

Abstract
We report the site-specific incorporation of a thiocyanate vibrational probe into the active site oxyanion hole of ketosteroid isomerase (KSI) to test the effect of hydrophobic steroid binding and solvent exclusion on the local electrostatic environment at this position. While binding of an uncharged ground state steroid analog shifts the observed –CN vibrational frequency by +0.4 cm−1 relative to unliganded KSI, binding of an intermediate steroid analog containing localized negative charge results in a +2.8 cm−1 shift. Based on a Stark tuning rate of 0.7 cm−1/(MV/cm), this shift indicates a fivefold larger change in the projection of the local electric field along the –CN bond in the presence of the charged ligand. Binding of a single ring phenolate with oxyanion charge localization equivalent to the intermediate steroid analog but lacking distal hydrocarbon rings results in an identical –CN peak shift. We conclude that solvent exclusion and replacement by hydrophobic steroid rings negligibly alter the electrostatic environment within the KSI oxyanion hole. Development of localized negative charge analogous to that of the dienolate intermediate during steroid isomerization dramatically increases the magnitude of the local electric field. This increase reflects field contributions from the localized negative charge itself as well as possible increased ordering of active site dipoles in response to charge localization.
The thousands of enzyme structures solved to date have consistently revealed that biological catalysis occurs within sequestered pockets containing complex interdigitations of polar and hydrophobic groups and from which water molecules are displaced upon substrate binding.1 This chemical complexity has sparked considerable controversy regarding the electrostatic nature of active sites and the role of substrate binding and solvent exclusion in shaping active site electrostatics.2–7 We report herein the site-specific incorporation of a thiocyanate vibrational probe8 into the active site of Pseudomonas putida ketosteroid isomerase (KSI) to directly and quantitatively test the effect of steroid binding and concomitant solvent exclusion on the local electrostatic environment.
KSI catalyzes a double-bond migration reaction in steroid substrates that involves formation of a dienolate intermediate within an active site oxyanion hole composed of Y16, protonated D103, and a preponderance of hydrophobic residues (Scheme 1A and Figure S1). Numerous physical changes occur upon steroid binding that might alter the local electrostatic environment within the KSI active site. In the free enzyme an ordered water molecule is positioned within hydrogen bonding distance of Y16 and D103.9 This and additional disordered water molecules present within the unliganded active site are displaced upon steroid binding (Scheme 1B) and substantially excluded by the dense constellation of hydrophobic residues that pack around the bound, hydrophobic steroid skeleton (Figure S1).9,10 Considering the ~250 Å3 excluded volume of a steroid, 8–10 water molecules within the active site are presumably displaced upon binding.11 Based on MD simulations, 2–4 water molecules may remain in the active site in the substrate- or product-bound ground states, but all waters appear to be excluded in the intermediate complex due to closer packing around the planar intermediate.9,12 Finally, ligand binding does not grossly alter the conformations of backbone and side chain groups observed in x-ray structures of free (1OPY) and bound (1OH0) KSI (0.5 Å RMSD). However, NMR and UV studies suggest that steroid binding restricts the motions of several active site groups, including Y16.10,13
Scheme 1.

(A) KSI Reaction Mechanism (B) Thiocyanate Probe Reports on Electrostatic Changes Upon Ligand Binding
To test the effect of these changes on the electrostatic character of the KSI active site, we incorporated a cyano (–CN) vibrational probe into the oxyanion hole. The nitrile stretching frequency is exquisitely sensitive to electric fields and this sensitivity, quantified by the vibrational Stark tuning rate, has been determined for thiocyanate probes in simple solvents and ribonuclease S as 0.7 cm−1/(MV/cm).8,14,15 We introduced the –CN probe into the oxyanion hole by engineering the M116C mutation into a cysteine-free variant of D40N KSI16 and converting the Cys-SH at position 116 to Cys-S-CN8 (referred to as KSI-CN). M116 is proximal to hydrogen bond donors Y16 and D103, ~3.7 Å from the oxygen of the bound intermediate analog equilenin (Figure S1), and surrounded by closely packed hydrophobic groups that sterically limit gross reorientation. Functional assays indicate that the M116C-CN modification decreases KSI catalysis (kcat/KM) and phenolate binding by less than 3-fold, as expected for minimal perturbation of the KSI active site (unpublished results).17
The FTIR spectrum of unliganded KSI-CN displays a narrow, symmetric transition (FWHM = 8 cm−1) centered at 2159.6 cm−1 (Figure 1), consistent with a single –CN conformation. This peak is 6 cm−1 narrower and 2.9 cm−1 lower in energy than the 2162.5 cm−1 peak (FWHM = 14 cm−1) observed for unfolded KSI-CN in 5.3 M urea (Figure S2), where the –CN probe is exposed to bulk water. These differences are similar to other examples of nitrile probes in proteins in which the folded protein interior sequesters the probe from the relatively disordered dipoles of liquid water, resulting in a 3–4 cm−1 narrowing of the inhomogeneous linewidth, and places the probe in an idiosyncratic local environment, giving peak shifts of 7–9 cm−1 to lower energy.8,18
Figure 1.
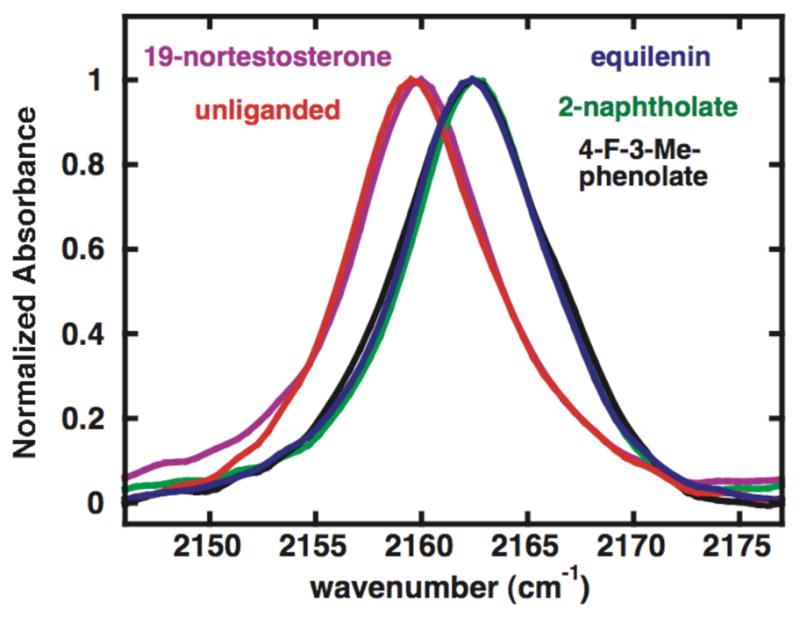
IR absorption spectra of free and ligand-bound KSI-CN, normalized to a maximum absorbance of 1.
To test the effect of ground state steroid binding and water displacement on the local electric field within the oxyanion hole, we recorded the spectrum of KSI-CN bound to the product analog 19-nortestosterone (19-NT) (Scheme 1B). The carbonyl oxygen of 19-NT and the oxygen of the ordered water molecule it replaces have similar calculated negative electrostatic potentials,19 leading to the expectation that this exchange alone might contribute only a small perturbation to oxyanion hole electrostatics superimposed on any effect from exclusion of the remainder of the water molecules. Observation of a large peak shift upon 19-NT binding, therefore, would strongly suggest a substantial electric field change from replacing disordered waters within the active site with the hydrophobic skeleton of 19-NT. The peak position of 2160.0 cm−1 observed for KSI-CN•19-NT is only 0.4 cm−1 higher in energy than free KSI-CN (Figure 1). Based on a Stark tuning rate of 0.7 cm−1/(MV/cm), this peak shift indicates that the net electric field experienced along the –CN bond axis has increased by 0.6 MV/cm, a minor change compared to reports of electric field changes in proteins of 10 MV/cm or higher upon mutation.20–23 This minor change suggested, most simply, a minimal contribution to local field from replacing active site disordered waters with hydrophobic steroid rings, a conclusion we directly tested as described below.
During steroid isomerization negative charge localizes on the substrate oxygen of the intermediate (Scheme 1A). This charge localization is accompanied by lengthening of the substrate C-O bond from 1.2 Å in the carbonyl ground state to 1.3 Å in the dienolate intermediate, presumably extending the oxyanion deeper into the oxyanion hole. To test whether negative charge localization and any accompanying active site changes alter the local electric field within the oxyanion hole, we acquired the spectrum of KSI-CN bound to the intermediate analog equilenin (Scheme 1B). We observed a single peak at 2162.4 cm−1 shifted 2.4 cm−1 higher in energy than that observed for 19-NT (Figure 1). This peak shift corresponds to a 3 MV/cm larger projection of the local electric field along the –CN bond, a fivefold larger change than the 0.6 MV/cm change observed upon 19-NT binding. The sign and magnitude of this difference are consistent with the close approach (~4 Å) of the localized oxyanion of equilenin to the probe and possible increased ordering of active site dipoles in response to increased charge localization in the oxyanion hole.24
The minimal peak shift upon 19-NT binding together with the much larger shift upon equilenin binding strongly suggested a substantial role for oxyanion charge localization in shaping local electrostatics and a negligible role for replacement of active site waters with hydrophobic steroid rings. To dissect these possible contributions, we recorded spectra of KSI-CN bound to 2-naphtholate and 4-F-3-Me-phenolate (Scheme 1B), intermediate analogs with planar A ring geometries and negative charges like equilenin but lacking distal steroid rings. Substituted phenolates bind in the KSI active site in an orientation nearly superimposable with that of equilenin,24 indicating that distal rings are not required for proper positioning. Furthermore, 2-naphtholate and 4-F-3-Me-phenolate are expected to have oxyanion charge localizations equivalent to equilenin, based on their nearly identical C-O bond lengths and aqueous pKa’s (Table 1).25 The –CN peak positions observed for the KSI-CN•2-naphtholate and KSI-CN•4-F-3-Me-phenolate are identical within error to that observed for bound equilenin (Figure 1).26 This result indicates that the projection of the local electric field within the oxyanion hole along the axis of the –CN probe is not significantly altered upon replacement of active site waters by the distal steroid rings. Furthermore, the identical change in local electric field in response to binding a full steroid and single ring phenolate of equivalent geometry and oxyanion charge localization implies that electrostatic interaction energies within the oxyanion hole are the same for these ligands.
Table 1.
KSI-CN Ligands, Their Aqueous pKa’s, C-O Bond Lengths, and the Observed –CN Vibrational Frequencies
| KSI ligand | pKaa | C-O length (Å) | –CN stretch (cm−1) b | Δ (cm−1)c |
|---|---|---|---|---|
| unliganded | – | – | 2159.6 | – |
| 19-nortestosterone | – | 1.22d | 2160.0 | 0.4 |
| equilenin | 9.7 | 1.31e | 2162.4 | 2.8 |
| 2-naphtholate | 9.6 | 1.32f | 2162.5 | 2.9 |
| 4-F-3-Me-phenolate | 9.8 | 1.33g | 2162.3 | 2.7 |
From Ref. 24.
Average of 3 or more replicates, standard deviation ± 0.1 cm−1.
Difference from unliganded KSI-CN.
Cambridge Structural Database (CSD): NOTEST01.
1.1 Å KSI•equilenin structure (PDB: 1OH0).
CSD: GAWHAO.
CSD: PIPGEA.
We conclude that solvent exclusion and replacement by the remote hydrophobic steroid rings negligibly alter the electrostatic environment within the KSI oxyanion hole. Development of localized negative charge analogous to that present in the dienolate intermediate, however, dramatically increases the magnitude of the local electric field. This increase reflects field contributions from the localized negative charge itself as well as possible increased ordering of active site dipoles in response to charge localization. These results predict that the change in local electric field will depend strongly on the degree of oxyanion charge localization–a prediction we are currently testing. Further quantitative tests of active site electrostatics in this and other enzymes, including selective introduction of –CN probes at specified positions within the KSI active site, will be facilitated by the general –CN probe incorporation chemistry8 employed herein.
Supplementary Material
Acknowledgments
This work was funded by grants to SGB (NIH GM27738) and DH (NIH GM64798 and NSF MCB-0641393). PAS was supported in part by an HHMI Predoctoral Fellowship.
Footnotes
Supporting Information Available: Experimental methods and Figures S1 and S2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fersht AR. Structure and Mechanism in Protein Science. 2. W. H. Freeman and Company; New York: 1999. [Google Scholar]
- 2.Quiocho FA, Sack JS, Vyas NK. Nature. 1987;329:561. doi: 10.1038/329561a0. [DOI] [PubMed] [Google Scholar]
- 3.Lockhart DJ, Kim PS. Science. 1993;260:198. doi: 10.1126/science.8469972. [DOI] [PubMed] [Google Scholar]
- 4.Warshel A, Aqvist J, Creighton S. Proc Natl Acad Sci U S A. 1989;86:5820. doi: 10.1073/pnas.86.15.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perutz M. Proc R Soc London B Biol Sci. 1967;167:448. [Google Scholar]
- 6.Dewar MJ, Storch DM. Proc Natl Acad Sci U S A. 1985;82:2225. doi: 10.1073/pnas.82.8.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frey PA, Whitt SA, Tobin JB. Science. 1994;264:1927. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 8.Fafarman AT, Webb LJ, Chuang JI, Boxer SG. J Am Chem Soc. 2006;128:13356. doi: 10.1021/ja0650403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SW, Cha SS, Cho HS, Kim JS, Ha NC, Cho MJ, Joo S, Kim KK, Choi KY, Oh BH. Biochemistry. 1997;36:14030. doi: 10.1021/bi971546+. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Q, Li YK, Mildvan AS, Talalay P. Biochemistry. 1995;34:6562. doi: 10.1021/bi00019a038. [DOI] [PubMed] [Google Scholar]
- 11.Molecular volumes were calculated using http://www.molinspiration.com.
- 12.Mazumder D, Kahn K, Bruice TC. J Am Chem Soc. 2003;125:7553. doi: 10.1021/ja030138s. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Q, Abeygunawardana C, Mildvan AS. Biochemistry. 1996;35:1525. doi: 10.1021/bi9525381. [DOI] [PubMed] [Google Scholar]
- 14.Suydam IT, Boxer SG. Biochemistry. 2003;42:12050. doi: 10.1021/bi0352926. [DOI] [PubMed] [Google Scholar]
- 15.Andrews SS, Boxer SG. J Phys Chem A. 2000;104:11853. [Google Scholar]
- 16.The D40N mutation mimics protonated D40 present in the KSI-intermediate complex and yields tighter ligand binding. KSI also contained C69S/C81S/C97S, surface mutations that ensure unique labelling of M116C but do not affect activity. See: Kim SW, Joo S, Choi G, Cho HS, Oh BH, Choi KYJ. Bacteriol. 1997;179:7742. doi: 10.1128/jb.179.24.7742-7747.1997.
- 17.Activity was measured with KSI-CN containing the wildtype D at position 40.
- 18.Getahun Z, Huang CY, Wang T, De Leon B, DeGrado WF, Gai F. J Am Chem Soc. 2003;125:405. doi: 10.1021/ja0285262. [DOI] [PubMed] [Google Scholar]
- 19.Luque FJ, Illas F, Orozco M. Journal of Computational Chemistry. 1990;11:416–430. [Google Scholar]
- 20.Pearson JG, Oldfield E, Lee FS, Warshel A. J Am Chem Soc. 1993;115:6851. [Google Scholar]
- 21.Park ES, Thomas MR, Boxer SG. Journal of the American Chemical Society. 2000;122:12297–12303. [Google Scholar]
- 22.Park ES, Andrews SS, Hu RB, Boxer SG. J Phys Chem B. 1999;103 [Google Scholar]
- 23.Suydam IT, Snow CD, Pande VS, Boxer SG. Science. 2006;313:200. doi: 10.1126/science.1127159. [DOI] [PubMed] [Google Scholar]
- 24.Kraut DA, Sigala PA, Pybus B, Liu CW, Ringe D, Petsko GA, Herschlag D. PLoS Biol. 2006;4:e99. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gross KC, Waybold PG, Hadad CM. Int J Quant Chem. 2002;90:445. [Google Scholar]
- 26.Spectra of KSI-CN containing the wildtype Asp residue at position 40 (D40) with bound equilenin or 4-F-3-Me-phenolate also showed identical -CN peak positions (data not shown), ruling out a difference resulting from the D40N mutation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.