

Abstract
Collagen is the fundamental structural protein, comprising 25–35% of the total body protein, its rod-like triple helix providing support in many tissues. Our laboratory has synthesised 113 Toolkit peptides, each 63 residues long, covering the entirety of the homotrimeric helix sequence of collagen II and collagen III. These are used primarily to investigate protein–collagen interactions, from which biomedical applications are under development. Upon increasing the temperature of a Toolkit peptide solution, a novel low temperature transition (LTT) as well as a broadening of the helix unfolding higher temperature transition (HTT) was observed. Here, we hypothesized that unfolding of imperfect helices can account for the LTT. Peptides of various purities were isolated by HPLC or gel filtration, and their unfolding measured by polarimetry, CD, and DSC. The resulting temperature transitions were fitted to a kinetic unfolding equation, allowing comparison of the data, and explanation of the observed melting curve complexity as due to peptide imperfections. Finally, using a mathematical model, this data can be replicated by setting a parameter that quantifies the mutual stabilization conferred by helices on each side of a peptide defect within a triple helix.
Keywords: Collagen structure, Peptide, Thermally responsive material, Tissue engineering, Polarimetry, Calorimetry
Abbreviations: LTT, low temperature transition; HTT, high temperature transition; DSC, differential scanning calorimetry; DMF, N,N-Dimethylformamide; CRP, Collagen-related peptide (sequence in Table 1); MALDI, Matrix-assisted laser desorption/ionization mass spectroscopy; TCEP, Tris(2-carboxy-ethyl)phosphine hydrochloride
1. Introduction
Proteins of the diverse collagen family form the primary structural scaffolds of the body. Accordingly, they bind a wide variety of molecules including adhesion proteins, lipoproteins, receptor proteins, glycosaminoglycans, and nucleic acids. Entire families of proteases are devoted to collagen breakdown, and collagen has its own protein chaperones to aid its folding and assembly into structural elements so it can be remodelled after injury and during growth [1]. Model collagen peptides have become essential tools for collagen research [2–4]. As shorter representations of whole collagen, they have been used to locate binding sites on collagen [5–12], for tissue engineering [13–15], for diagnostics [16], as ligands for platelet and cellular receptors [5,10], as tools for investigating chemical kinetics [17], and such peptides are well characterised by structural biology studies [9,18–30].
As part of this effort, our laboratory has synthesised peptides varying in length, between the minimal 21-residue sequence required for stable collagen helix formation at room temperature, up to Toolkit peptides 63 residues long. Toolkit peptides have a 27-residue “guest” collagen sequence framed between the helix-inducing “host” sequences GPC(GPP)5- and -(GPP)5GPC, and their helicity has been shown to be required for location of protein binding sites on collagen. This Toolkit set comprises over 150 peptides, covering all of the triple-helical COL domains of collagens II and III and some of collagens I and IV, and its diversity of sequence supports all the biomaterial applications above.
Collagen helix formation requires a repeating G-X-X′ triplet, where G is glycine, X is commonly proline (P), and X′ is commonly hydroxyproline (O). To check peptide helicity, optical or calorimetric changes are measured upon helix unfolding as the temperature is increased. For polarimetry [7] and CD [17], the ideal unfolding signal for a homogeneous helix population is a single sigmoidal curve. Likewise, the ideal DSC unfolding signal approximates to a single Gaussian curve [27]. However, analysis of Toolkit peptides revealed more complex signals. The expected “high temperature transition” (HTT) was observed (between 35 °C and 55 °C), but this was preceded by a broad “low temperature transition” (LTT) event occurring over a range of up to 20 °C. Both transition signals have sigmoidal (CD/polarimetry) or Gaussian-like (DSC) characteristics similar to helix unfolding. The LTT could be due to peptide self-association into a higher order structure [3,21,23], helix strand misalignment [24], structural distortions caused by peptide imperfections [18,20,25,26], a change in helix conformation [22], unfolding of a less stable part of the helix [31], or might have other unknown origins.
Here, we hypothesize that unfolding of imperfect helices can account for the LTT, and that only one peptide strand of a triple helix needs to be imperfect in order to destabilise its structure. These imperfections, as quantified by mass spectroscopy, are impure peptides where the canonical G-X-X′ sequence required for collagen helicity is disrupted by additions or deletions of an amino acid. To investigate this hypothesis, empirical unfolding data from Toolkit peptides at different stages of purification was collected. The hypothesis was also tested using the 36-residue collagen-related peptide (CRP), GCO(GPO)10GCOG-NH2.
Work on these shorter collagenous peptides, up to 36 residues, dominates the field [1]. They offer fewer permutations for possible misalignment during helix assembly and are easier to synthesise at high purity, minimising two of the possible causes of the LTT listed above. Even so, this LTT is observable in some previous reports [19,32], but has remained largely unremarked upon, possibly due to its apparently small magnitude when viewing primary rather than first-derivative CD or polarimetry data. Few papers provide mass spectroscopy data, e.g [19]. and Fig. 2 [32], rendering purity difficult to assess retrospectively, and often there are no remarks made over the impurities (e.g. minus proline) shown in a mass-spectroscopy plot [24]. In summary, there may be several reasons why literature on the LTT is sparse.
Fig. 2.

Investigating five Toolkit peptides using different techniques. Raw (DSC) and derivative (polarimetry and CD) data illustrate helical unfolding transitions. The scale bar for derivative data is 10−3 deg cm−1 K−1 or 0.22 deg cm2 dmol−1 K−1 polarimetric rotation units. In CD[θ]225 units, it is 1 deg cm−1 K−1 or 218 deg cm2 dmol−1 K−1. In DSC units, it is 1 J g−1 K−1 or 5.5 kJ mole−1 K−1. Data is curve fitted using the D/z equation in order to demonstrate signals due to the LTT and HTT. Approximate peptide purities from mass spectroscopy for each peptide are quoted to the left.
Finally, we discuss the extent to which a mathematical model can account for this LTT by defining known quantities of imperfect peptide as a parameter, and adjusting other parameters in order to match closely our experimental results. From this, we propose a two-fit unfolding analysis for use in characterising the HTT and LTT of peptides in future studies.
2. Procedures
2.1. Peptide synthesis
Peptides were synthesized as C-terminal amides on TentaGel R-Ram resin using an Applied Biosystems Pioneer solid phase peptide synthesiser. Fmoc-amino acids (4 eqv.) were activated with O-(1H-6-Chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate (4 eqv.) in the presence of N-ethyldiisopropylamine (8 eqv.). C-terminal cysteine was coupled as Fmoc-Cys(trityl) pentafluorophenyl ester (4 eqv.) in the presence of N-Hydroxybenzotriazole (HOBt, 4 eqv.). Fmoc deprotection was effected with 2% (v/v) piperidine and 2% (v/v) 1,8-diazabicyclo- [5,4,0]undec-7-ene in N,N-Dimethylformamide (DMF). When glycine is followed by aspartate, it was introduced into the sequence as Fmoc(FmocHmb)Gly-OH to avoid aspartimide formation, and all subsequent deprotections were effected with 20% (v/v) piperidine in DMF with 0.1 M HOBt. All peptides were unblocked at the N-terminus to aid solubility. Their sequences and abbreviations are given in Table 1.
Table 1.
Peptides used in this study and their mass.
| Peptide |
Sequence |
Mass (Da) |
|---|---|---|
| CRP | GCO-(GPO)10-GCOG-NH2 | 3294 |
| II-05 | GPC-(GPP)5-GARGFOGTOGLOGVKGHRGYOGLDGAK-(GPP)5-GPC-NH2 | 5710 |
| II-40 | GPC-(GPP)5-GRVGPOGSNGNOGPOGPOGPSGKDGPK-(GPP)5-GPC-NH2 | 5525 |
| III-10 | GPC-(GPP)5-GQPGPOGPOGTAGFOGSOGAKGEVGPA–(GPP)5-GPC-NH2 | 5432 |
| III-24 | GPC-(GPP)5-GPKGNDGAOGKNGERGGOGGOGPQGPO-(GPP)5-GPC-NH2 | 5500 |
| III-30 | GPC-(GPP)5-GAOGLRGGAGPOGPEGGKGAAGPOGPO-(GPP)5-GPC-NH2 | 5324 |
| III-53 | GPC-(GPP)5-GIKGHRGFOGNOGAOGSOGPAGQQGAI-(GPP)5-GPC-NH2 | 5573 |
Cleavage of the peptides from the resin and simultaneous side-chain deprotection was done by treatment of the peptide-resin with a trifluoroacetic acid, water and triisopropylsilane mixture (95:2.5:2.5 v/v, 10 mL) containing DL-dithiothreitol (0.25 g), for 3 h. The resin was filtered and the filtrate concentrated under reduced pressure to ca. 1 mL volume, after which the crude peptides were precipitated with ice-cold ether. The filtered crude peptides were ether-washed (twice), dissolved in 5% acetonitrile in water containing 0.1% trifluoroacetic acid and then lyophilized.
Crude products were purified by reverse phase HPLC on an ACE phenyl 300 (10 μm) column using a linear gradient of acetonitrile in water containing 0.1% trifluoroacetic acid. Fractions of peptides III-10 and III-30 containing the purest product were identified by mass spectroscopy and by analytical HPLC on an ACEphenyl300 (5 μm) column. They were pooled and freeze dried. This purification was however unsatisfactory for peptide III-30, and we also needed purest possible III-10 for comparative purposes. Therefore, we HPLC re-purified (same methodology) these peptides, accepting low yields to attain higher purity. For the CRP preparation (see Table 1), all fractions containing enough peptide for analysis were individually freeze dried, except for the very similar and purest fractions 4–6, which were pooled. The freeze dried CRP fractions were all analyzed using polarimetry.
The production of long peptides in sufficient quantity (0.1 g scale) for widespread use is necessarily a compromise between yield and purity. This is because an imperfect peptide usually differs by only one added or deleted residue in 63, rendering the separations above difficult.
2.2. Mass spectroscopy
Peptide samples were desalted by adsorbing to pre-conditioned μC18 Ziptip (Millipore) before elution and use. For Matrix-assisted laser desorption/ionization mass spectroscopy (MALDI), samples were washed with 0.1% trifluoroacetic acid and eluted with matrix solution. For Electrospray, samples were eluted with 70% MeOH/0.2% formic acid. When run on the Waters MALDI MicroMX, samples were mixed with ferulic acid matrix (10 mg/ml in 50% acetonitrile, 0.1% trifluoroacetic acid), dried and washed with 0.1% trifluoroacetic acid. Data was collected at laser power just above threshold level, continually moving the laser target position after 1–2 shots in order to minimise fragmentation. All peptides were checked for their expected (reduced) mass by mass spectroscopy, and the signal strength for that peak was assigned a value of 100. We considered masses of +23, +18, and +39 Da to be pure peptides with additional sodium, water, or potassium bound non-covalently. Most other peaks could be accounted for by additions or deletions of glycine, proline, or cysteine at some unknown, presumably random, point in the peptide. Imperfect peptides were assumed to ionize in a similar manner to the pure peptide, with similar signal strength per molecule, allowing us to compile tables showing the relative abundance of pure and imperfect peptides, and estimate overall purity.
2.3. Gel filtration separation of peptide helices from monomeric component chains and TCEP
Peptide samples were prepared by diluting 200 μl of 1 mg/ml peptide with 600 μl of cold 10 mM phosphate buffered saline (pH 7.4), and Tris(2-carboxy-ethyl) phosphine hydrochloride (TCEP) added to a final concentration of 2 mM where appropriate. Samples initially at 4 °C were loaded onto a Bio-sep Sec-S2000 Gel filtration column (300 × 21.2 mm, 5 μm bead size, 14.5 ± 1.5 nm pore size) equilibrated at 10 °C in the same buffer. Running isocratically, the eluant was monitored at 214 nm. Peaks were fitted mathematically using a combination of the Rankin equation to model the peak broadening effects of variable pore sizes within any one gel filtration resin [33], and work to model the peak broadening effects of diffusion and inhomogeneity of flow [34].
2.4. Helical unfolding transitions determined by polarimetry, CD, NMR, and DSC
Each peptide was dissolved at 2.5 mg/ml in phosphate buffered saline pH 7.0 with 2 mM Tris(2-carboxy-ethyl)phosphine hydrochloride (TCEP). They were heated at 60 °C (90 °C for CRP) for 15 min to dissociate the triple-helical form and then kept at 4 °C overnight (10–18 h) to refold. Peptide solutions were heated from 8 °C to 80 °C (90 °C for CRP) at 1 °C/min. Observations were made every 16 s when using a Microcal VP-DSC, every 15 s when using an Autopol III polarimeter, or every 60 s when using a Aviv215 CD spectrophotometer at 225 nm. For polarimetry and CD, the change in optical rotation or ellipticity every 2.5 °C was plotted against temperature, giving transition curve similar to those observed by DSC. We did not observe any transitions related to the presence of TCEP.
For NMR, after allowing helical refolding for 12–18 h, peptide solutions were initially probed at 8 °C using a Brucker AMX 600 spectrophotometer (128 scans, ∼170 s). The temperature was raised to 13 °C over 30 s and allowed to equilibrate for 30 s. The coils were re-shimmed and water suppression parameters adjusted over the next 90 s so as to recommence data acquisition at 13 °C as close as possible to 5 min later than it was commenced at 8 °C. The procedure was repeated up to 58 °C in 5 °C increments, resulting in an overall rate of temperature increase of 1 °C/min. The machine was restored to 8 °C as rapidly as possible, the original parameters reset, and a final probe made to obtain a non-helical spectrum at 8 °C, before the peptide could re-fold.
2.5. Deconvolution of polarimetry, circular dichroism, and differential scanning calorimetry data
The D/z kinetic equation was fitted to these transition curves in order to investigate peptide helix unfolding. This equation is reproduced below [27].
The assumption that ln(k(unfold)) increases linearly with temperature has been proven for the unfolding of collagen helices [27]. The equation can be rearranged to the following, relating Tmax with the D and z parameters.
Here, Tmax is the temperature at which the maximal rate of helix unfolding occurs; r = scanning rate (1 °C/min); Te = the temperature at which the experiment is currently running; De = the time at temperature Te for the number of folded helices to reduce by a factor of 10; and z = the increase in temperature required for the value of D to decrease tenfold. When Te = Tmax, this equation simplifies to give D(Te = Tmax) = z/r, a direct relationship between D and z dependent on the scanning rate. So, when the scanning rate is 1 °C min−1, as used here, D(Tmax) has the same numerical value as z, with D(Tmax) expressed in minutes and z in °C.
For general quality control of peptides in our laboratory, we use the Excel solver algorithm to optimize two sets of three parameters from this equation (Tmax, z (=D), and peak intensity), describing the two kinetic events of the HTT and the LTT (Fig. 2). The optimization was done by minimizing the residual signal not accounted for by these two events. Tmax is the temperature where the derivative’s signal at its greatest, reflecting the point of fastest helix unfolding, z describes the peak width, while the equation form sets the peak shape.
When we obtained multiple datasets from peptide fractions of varying impurity (Figs. 1,4 and 5), we generated a model peak to fit the HTT, initially from the purest peptide preparation. We estimated the proportion of the total unfolding signal that could be accounted for by this main transition for each sample, considering it the “percentage signal accounted for by the high temperature transition”.
Fig. 1.

Purification and analysis of Toolkit peptide III-10. (a) Mass spectra of crude III-10 peptide (left), with accompanying polarimetry denaturation data (top right) from a 2.5 mg/ml solution in 10 mM phosphate buffered saline (pH 7.4) which was allowed to fold at 8 °C for 12 h, upscanning at 1 °C min−1. Inset (middle) is the 5 + ion section of the original mass spectrum from which the line spectra at left was derived. (b) and (c). As (a), except for HPLC-purified and HPLC re-purified III-10 peptide respectively. (d–f). Derivative polarimetry analysis for samples (a–c) respectively. Peaks representing the HTT were modelled (thin lines) using the D/z rate equation as described in materials and methods. The area under these peaks was considered to be the proportion of the total signal (thick lines) which could be ascribed to the HTT (Table 3a). The scale bar for the derivative data is 1 × 10−3 deg cm−1 K−1. Multiplying by 218 converts polarimetric rotation in deg. to rotation in deg cm2 dmol−1.
Fig. 4.

Polarimetry traces from HPLC- purified CRP and impure fractions. Raw data from just fractions 4–6, 8, and 13 (4–6 being pooled) are shown in panel (a) for clarity, and derivatives of all data are in panel (b), best-fitted to a 76.2 °C HTT that fitted pure peptide data. The fraction of signal that could be accounted for by the 76.2 °C HTT was calculated and detailed in Table 3b. This is lower than that assigned to the HTT if the data was all handled independently (Table 4). For panel (a), the scale can be converted to rotation in deg cm2 dmol−1 by multiplying by 132. The bar in panel (b) is 10−3 deg cm−1 K−1 or 0.13 deg cm2 dmol−1 K−1.
Fig. 5.

Purification of Toolkit peptide III-30 by Gel filtration. The gel filtration of 29%-pure HPLC-purified fraction in trace (a) shows the separation of the higher mass helical component of III-30 away from the reducing agent TCEP and single chain peptide that did not fold to a helix. Polarimetry derivative traces are given for the same sample before separation (b), for re-purified HPLC III-30 (c), for Gel filtration-separated helix (d) and for single strand (e) samples after they had been given time to refold at 4 °C for 12 h. The scale bar is as for Fig. 1.
2.6. Modelling effects of peptide imperfections upon a helix unfolding transition
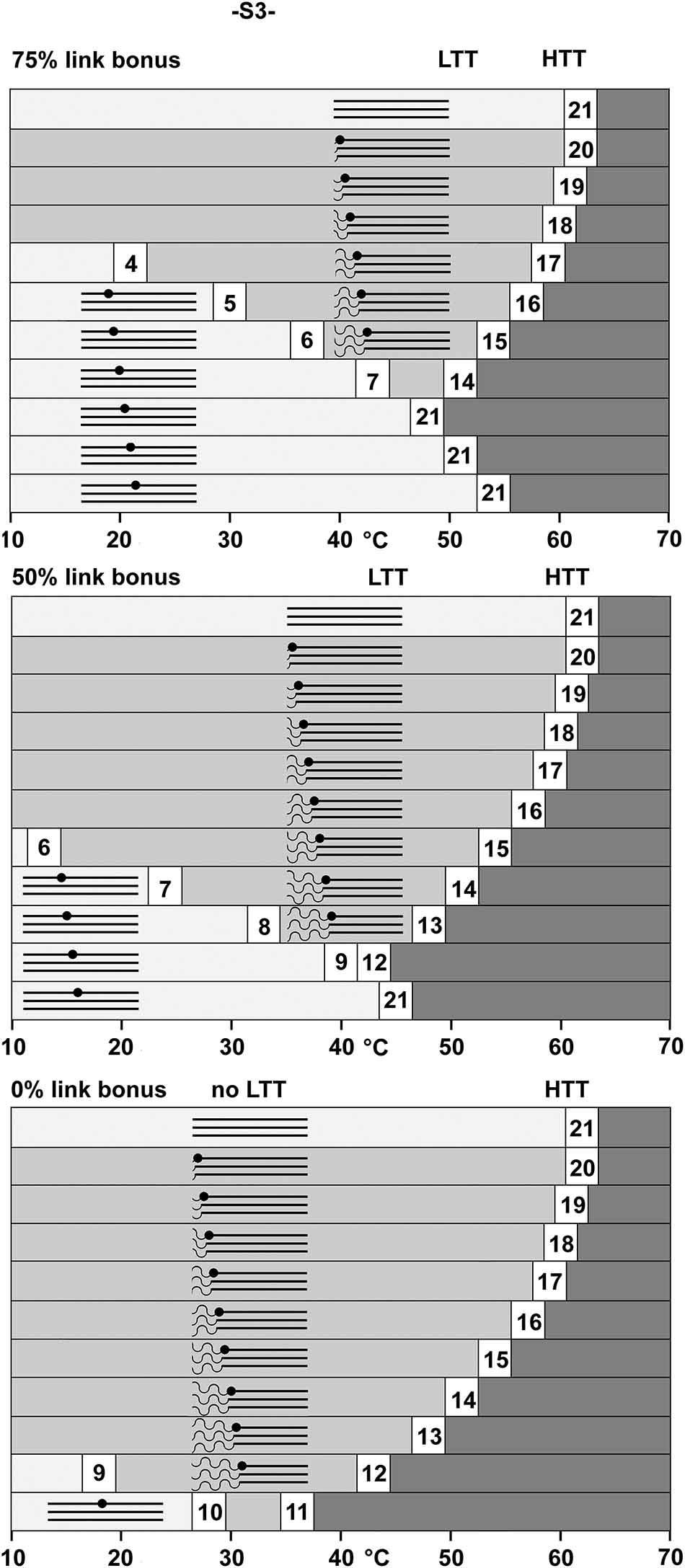
Dr A. Persikov [28] kindly supplied helical unfolding Tmax values for a test set of (GPP)n and (GPO)n peptides. These data have been used to predict Tmax depending on the peptide sequence, its length, and whether it is amide blocked on the carboxyl terminal or not (http://compbio.cs.princeton.edu/csc/). Table 2 shows calculated Tmax values of (GPP)n using this data. Peptide imperfections should then contribute to a population of helices with a decreased Tmax, because addition or deletion of an amino acid in collagen disrupts the canonical Gly-X-X′ repeating unit required for helix stability. At one extreme, imperfection might cause total disruption, in that the residues either side of the imperfection fold to form two thermodynamically separate helices that make no contribution to each other’s stability. For example, if a 21-triplet (GPP)n helix with a Tmax of 62 °C when pure had an imperfection between the 9th and 10th triplet, this might result in two independent helices of 9 and 12 triplets, with melting temperatures of 18 °C and 43 °C respectively. At the other extreme, the imperfection, being on only one strand of the helix, might have no demonstrable effect, and the whole population of helices, imperfect or not, unfolds at 62 °C. Between these two extremes a helix may form on one side of the imperfection and induce helicity on the other side to some degree, contributing a link bonus to helix stability.
Table 2.
Imperfection model predicting helical stabilities within a (GPP)21 helix Top: Helices 5 21 triplets long represent the length of helix on one side of a helical imperfection, length 21 being the full (GPP) 21 helix. 2nd row: The expected Tmax for these helix lengths if they were independent helices and amide blocked on C-terminus. 3rd row: The maximal stability increase conferred by the remaining helix ranges from 0 to 138 °C here if the imperfection has no effect. (100% link bonus, Tmax = 62 °C as for a 21 triplet helix). 5-6th row: Helical stabilities when the imperfection has an effect, with link bonuses of 50% and 75%. Bottom two rows: The link bonus requires a helix on the other side of the imperfection, so the final stabilities of each section on either side of an imperfection is shown. Bold numbers show stabilities or derived stabilities predicted by the algorithm as if water did not freeze. These are of course impossible, but can give some handle on how the model may operate when a stability bonus is conferred by an adjacent helix to raise the predicted stability above 0 °C. Underlined numbers show helix fragments that have benefitted in stability from a helix on the other side of the imperfection. Numbers in grey surround show a patch were a number of helical sections all unfold at temperatures close to each other.
| Length of fragment, n, (GPP)n | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 |
| Tmax°C | −66 | −37 | −14 | 4 | 18 | 28 | 36 | 43 | 48 | 51 | 54 | 57 | 59 | 60 | 61 | 62 | 62 |
| Max. link Bonus in °C (+100%) | 138 | 99 | 76 | 58 | 44 | 34 | 26 | 19 | 14 | 11 | 8 | 5 | 3 | 2 | 1 | 0 | 0 |
| Tmax°C including link bonus: | |||||||||||||||||
| Link bonus +50% | −2 | 13 | 24 | 33 | 40 | 45 | 49 | 52 | 55 | 57 | 58 | 59 | 60 | 61 | 62 | 62 | 62 |
| Link bonus +75% | 30 | 37 | 43 | 48 | 51 | 54 | 56 | 57 | 59 | 60 | 60 | 61 | 61 | 61 | 62 | 62 | 62 |
| Actual Tmax °C for helix fragment | |||||||||||||||||
| Link bonus +50% | −2 | 13 | 24 | 33 | 40 | 45 | 45 | 43 | 48 | 51 | 54 | 57 | 59 | 60 | 61 | 62 | 62 |
| Link bonus +75% | 30 | 37 | 43 | 48 | 51 | 54 | 54 | 51 | 48 | 51 | 54 | 57 | 59 | 60 | 61 | 62 | 62 |
Continuing the above example, if imperfection does not affect stability, both peptide sections on either side of the imperfection unfold simultaneously at 62 °C as if they were a perfect 21-triplet helix. The link bonus then increases the stability of the 9-triplet helix by +44 °C (62 °C–18 °C) and the 12-triplet helix by +19 °C (62 °C–43 °C). If half the maximum link bonus is conferred across the imperfection, the 9-triplet section would unfold at 40 °C (18 °C + 22 °C link bonus). With the 9-triplet section unfolded, the 12-triplet section then has no link bonus, so it unfolds at 43 °C. This concept can generate complex effects. For instance, if the link bonus is again half of the maximum, an imperfection between the 7th and 8th triplet means that the longer 14 residue-helix on one side of the imperfection, with a Tmax value of 51 °C, can induce the 7 residue-helix on the other side of the imperfection to fold with a Tmax of 24 °C (−14 °C + 38 °C link bonus). Under this imperfection model, when this molecule is heated, 7 triplets of the helix unfold with a Tmax of 24 °C, and the rest with a Tmax of 51 °C. Instruments will record the corresponding result, displaying both a low and high temperature transition.
Melting temperatures expected for helix sections of 5–21 triplets with 50% or 75% link bonuses are shown in the final row of Table 2, where the remaining 16–0 triplets of a 21 triplet Toolkit helix are on the other side of the imperfection. When the imperfection is towards the middle of the helix, both helix fragments on either side of it can stabilize each other, resulting in a relatively small temperature range (Table 2, numbers highlighted behind in grey) within which most of the imperfect helices unfold.
Instrumental recordings were then compared with melting curves generated using this imperfection model. The model does not give a thermodynamic description: its value is to verify whether imperfect peptides could account for the observed melting curve complexity. Imperfections were assumed to occur randomly along the length of the peptide, and thus there are many minor contributions to a full unfolding profile from each possible location of an imperfection along a helix. The model has three parameters, the impurity percentage, estimated from mass spectroscopy, the link bonus conferred by adjacent helices across the imperfection (0–100%), and the intrinsic peak width (°C) of any one molecular species when unfolding, defined as the parameter z (Equations (1) and (2)). As the purity is known, and the intrinsic peak width (z) is similar for all peptides of similar length, the link bonus can be fitted such that a model unfolding curve of an impure peptide mixture approximates to empirical data. This also allows the investigator to estimate the effects of planned helix interruptions [35] upon the unfolding profiles of collagen peptides before synthesis of the peptide.
3. Results
3.1. Status of Toolkit peptide III-10 after peptide synthesis and HPLC purification
Fig. 1 shows mass-spectroscopy data of crude (1a), HPLC-purified (1b), and HPLC re-purified (1c) peptide III-10 (Table 1). The main impurities were found to differ from the pure peptide M/z peak by only one residue (minus Cysteine or plus Glycine). We refer to these impurities as imperfect peptides. We observed progressive purity improvements upon HPLC purification and re-purification, from 69% (crude), to 75%, and then to 88% (Table 3a). Unfolding of the III-10 samples was initially observed by polarimetry (Fig. 1 a–c) and derivatives plotted (Fig. 1d–f). In Fig. 1d–f, a Tmax (HTT) was fitted to the purest sample, being 78% of the total signal, (Fig. 1f), and then its parameters were held in order to see how much of the total signal could be accounted for by this HTT in less pure samples, being 67% and 55% of the total signal in Fig. 1e and d respectively.
Table 3.
Analysis of peptide preparations of peptides III-10, CRP, and III-30 3a) Major mass components of crude, purified, and re-purified III-10 are recorded to give an electrospray mass-spectroscopy purity estimate (n = 2, different days). In darker grey, impurities deriving from deletions or additions of Cysteine, Alanine, Proline and Glycine are listed with their effect on the M/z. Reading the numbers in light grey cells across from each sample listed at left, the pure peptide M/z peak has been defaulted to an intensity of 100, and the intensities of impurities given relative to this. The M/z purity estimate is calculated by dividing the intensity of the pure peptide peak (and its ion/water derivatives) by the row’s sum of all the intensities listed in light grey cells. The percentage of pure helices within a population of folded helices is then estimated in brackets (but see discussion). The proportion of the total signal that could be accounted for by a single high temperature transition (HTT), is given in the right hand column and estimated by fitting to a D/z unfolding transition (equations (1) and (2)). The total polarimetric rotation change from 8° to 60 °C for a 2.5 mg/ml solution in PBS is given in square bracket (10 cm path length). 3b) Analysis is as for Table 3a, but for HPLC purification of CRP (Fig. 4a,b), where fractions 4–6 were considered “pure”, and later fractions impure. Fraction purity was estimated from the mass spectrum, while polarimetry was used to estimate the proportion of total signal from a high temperature transition. The total polarimetry signal change from 8° to 90 °C at 1 mg/ml in 200 mM AcOH across all the datasets was +0.143°±°0.012° over a 10 cm path length, from a starting signal of −0.428°±°0.005. 3c) Analysis is as for Table 3a. The mass spectrum preparation of HPLC-purified III-30 meant it was still unusable. However, this sample could be purified by Gel filtration to give “strand” and “helix” fractions (Fig. 5a) that were probed by polarimetry (Fig. 3b–d), or further re-purified by HPLC if one was prepared to accept significant losses.
| 3a - III-10 |
Pure Peptide |
Significant impurities |
M/s (helical) Purity Estimate % | % signal from HTT, Polarimetry | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| State | M/z 5432 | -H20, +[O], +Na/K+ | Cys |
Pro |
Gly |
Gly |
Ala |
Pro |
Other M/z > 5200 | ||
| −103 | −97 | −57 | +57 | +71 | +97 | ||||||
| Crude | 100 | 40 | 12 | 6 | 1 | 19 | 6 | 5 | 14 | 69 (33) | 55 [0.337] |
| Purified | 100 | 29 | 10 | 5 | 3 | 10 | 2 | 3 | 8 | 75 (41) | 67 [0.379] |
| High purity | 100 | 28 | 1 | 2 | 3 | 4 | 2 | 3 | 3 | 88 (68) | 78 [0.410] |
| 3b - CRP |
Pure peptide |
Significant impurities |
M/s (helical) Purity Estimate % | % signal from HTT, Polarimetry | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R-phase column fraction | M/z 3294 | +H2O + Na+ + K+ | Gly Pro |
Pro |
Gly |
Gly |
Pro |
Cys |
2Gly |
Other M/z > 2500 | ||
| −154 | −97 | −57 | +57 | +97 | +103 | +114 | ||||||
| 4 to 6 | 100 | 18 | – | – | – | 3 | – | – | – | – | 97 (91) | 91 |
| 7 | 100 | 5 | – | – | – | 33 | – | – | – | – | 76 (44) | 68 |
| 8 | 100 | 24 | – | – | – | 48 | – | – | – | – | 72 (37) | 53 |
| 9 | 100 | – | 23 | 49 | 15 | 67 | – | 41 | – | 123 | 24 (01) | 34 |
| 10 | 100 | 23 | – | 40 | – | 73 | 42 | – | – | 43 | 38 (05) | 35 |
| 11 | 100 | – | – | – | – | 108 | 139 | – | – | – | 29 (02) | 31 |
| 12 | 100 | – | – | – | – | 161 | – | – | 26 | – | 35 (04) | 47 |
| 13 | 100 | – | 125 | – | 38 | – | – | – | 63 | – | 31 (03) | 39 |
| 3c - III-30 |
Pure Peptide |
Significant impurities |
M/s (helical) Purity Estimate % | % signal from HTT, Polarimetry | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gel filtration fraction | M/z 5324 | +H2O + Na+ + K+ | 2Pro |
2Gly Pro |
Gly Pro |
Pro |
Gly |
Gly |
Pro |
Other M/z > 5000 | ||
| −194 | −211 | −154 | −97 | −57 | +57 | +97 | ||||||
| HPLC-purified | 100 | 5 | 15 | 11 | 20 | 51 | 31 | 31 | 5 | 89 | 29 (02) | 26 [0.242] |
| Strand | 100 | 12 | 18 | 12 | 22 | 67 | 40 | 59 | 7 | 103 | 25 (02) | 20 [0.246] |
| Helix | 100 | 6 | 10 | 0 | 10 | 30 | 23 | 16 | 0 | 25 | 48 (11) | 80 [0.326] |
| HPLC-Repuriified | 100 | 7 | 2 | 0 | 0 | 4 | 13 | 6 | 3 | 5 | 77 (46) | 69 [0.334] |
To compare these data with results from other techniques, we investigated the HPLC-purified sample also using CD and DSC (Fig. 2), and 67% of the total polarimetry rotation change between 8 °C and 70 °C, 58% of the CD ellipticity change and 63% of the DSC endotherm was due to the HTT. In terms of signal, this is 0.337° (82.2 deg cm2 dmol−1) for the polarimeter, −41 millidegrees cm−1 (−8.94 deg cm2 dmol−1,[θ]225) for CD, and 63 J g−1 K−1 (347 kJ mol−1 K−1) for DSC. All techniques yielded similar Tmax values (Fig. 2). Therefore, only a proportion of the data, 58–67% for the HPLC-purified peptide fraction across all three techniques, could be attributed to the HTT. From the polarimetry results, this improves to 78% when the peptide is re-purified (Fig. 1f and Table 3a), but is merely 55% before purification (Fig. 1d and Table 3a).
The simplest model, in which perfect and imperfect peptides are assumed to associate randomly to form helices, gives estimates of helix purity much lower than peptide purity (Table 3a), since only one imperfect peptide strand is required to render each helix imperfect. These estimates of helix purity are lower than the proportion of signal attributable to the HTT (Table 3a). However, loss or gain of a residue close to the end of a peptide strand, such as minus Cys, is unlikely to affect helix formation. If one makes that assumption, the respective helical purities of crude, HPLC-purified, and HPLC re-purified peptides are estimated by mass spectroscopy as 41%, 53%, and 70%. In reality, helical purities might be expected to be even greater than these latter values, as imperfect peptides with interruptions in the canonical G-X-X′ repeat are intuitively less likely to fold into helices.
3.2. Using four techniques to compare III-10 data with four other Toolkit peptides
To ensure that the above results were not peculiar to peptide III-10, and to further compare polarimetry with DSC and CD, we investigated four other peptides. These peptides displayed transitions at different temperatures, and had different proportions of total signal attributable to the HTT. Comparative data with approximate peptide purities (minus Cysteine counted as pure for these purposes) are shown in Fig. 2.
Within Fig. 2, we show two fitted peaks as for an HTT and LTT analysis based on a single dataset. An LTT is observed in all peptides, and the data for all three techniques are sufficiently similar to be used interchangeably. The lower signal of II-5 may be due to slow refolding, a process for investigation in future.
As the LTT is a new observation that can be of similar magnitude to the HTT, we probed it by NMR in case it represented an unknown rearrangement within the molecule. We also required additional proof that the HTT was indeed attributable to helix unfolding. In Fig. 3, Helix-specific 1H NMR peaks were identified in two ways. First, an NMR spectrum at 8 °C after 12–18 h of refolding was compared with another spectrum where the peptide had been cooled from 60 °C to 8 °C without allowing time for helix refolding. Second, we tracked the chemical shift (ppm) of Glycine-NH protons with temperature. Values below ∼5 ppb K−1 indicate that the proton is protected from solvent, which is only the case in helical peptides [29]. These peaks are highlighted in Fig. 3 and their identities and ppm shifts are given in supplementary Table 1. The disappearance of the helix peaks correlated with the completion of the HTT recorded using the other three instruments. These experiments prove that the HTT involves helix unfolding, but could not verify whether there was helix unfolding during the LTT because peak broadening prevented quantitative analysis of the peaks. There was no evidence to suggest that the LTT represents a change from one helical form to another, such as a change in the helical pitch.
Fig. 3.

NMR temperature spectra for Toolkit peptides. Each stack of 11 spectra show data taken from 8 °C to 58 °C in 5 °C increments. Below each stack is a spectrum taken at 8 °C immediately after the 58 °C spectrum, while the peptide is still non-helical. Single strand Glycine-NH peaks are highlighted in light grey and proton peaks that could be unambiguously assigned and attributed to the helix are highlighted in darker grey.
3.3. Analysis of CRP fractions after HPLC purification
We now show data from a shorter 36-residue peptide. Fig. 4a shows polarimetry traces of samples encompassing fractions 4–6, 8, and 13 of an HPLC purification of crude CRP (Table 1). These data and those from the remaining fractions 7, 9, 10, 11, and 12 are derivatized to give the traces in Fig. 4b. The mass spectrum of each fraction and its estimated purity, with a calculated resulting helical purity, is given in Table 3b. The purest fractions 4–6 had a narrow HTT signal with a Tmax at 76.2 °C. This HTT accounts for a progressively smaller amount of the total signal as peptide imperfections increase across fractions 7–13 (Table 3b). The reduction in the HTT was estimated by measuring the maximum proportion of the total transition from those datasets that could be accounted for by a narrow HTT identical to that observed from the purest fractions 4–6.
Some peptide fractions contained peptide with two or more imperfections, and very high impurity levels. For instance, fraction 13 has just 31% purity, with12% of the peptide having 1 imperfection, and 57% of the peptide having 2 imperfections. If helix formation is random and equally likely, this would mean that just 2.9% of its helices are pure, with others having up to six imperfections in relative proportions of 3.2%, 17.5%, 12.5%, 32.9%, 11.6%, and 19.2% successively. Despite this, the total signal as reported by polarimetry is similar for all the different CRP fractions, implying that many imperfect CRP strands can still fold to form less stable helices. This is because CRP, a GPO polymer, has a near-perfect sequence for collagen helix formation. Even clipping it entirely into two equal parts yields helices with a Tm of approximately 7–8 °C [28], so multiple imperfections are required to prevent folding entirely. Some imperfections must have little effect: for fractions 9–13, 30–40% of the total signal can be accounted for by an HTT of 76.2 °C, far in excess of the expected proportion of pure helices of just 1–5%.
This shows that even with shorter peptides, if the folding temperature is low enough (e.g. 4 °C) for a LTT to be observed, and the stability of the pure helix high enough [35], most of the imperfect peptide still forms a helix.
3.4. Analysis of gel filtration fractions from low purity III-30 peptide
Gel filtration provides a method which can purify a peptide in triple-helical state, different from HPLC purifications that operate on an unfolded peptide. Initial purification of III-30 using HPLC yielded a sample of just 29% purity, unacceptably low (Table 3c). We improved this in two ways:
First, a portion of the refolded HPLC-purified sample prior to polarimetry was loaded onto the gel filtration column (Fig. 5a). This data was deconvolved using existing methodology [33,34] to show the three different components, where the highest mass helical component eluted first, and was collected separately from the TCEP and single-strand components. The helical and TCEP/single-strand components were concentrated, heated and refolded 12 h at 8 °C prior to polarimetry (Fig. 5d, e), for comparison with the sample as it was prior to gel filtration. In the helical component, the proportion of signal due to the HTT increased to 80% compared to a prior 26%, and the fraction was 48% pure. In the TCEP/strand component, the proportion of the HTT decreased to 22%, the fraction being 25% pure.
Second, we re-purified the peptide by HPLC as described in materials and methods, retaining only the purest eluted fraction. This achieved a purity of 77% by mass spectroscopy (Table 3c). To check the effect of this increased purity upon helix formation, the HPLC-purified and HPLC re-purified samples were concentrated, heated and refolded for 12 h at 8 °C prior to analysis by polarimetry (Fig. 5b, c). The proportion of signal due to the HTT increased from 26% to 69% (Table 3c). This HPLC re-purification provided a III-30 sample that could be usefully compared to the helix-fraction sample taken from gel filtration.
4. Discussion
4.1. All techniques for measuring peptide unfolding yield consistent data
Several lines of argument lead us to conclude that the LTT is a helix unfolding event, akin to the HTT, rather than some other molecular rearrangement.
First, CD, DSC, and polarimetry all report similar HTTs-to-LTT ratios for a given peptide, suggesting that the underlying processes are fundamentally related (Fig. 2).
Second, we discount dispersal of peptide aggregates prior to unfolding as the cause of the LTT, because we did not observe NMR peak broadening as the temperature was raised through the LTT temperature range, instead the peak simple disappeared over the HTT temperature range. Light scattering experiments also provide no evidence for aggregation of a sample Toolkit peptide (data not shown). Moreover, helix aggregation might be expected to cause a higher temperature transition as a shoulder to the main HTT, as it should stabilize the peptide helix in the same manner that aggregation or fibril formation stabilizes whole collagen [15,36,37]. This was not observed.
Third, if the LTT was due to some rearrangement of the helix, it might be expected to have a similar, fixed signal ratio to the HTT, given the similarities within Toolkit peptides, as the amount of helix rearrangement should directly correlate with the amount of helix. It might also be expected to happen at some predictable temperature relative to the HTT. Neither of these is the case.
Fourth, no known transition can account for the LTT except helical unfolding. While some helix rearrangement prior to heat-denaturation has been reported in previous work [22], it is a subtle change in proline ring pucker, with little else observable by NMR. This is unlikely to account for the significant enthalpy (DSC) and rotation signals we observe here from the LTT. As the LTT can account for more than 50% of the total signal change between 8 °C and the fully denatured state (∼70–90 °C), it must least involve significant changes to hydrogen bonding or helical twist if it were not helix unfolding itself.
Fifth, if no time is allowed for helix to refold at 8 °C after heat-denaturation, only a trace signal is observed across the temperature spectrum produced by DSC, polarimetry, and CD (data not shown). The LTT must therefore derive from either secondary structure changes or aggregation.
Last and most significantly, the CD signal at 225 nm and the DSC endotherm have already been correlated to helix unfolding, e.g [38].
To conclude, all the evidence points towards the LTT being a helix unfolding event.
4.2. Gel filtration separations based on helical stability
The 36-mer CRP from HPLC-fractions 4–6 was 97% pure. Similarly-purified Toolkit peptides III-24 (82%), II-40 (83%) and III-53 (87%) reflect the quality we would expect to achieve routinely in such peptides, 63-mers. We regard Toolkit peptides III-10 (81%) and the re-purified III-30 (77%) as satisfactory, whilst II-5 (70%) proved more difficult to synthesise and purify in useful yield. The use of the lower purity (29%) preparations of III-30 is confined to this study.
From an initial purity of 29% for III-30, the mass spectra data in Table 3c shows that the purity of the gel-filtered helical fraction was improved to 48%, whereas HPLC re-purification improved purity to 77%. However, when these were compared by polarimetry (Fig. 5c, d), 80% of the total signal could be accounted for by HTT from the gel filtration purification, higher than the 69% observed from the HPLC re-purified fraction. It follows that imperfections in peptides within the gel filtration helix fraction have less impact upon helical stability. We suggest that this helical fraction has proportionally fewer peptides with imperfections in the centre of the peptide, and more located near the ends of the peptide, as end-imperfect peptides will destabilise the helix less. Some doubly imperfect “minus 2 Pro” or “minus ProGly” peptides were found in this fraction, perhaps implying that double deletions between the same two residues can occur near the end of the helix.
Conversely, from the same initial purity of 29%, the mass spectra data show that the purity of the gel-filtered single-strand III-30/TCEP fraction was reduced to 25%, an increase in the number of peptide imperfections. As helix and single-stranded peptides are in equilibrium, “removing” the helical component from an equilibrated solution by gel filtration shifts the equilibrium such that single strands may now fold over 12 h to form more helix, observable by polarimetry. However, the resulting polarimetry trace for this fraction had just 20% of its signal accountable for by an HTT (Table 3c), lower than the initial 26% (Table 3c), and the LTT Tmax was visibly shifted to ∼28 °C (Fig. 5e) as opposed to an initial ∼35 °C (Fig. 5b). Therefore, peptides with helix-disrupting imperfections were concentrated in the single-strand/TCEP fraction of III-30.
This data illustrates that the destabilisation effect of imperfect peptides upon a helix is dependent upon the location of the imperfection, and that if helicity is the goal, removal of helix-disrupting imperfections by gel filtration may be more effective than direct purification of an unfolded peptide.
4.3. Even very low purities have little effect upon the Tmax of the high temperature unfolding transition
So far, we have mathematically fitted an HTT curve to the data from the purest sample and calculated the proportion of signal that is accountable by this HTT. We have then taken the same parameters for Tmax and z and attempted to fit the “pure” HTT to samples that are less pure to give the results in Table 3a–c, estimating the percentage of the signal in each trace due to the HTT, standardized on the purest peptide we can obtain.
Most unfolding analysis in our laboratory is performed only once as a quality control on each peptide, and the purity may vary between peptides. The question, then, is how the observed HTT, determined by a 2-part transition analysis, varies from that of the pure peptide. Analysis of the data from Figs. 1,2 and 4, and 5 yielded the results in Table 4. The 97% pure CRP from fractions 4–6 was found to have an HTT of 76.2 °C (Table 4). Surprisingly, the less pure fractions also yielded HTTs between 74.0 °C and 76.4 °C (Table 4) showing that even quite impure peptide yields a good estimate of the intrinsic HTT. However, the unexpectedly high HTT from fraction 12 was also the broadest peak of the set, with z = 15.1, suggesting that its 76.4 °C Tmax was less accurate.
Table 4.
Using the imperfection model to explain helical unfolding data. Real data is analyzed from Figs. 1 and 4, and 5 as detailed in the left column. Parameters used in the “imperfection model” are shaded in grey and used to generate the traces in Fig. 6 (left column). Real and model-generated data can both be interpreted using the D/z equation to give two fits, one for each of the LTT and the HTT. As the imperfection model generates traces with a total signal of 1, the residual sum of squares remaining after fitting the HTT and LTT are comparable. Model parameters given here that generated the traces 6f-h had been adjusted to try an mimic the data shown in Figs. 1f, 4b (fracs 4–6), and 5b respectively. ∗ allowed to vary from 97% as no good fit was possible (see section 4.5).
| Figure | Peptide | Empirical data type | Two-fit data interpretation |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HTT |
LTT |
Residual sum of squares | ||||||||||
| Tmax | z | % total | Tmax | z | % total | |||||||
| 1d | III-10 | Polarimeter (crude peptide) | 53.0 | 9.3 | 64 | 39.0 | 16.4 | 36 | n/a | |||
| 1e, 2 | III-10 | Polarimeter (HPLC-purified) | 53.3 | 8.0 | 67 | 40.7 | 14.5 | 33 | n/a | |||
| 2 | III-10 CD | CD (HPLC-purified) | 54.2 | 6.9 | 51 | 42.0 | 19.8 | 49 | n/a | |||
| 2 | III-10 DS | DSC (HPLC-purified) | 53.1 | 6.2 | 58 | 42.5 | 22.8 | 42 | n/a | |||
| 1f | III-10 | Polarimeter (HPLC re-purified) | 52.7 | 8.4 | 78 | 37.0 | 13.8 | 22 | n/a | |||
| 4b | CRP | Polarimeter (HPLC-fractions 4–6) | 76.2 | 11.1 | 87 | 49.8 | 25.7 | 13 | n/a | |||
| 4b | CRP | Polarimeter (HPLC fraction 7) | 75.9 | 11.0 | 68 | 49.2 | 77.0 | 32 | n/a | |||
| 4b | CRP | Polarimeter (HPLC fraction 8) | 74.8 | 12.2 | 65 | 40.4 | 63.8 | 35 | n/a | |||
| 4b | CRP | Polarimeter (HPLC fraction 9) | 74.0 | 12.5 | 26 | 55.1 | 47.4 | 74 | n/a | |||
| 4b | CRP | Polarimeter (HPLC fraction 10) | 75.3 | 13.2 | 40 | 49.5 | 35.9 | 60 | n/a | |||
| 4b | CRP | Polarimeter (HPLC fraction 11) | 74.8 | 12.1 | 22 | 60.7 | 43.2 | 78 | n/a | |||
| 4b | CRP | Polarimeter (HPLC fraction 12) | 76.4 | 15.1 | 53 | 45.4 | 52.4 | 47 | n/a | |||
| 4b | CRP | Polarimeter (HPLC fraction 13) | 75.2 | 12.0 | 30 | 57.1 | 49.2 | 70 | n/a | |||
| 5b | III-30 | Polarimeter (HPLC-purified) | 43.9 | 15.2 | 72 | 31.7 | 14.8 | 28 | n/a | |||
| 5c | III-30 | Polarimeter (HPLC re-purified) | 47.7 | 6.6 | 69 | 35.6 | 11.0 | 31 | n/a | |||
| 5d | III-30 | Polarimeter (helix fraction) | 47.8 | 8.4 | 80 | 30.9 | 12.7 | 20 | n/a | |||
| 5e | III-30 | Polarimeter (strand fraction) | 45.1 | 10.8 | 36 | 30.0 | 15.0 | 64 | n/a | |||
| Figure | Modelled Peptide | Imperfection model parameters. |
HTT |
LTT |
Residual Sum of Squares | |||||||
| Tmax | z | Helical purity (%) | %Link bonus | Tmax | z | % total | Tmax | z | % total | |||
| 6a | (GPP)21 | 62.2 | 5.0 | 100 | n/a | 62.2 | 5.0 | 100 | – | – | – | 0.000 |
| 6b | (GPP)21 | 62.2 | 5.0 | 40 | 0 | 61.5 | 5.9 | 68 | 52.5 | 26.3 | 32 | 0.039 |
| 6c | (GPP)21 | 62.2 | 5.0 | 40 | 50 | 61.5 | 6.1 | 69 | 47.7 | 17.3 | 31 | 0.047 |
| 6d | (GPP)21 | 62.2 | 5.0 | 40 | 75 | 61.6 | 5.8 | 66 | 52.1 | 9.9 | 34 | 0.014 |
| 6e | Toolkit | 48.6 | 5.0 | 40 | 75 | 48.4 | 5.7 | 65 | 39.1 | 8.7 | 35 | 0.013 |
| 6f | III-10 | 54.4 | 9.1 | 43 | 55 | 52.6 | 10.2 | 69 | 39.7 | 15.8 | 31 | 0.005 |
| 6g | III-30 | 47.4 | 12.5 | 02 | 60 | 46.3 | 12.5 | 46 | 33.5 | 15.6 | 54 | 0.016 |
| 6h | CRP | 76.6 | 10.5 | 91∗ | 65 | 76.6 | 10.5 | 97 | 53.5 | 29.8 | 03 | 0.000 |
This conclusion was reinforced by analysis of the Toolkit peptides III-10 and III-30. Tmax of the HPLC-purified peptide III-10 was 53.5 ± 0.3 °C and its peak width was z = 8 or less (3 techniques) compared to the 53.0 °C estimated from the crude peptide, which had the higher value of z of 9.3 (Table 4). Likewise, Tmax of the helical fraction from gel filtration of III-30 was 47.8 °C and z = 8.4, and Tmax of the re-purified III-30 HPLC fraction was 47.7 °C and z = 6.6, whereas the initial, poor quality, HPLC-purified fraction had a Tmax of just 43.9 °C, whilst its z = 15.2 peak width was an indicator of poor accuracy. We conclude that this “two temperature transition” analysis gives an HTT Tmax generally within 1–2 °C of the true Tmax, the level of accuracy varying inversely with the z value.
4.4. The width (z) of an unfolding transition is indicative of the heterogeneity of the helices that are unfolding
The observed Tmax of the LTT from the same peptide varied greatly depending on the purity of the sample. For the impure CRP fractions 7–13, Tmax values were recorded between 40.4 and 60.6 °C, with the LTT Tmax of the pure fraction being 49.8 °C. The corresponding z values were between 36 and 77 (Table 3) and 26. Impure CRP peptide HTT z values were far lower, ranging between 11.0 and 15.1, with z = 11.1 for the HTT of the purest CRP helix we could form. Therefore, the typically large z reported from CRP LTTs must reflect the unfolding of a mixture of different helices with varying imperfections.
This is observed to a lesser extent with Toolkit peptide LTT Tmax values. The purest peptide III-10 and III-30 fractions report LTT Tmax values of 37.0 °C and 35.6 °C respectively, with impure fractions reporting values up to 5 °C different. The accompanying LTT z values are 13.8 and 11.0, with impure fractions yielding z = 16.4 and 14.8 respectively. These values are larger than the HTT z values of the purest III-10 and III-30, those being 8.4 and 6.6 respectively.
The analysis of the LTT in Table 4 is therefore more useful as an indicator of peptide heterogeneity, rather than a measure of the Tmax of an imperfect helix, since it attempts to fit the unfolding of multiple helix species as though they were just one species. Any value of z over 12 for the shorter CRP peptide, or over 9 for the longer Toolkit peptides, would suggest that the unfolding transition is due to the presence of more than one type of helix.
4.5. Equilibrium-based investigations to calculate Tm detect impure helices poorly
Even for shorter peptides, mass spectra have been published where a 5–8% “minus Pro” imperfection within (POG)10 was considered acceptable [24]. The occurrence of imperfection will increase with peptide length. Equilibrium or near-equilibrium unfolding studies cannot easily detect different helical forms. As temperature increases, the 15–25% of helices containing an imperfection, assembled from a peptide that is 5–8% impure, will unfold first, and the 2 pure strands out of 3 released from those helices may then refold slowly to a new equilibrium, where they are again helical while the imperfect strands do not refold. The LTT will be hard to detect, being 1/3 of the amplitude of that seen in kinetic unfolding systems and representing the unfolding only of imperfect strands; this is possibly why LTTs have remained unreported for so long. Equilibrium measurement of such peptides also presents a problem for those calculating ΔS° or ΔH° values from CD or polarimetry data. The impurity will reduce the fraction folded, and cause both ΔS° and ΔH° to be less negative, resulting in a slight increase in calculated ΔG° values for temperatures below the unfolding temperature.
4.6. Using the imperfection model to explain the apparent low temperature signal
One enduring feature of the LTT is that it is separable from the HTT. It normally appears as a well-defined shoulder on the side of the HTT, but can also be present as a distinct peak (see III-24, Fig. 2). However, we had expected imperfect helices to have apparently random Tmax values lower than that of the HTT of the pure peptide, displaying no defined peak.
The above data showed that impurities can cause the LTT, so we attempted to formulate a mathematical model (materials and methods) which could describe the unfolding of all the helical populations within a sample, and generate a well-defined LTT derivative signal as it might be observed by DSC, CD, or polarimetry. Starting from pure (GPP)21 peptide that is expected to have a HTT Tmax of 62 °C, we generated an expected transition of unity area using a z value of 5.0 for peptides of 21 triplets to give the trace in Fig. 6a, where z = 5 is the lowest value we have observed upon analysis of the full Toolkit peptide set (data not shown). We then simulated the unfolding of the same peptide when its helical purity is just 40%. If an imperfection in the helix has no effect, then the stimulated transition is identical to Fig. 6a. The opposite is shown in Fig. 6b, where the helices on either side of the imperfection are assumed to fold and unfold entirely independently. There, the contribution of the pure helix is filled in grey, while that of the impure helices is in white. The additional two fitted curves show how the generated data may be interpreted by a two-transition HTT and LTT analysis, for which the parameters are given in Table 4. This type of transition in Fig. 6b is generally not observed for Toolkit peptides (see Fig. 2), so Fig. 6c and d show overall unfolding transitions when the link bonus is set at 50% and 75% respectively (see materials and methods). With these parameters in place, a broad peak becomes evident, with a Tmax of ∼45 °C and ∼52 °C in Fig. 6c–d respectively, similar to the LTT observed in real data. The effects of the link bonus are described in detail in supplementary Fig. 1.
Fig. 6.

Helical transition profiles generated by the peptide imperfection model. Trace (a) shows a single transition event (peak width parameter z = 5) as expected for a 100% pure (GPP)21 peptide with a Tmax of 62.2 °C. Upon reducing the purity to 40% (b–d), the pure proportion (grey) still unfolds in the same manner. The remaining signal which makes the total, the thick line, is from helices with lower melting temperatures containing peptides with various imperfections. Trace (b) assumes that any imperfection completely disrupts the helix, while traces (c) and (d) assume that 50% or 75% (respectively) of the stability bonus due to a proximal helix is conveyed across the imperfection. Trace (e) shows how trace (d) would look for a typical Toolkit peptide of lower stability than (GPP)21. Traces (f–h) are generated from parameters (see Table 4) designed to mimic the real results seen for II-10 (f), III-30 (g), and CRP (h). Each trace has 2 other component curves from an analysis that assumes that there are just high temperature and low temperature unfolding elements to the data (Table 4), with only small residuals. This latter 2-component analysis is simpler to do routinely and makes no assumptions other than being a convenient way to describe the data.
The model also predicts LTT transitions for Toolkit peptides when parameters are adjusted according to their sequences and HTT Tmax values (Fig. 6e). To further test this, we allowed these parameters to take optimal values in order to mimic data derived from III-10 (Fig. 6f, mimicking Fig. 1f), III-30 (Fig. 6g, mimicking Fig. 5b), and CRP (Fig. 6h, mimicking Fig. 4b, fractions 4–6). In each case, the helical purity had been calculated from the peptide purity as determined by mass spectroscopy, and was not initially allowed to vary. Only Tmax, z, and the link bonus were altered to fit the data.
While the model was able to provide transitions that were visibly close to real data, it remains imperfect on three counts. First, the calculated link bonus differs between traces 6f-h (Table 4) despite similarities in how the helix is interrupted, and this value significantly affects both amplitude and Tmax of the modelled LTT. Second, we were unable to obtain a good fit to the CRP data. With 97% pure CRP peptide coming from fractions 4–6, the model predicts that ∼97% of the total signal should be in the HTT as opposed to the observed 87%, suggesting that there is some other factor that can induce an LTT within CRP and thereby possibly within Toolkit peptides also. Third, the notion that a 10% peptide imperfection will cause 30% of the resultant folded helices to be imperfect is simplistic as impure peptides will not fold into helices as fast as pure peptides, and thus 30% will be an over-estimate, with the model slightly exaggerating the LTT.
5. Conclusion
For both CRP and Toolkit peptides, we have demonstrated that peptide imperfection is one variable that can account for the majority of the low temperature transition observed by polarimetry, DSC, and CD. This transition is caused by the unfolding of imperfect peptide helices. The LTT is observed in the CRP due to its almost optimal helix-inducing sequence, and in Toolkit peptides due to their length. Gel filtration and the imperfection model show that the helix-destabilising effect of an imperfection is dependent upon its location within the peptide. Unfolding of helices with imperfections towards their ends contribute to the HTT, whilst helices with imperfections in the middle cause an LTT. Such helix heterogeneity broadens the HTT without significantly changing Tmax. The Tmax of the far broader LTT varies much more, where z becomes a parameter that describes the heterogeneity of the helical mixture. We have demonstrated that these data can be replicated closely using a mathematical model that uses a link parameter describing the effects of an imperfection upon helix stability. This strengthens the interpretation that the LTT can be caused by peptide imperfections. In published work where an LTT was observed and commented on, it was interpreted as a partial unfolding of the helix. Given the work reported here, and the lack of an accompanying mass spectrum, it is possible that those effects were due to imperfect peptide helices.
Acknowledgements
This work was supported by the British Heart Foundation. [PG/08/011/24416].
Footnotes
Supplementary data related to this article can be found online at doi:10.1016/j.biomaterials.2011.05.025.
Appendix. Supplementary data
The following are the Supplementary data related to this article:
{kind=link}
References
- 1.Fields G.B. Synthesis and biological applications of collagen-model triple-helical peptides. Org Biomol Chem. 2010;8(6):1237–1258. doi: 10.1039/b920670a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boudko S.P., Engel J., Okuyama K., Mizuno K., Bachinger H.P., Schumacher M.A. Crystal structure of human type III collagen gly991-gly1032 cystine knot-containing peptide shows both 7/2 and 10/3 triple helical symmetries. J Biol Chem. 2008;283(47):32580–32589. doi: 10.1074/jbc.M805394200. [DOI] [PubMed] [Google Scholar]
- 3.Kar K., Amin P., Bryan M.A., Persikov A.V., Mohs A., Wang Y.H. Self-association of collagen triple helical peptides into higher order structures. J Biol Chem. 2006;281(44):33283–33290. doi: 10.1074/jbc.M605747200. [DOI] [PubMed] [Google Scholar]
- 4.Miles C.A. Kinetics of the helix/coil transition of the collagen-like peptide (pro-hyp-gly)10. Biopolymers. 2007;87(1):51–67. doi: 10.1002/bip.20787. [DOI] [PubMed] [Google Scholar]
- 5.Farndale R.W., Lisman T., Bihan D., Hamaia S., Smerling C.S., Pugh N. Cell-collagen interactions: the use of peptide Toolkits to investigate collagen-receptor interactions. Biochem Soc Trans. 2008;36(Pt 2):241–250. doi: 10.1042/BST0360241. [DOI] [PubMed] [Google Scholar]
- 6.Lebbink R.J., Raynal N., de Ruiter T., Bihan D.G., Farndale R.W., Meyaard L. Identification of multiple potent binding sites for human leukocyte associated Ig-like receptor LAIR on collagens II and III. Matrix Biol. 2009;28(4):202–210. doi: 10.1016/j.matbio.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Raynal N., Hamaia S.W., Siljander P.R., Maddox B., Peachey A.R., Fernandez R. Use of synthetic peptides to locate novel integrin alpha2beta1-binding motifs in human collagen III. J Biol Chem. 2006;281(7):3821–3831. doi: 10.1074/jbc.M509818200. [DOI] [PubMed] [Google Scholar]
- 8.Giudici C., Raynal N., Wiedemann H., Cabral W.A., Marini J.C., Timpl R. Mapping of SPARC/BM-40/osteonectin-binding sites on fibrillar collagens. J Biol Chem. 2008;283(28):19551–19560. doi: 10.1074/jbc.M710001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emsley J., Knight C.G., Farndale R.W., Barnes M.J. Structure of the integrin alpha2beta1-binding collagen peptide. J Mol Biol. 2004;335(4):1019–1028. doi: 10.1016/j.jmb.2003.11.030. [DOI] [PubMed] [Google Scholar]
- 10.Jarvis G.E., Raynal N., Langford J.P., Onley D.J., Andrews A., Smethurst P.A. Identification of a major GpVI-binding locus in human type III collagen. Blood. 2008;111(10):4986–4996. doi: 10.1182/blood-2007-08-108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leo J.C., Elovaara H., Bihan D., Pugh N., Kilpinen S.K., Raynal N. First analysis of a bacterial collagen-binding protein with collagen Toolkits: promiscuous binding of YadA to collagens may explain how YadA interferes with host processes. Infect Immun. 2010;78(7):3226–3236. doi: 10.1128/IAI.01057-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu H., Raynal N., Stathopoulos S., Myllyharju J., Farndale R.W., Leitinger B. Collagen binding specificity of the discoidin domain receptors: binding sites on collagens II and III and molecular determinants for collagen IV recognition by DDR1. Matrix Biol. 2011;30(1):16–26. doi: 10.1016/j.matbio.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stahl P.J., Romano N.H., Wirtz D., Yu S.M. PEG-based hydrogels with collagen mimetic peptide-mediated and tunable physical cross-links. Biomacromolecules. 2010;11(9):2336–2344. doi: 10.1021/bm100465q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wojtowicz A.M., Shekaran A., Oest M.E., Dupont K.M., Templeman K.L., Hutmacher D.W. Coating of biomaterial scaffolds with the collagen-mimetic peptide GFOGER for bone defect repair. Biomaterials. 2010;31(9):2574–2582. doi: 10.1016/j.biomaterials.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walton R.S., Brand D.D., Czernuszka J.T. Influence of telopeptides, fibrils and crosslinking on physicochemical properties of type I collagen films. J Mater Sci Mater Med. 2009;21(2):451–461. doi: 10.1007/s10856-009-3910-2. [DOI] [PubMed] [Google Scholar]
- 16.Jones C.I., Bray S., Garner S.F., Stephens J., de Bono B., Angenent W.G. A functional genomics approach reveals novel quantitative trait loci associated with platelet signaling pathways. Blood. 2009;114(7):1405–1416. doi: 10.1182/blood-2009-02-202614. [DOI] [PubMed] [Google Scholar]
- 17.Mizuno K., Boudko S.P., Engel J., Bachinger H.P. Kinetic hysteresis in collagen folding. Biophys J. 2010;98(12):3004–3014. doi: 10.1016/j.bpj.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baum J., Brodsky B. Folding of peptide models of collagen and misfolding in disease. Curr Opin Struct Biol. 1999;9(1):122–128. doi: 10.1016/s0959-440x(99)80016-5. [DOI] [PubMed] [Google Scholar]
- 19.Cejas M.A., Kinney W.A., Chen C., Vinter J.G., Almond H.R., Jr., Balss K.M. Thrombogenic collagen-mimetic peptides: self-assembly of triple helix-based fibrils driven by hydrophobic interactions. Proc Natl Acad Sci U S A. 2008;105(25):8513–8518. doi: 10.1073/pnas.0800291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y.S., Chen C.C., Horng J.C. Thermodynamic and kinetic consequences of substituting glycine at different positions in a pro-hyp-gly repeat collagen model peptide. Biopolymers (Pept Sci) 2011;96:60–68. doi: 10.1002/bip.21470. [DOI] [PubMed] [Google Scholar]
- 21.Hwang E.S., Thiagarajan G., Parmar A.S., Brodsky B. Interruptions in the collagen repeating tripeptide pattern can promote supramolecular association. Protein Sci. 2010;19(5):1053–1064. doi: 10.1002/pro.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kai T., Uchiyama S., Nishi Y., Kobayashi Y., Tomiyama T. Two states of the triple helix in the thermal transition of the collagen model peptide (pro-pro-gly)10. J Biomol Struct Dyn. 2004;22(1):51–58. doi: 10.1080/07391102.2004.10506980. [DOI] [PubMed] [Google Scholar]
- 23.Kar K., Ibrar S., Nanda V., Getz T.M., Kunapuli S.P., Brodsky B. Aromatic interactions promote self-association of collagen triple-helical peptides to higher-order structures. Biochemistry. 2009;48(33):7959–7968. doi: 10.1021/bi900496m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khew S.T., Tong Y.W. Characterization of triple-helical conformations and melting analyses of synthetic collagen-like peptides by reversed-phase HPLC. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;858(1–2):79–90. doi: 10.1016/j.jchromb.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 25.Li Y., Brodsky B., Baum J. NMR conformational and dynamic consequences of a gly to ser substitution in an osteogenesis imperfecta collagen model peptide. J Biol Chem. 2009;284(31):20660–20667. doi: 10.1074/jbc.M109.018077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Long C.G., Braswell E., Zhu D., Apigo J., Baum J., Brodsky B. Characterization of collagen-like peptides containing interruptions in the repeating gly-x-y sequence. Biochemistry. 1993;32(43):11688–11695. doi: 10.1021/bi00094a027. [DOI] [PubMed] [Google Scholar]
- 27.Miles C.A., Bailey A.J. Studies of the collagen-like peptide (pro-pro-gly)10 confirm that the shape and position of the type I collagen denaturation endotherm is governed by the rate of helix unfolding. J Mol Biol. 2004;337(4):917–931. doi: 10.1016/j.jmb.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 28.Persikov A.V., Ramshaw J.A., Brodsky B. Prediction of collagen stability from amino acid sequence. J Biol Chem. 2005;280(19):19343–19349. doi: 10.1074/jbc.M501657200. [DOI] [PubMed] [Google Scholar]
- 29.Slatter D.A., Miles C.A., Bailey A.J. Asymmetry in the triple helix of collagen-like heterotrimers confirms that external bonds stabilize collagen structure. J Mol Biol. 2003;329(1):175–183. doi: 10.1016/s0022-2836(03)00380-2. [DOI] [PubMed] [Google Scholar]
- 30.Carafoli F., Bihan D., Stathopoulos S., Konitsiotis A.D., Kvansakul M., Farndale R.W. Crystallographic insight into collagen recognition by discoidin domain receptor 2. Structure. 2009;17(12):1573–1581. doi: 10.1016/j.str.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sacca B., Renner C., Moroder L. The chain register in heterotrimeric collagen peptides affects triple helix stability and folding kinetics. J Mol Biol. 2002;324(2):309–318. doi: 10.1016/s0022-2836(02)01065-3. [DOI] [PubMed] [Google Scholar]
- 32.Henkel W., Vogl T., Echner H., Voelter W., Urbanke C., Schleuder D. Synthesis and folding of native collagen III model peptides. Biochemistry. 1999;38(41):13610–13622. doi: 10.1021/bi9905157. [DOI] [PubMed] [Google Scholar]
- 33.Ackers G.K. Molecular exclusion and restricted diffusion processes in molecular-sieve chromatography. Biochemistry. 1964;3:723–730. doi: 10.1021/bi00893a021. [DOI] [PubMed] [Google Scholar]
- 34.Ackers G.K. Molecular sieve methods of analysis. In: Neurath H., Hill R.L., editors. The proteins. 1975. pp. 2–94. [Google Scholar]
- 35.Bella J., Liu J., Kramer R., Brodsky B., Berman H.M. Conformational effects of gly-x-gly interruptions in the collagen triple helix. J Mol Biol. 2006;362(2):298–311. doi: 10.1016/j.jmb.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 36.Leikina E., Mertts M.V., Kuznetsova N., Leikin S. Type I collagen is thermally unstable at body temperature. Proc Natl Acad Sci U S A. 2002;99(3):1314–1318. doi: 10.1073/pnas.032307099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miles C.A., Ghelashvili M. Polymer-in-a-box mechanism for the thermal stabilization of collagen molecules in fibers. Biophys J. 1999;76(6):3243–3252. doi: 10.1016/S0006-3495(99)77476-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mizuno K., Peyton D.H., Hayashi T., Engel J., Bachinger H.P. Effect of the gly-3(S)-hydroxyprolyl-4(R)-hydroxyprolyl- tripeptide unit on the stability of collagen model peptides. FEBS J. 2008;275(23):5830–5840. doi: 10.1111/j.1742-4658.2008.06704.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.