Figure 3.
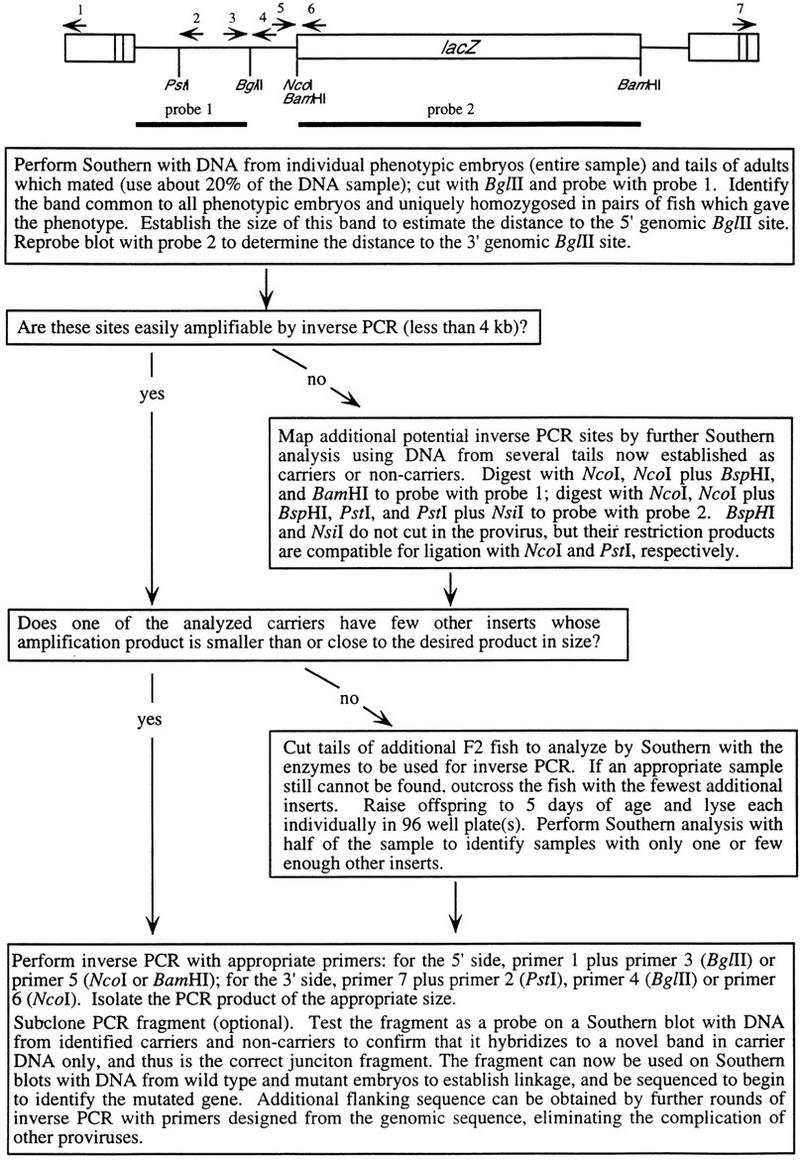
Isolation of genomic sequence flanking mutagenic insertions from multiple insert families. The schematic at top indicates the structure of the provirus along with the position of Southern blot probes and PCR primers.
Phenotypes of insertional mutants
From the first 92 F2 families screened, we obtained 9 recessive embryonic lethal mutations. Each F2 family contains on average 9.6 unique proviral insertions. We obtained on average 8 pair matings/family; therefore, we estimate that 760 insertions were screened and calculate our mutant frequency for these families at ∼85 insertions per embryonic lethal mutation, or 1 embryonic mutation per 9 families screened. In large chemical mutagenesis screens, the frequency was ∼1 embryonic lethal mutation per F2 family. We also identified two dominant mutations induced by F5 virus.
The phenotypes of the nine recessive embryonic lethal mutants obtained in the large screen to date are shown in Figure 4 and summarized in Table 2. As found in large chemical mutagenesis screens, many embryonic lethal mutations are relatively nonspecific. At least some of these may be caused by mutations in genes expressed widely in the embryo and required for cell survival or growth. Some mutants display highly specific defects. For example, nearly normal mutant embryos appear normal at 5 days of age, except that in the majority of homozygotes, the swim bladder fails to inflate and the embryos die. As discussed below, this very specific phenotype results from a mutation in a widely expressed housekeeping type gene EF1γ. Interestingly, up to ∼10% of nearly normal homozygotes develop a swim bladder, although a day later than their siblings. If these fish are raised, they are small in relation to their siblings and weigh only one-fourth as much at 2 months of age (Fig. 4). bleached blond mutant embryos have striking specific defects in the appearance of pigmentation in their melanocytes and in the pigmented epithelium in the retina, apparently due to a mutation in the gene encoding the Ac45 subunit of the vacuolar ATP synthase. They must have other defects as well, however, because the majority of embryos fail to develop a swim bladder and those that do develop one fail to thrive, although they can survive for many days. hi43 mutant embryos are generally normal, for example, head and jaw structures appear well developed, unlike most pleiotropic mutants; however, the liver is clearly abnormal, appearing dark and lacking circulation, and the gut appears compressed, although it is unclear whether or not this is a consequence of the degree of unconsumed yolk.
Figure 4.
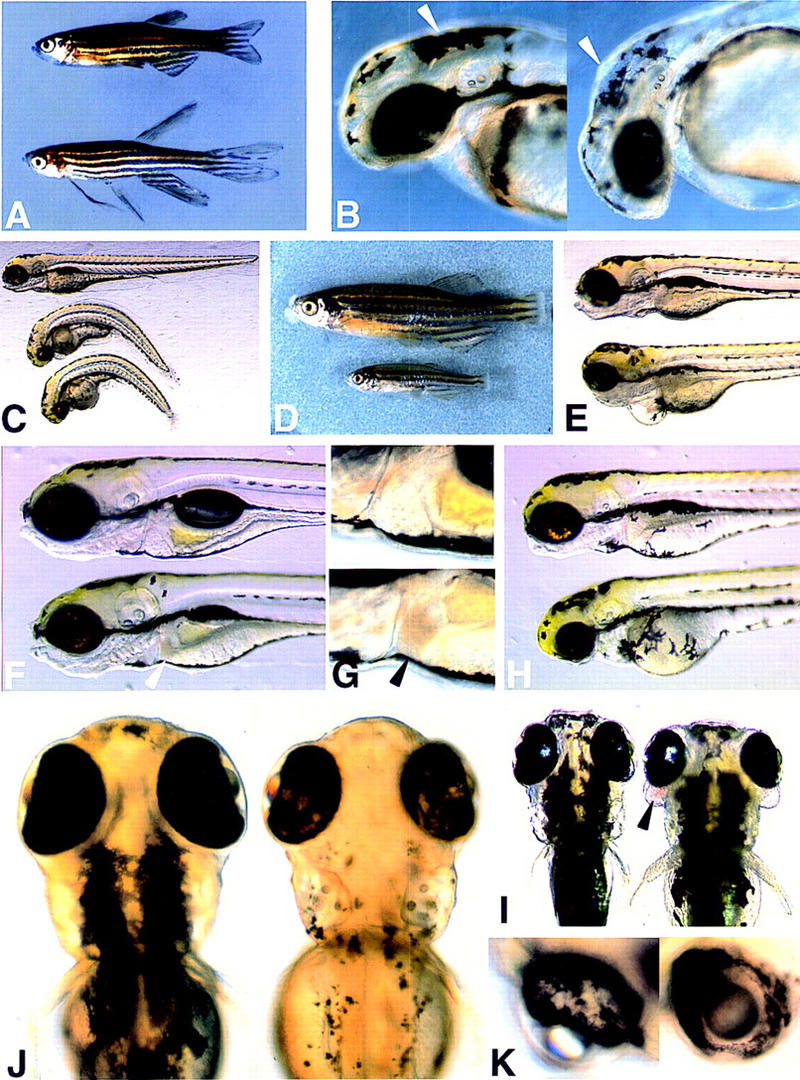
Photographs of wild-type vs. mutant embryos or adult fish for 9 of the 11 insertional mutants described. (A) Ten-week-old wild-type (top) vs. dominant mutant hiD862 with long fins. (B) Wild-type (left) vs. bubble brain at day 2. (Arrowhead) Region of enlarged ventricle in mutants. (C) Wild-type (top) vs. two no knack mutant embryos at day 4. (D) Nine-week-old wild-type (top) vs. a nearly normal sibling, one of ∼10% of the homozygotes that survived. (E) Wild-type (top) vs. hi37 mutant at day 4. (F) Wild-type (top) vs. hi43 at day 5. (Arrowhead) Liver that is abnormal in the mutant. (G) Closer view of wild-type (top) vs. hi43 liver region. (H) Wild-type (top) vs. hi63 mutant at day 3. (I) Wild-type (left) vs. hi96 mutant embryo at day 4. (Arrowhead) Unusual edema with pooled blood around eye. Edema around body of mutant is also visible. (J) Wild-type (left) vs. bleached blond mutant at day 4. (K) Closer view of eyes of bleached blond at day 4 showing mottled appearance.
Table 2.
Phenotype of insertional mutants isolated in the large-scale screen
| Name
|
Allele
|
Phenotype
|
Visible onset
|
|---|---|---|---|
| Recessive Insertional mutants | |||
| no knack | nokhi5 | reduced circulation, edema around heart, bent body, jaw does not form, sb− | day 3 |
| bubble brain | bbrhi10 | enlarged brain ventricle; yolk disappears from yolk extension, and later development is impaired; embryo dies by day 4–5 | late day 1 |
| nearly normal | nanhi14 | appears normal except swim bladder does not inflate; not completely penetrant; embryos that develop a swim bladder grow up small | day 5 |
| hi37 | reduced head structures; eyes look wide apart, reduced circulation, edema around heart at day 4; jaw does not form; sb− | day 3 | |
| hi43 | liver structure is defective, blood does not appear to circulate normally through liver, and it darkens; gut tube abnormal; swim bladder fails to inflate but embryos are still alive at 1 week; sb− | day 4 | |
| hi61 | generally underdeveloped; sb− | day 5 | |
| hi63 | small head structures including eye and ear; jaw does not develop; sb− | late day 3 | |
| hi96 | multiple and unusual edemas including around eyes; heart and finally embryo appears as if atop a huge ball; reduced circulation; sb− | day 4 | |
| bleached blond | bblhi112 | pigmentation is defective in both the eyes and the melanocytes; eyes appear mottled; sb+/− | day 2 |
| Dominant insertional mutants | |||
| hiD862 | all fins are abnormally long | young adult | |
| Hagoramo | HaghiD2 | stripes are interrupted in body region; this is second insertional allele in this gene | young adult |
sb−, lacks swim bladder at day 5; sb+/−, some embryos develop a swim bladder, but most do not.
Interestingly, one of the two dominant mutations, Hagoramo, induced by F5 virus represents a second insertional allele of a gene mutated in our pilot screen. The insertions cause a disruption of the stripe pattern in adult fish. K. Kawakami and N. Hopkins (unpubl.) cloned the gene responsible and showed that the insertions lie within 10 kb of each other within the fifth intron of a gene that would encode a protein of unknown function containing WD repeats. The possible implications of this result for the randomness of proviral integration is discussed below. The second dominant mutation we identified displays long fins in adult fish (Fig. 4A), a phenotype identified many years ago in the zebrafish and reisolated in large screens, also as dominant mutations.
Identification of genes mutated in four of nine recessive embryonic lethal insertional mutants and one of two dominant mutants
The proviral insertion responsible for each mutation was identified as described above, except in the case of long fin. Only 3 (of 30) F1 fish with the long fin phenotype were found initially, and each had multiple insertions that yielded closely migrating bands on Southern blots, complicating the identification of a candidate insert. Thus, we outcrossed these fish, raised their progeny, and at 8 weeks of age identified a common insertion in the 50% of the fish that displayed the long-fin phenotype.
Following the protocol outlined in Figure 3, we readily obtained a junction fragment linked to each of the insertional mutants, and following protocols established in our pilot screen, we cloned the genes from 5 of the 11 mutants described here (see Table 3). In Table 3, we have also included the 7 mutants obtained in our pilot screen and have grouped all 18 insertional mutants studied to date in our laboratory on the basis of the type of gene function underlying the mutant phenotypes, if the function of the gene is known. As expected, the genes involved represent diverse categories of functions. A number of the genes are novel.
Table 3.
Insertional mutants and their genes
| Mutant
|
Allele
|
Phenotype
|
Gene
|
|---|---|---|---|
| Transcription | |||
| not really finished | nrlhi399a | photoreceptor layer in eye dies | transcription factor (NRF-1b/IBRc) |
| no arches | narhi1d | multiple defects, apoptosis | polyadenylation factore/RNasef |
| Translation | |||
| bubble brain | bbrhi10 | enlarged ventricle in brain | 60S ribosomal protein–L44g |
| nearly normal | nanhi14 | ∼90% no swim bladder, ∼10% small adult | translation factor–EF1γn |
| Other cell biology | |||
| no knack | nokhi5 | edema, poor circulation | protein sorting factor–αNACi |
| bleached blond | bblhi112 | pigment defects in eye and body | vacuolar ATP synthase, Ac45 subunitj |
| Novel EST (expressed sequence tag) | |||
| pescadillo | peshi2k | some organs smaller | BRCT mofitl |
| not really started | nrshi891m | yolk degenerates | possible transporter |
| dead eye | dyehi4k | CNS apoptosis | possible nuclear pore proteinn |
| Hagoramo | HaghiD1,D2o | stripes interrupted in body region (dominant) | F-box/WD-40 repeat proteinp |
| Not yet cloned | |||
| hiD862 | long fins (dominant) | ||
| hi37 | reduced head structures | ||
| hi43 | liver defects | ||
| hi61 | lethal | ||
| hi63 | little head, no jaw | ||
| hi96 | multiple edemas | ||
| hi80A | multiple defects |
Becker et al. (1998); bEvans and Scarpulla (1990); cGomez-Cuadrado et al. (1995); dGaiano et al. (1996b); eBarabino et al. (1997); fBai and Tolias (1996); gDavies et al. (1986); hCormier et al. (1991); iWiedmann et al. (1994); jSupek et al. (1994); kAllende et al. (1996); lBork et al. (1997); mM. Allende, H. Wang, S. Marty, S. Lin, and N. Hopkins, (unpubl.); nGrandi et al. (1993); oK. Kawakami and N. Hopkins (unpubl.); pBai et al. (1996).
Analysis of the RNA expression of the candidate genes mutated by insertion in each of the four recessive mutants described above, by RT–PCR (no knack, nearly normal), Northern blot (bubble brain, bleached blond), and/or in situ hybridization analysis (no knack, nearly normal), shows that expression is either significantly reduced or undetectable in homozygous mutant embryos.
In the case of bleached blond, there were two insertions linked to the mutation. One of them is inserted in the gene for a vacuolar ATPase subunit. Although we cannot prove that the other insertion does not also contribute to the mutation, the fact that expression of the V-ATPase gene is knocked out in mutant embryos implies that the mutation is caused by the disruption of this gene.
Discussion
We have described the production of transgenic zebrafish, which together harbor ∼500,000–1,000,000 proviral insertions in their germ lines, and a protocol for breeding ∼100,000 of these insertions to homozygosity. Although difficult to set up initially, our screen runs smoothly now. Barring problems of fish husbandry, we should achieve our goal within a total of 3.5 years, completing the task ∼2 years from the date of this report. The total number of insertions we will screen and the number of mutants we will obtain remain uncertain. As noted above, the results in this report pertain to fish derived from founders made with F5 virus. About 60% of the founders were made with GT virus. These founders had higher embryo assay values on average and, as discussed below, GT virus also contains a gene trap sequence (Gossler et al. 1989; Friedrich and Soriano 1991). How these two differences will impact the mutagenic frequency of GT virus relative to F5 virus remains to be seen.
Despite the labor-intensive nature of this screen, the extraordinary ease of cloning the mutated genes could make the method a powerful complement to chemical mutagenesis and even potentially an attractive replacement. The most important question in deciding the ultimate utility of this technology is whether retroviral proviruses integrate at random into the fish genome. P elements, for example, can readily mutate only about one-third to one-half of the genes required for embryonic development of the fly (Kidwell 1986; Spradling et al. 1995). Although still extraordinarily useful, this precludes the use of insertional mutagenesis in flies to achieve saturation.
Many experiments have addressed the question of whether proviral insertions occur randomly. Some results argue for, and some against, random integration, although the bulk of the evidence suggests that whereas many sites are available for integration, the frequency with which viruses integrate into different sites varies (Withers-Ward et al. 1994). Evidence for integration hot spots and for preferred integrations into the 5′ ends of actively transcribed genes have been obtained (Vijaya et al. 1986; Rohdewold et al. 1987; Mooslehner et al. 1990; Scherdin et al. 1990). We obtained two hits in the Hagoramo gene and the two insertions are 10 kb apart. These insertions cause dominant mutations and are seen in F1 fish. This seemed surprising at first; however, the probability of obtaining a second insertion within 10 kb of one already in hand is 1 in 80,000, and ∼60,000 F1 fish, representing perhaps 50,000–60,000 insertions, have already passed through our laboratory. In any case, insertional hot spots, like hot spots for chemical mutagens, are not problematic. Rather, it would be the inability to hit a substantial fraction of genes that could limit the usefulness of this approach. However, given the difficulty of achieving saturation in a vertebrate with any mutagen, if retroviruses integrate into a substantial fraction of the genes, insertional mutagenesis should still be at least a powerful complement to chemical mutagenesis.
Prospects for improving the efficiency of this method
Technical innovations could lead to significant increases in the efficiency of this methodology. The simplest to achieve are probably improvements to the viral mutagen. We have already improved our technology considerably, primarily by obtaining higher-titer, less-toxic virus stocks. When we first observed transmission of retroviral proviruses through the fish germ, the number of insertions transmitted to the F1 generation averaged one for every seven founder fish (Lin et al. 1994). With F5 virus, we estimate that every founder fish transmits on average 20 insertions to its F1 progeny. With GT virus, the number may prove to be higher. Furthermore, GT virus stocks were less toxic than F5 stocks. Higher titer and reduced toxicity reduced the total time needed to generate founder fish with an estimated 500,000–1,000,000 insertions from 14 months when we began this experiment, to about 2 months had we used GT virus from the start. If increased transgenesis rates with GT virus allow the production of F2 families with 15–20 unique inserts instead of 10 as obtained with F5 virus, the number of F2 families needed to isolate a given number of mutants should drop significantly as well.
In mice, proviral insertions induce mutations at a frequency of ∼1 per 20 insertions (Jaenisch 1988). Our frequency of 1 mutation per 85 insertions is considerably lower. An increase in mutant frequency per insert would have a dramatic impact on the efficiency of our method. Given the smaller size of the fish genome, the lower mutation frequency we observe was unexpected and the reasons for it remain unknown. It is possible that fewer genes are required to produce a viable 5-day-old fish embryo than a mouse that survives through birth. It is also possible that the genome duplication observed in teleosts (Amores et al. 1998; Aparicio 1998) could have resulted in fish having more loci with functional redundancy than mice. Another possibility is that retroviral proviral insertions occur preferentially into genes in the mouse but not in the fish, increasing the mutant frequency per insert in mice. Another possibility is that the particular viral vectors we have used are not as mutagenic as those that have been used in mice. For example, in Hagoramo mutants, proviral integrations in a large intron and in the same transcriptional orientation as the gene fail to significantly perturb detectable transcripts, and the mechanism by which the insertion causes the mutation remains unclear (K. Kawakami, unpubl.). Furthermore, the proviral insertions that induced a number of our embryonic lethal mutations, and that do impact transcription dramatically, reside in the first intron of the genes they mutate. This could reflect a bias in integration site, or it could be that insertions in the first intron are more likely to result in a strong mutant phenotype than insertions that occur in subsequent introns, as in the case of Hagoramo. GT virus contains a gene trap sequence, and this might influence its impact on gene expression when integrated into a wider array of introns and hence impact its mutagenic frequency.
The availability now of transgenic zebrafish with GFP-labeled organs (Higashijima et al. 1997; Long et al. 1997), and our ability to generate 1,000,000 proviral insertions in the zebrafish germ line in only ∼8–12 weeks, suggest that specific and relatively rapid, large-scale insertional mutagenesis screens may now be possible. Haploid screens of F1 embryos from founders would allow one to screen a very large number of different insertions, although the clone size for most insertions would range from ∼3% to ∼20%. Possibly such screens could allow one to process a saturation number of insertions and this might be achieved more easily than the diploid screen described here.
Biological goals of this experiment
Because of the demanding nature of this experiment, we have chosen not to incorporate more sophisticated screening into the protocol for now but, instead, to limit the screen to visual observation of F3 embryos in a dissecting microscope. We plan to keep all of the mutants we isolate and to rescreen them with a battery of probes and assays to look for specific defects that might be missed otherwise, an approach known as a shelf screen. Shelf screens have proven fruitful in flies and should prove useful in fish. Consistent with this statement, in the zebrafish, when more sophisticated screens have been used to identify mutants, the majority of mutants found have proven to be lethal. For example, most mutants found by in situ hybridization screening of embryos (D. Yelon and D. Stainier; N. Trede and L. Zon; both pers. comm.), by screening for axons with aberrant projections from retina to tectum (Baier et al. 1996), or by screening for larvae with vision defects (Brockerhoff et al. 1995) have proven to be lethal, usually by 5–6 days of age, and therefore would be detected and kept in our screen. Thus, whereas many fascinating mutants will undoubtedly be missed, the vast majority of mutants that have ever been found in zebrafish screens (and many others like them that have been missed) would be found with our simple strategy. In gathering a large collection of embryonic visible and lethal mutations, one is in reality carrying out a collection of specific screens. The results of the large chemical mutagenesis screens suggest that among the 1000 mutants we hope to obtain, we might expect ∼25%–30% to have specific defects in the development of particular organ systems.
Although we have isolated only a small number of insertional mutants so far, several appear biologically interesting. not really finished, bleached blond, hi43, and the dominant mutants Hagoramo, and the one with the long fin phenotype would be kept in screens for specific developmental mutations of the zebrafish. The finding that a small fraction of EF1γ−/− fish survive and grow slowly to be miniature adults is of interest in terms of whole animal growth control. Among the hundreds of mutants we hope to isolate, there should be many more that will reveal novel and important aspects of vertebrate development including some with relevance to understanding human development and possibly human diseases. In addition, given the size of our screen, we should obtain groups of mutants with defects in any particular developmental processes, and analysis of these should help to elucidate the molecular basis of organogenesis for many of the embryonic organs.
Materials and methods
Animals
Zebrafish were raised and maintained as described previously (Culp et al. 1991), with the following exceptions. Synchronized eggs for injection were obtained by placing four females and two males (which had been separated the night before) in a 4-liter mating chamber for 10–15 min. Pair matings for raising F1 and F2 fish and for screening F3 embryos were performed in 1-liter mating chambers as in Mullins et al. (1994). Paramecia fed to fry were counted and delivered in measured amounts three times a day; a total of ∼400 paramecia per fry a day were required between days 5 and 7 and 800 paramecia per fry a day between days 8 and 11 to allow fish to reach a size at which they could eat brine shrimp.
Founder fish were generated from embryos from either of two lethal-free lines that were obtained as follows: We crossed outbred fish originally from Tübingen but carried in our laboratory for ∼6 years with AB*, a line selected by C. Walker (University of Oregon) as highly suitable for use in haploid and early pressure screens. We raised families from each of 15 pair matings. Sibling matings within each family were performed to identify families with no embryonic lethal mutations. Two lines, designated TAB-5 and TAB-14, were identified (no embryonic mutations seen in 18 matings from TAB-5 or in 22 matings from TAB-14) and used to obtain embryos for virus injections.
Virus preparation and injection
A packaging cell line 293 gp/bsr (Miyoshi et al. 1997), grown in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum, penicillin, streptomycin, and fungisome, was infected with SFGnlslacZ virus (Gaiano et al. 1996a) at three multiplicities of infection (M.O.I.s), 0.05, 0.5, and 5. Four days later, cells were trypsinized and stained with the vital stain fluorescein di-β-d-galactopyranoside (FDG; Nolan et al. 1988), and passed through a cell sorter. Moderate and highly fluorescent cell populations from each of the three cell populations were selected, grown for 1 week, and then cloned. A total of 46 clones were screened to identify the one capable of producing the highest titer of virus following calcium phosphate-mediated transfection with the plasmid pHCMV-G (Yee et al. 1994), which encodes the envelope protein of vesicular stomatitis virus. Medium was changed 24 hr after transfection, and collections of supernatant were made at 48, 72, 96, and 120 hr and titered on mouse 3T3 cells. Titers ranged from 0 to 5.4 × 106 CFU/ml. The 10 best lines were selected and viral supernatants titered on a fish cell line PAC2. Two lines gave higher titers than the others, but one line proved difficult to work with. The F5 line, derived from an infection at M.O.I. = 5 and harboring a single proviral genome, was selected and used to produce F5 virus for the experiment. Large virus stocks were prepared by calcium phosphate transfection of F5 cells that had been seeded 1 day earlier on fifteen 15-cm tissue culture plates treated with 0.01% poly-l-lysine. A total of 50 μg of pHCMV-G DNA per plate were used in the transfection. Media were changed at 24-hr post-transfection. Media collected at 48, 72, and 96-hr post-transfection were filter sterilized (0.2 μm filter) and concentrated by centrifugation at 21,000 rpm with a SW28 rotor for 1.5 hr at 4°C (Burns et al. 1993). Viral pellets were resuspended in 30 μl of PBS, titered, and used fresh or frozen at −80°C for future use. Virus was injected as described previously. We estimate that ∼250,000 embryos were injected on ∼230 days over a period of 12 months.
Embryo assay
To determine whether viral stocks had high titers on embryos and to ensure that founder fish that were raised were efficiently infected, we determined the proviral DNA content of several injected embryos from every batch injected using an assay designated the embryo assay.
For the bulk of the project, this assay was performed by quantitative Southern analysis. Ten injected embryos were lysed as five pools of two at 3–5 days of age in 100 mm Tris (pH 8.3), 200 mm NaCl, 5 mm EDTA, 0.4% SDS, 100 μg/ml proteinase K and lysed overnight. DNA was precipitated with ethanol, resuspended, and digested with PvuII, which cuts several times in the viral sequence. The samples were then electrophoresed through 0.8% agarose and Southern blotted along with a reference control from a fish with one proviral insert. Blots were then hybridized with probes to the provirus and to the zebrafish RAG2 gene. Bands corresponding to each probe were quantitated with a Molecular Dynamics PhosphorImager, the virus/RAG2 ratio calculated and normalized to the internal reference = 1. Subsequently, the embryo assay was performed by real time quantitative PCR. Single embryos were lysed at 2 days of age and processed as described below for the fin clips.
Identification of multi-insert F1 fish
We raised 30 fish per F1 family. To identify fish with at least three unique proviral inserts, we proceeded as follows. At 8–10 weeks of age, fish were anesthetized and placed on a small piece of parafilm, and the end of their caudal fins were amputated with a scalpel and placed in wells of a 96-well plate. The fish were stored in disposable 16-oz (473 ml) cups while the fin clips were processed. DNA was extracted by incubation in 50 μl of ELVIS lysis buffer (50 mm KCl, 10 mm Tris at pH 8.5, 0.01% gelatin, 0.45% NP-40, 0.45% Tween 20, 5 mm EDTA, 200 μg/ml proteinase K) for at least 2 hr at 55°C. Proteinase K was then inactivated by placing the samples at 96°C for 15 min.
Approximately 1 μl from each sample served as template for real time quantitative PCR with a Perkin-Elmer 7700 Sequence Detector (Heid et al. 1996). Primers and the probe used to amplify viral sequences are as follows: SFG F, 5′-CGCTGGAAAGGACCTTACACA-3′; SFG R, 5′-TGCGATGCCGTCTACTTTGA-3′, and SFG probe, 5′-FAM-CTGCTGACCACCCCCACCGC-TAMRA-3′. A separate primer/probe combination was utilized for an internal reference amplicon to amplify the RAG1 locus (RAG F, 5′-ATTGGAGAAGTCTACCAGAAGCCTAA-3′; RAG R, 5′-CTTAGTTGCTTGTCCAGGGTTGA-3′, RAG probe, 5′-JOE-GCGCAACGGCGGCGCTC-TAMRA-3′). The SFG primers and RAG primers were used at final concentrations of 74 and 150 nm, respectively, whereas both RAG and SFG probes were used at 200 nm. Each reaction was carried out in a final volume of 12.5 μl with Perkin-Elmer Master Mix. The cycling profiles were 2′@50°C, 10′@95°C, and 30× (15‘@95°C, 1′@60°C). Each 96-well run contained six wells of a reference control from a fish with six inserts. At the end of each run, the RAG and SFG Cts (threshold cycle; the cycle at which the amount of product passed a certain threshold in the linear amplification range) were calculated for each sample and a ΔCt value was defined by subtracting the SFG Ct from the RAG Ct. The larger the ΔCt, value the greater the number of viral insets for any given sample. By subtracting the average six-insert fish ΔCt from each sample’s ΔCt, we calculate the ΔΔCt, which can then be used in the following formula to estimate the number of inserts per fish: n = 6 × 2ΔΔCt.
The top eight fish (i.e., the eight with the highest ΔCt values) from each F1 family were further analyzed by Southern blot to allow selection of fish with the most unique inserts relative to one another. The remainder of the fin clip samples for these fish were digested with BglII, which cuts only once in the provirus, and electrophoresed through 0.8% agarose for ∼1200 volt hr, and Southern blotted. Blots were probed so as to yield one band per insert. Only fish with at least three unique inserts were kept and used to generate F2 families.
Identification of mutagenic insertions and gene cloning
The protocol shown in Figure 3 was followed to identify the mutagenic insert and determine the correct digest(s) to use for inverse PCR. Inverse PCR was carried out as described in Allende et al. (1996). Sequences were compared with the public database with BLAST (Altschul et al. 1990), and when significant homologies were found (usually to zebrafish ests), expression of those genes were analyzed in mutant and wild-type embryos by Northern analysis, RT–PCR, and/or in situ hybridization as described in Allende et al. (1996).
Acknowledgments
This research was supported by grants from Amgen and from the National Center for Research Resources. We thank Bob Bosselman for his support and interest, which made this project possible. We thank a number of individuals who made very significant contributions to this work including Marcello Antonelli, who performed in situ hybridization on no knack and nearly normal mutant embryos; Paul de Figueiredo, who performed all of the lacZ titering; Craig Nelson, who did the experiments that first showed that quantitative PCR could be used to distinguish fish with multiple insertions; Mary Laplante for her ability to collect newly fertilized eggs day after day; and Wade Huber for helping to design methods for storing individual fish and for other technical support. Our fish are maintained by an exceptional staff including Tom Such, Jennifer Mugg, Jason Anderson, Tim Angelini, Kate Coughlin, Sean Fuss, and Megan Geotis. We thank our skillful tail-clipping team of Erin Flynn and Jennifer Montgomery, Mr. Ellis London of Tropic Isle for his excellent advice on maintaining our fish facilities, and Katie Douglas and Bethany Sutton of Amgen for their speedy sequencing assistance. The support of our staff, Julie Barrett and, more recently, Sarah Heany, is most gratefully acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
As of publication date we are isolating mutants at a pace of 5/week and have accumulated approximately 45 insertional mutants to date.
Footnotes
E-MAIL nhopkins@mit.edu; FAX (617) 258-0258.
References
- Allende M, Amsterdam A, Becker T, Kawakami K, Gaiano N, Hopkins N. Insertional mutagenesis in zebrafish identifies two novel genes, pescadillo and dead eye, essential for embryonic development. Genes & Dev. 1996;10:3141–3155. doi: 10.1101/gad.10.24.3141. [DOI] [PubMed] [Google Scholar]
- Altschul S, Gish W, Miller W, Myers E, Lipman D. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wnag YC, Westerfield M, Ekker M, Postlethwait JH. Zebrafish hox clusters and vertebrate genome evolution. Science. 1998;282:1711–1714. doi: 10.1126/science.282.5394.1711. [DOI] [PubMed] [Google Scholar]
- Aparicio S. Exploding vertebrate genomes. Nat Genet. 1998;18:301–303. doi: 10.1038/ng0198-301. [DOI] [PubMed] [Google Scholar]
- Bai C, Tolias PP. Cleavage of RNA hairpins mediated by a developmentally regulated CCCH zinc-finger protein. Mol Cell Biol. 1996;16:6661–6667. doi: 10.1128/mcb.16.12.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Baier H, Klosterman S, Trowe T, Karlstrom RO, Nusslein-Volhard C, Bonhoeffer F. Genetic dissection of the retinotectal projection. Development. 1996;123:415–425. doi: 10.1242/dev.123.1.415. [DOI] [PubMed] [Google Scholar]
- Barabino SM, Hubner W, Jenny A, Minvielle-Sebastia L, Keller W. The 30-kD subunit of mammalian cleavage and polyadenylation factor and its yeast homolog are RNA-binding zinc figer proteins. Genes & Dev. 1997;11:1703–1716. doi: 10.1101/gad.11.13.1703. [DOI] [PubMed] [Google Scholar]
- Becker TS, Burgess S, Amsterdam A, Allende ML, Hopkins N. not really finished is crucial for development of the zebrafish outer retina and encodes a transcription factor highly homologous to human Nuclear Respiratory Factor-1 and avian Initiation Binding Repressor. Development. 1998;125:4369–4378. doi: 10.1242/dev.125.22.4369. [DOI] [PubMed] [Google Scholar]
- Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- Brockerhoff SE, Hurley JB, Janssen-Bienhold U, Neuhauss SC, Driever W, Dowling JE. A behavioral screen for isolating zebrafish mutants with visual system defects. Proc Natl Acad Sci. 1995;92:10545–10549. doi: 10.1073/pnas.92.23.10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JC, Friedmann T, Driever W, Burrascano M, Yee J-K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci. 1993;90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley L, Kelley R, Spradling A. Insertional mutagenesis of the Drosophila genome with single P elements. Science. 1988;239:1121–1128. doi: 10.1126/science.2830671. [DOI] [PubMed] [Google Scholar]
- Cormier P, Osborne HB, Morales J, Bassez T, Pouhle R, Mazabraud A, Mulner-Lorillon O, Belle R. Molecular cloning of Xenopus elongation factor 1 gamma, major M-phase promoting factor substrate. Nucleic Acids Res. 1991;19:6664. doi: 10.1093/nar/19.23.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culp P. Random DNA integrations as an approach to insertional mutagenesis in the zebrafish (Brachydanio rerio) PhD. thesis. Cambridge, MA: Massachusetts Institute of Technology; 1994. [Google Scholar]
- Culp P, Nüsslein-Volhard C, Hopkins N. High-frequency germ-line transmission of plasmid DNA sequences injected into fertilized zebrafish eggs. Proc Natl Acad Sci. 1991;88:7953–7957. doi: 10.1073/pnas.88.18.7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MS, Henney A, Ward WH, Craig RK. Characterization of an mRNA encoding a human ribosomal protein homologous to the yeast L44 ribosomal protein. Gene. 1986;45:183–191. doi: 10.1016/0378-1119(86)90253-2. [DOI] [PubMed] [Google Scholar]
- Driever W, Solnica-Krezel L, Schier A, Neuhauss S, Maliki J, Stemple D, Stainier D, Zwartkruis F, Abdelilah S, Rangini Z, Belak J, Boggs C. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46. doi: 10.1242/dev.123.1.37. [DOI] [PubMed] [Google Scholar]
- Emi N, Friedmann T, Yee J-K. Pseudotype formation of murine leukemia virus with the G protein of vesicular stomatitis virus. J Virol. 1991;65:1202–1207. doi: 10.1128/jvi.65.3.1202-1207.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MJ, Scarpulla RC. NRF-1: A trans-activator of nuclear-encoded respiratory genes in animal cells. Genes & Dev. 1990;4:1023–1034. doi: 10.1101/gad.4.6.1023. [DOI] [PubMed] [Google Scholar]
- Friedrich G, Soriano P. Promoter traps in embryonic stem cells: A genetic screen to identify and mutate developmental genes in mice. Genes & Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- Gaiano N, Allende M, Amsterdam A, Kawakami K, Hopkins N. Highly efficient germ-line transmission of proviral insertions in zebrafish. Proc Natl Acad Sci. 1996a;93:7777–7782. doi: 10.1073/pnas.93.15.7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiano N, Amsterdam A, Kawakami K, Allende M, Becker T, Hopkins N. Insertional mutagenesis and rapid cloning of essential genes in zebrafish. Nature. 1996b;383:829–832. doi: 10.1038/383829a0. [DOI] [PubMed] [Google Scholar]
- Gomez-Cuadrado A, Martin M, Noel M, Ruiz-Carrillo A. Initiation binding receptor, a factor that binds to the transcription initiation site of the histore h5 gene, is a glycosylated member of a family of cell growth regulators. Mol Cell Biol. 1995;15:6670–6685. doi: 10.1128/mcb.15.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossler A, Joyner AL, Rossant J, Skarnes WC. Mouse embryonic stem cells and reporter constructs to detect developmentally regulated genes. Science. 1989;244:463–465. doi: 10.1126/science.2497519. [DOI] [PubMed] [Google Scholar]
- Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Grandi P, Doye V, Hurt EC. Purification of NSP1 reveals complex formation with 'GLFG' nucleoporins and a novel nuclear pore protein NIC96. EMBO J. 1993;12:3061–3071. doi: 10.1002/j.1460-2075.1993.tb05975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gridley T, Soriano P, Jaenisch R. Insertional mutagenesis in mice. Trends Genet. 1987;3:162–166. doi: 10.1101/gad.1.4.366. [DOI] [PubMed] [Google Scholar]
- Grunwald DJ, Streisinger G. Induction of recessive lethal specific locus mutations in the zebrafish with ethylnitrosourea. Genet Res. 1992;59:103–116. doi: 10.1017/s0016672300030317. [DOI] [PubMed] [Google Scholar]
- Grunwald DJ, Kimmel CB, Westerfield M, Walker C, Streisinger G. A neural degeneration mutation that spares primary neurons in the zebrafish. Dev Biol. 1988;126:115–128. doi: 10.1016/0012-1606(88)90245-x. [DOI] [PubMed] [Google Scholar]
- Haffter P, Granato M, Brand M, Mullins M, Hammerschmidt M, Kane D, Odenthal J, van Eeden F, Jiang Y, Heisenberg C-P, et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 1996;123:1–36. doi: 10.1242/dev.123.1.1. [DOI] [PubMed] [Google Scholar]
- Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Higashijima S, Okamoto H, Ueno N, Hotta Y, Eguchi G. High-frequency generation of transgenic zebrafish which reliably express GFP in whole muscles or the whole body by using promoters of zebrafish origin. Dev Biol. 1997;192:289–299. doi: 10.1006/dbio.1997.8779. [DOI] [PubMed] [Google Scholar]
- Jaenisch R. Transgenic Animals. Science. 1988;240:1468–1474. doi: 10.1126/science.3287623. [DOI] [PubMed] [Google Scholar]
- Kidwell MG. P-M mutagenesis. In: Roberts DB, editor. Drosophila: A practical approach. Oxford, UK: IRL Press; 1986. pp. 59–81. [Google Scholar]
- Kimmel CB. Genetics and early development of zebrafish. Trends Genet. 1989;5:283–288. doi: 10.1016/0168-9525(89)90103-0. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Kane DA, Walker C, Warga RM, Rothman MB. A mutation that changes cell movement and cell fate in the zebrafish embryo. Nature. 1989;337:358–362. doi: 10.1038/337358a0. [DOI] [PubMed] [Google Scholar]
- Lin S, Gaiano N, Culp P, Burns J, Friedmann T, Yee J-K, Hopkins N. Integration and germ-line transmission of a pseudotyped retroviral vector in zebrafish. Science. 1994;265:666–669. doi: 10.1126/science.8036514. [DOI] [PubMed] [Google Scholar]
- Long Q, Meng A, Wang H, Jessen JR, Farrell MJ, Lin S. GATA-1 expression pattern can be recapitulated in living transgenic zebrafish using GFP reporter gene. Development. 1997;124:4105–4111. doi: 10.1242/dev.124.20.4105. [DOI] [PubMed] [Google Scholar]
- Meisler MH. Insertional mutation of 'classical' and novel genes in transgenic mice. Trends Genet. 1992;8:341–344. doi: 10.1016/0168-9525(92)90278-c. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Takahashi M, Gage FH, Verma IM. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc Natl Acad Sci. 1997;94:10319–10323. doi: 10.1073/pnas.94.19.10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooslehner K, Karls U, Harbers K. Retroviral integration sites in transgenic Mov mice frequently map in the vicinity of transcribed DNA regions. J Virol. 1990;64:3056–3058. doi: 10.1128/jvi.64.6.3056-3058.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JH, Lee-Tischler MJ, Kwon H-Y, Schrick JJ, Avner ED, Sweeney WE, Godfrey VL, Cacheiro NLA, Wilkinson JE, Woychik RP. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science. 1994;264:1329–1333. doi: 10.1126/science.8191288. [DOI] [PubMed] [Google Scholar]
- Mullins M, Hammerschmidt M, Haffter P, Nüsslein-Volhard C. Large-scale mutagenesis in the zebrafish: In search of genes controlling development in a vertebrate. Curr Biol. 1994;4:189–202. doi: 10.1016/s0960-9822(00)00048-8. [DOI] [PubMed] [Google Scholar]
- Nolan GP, Fiering S, Nicolas J, Herzenberg LA. Fluorescence-activated cell analysis and sorting of viable mammalian cells based on β-D-galactosidase activity after transduction of Escherichia coli lacZ. Proc Natl Acad Sci. 1988;85:2603–2607. doi: 10.1073/pnas.85.8.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Gerber AS, Hartl DL. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988;120:621–623. doi: 10.1093/genetics/120.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD, Brinster RL. Germ-line transformation of mice. Annu Rev Genet. 1986;20:465–499. doi: 10.1146/annurev.ge.20.120186.002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinchik EM. Chemical mutagenesis and fine-structure functional analysis of the mouse genome. Trends Genet. 1991;7:15–21. doi: 10.1016/0168-9525(91)90016-j. [DOI] [PubMed] [Google Scholar]
- Rohdewohld H, Weiher H, Reik W, Jaenisch R, Breindl M. Retrovirus integration and chromatin structure: Moloney Murine Leukemia proviral integration sites map near DNase I-hypersensitive sites. J Virol. 1987;61:336–343. doi: 10.1128/jvi.61.2.336-343.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherdin U, Rhodes K, Breindl M. Transcriptionally active genome regions are preferred targets for retrovirus integration. J Virol. 1990;64:907–912. doi: 10.1128/jvi.64.2.907-912.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlovsky A, Guenet JL, Johnson LL, Dove WF. Induction of recessive lethal mutations in the T/t-H-2 region of the mouse genome by a point mutagen. Genet Res. 1986;47:135–142. doi: 10.1017/s0016672300022977. [DOI] [PubMed] [Google Scholar]
- Solnica-Krezel L, Schier A, Driever W. Efficient recovery of ENU-induced mutations from the zebrafish germline. Genetics. 1994;136:1401–1420. doi: 10.1093/genetics/136.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Stern DM, Kiss I, Roote J, Laverty T, Rubin GM. Gene disruptions using P transposable elements: An integral component of the Drosophila genome project. Proc Natl Acad Sci. 1995;92:10924–10830. doi: 10.1073/pnas.92.24.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Supekova L, Mandiyan S, Pan Y-CE, Nelson H, Nelson N. A novel accessory subunit for vacuolar H+-ATPase from chromaffin granules. J Biol Chem. 1994;269:24102–24106. [PubMed] [Google Scholar]
- Varmus HE. Form and function of retroviral proviruses. Science. 1982;216:812–820. doi: 10.1126/science.6177038. [DOI] [PubMed] [Google Scholar]
- Vijaya S, Steffen D, Robinson H. Acceptor sites for retroviral integrations map near DNase I-hypersensitive sites in chromatin. J Virol. 1986;60:683–692. doi: 10.1128/jvi.60.2.683-692.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–725. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The zebrafish book. Eugene, OR: University of Oregon Press; 1995. [Google Scholar]
- Wiedmann B, Sakai H, Davis TA, Wiedmann M. A protein complex required for signal-sequence-specific sorting and translocation. Nature. 1994;370:434–440. doi: 10.1038/370434a0. [DOI] [PubMed] [Google Scholar]
- Willett CE, Cherry JJ, Steiner LA. Characterization and expression of the recombination activating genes (rag1 and rag2) of zebrafish. Immunogenetics. 1997;45:394–404. doi: 10.1007/s002510050221. [DOI] [PubMed] [Google Scholar]
- Withers-Ward ES, Kitamura Y, Barnes JP, Coffin JM. Distribution of targets for avian retrovirus DNA integration in vivo. Genes & Dev. 1994;8:1473–1487. doi: 10.1101/gad.8.12.1473. [DOI] [PubMed] [Google Scholar]
- Yee J-K, Miyanohara A, LaPorte P, Bouic K, Burns JC, Friedmann T. A general method for the generation of high titer pantropic retroviral vectors: Highly efficient infection of primary hepatocytes. Proc Natl Acad Sci. 1994;91:9564–9568. doi: 10.1073/pnas.91.20.9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon C, Kawakami K, Hopkins N. Zebrafish vasa homologue RNA is localized to the cleavage planes of 2- and 4-cell-stage embryos and is expressed in the primordial germ cells. Development. 1997;124:3157–3166. doi: 10.1242/dev.124.16.3157. [DOI] [PubMed] [Google Scholar]
- Zavada J. Pseudotypes of vesicular stomatitis virus with the coat of murine leukemia and of avian myeloblastosis virus. J Gen Virol. 1972;125:183–191. doi: 10.1099/0022-1317-15-3-183. [DOI] [PubMed] [Google Scholar]