

Abstract
DNA double-strand-break repair (DSBR) is, in many organisms, accomplished by homologous recombination. In Escherichia coli DSBR was thought to result from breakage and reunion of parental DNA molecules, assisted by known endonucleases, the Holliday junction resolvases. Under special circumstances, for example, SOS induction, recombination forks were proposed to initiate replication. We provide physical evidence that this is a major alternative mechanism in which replication copies information from one chromosome to another generating recombinant chromosomes in normal cells in vivo. This alternative mechanism can occur independently of known Holliday junction cleaving proteins, requires DNA polymerase III, and produces recombined DNA molecules that carry newly replicated DNA. The replicational mechanism underlies about half the recombination of linear DNA in E. coli; the other half occurs by breakage and reunion, which we show requires resolvases, and is replication-independent. The data also indicate that accumulation of recombination intermediates promotes replication dramatically.
Keywords: DNA replication, DNA repair, double-strand break-repair, recombination, Escherichia coli, RuvABC, RecG, Holliday junction
DNA double-strand breaks (DSBs) are common lesions that occur in all cells. They result from DNA damage from processing of arrested replication forks (Seigneur et al. 1998) and are hypothesized to occur as normal intermediates in DNA replication, (e.g., Skalka 1974; Kuzminov 1995). Because DSB accumulation is toxic to cells, multiple mechanisms have evolved for their repair. Homologous recombination may be the exclusive mechanism for DSB repair (DSBR) in Escherichia coli, is the dominant mechanism in some eukaryotes including baker's yeast, and is one alternative in mammals including humans (e.g., Haber 1999). Simple ligation of DNA ends (nonhomologous end joining) is a major alternative repair route in mammals that can result in loss of genetic material and gross chromosome changes (Tsukamoto and Ikeda 1998; Haber 1999). DSBR via recombination is conserved in evolution, as are its important proteins, and it is required for the normal functions of cells (for review, see Kanaar and Hoeijmakers 1998; Haber 1999). Aberrant DSBR could underlie the excessive recombination linked to phenotypes of genetic instability, premature aging, and cancer (e.g., Ellis et al. 1995; Yu et al. 1996).
In addition to its roles in the maintenance of genomic stability, homologous recombination creates new cellular and organismal combination of alleles and ensures proper segregation of chromosomes during meiosis. In E. coli, the RecBCD recombination system both provides nearly all DSBR (Kowalczykowski et al. 1994; Myers and Stahl 1994) and catalyzes recombination of the linear DNA intermediates in conjugation and phage-mediated transduction, two important avenues of genetic exchange between bacterial cells (Clark and Sandler 1994; Lloyd and Low 1996; Rosenberg and Motamedi 1999). DSBR is also the major sexual recombination route in yeast meiosis (e.g., Haber 1998; Smith and Nicolas 1998).
Possible styles of recombination can be defined based on the proposed involvement of DNA replication (Meselson and Weigle 1961; Fig. 1): Break–join recombination models use no replication. Parental DNAs are cut and rejoined, producing recombinant molecules made entirely of parental DNA. Break–copy models use a fragment from one parental molecule to prime replication from a homolog, thereby producing recombinant molecules with DNA material from one parent joined to new DNA carrying information from the other. A paradox for the RecBCD system is that the only direct physical evidence bearing on whether recombined DNA is replicated has demonstrated clearly the existence of break–join recombinants (see below). However there is mounting suggestive, but indirect, evidence that would be unified by the existence of a break–copy pathway.
Figure 1.
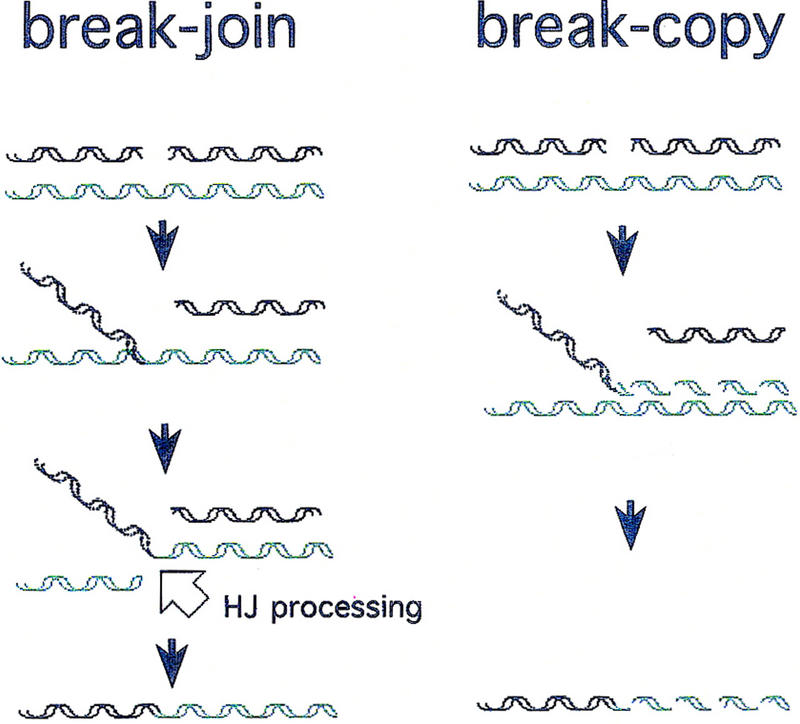
Two early general models for homologous recombination (adapted from Meselson and Weigle 1961). Dashed helices represent newly synthesized DNA. Solid helices represent “old” parental DNA. HJ processing (open arrow) indicates action of HJ resolution proteins, including an endonucleolytic cleavage (such as RuvC performs) to break the invaded (green) molecule and allow its ligation to the black fragment. No strand polarities are shown because specific polarities are not implied by either model (see Discussion).
The direct evidence for break–join recombination was obtained using techniques in which phage λ DNA is used as a substrate for the bacterial recombination system (Meselson and Weigle 1961; Meselson 1964; Stahl and Stahl 1971; McMilin and Russo 1972). [λ lacking its own recombination genes recombines exclusively via a RecBCD-dependent mechanism (Lam et al. 1974; Henderson and Weil 1975). λ is the molecule with which the RecBCD system's recombination hot-spot sequence Chi was discovered.] Using density-labeled λ (13C and 15N) that were allowed to recombine in unlabeled E. coli, these groups separated unreplicated from replicated λ progeny in cesium formate equilibrium density gradients. They determined that recombinants were present among fully unreplicated molecules and could even occur under a full block to replication, thereby providing direct physical evidence for RecBCD-mediated break–join recombination in E. coli (McMilin and Russo 1972; McMilin et al. 1974).
Although break–copy mechanisms were not excluded (see Siegel 1974), break–join was considered to be the major route for RecBCD-mediated recombination (e.g., Thaler and Stahl 1988; West 1992; Kowalczykowski et al. 1994). The apparent dominance of break–join was bolstered by the discoveries of endonucleases specific for the strand-exchange junctions [such as Holliday junctions (HJs)], which connect recombining molecules (Kemper et al. 1984; Connolly et al. 1991; Sharples et al. 1998), and by the demonstration of a requirement for such enzymes for conjugational and transductional recombination in E. coli (Lloyd 1991). Such endonucleases are expected to be required for completion of break–join events, for example, for breaking the molecule indicated by the open arrow in Figure 1.
More recently, good arguments for why replication should be a possible consequence of RecBCD-mediated recombination and DSBR in E. coli have been advanced, (e.g., Smith 1991). However, much of the evidence in apparent support of break–copy models has been obtained under special circumstances, and all of it to date has been indirect (for review, see Discussion) in that replication and recombination were not demonstrated to have occurred in the same DNA molecules.
Here, we present physical evidence that replicational recombination is a major route to DSBR in E. coli, in addition to the established break–join mechanism. We used phage λ DNA (lacking the λ recombination functions) as the substrate for RecBCD-mediated recombination because well-established, sensitive methods allow DNA labeling and physical detection of new DNA. λ has the advantages that all RecBCD-mediated DSBR uses a known, defined break-site—the packaging origin, cos, which is cleaved during DNA packaging (Kobayashi et al. 1982, 1983; Thaler and Stahl 1988)—and that recombinant DNAs are packaged into phage particles selectively. Using physical analysis of the recombined DNAs, we find that about half of all RecBCD-mediated recombination of λ DNA occurs by a break–join mechanism. We show that the HJ processing proteins of E. coli are required for that mechanism, whereas the major replicative polymerase, DNA polymerase III (Pol III), is not. We report the discovery of a second RecBCD-mediated recombination mechanism that is independent of the HJ processing proteins and requires DNA Pol III. This recombination occurs only when DNA replication is permitted and produces recombinant molecules that all contain some newly synthesized DNA, demonstrating a direct physical association of recombination with replication in the same DNA molecules. The extent of the new DNA synthesis is compatible with break–copy models (alternatives discussed below). This replicational recombination mechanism accounts for about half of all RecBCD-mediated recombination of λ DNA.
The results demonstrate a replicational recombination route in the RecBCD system of DSBR recombination in E. coli, showing the existence of the replicated recombinant molecules directly. We also show that the two mechanisms, replicational and break–join recombination, can be separated.
Results
Strategy for blocking break–join recombination
We sought to determine whether a replicational mechanism of recombination occurs in the RecBCD system, in addition to the established break–join process. Because any putative replicational recombination might be easier to detect in the absence of break–join events, we attempted to block break–join recombination. We hypothesized that break–join recombination might have a unique requirement for the proteins that process branched molecules resulting from strand exchange (including HJs) in E. coli. In Figure 1, note that break–join recombination actually requires two DSBs: one to initiate attachment of the broken molecule to a homolog and another (open arrow) to break the homolog (green molecule) so that it can ligate with the DNA fragment that invaded it. This second break occurs in a strand-exchange junction (Fig. 1). A Holliday junction cleaving protein, such as the E. coli RuvC endonuclease (Connolly et al. 1991), might be expected to make this second break in vivo. Because the E. coli RecBCD system appears to use either of two systems, RuvABC or RecG (Lloyd 1991), for processing branched intermediates, we attempted to detect RecBCD-mediated recombination of phage λ DNA in the absence of both systems, in ruv recG double mutant cells. In this paper, all of the possible branched intermediates will be referred to as HJ for Holliday junctions and other branched intermediates.
λ red gam mutants form plaques on E. coli ruv recG strains
One measure of λ recombination in the RecBCD system is the ability of λ recombination-defective strains (λ red gam) to form plaques on RecBCD+ E. coli (for review, see Smith and Stahl 1985). In RecBCD+ E. coli, rolling circle replication does not occur detectably because RecBCD destroys rolling circles. The monomeric λ chromosomes produced by bidirectional (υ) replication must recombine to form packageable substrates [dimers and multimers are packageable, whereas monomers are not (Feiss and Becker 1983; Rosenberg et al. 1985), but see Thomason et al. (1997)]. Because only the host RecBCD pathway is available for recombination, λ red gam cannot form plaques on cells that are recombination-defective such as recA null mutant strains. The data in Table 1 reveal that unlike recA strains, ruvA recG and ruvC recG double mutant cells allow plaque formation of three different λ red gam strains. This is observed for ruv recG combinations constructed in two different E. coli genetic backgrounds (Table 1; Materials and Methods). Plaques were about the same size as those on isogenic rec+ parents and did not form on recA control strains (not shown). These data suggest that, unlike recA strains, ruv recG double mutants allow RecBCD-mediated recombination of phage λ DNA. To be sure that this plaque formation reflected recombination-proficiency, we measured the frequencies of RecBCD-mediated λ recombination in the absence of Ruv and RecG functions using a quantitative assay.
Table 1.
Efficiency of plating of λ red gam on ruv recG-deficient E. coli strains
|
E. coli strains
|
eop ± sdc
|
||
|---|---|---|---|
| λ Chi+d
|
λ Chioe
|
λ nin−f
|
|
| rec+a | 1.0 | 1.0 | 1.0 |
| ruvA recGa | 1.1 ± 0.3 | 0.98 ± 0.2 | 0.96 ± 0.1 |
| ruvC recGa | 1.0 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 |
| rec+b | 1.0 | 1.0 | 1.0 |
| ruvA recGb | 0.91 ± 0.3 | 0.88 ± 0.2 | 0.91 ± 0.1 |
| ruvC recGb | 0.95 ± 0.1 | 0.87 ± 0.5 | 0.86 ± 0.1 |
Isogenic derivatives of strain FC40 published previously (Harris et al. 1996) (Table 2).
Isogenic derivatives of strain SMR632 (Materials and methods, Table 2).
Efficiency of plating (eop) for each strain was determined by dividing the λ titer on the ruv recG strain by its titer on the rec+ strain. This number was then corrected for the viability of the cultures of cells on which the plaques were assayed by dividing by the relative viability of the strain. The relative viability of each strain was determined as follows: (viable cell count of ruv recG/total cell count of ruv recG) + (viable cell count of rec+ total cell count of rec+). Each determination is a mean (± standard deviation) of 3 independent experiments in which hundreds of plaques were counted. The absolute viability of the rec+ strains were a0.9 ± 0.04, and b0.7 ± 0.1 cfu/cell counted (mean ± SE for the 3 experiments reported) and the relative viabilities were a0.4 ± 0.1, and b0.4 ± 0.1 for their ruvA recG derivatives, and a0.4 ± 0.1, and b0.4 ± 0.7 for their ruvC recG derivatives. These values are as reported (Lloyd 1991).
λ Δb1453 cl857 Chi+ C (The Δb1453 deletion removes int, red, and gam.)
λ Δb1453 cl857 cll68
λ bio1 Δnin5 (the bio1 substitution removes int, red, and gam. Δnin5 removes λ analogs of E. coli recombination genes, discussed in the text.)
Assays for the frequency of RecBCD-mediated recombination
A standard assay was used to measure the frequency of RecBCD-mediated recombination of λ DNA (Fig. 2). As with the experiments reported above (and in all experiments in this paper) the λ used are red gam so that recombination is exclusively via the host RecBCD system. Also, as described above, this means that all progeny must contain recombinant chromosomes (whether these are detectably recombinant, or occurred between DNAs of the same genotype). To measure the frequency of homologous recombination in the face of this requirement for recombination, one can provide an alternative route to dimerization (and packaging) so that any homologous recombination events are gratuitous and quantifiable. In the assay used here [(Razavy et al. 1996) modified from Thaler et al. (1989)] dimerization is achieved via the λ Int system of site-specific recombination, and gratuitous homologous crossovers (splices) are measured only from among the site-specific recombinants. This assay is sensitive and the results correlate well with standard P1 transductional recombination assays (Razavy et al. 1996; Razavy 1997).
Figure 2.

Design of λ crosses used to measure the frequency of recombination in rec+ and ruv recG cells. (A) The strategy for this assay is described in the text (and Razavy et al. 1996). This general diagram shows all of the relevant genetic markers used. The open box (left) represents either of two different deletions (Δb527 or Δb2; Materials and Methods) both starting from the core att site and removing DNA to its left. The solid box represents a deletion/substitution (bio1) starting from the core att site and removing DNA to its right, resulting in a net loss of ∼2 kb of DNA. The arrow indicates the direction of the Chi sequence; + indicates the wild-type copy of the S gene; the other parent carries Sam7. Crosses performed (Fig. 3) varied the presence/absence of the Chi site, Chi+C, and of the nin deletion, Δnin5. All phage are red gam, the top phages by carrying red3 gam210 mutant alleles and the bottom phages by virtue of the bio1 substitution. (B) A representative cesium formate equilibrium density gradient of a cross progeny showing the denser peak formed by site-specific recombination, which contains neither Δ nor bio1 net deletions (fractions 15–20). The next lighter peak (fractions 21–25) includes the top parental phage (A) plus its S+ recombinant derivatives. (□,●) “Total phage” (Sam7 and S+) and S+ recombinants, respectively. These plaques were assayed on SuIII+ recA (for total phage) and SuII+ recA (for S+ recombinants) cells, which do not allow plaque formation of phage with the bio substitution but do allow the gam (amber)210 carriers to form plaques (Materials and Methods). Thus, we do not see the double-deletion (Δbio1) site-specific recombinant peak. To calculate the frequency of λS+ homologous recombinants among site-specific recombinants, the titer of λS+ in each fraction (15–20) is divided by the total titer in that fraction, and the mean ± s.d. for all the fractions in the peak is expressed as a percentage (Fig. 3). The data shown are from a cross in rec+ SMR632 cells using Chio nin+ phage (Materials and Methods).
In Figure 2A, note that site-specific Int-dependent recombination occurs between two half att sites of the recombining λ molecules. These sites have too little sequence identity for homologous recombination. One parent is deleted from the att site leftward (λ), whereas the other carries a deletion–substitution (bio1) from att rightward. These net deletions decrease the size of the λ chromosome but do not alter the size of the phage capsid, so that phages carrying the Int-mediated recombinant with no net deletions are denser than either parent (more DNA in the same size capsid). The denser site-specific recombinant can be separated from both single deletion (parental) phages in a cesium formate equilibrium density gradient (Razavy et al. 1996) (Fig. 2B), and homologous exchanges quantified from among them.
RecBCD-mediated λ recombination is equally efficient in rec+ and ruv recG cells
The amount of λ recombination in ruvC recG cells was quantified using the assay described above using a set of phages as illustrated in Figure 2. In Figure 3 (left), results from three independent experiments performed in rec+ and ruvC recG strains are summarized in the nin+ panel (the significance of nin is discussed below). We observed no significant difference in the percentage of λ recombinants between crosses performed in rec+ and isogenic ruvC recG cells.
Figure 3.

RecBCD-pathway recombination of λ in the absence of RuvC and RecG. The three graphs summarize the results of three different experimental designs, measuring the efficiency of λ recombination in rec+ and ruvC recG cells. For each design, we used a different set of phages: nin+ (left), Δnin (middle), and Δnin Chi+/o (right). Each bar represents the mean percentage of homologous recombination among site-specific Int-mediated recombinants (±s.d.; calculated as described in Fig. 2). Three independent experiments were performed for nin+ and Δnin crosses. Two experiments were performed for the Δnin Chi+/o cross. The deletion Δnin shortens the DNA segment whose recombination is assayed (see Fig. 2) and therefore necessarily decreases the percent recombination relative to nin+ crosses. Thus, the important comparison for both nin+ and Δnin crosses is between presence or absence of RuvC RecG in each.
The λnin region encodes a function(s) responsible for approximately half the recombination in ruvC recG cells
A possible reason for the high efficiency of λ recombination in ruv recG cells could be that a λ-encoded HJ processing protein substitutes for the E. coli Ruv or RecG proteins. The nonessential λ nin region encodes analogs of E. coli recombination proteins including a demonstrated resolvase, Rap (Sharples et al. 1998). We performed similar λ crosses with phages deleted for this region. The results of three independent sets of experiments are displayed in Figure 3 (Δnin panel). We found that when the nin region is deleted, λ recombination is decreased by approximately half in ruvC recG cells compared with the rec+ controls. This supports the hypothesis that a λ-encoded function can resolve recombination intermediates in vivo. However, it does not address how the remaining half of λ recombination works in ruv recG cells. (This remaining half is also independent of E. coli rusA, recE, and recT genes, data not shown). To explore the mechanism of recombination in the absence of these known HJ processing proteins, and more specifically, to test whether it is replicational, all of the remaining experiments presented were conducted using λ phage carrying the deletion Δnin5.
Chi stimulates recombination normally in the absence of nin, RuvC, and RecG: the RecBCD system performs apparent resolvase-independent recombination
The Chi site (5′-GCTGGTGG) promotes RecBCD-mediated recombination and DSBR specifically (Kowalczykowski et al. 1994; Myers and Stahl 1994; Eggleston and West 1996). It is the DNA recognition sequence of the RecBCD enzyme and promotes RecBCD-mediated recombination in its own vicinity, acting as a recombination hot spot. To test whether the apparently resolvase-independent recombination of λ in ruvC recG cells is normal RecBCD-mediated recombination, we tested whether Chi stimulates recombination normally in the absence of RuvC and RecG. The frequency of recombination was quantified from λ crosses performed in parallel with Chi+ and Chio phages in rec+ and ruvC recG cells. The data in Figure 3 (Chi+/o Δnin panel) show that Chi promotes recombination as well in the absence of RuvC and RecG as in their presence. Chi activity (recombination frequency in the Chi+ cross/recombination frequency in the Chio cross) was 3.3- and 3.3-fold in rec+ (experiments 1 and 2) and 3.8- and 3.9-fold in ruv recG cells (experiments 1 and 2, respectively). [These are typical Chi values for recombination in the large DNA interval measured (Razavy et al. 1996)]. We conclude that Chi stimulates RecBCD-mediated recombination normally in the absence of the known HJ processing proteins. This RecBCD-mediated recombination is replicational, as shown below.
RecBCD-mediated recombination in ruvC recG cells is replication dependent and requires DNA Pol III
We hypothesized that replication may help to resolve recombination intermediates, perhaps by making endonucleolytic cleavage unnecessary, as illustrated for break–copy recombination in Figure 1 (see also Morgan and Severini 1990). If this were the case, the recombination in the absence of known HJ processing proteins would be replication dependent. We therefore assayed λ recombination in the absence of the known HJ processing proteins (RuvC, RecG, and nin-encoded Rap) and DNA replication. DNA replication was blocked using a temperature-sensitive allele of dnaE encoding the core enzyme of DNA Pol III, the major replicative polymerase of E. coli (dnaEts486; see Materials and Methods), and shifting the cells to restrictive temperature for the λ infections. Because the Int site-specific recombination system (Figs. 2 and 3) is temperature sensitive, and therefore inappropriate, we used a different assay for recombination proficiency in these replication-blocked experiments (modified from Stahl et al. 1972; see Materials and Methods). As discussed, λ progeny formation requires recombination. Because λ DNA multimers are required for packaging in RecBCD+ cells, the only route to multimerization, and therefore progeny formation, of these Int− phage is via homologous recombination of monomers. Therefore, λ infections yield phage progeny only if cells are recombination proficient. Thus, if replication is required for recombination when the resolvases are absent, no progeny should be detected in the absence of replication in ruv recG cells.
Int− λ phages density labeled with 13C and 15N were infected into unlabeled E. coli cells that carry the dnaEts486 allele. A complete replication block was achieved by performing the experiments at high temperature (43.5°C, Fig. 4; Materials and Methods). Any new DNA synthesis would incorporate light nucleotides. This can be detected in a cesium formate density gradient of the phage progeny (Fig. 4).
Figure 4.

λ progeny formation in the absence of DNA replication requires RuvC/RecG. λ progeny formation was used to assay recombination. Replication was blocked by infecting cells that carry a temperature sensitive allele of the dnaE gene with density-labeled λ (λSR27) at 43.5°C, at which temperature we obtain a complete replication block. These graphs show the titers of λ in the fractions of a density gradient obtained following each infection. The densest fractions are to the left on each graph. The first peak in all gradients con-tains unadsorbed λ. These phage carry heavy protein coats and HH DNA. They did not enter the light cells and, therefore, are not part of the λ progeny. These serve as a density reference marker. (A) Density gradient of infection in rec+ cells. Two peaks are apparent. The second peak contains λ progeny that have entered the cell, recombined, and packaged. These carry light protein coats and HH DNA. No other peaks are detected because of the replication block. (B) ruvC recG cells. Few or no λ progeny are detected. (C) recA cells. Few or no λ progeny are detected relative to the unadsorbed phage peak.
In Figure 4A, note the two peaks of phage that emerge from infection of rec+ cells. The denser peak represents phage that possess heavy protein coats in addition to their fully heavy (HH) DNA. These are unadsorbed phage that did not enter the light E. coli and are not part of the progeny. These serve as a density reference. The less-dense peak represents phage with light capsids and unreplicated (HH) DNA. These are phage progeny resulting from break–join recombination events (this point is confirmed below and in Fig. 6, see below). Because these phage have no Int (site-specific recombination) system operative (Materials and Methods) they are inferred to have resulted from RecBCD-mediated break–join recombination. This is confirmed in a parallel infection of recA recombination-defective cells (Fig. 4C), in which few or no λ progeny are produced (because recombination is required for packaging). The absence of lighter peaks confirms that the replication block was complete.
Figure 6.

Extent of DNA replication in central and right-end λ recombinants in crosses with some replication allowed in rec+ and ruv recG cells. These crosses were conducted under partial replication block (Materials and Methods) to allow visualization of any break–copy recombinants. [If full replication block is used, no HH peak is visible for ruv recG (Fig. 4B).] (A) The relevant genotypes of phages used in this experiment. These phage (Sawitzke and Stahl 1997, Materials and Methods) carry the nin5 deletion and are marked to allow selection of J+ S+ recombinants from which central (J+ cI S+, clear, ●) and right end (J+ cI+ S+, turbid, ○) recombinants are enumerated. (B,C) Density-labeled phages were allowed to recombine under partial replication block and the progenies centrifuged to equilibrium in cesium formate density gradients, which were fractionated. Note that the progenies band into unreplicated, HH, and replicated, HL and LL peaks. Total λ (□) and J+ S+ recombinants were assayed (Materials and Methods), and central (●) and right end (○) recombinants were counted. The first peak (leftward) in these experiments represents unadsorbed phage (heavy coats and HH DNA), which are not part of the λ progeny. (B) Density gradient of the λ cross in rec+ cells. (C) Density gradient of the λ cross in ruvC recG cells.
Importantly, we recovered few or no phage progeny from ruvC recG cells when replication was fully blocked (Fig. 4B). These data indicate that recombination in ruvC recG cells requires DNA replication. Because DNA replication was blocked by use of dnaEts, a mutation of the structural gene encoding Pol III, the data also identify DNA Pol III as the polymerase required for this replication. Thus, the data imply that recombination in the absence of RuvC and RecG is replicational. We hypothesize that unresolved recombination intermediates in the ruv recG cells initiate replication forks, as in break–copy models (Figs. 1 and 5) and that DNA replication to the end of the chromosome can produce recombinant molecules.
Figure 5.

Predictions of break–copy and break–join recombination models. The thick lines represent parental DNA (black and gray). The thin broken lines represent newly synthesized DNA. Specific strand polarities are not indicated because no specificities are implied by either model depicted (see Discussion). (A) The phage λ DNA molecule is linearized during DNA packaging by the endonuclease terminase (○), which remains bound to the λ left end after DNA cleavage (Kobayashi et al. 1982, 1983). [Hexagon represents the phage prohead attached to terminase during packaging and concurrent DSBR recombination (Kobayashi et al. 1984).] Only the right end is available for DSBR (Kobayashi et al. 1982, 1983), which begins with degradation leftward by RecBCD exonuclease (for review, see Kowalczykowski et al. 1994; Myers and Stahl 1994). [Note that Chi sites (not shown) are recombination hot spots in this pathway because when RecBCD reaches Chi, Chi decreases RecBCD nuclease activity allowing the DNA there to recombine (for review, see Myers and Stahl 1994; Rosenberg and Motamedi 1999)]. In a break–copy process (B,C), the degraded right end initiates a replication fork. Semiconservative replication of density-labeled DNA to the end of the chromosome followed by the conservative segregation of the new strands (shown) would produce recombinant molecules with the following densities: (C) End recombinants inherit mostly parental DNA and would be expected to band in or near the HH peak in a density transfer experiment (see Fig. 6B,C). (B) Central recombinants would contain roughly half parental and half newly synthesized DNA and would band in the HL peak in a density transfer experiment (see Fig. 6B,C). (D) In break–join, recombination intermediates are resolved by the HJ processing systems. The recombinant molecules inherit only atoms from parental DNA; no new synthesis is required to complete the recombination reaction. These central recombinants would fall into the first few fractions of the HH peak in a density transfer experiment (see Fig. 6B).
Physical evidence for a break–copy mechanism
Figure 5 outlines some specific predictions of break–copy recombination models. If recombination occurs between density-labeled DNAs (thick solid lines, Fig. 5B–D) in unlabeled cells, then break–copy recombinants that occur in the center of the chromosome should contain both heavy, unreplicated parental DNA (solid lines, Fig. 5B) and newly replicated, light DNA (dashed lines, Fig. 5B). End recombinants formed by break–copy could contain almost all heavy DNA with just a little new, light DNA (Fig. 5C). This contrasts with the prediction for break–join recombination, in which even central recombinants should be fully unreplicated, composed of fully HH DNA (Fig. 5D).
Our results above suggested that in rec+, break–join (HH central recombinants) should be present (and break–copy recombinants might too), but that in ruv recG, there would be no fully HH central recombinants. We tested these predictions using a λ recombination assay (Meselson 1964; Stahl et al. 1972; Sawitzke and Stahl 1997) in which density-labeled phages recombine in the presence of light isotopes in E. coli in which a small amount of DNA replication is permitted. The partial replication block was achieved as described (Sawitzke and Stahl 1997) with the addition that a special allele of the E. coli dnaB replication helicase gene was used (grpD55) that blocks use of the λ replication origin by DnaB, but allows normal E. coli replication (Bull and Hayes 1996) (Materials and Methods). The phages (Sawitzke and Stahl 1997) are marked such that recombination events occurring in the center of the chromosome (between the J and cI genes, Fig. 6A) can be measured separately from recombination events occurring at the right end of λ chromosome (between the cI and S genes, Fig. 6A). The λ Int (site-specific) system is inactivated by mutation such that only RecBCD-mediated homologous recombinants are measured (Sawitzke and Stahl 1997) (Materials and Methods).
Progeny phage can be separated physically from (parental) unadsorbed phage based on their densities. The unadsorbed phage occupy the densest peak of cesium formate density gradients (Fig. 6B,C). The cross progenies are further separated based on the extent of DNA synthesis in each packaged DNA molecule. Mostly or completely unreplicated (HH) and replicated (heavy–light, HL, and light–light, LL) progeny are distinguished physically in this assay (Fig. 6B,C). Intermediate densities are also seen. The amount of central and right end recombination is assayed for each gradient fraction (Fig. 6A, Materials and Methods).
Representative data presented in Figure 6, B and C (and numerous independent experiments that repeated these results), allow the following conclusions:
The HH peak from the rec+ infection contains central recombinants (Fig. 6B, solid circles). We conclude that these have arisen via a break–join mechanism, without extensive synthesis of DNA.
Note that the number of central recombinants (solid circles) exceeds the number of end recombinants (open circles) in the HH peak in rec+ (Fig. 6B, fractions 26, 27). This presumably reflects the larger size of the central interval (between 18–22 kb) than of the end interval (4.8 kb). [We express the central interval as a range because the exact position of the Jts allele is not known; 18 and 22 kb are the distances between the ends of the J gene and the cI marker (Daniels et al. 1983)].
There are essentially no central recombinants (solid circles) in the heaviest fractions of the HH peak in the ruvC recG infection (Fig. 6C). Note that in ruvC recG, there are more end (open circles) than central recombinants in the HH peak (Fig. 6C, fractions 24, 25). These data indicate that break–join recombination yielding HH central recombinants does not occur appreciably in the absence of RuvC and RecG. This supports the conclusions from results shown in Figure 4, in which no recombinant progeny were produced when replication was completely blocked in a ruvC recG strain. The presence of even a small number of end recombinants in the ruvC recG HH peak (Fig. 6C) may seem inconsistent with the absence of any recombinants at all in ruv recG cells when replication is completely blocked (Fig. 4). We suggest that the end recombinants in the HH peak have probably experienced a small amount of replication but not enough to separate them from the HH peak (see Fig. 5C).
The central recombinants in ruvC recG, which are absent from the HH peak, are seen here in the HL peak (Fig. 6C). Note that almost all of the central recombinants in ruvC recG are in the HL peak. This excess of central recombinants in the HL peak is expected if the central recombinants are formed by replication, suggesting that recombination reactions initiated at the center are completed by replicating out to the end of the chromosome (Fig. 5B). This result supports break–copy models (see Fig. 5, other possibilities discussed below) and demonstrates directly that the recombinant molecules formed in the absence of Ruv and RecG are replicated.
Physical evidence for break–copy and break–join recombination pathways in rec+ cells
As discussed above, accumulation of central recombinants in the HL peak of ruvC recG cross is expected if replication is used to form central recombinants (as seen in Fig. 5B). Informatively, we also see this accumulation of central recombinants in the HL peak of rec+ crosses (Fig. 6B, compare the ratio of central/end recombinants in the HL peak with the HH peak). This is the first physical demonstration of replicative recombination in the RecBCD pathway in rec+ cells, that is, the replicated DNA is present in the same DNA molecules that have recombined (other evidence reviewed below). Previous direct evidence bore on the existence of the break–join mechanism only (McMilin and Russo 1972; Lam et al. 1974; also Fig. 6B, HH peak). These data show that a significant fraction of recombination in wild-type E. coli occurs via a replicative mechanism even when Ruv and RecG functions are present.
Amounts of replicative and break–join recombination in rec+ cells
In rec+ (Fig. 6B), the ratio of central/end recombinants in the HL peak is 5.3, or about twice that seen in the HH peak (2.5), thus implying that about half of the recombination in rec+ is replicative. The rough equality of replicative and break–join recombination was also inferred from the observation that, in ruv recG cells, recombination frequency drops to half that seen in rec+ (Fig. 3, Δnin), in which no break–join events can occur (Figs. 4 and 6C), and that all recombination is replication dependent (Fig. 4).
Estimation of the amount of DNA replication associated with recombination
A rough estimation of the amount of newly synthesized DNA associated with recombination in the cross displayed in Figure 6C can be made as follows. The number of fractions between the fully HH and fully LL shows that each fraction accounts for a change of ∼8.3% in the proportion of the DNA that is heavy or light. If the segregation of old and newly synthesized strands following recombination is conservative (see Fig. 5), then a change of one fraction also represents a change of 8.3% of the length of the λ genome from heavy to light. For ruvC recG, the fractions with an excess of central recombinants (27–32, Fig. 6C) correspond to 17%–58% of the genome being new (the most abundant fraction having ∼50% new DNA). This is a remarkable correspondence with the distance of the central recombination events (recombination between J and cI) to the λ right end. J is between 59% and 66% of the λ genome from the right end (the position of the Jts marker is unknown), whereas cI is 17% from the right end. This observation is compatible with break–copy models with a conservative segregation of new strands as proposed in Figure 5. Semiconservative segregation would produce half as much new DNA. These data show that not only is new synthesis present directly in the same DNA molecules that recombined, but also that the amount of synthesis corresponds to that expected from the cross-over point to the end of the chromosome (Fig. 5B) as in break–copy models (alternative discussed below).
Absence of RuvC and RecG promotes replication of λ
An unexpected but highly informative result was seen in the experiments performed in parallel, shown in Figure 6, B and C. Although the experiment was performed under the same conditions in rec+ and ruvC recG cells, we observed ∼135× more phage with replicated DNA when the E. coli Ruv and RecG resolution systems were absent. This was calculated by dividing the area under the LL peak of the ruvC recG graph with the LL peak for rec+. (This difference is especially apparent in the LL peaks of the rec+ and ruvC recG gradients shown in Figure 6, in which the titer of LL phage is 8.7 × 103 and 1.2 × 106 for rec+ and ruvC recG, respectively. We excluded the HL peaks from these calculations because in rec+, some HL recombinants will be break–join events between HH and LL molecules.) This result was repeated in two additional experiments in which the extent of phage with replicated DNA in ruvC recG was 108× and 74× greater than in rec+ cells. These data suggest that strand-exchange intermediates, which accumulate in the absence of Ruv and RecG HJ processing proteins, promote replication (see also Harris et al. 1996).
Discussion
The data shown here demonstrate the following:
RecBCD-mediated λ recombination in the absence of the E. coli Ruv and RecG HJ resolution systems is dependent on either a nin-encoded function(s) or DNA replication. Each accounts for approximately half of the total recombination in these cells (Figs. 3, 4, and 6A, see above). The nin-encoded function responsible has not been identified but is likely to be the Rap HJ resolvase (Sharples et al. 1998), which facilitates some kinds of recombination events in vivo (Hollifield et al. 1987; Stahl et al. 1995).
λ recombination in the absence of the known HJ processing systems requires the major replicative polymerase, DNA Pol III (Fig. 4).
Direct physical analysis of recombined DNA for incorporation of new (light) isotopes revealed that break–join recombination occurs in wild-type cells (Figs. 4 and 6; McMilin and Russo 1972; Stahl et al. 1972; McMilin et al. 1974) and absolutely requires HJ processing proteins such as Ruv, RecG, or the nin function (Figs. 4 and 6).
Both classes of recombination utilize Chi sites efficiently, so we suggest that there are two pathways (and basic mechanisms) of E. coli RecBCD-mediated recombination and DSBR: a break–join pathway that requires HJ resolvases (e.g., see Fig. 1) and a replicative pathway that can operate independently of resolvases and requires DNA Pol III. We suggest that these are alternative fates of strand-exchange intermediates (e.g., Fig. 5).
In the absence of resolvases, essentially all of the central recombinants contain newly replicated DNA, indicating that they originated by a replicational recombination mechanism (Fig. 6C).
Physical analysis of recombinants in wild-type cells also revealed a substantial fraction of replicational recombination (excess of HL over HH central recombinants) even when the HJ processing proteins are present (Fig. 6B). Therefore we conclude that the replicational recombination pathway is a normal part of RecBCD-mediated λ recombination, not a special mechanism that occurs only in ruvC recG-defective cells. In the rec+ cells, the excess of putative break–copy (HL central) recombination relative to end recombinants in the HL peak is twofold over that seen in the HH (unreplicated, break–join) peak (Fig. 6B). This provides independent evidence that about half of RecBCD-dependent DSBR is break–join and the other half replicative.
The extent of new DNA synthesis in the replicational recombination observed corresponds to the fraction of the λ genome from the cross-over point to the λ right end, in support of conservative break–copy models (Fig. 5, alternatives discussed below).
DNA replication is promoted dramatically in the absence of RuvC and RecG HJ processing proteins, suggesting that strand-exchange recombination junctions (HJs or other) may act as assembly sites for replication forks (this proposal was made previously based on data on recombination-dependent stationary-phase mutation, Harris et al. 1996).
The results summarized above provide a physical demonstration (via detection of replicated recombinant molecules) of a replicational recombination route in the RecBCD system of DSBR recombination in E. coli. The data also show that the replicational and break–join mechanisms can be separated: Replicational recombination is the only mechanism in ruv recG cells (Figs. 4 and 6) whereas break–join is the sole route when resolvases are present and replication is blocked (Fig. 4). These findings will greatly aid further dissections of both RecBCD-mediated DSBR mechanisms.
Previous evidence
Groundbreaking previous work led to the proposal of replicational recombination in E. coli. First, the discovery and characterization of a DNA replication mode that is replication origin independent and recombination protein dependent (stable-DNA replication, or SDR) is most easily understood by the postulate that recombination intermediates initiate replication, as in break–copy models (Kogoma 1997). The evidence is voluminous, important, and highly suggestive but indirect. Recombination-related genetic requirements were demonstrated, but DNA molecules that were both recombined and replicated were not. SDR is not a general process because it is seen only in RNase H-deficient mutants, or during an SOS (DNA damage) response (Kogoma 1997).
SDR-like replication was also observed very recently using phage λ. One λ DNA molecule was shown to be replicated at enhanced levels when a coinfecting λ molecule was linearized (“cut”), and the enhancement required recombination proteins (Kuzminov and Stahl 1999). The results demonstrate replication that is enhanced by recombination proteins and DNA damage. As with SDR, the evidence for association of replication and recombination is indirect for three reasons: (1) The replicated DNA was not shown to have recombined, and recombined DNA showed no evidence of having been replicated (Kuzminov and Stahl 1999); (2) no requirement for homology between the cut molecule and the replicated molecule was reported; and (3) all of the recombination proteins implicated (RecA, RecB, RecF) function dually—in recombination and in induction of the SOS response (Walker 1996). Thus, whether this is SOS-promoted or recombination-promoted replication is unclear. Recombination and replication might not have been associated directly in the same DNA molecules.
Second, the existence of a recombination protein-dependent mutation mechanism operating in stationary-phase E. coli cells (Harris et al. 1994, 1996; Foster et al. 1996) and requiring DNA Pol III (Foster et al. 1995; Harris et al. 1997) is also most easily accommodated by models in which RecBCD-mediated DSBR can prime replication that leads to polymerase error and mutation (Harris et al. 1994; Rosenberg 1997; Lombardo and Rosenberg 1999). Here too, a direct demonstration of replicated recombinants has not yet been made.
Third, replication primosome assembly protein PriA is important for replication and is partially required for conjugational and transductional recombination. Its absence causes a roughly two-thirds reduction in recombination (Kogoma et al. 1996). This result is easily understood if replication is required for about two-thirds of RecBCD-mediated recombination, but this did not distinguish this hypothesis from the possibility that PriA, a DNA-binding protein, enhances recombination independently of its action in promoting replication. Although the biochemistry of PriA is consistent with a role in promoting replication during recombination (Liu et al. 1999), it is not yet known whether that is the role of PriA in recombination in vivo.
Other good arguments have been advanced (e.g., Smith 1991; Kuzminov 1995; Courcelle et al. 1997).
The mechanism of the replicational DSBR recombination in E. coli
Break–copy mechanisms for the replicational DSBR such as the one shown in Figure 5 are supported by the results reported here. There is a close correspondence between the amount of newly synthesized DNA in the replicational recombinants with the distance from the cross-over point to the end of the λ chromosome. This observation is compatible with and supportive of break–copy models in which the new strands segregate conservatively (Fig. 5). However, alternatives are possible.
Alternative interpretations
First, in phage T4, a different mode of replicational recombination, called “join–cut–copy,” has been demonstrated (in addition to standard break–copy done by T4, Mosig 1998). The join–cut–copy events proceed only via leading-strand synthesis. An invading 3′ end synthesizes one new strand from the cross-over point rightward (in diagrams such as Fig. 5) and then a T4-encoded endonuclease cuts the template molecule on the opposite strand at the cross-over junction. The 3′ end from this nick primes leading-strand synthesis from the cross-over point leftward (see Mosig 1998). This odd mechanism produces a recombinant that contains one new strand from the cross-over point rightward and the other new strand from the cross-over point leftward. As yet, no recombination nuclease is known to have this function in E. coli (but see Chiu et al. 1997), but we cannot rule this mechanism out. Further experiments will be required to distinguish break–copy from join–cut–copy (and other possible) modes of replicational recombination and to address more directly models with conservative versus semiconservative segregation of new strands.
Second, DNA replication pausing has been shown to lead to double-strand breakage in E. coli, in a process that requires Ruv proteins (Seigneur et al. 1998). Could the role of replication in recombination reported here be in production of DSBs, which are necessary for RecBCD to load onto and recombine DNA? Three facts argue against this idea: First, such DSBs should not occur in cells lacking Ruv functions (Seigneur et al. 1998), whereas our requirement for replication in recombination is seen in Ruv− cells (Figs. 4 and 6). Second, in λ, the cos site is well documented to be the DSB site at which RecBCD loads and to be required even when DNA replication is allowed (Kobayashi et al. 1982, 1983, 1984) (see Fig. 5). Thus, it is most unlikely that the role of replication is to provide DSBs. Finally, this postulate does not predict the specific absence of break–join (central) recombinants among unreplicated molecules in ruv recG (Fig. 6C), whereas models such as break–copy do.
Strand polarity
Neither break–join nor replicative mechanisms bear particularly on the polarity of RecA-mediated strand-invasion that creates bimolecular strand-exchange intermediates (e.g., Fig. 5B,C,D). The possibility that both 5′ and 3′ single-strand DNA ends created by RecBCD can invade (Rosenberg and Hastings 1991) [supported by in vivo evidence (Hagemann and Rosenberg 1991; Miesel and Roth 1996; Razavy et al. 1996) and some biochemistry (Dutreix et al. 1991; Taylor and Smith 1995; Shan et al. 1997)], and the hypothesis that only 3′ ends can invade [as observed under different in vitro reaction conditions (e.g., Anderson and Kowalczykowski 1997) and in an unusual unimolecular reaction in vivo (Friedman-Ohana and Cohen 1998)] can both be accommodated by our observation of roughly equal break–join and replicative recombination. For example, Harris et al. (1996) suggested that 3′ end invasions might prime the replication in break–copy models whereas 5′ end invasions might lead only to break–join, in accordance with the rough equality (1:2) of 3′ and 5′ heteroduplex recombinants observed previously (Hagemann and Rosenberg 1991). These possibilities will require further study to address.
Replicational recombination in other organisms
The connection between recombination and replication is best established in bacteriophage T4, in which much of DNA replication requires homologous recombination functions (Dannenberg and Mosig 1981; Luder and Mosig 1982; Dannenberg and Mosig 1983; Formosa and Alberts 1986). Although no other system has yet provided as direct a demonstration of replicational recombination as the T4 system and the data for E. coli presented here, replicational recombination models are currently gaining support in multiple systems including yeast (e.g., Strathern et al. 1995; Morrow et al. 1997; Bosco and Haber 1998; Holmes and Haber 1999) and mammalian cells (Harris et al. 1999). Such replicational DSBR could be an important source of nonreciprocal translocations, loss of heterozygosity, telomere extension, and other genome rearrangements important in formation of human cancers and aging (e.g., Ellis et al. 1995; Yu et al. 1996; Nugent et al. 1998; Haber 1999).
Why is either Ruv or RecG required for conjugational and transductional recombination?
In the phage λ assay system, replication can, in effect, substitute for the Ruv and RecG recombination intermediate processing systems of E. coli. However, this is not observed for the E. coli chromosome. Double mutants of any ruv gene with recG are recombination-deficient for E. coli conjugational and transductional recombination (Lloyd 1991), as if the replicational RecBCD-mediated mechanism cannot substitute in these processes (see Lloyd 1991; Eggleston and West 1996; Harris et al. 1996 for views of the roles of Ruv and RecG in DSBR). Several explanations are possible for this apparent discrepancy. First, it is possible that conjugation and transduction are strictly nonreplicational events. Second, it is also possible that for some reason, DNA replication forks assembled at recombination junctions are less processive than those that start at a replication origin (Bosco and Haber 1998), such that the 48-kb λ genome can be replicated by recombination but the 4.5-Mb E. coli genome cannot. A more unifying class of explanation than either of these is presented in Figure 7.
Figure 7.

Break–copy models illustrated for RecBCD-mediated recombination of λ (A) and E. coli (B,C) genomes. The hexagon represents the prohead during λ DNA packaging from cos to cos. The ball represents the terminase protein that linearizes λ, binds the prohead, and packages the DNA.
The replication forks initiated at recombination intermediates should be different from those that start at a replication origin in that they are associated with an HJ behind the advancing fork (Fig. 7). The migration of HJ-containing replication bubbles around the E. coli chromosome might require branch migration proteins such as RecG or RuvAB (Fig. 7B,C). λ might escape this need either because the distance is shorter or because some other activity substitutes for Ruv/RecG-mediated branch migration of the replication bubble. For example, phage DNA packaging occurs concurrently with RecBCD-mediated recombination of the λ chromosome (see Fig. 7A) because the DSB made to initiate packaging is the same one for RecBCD loading (Kobayashi et al. 1984; Myers and Stahl 1994). The packaging apparatus travels in the same direction (rightward, Fig. 7A) as the branch migration that would be necessary to move the junction rightward. Perhaps the packaging apparatus can move the junctions at the forks for λ. Alternatively, because a replication bubble will not encounter any replication terminus in λ DNA (as it would in the E. coli chromosome), replication forks started at a recombination intermediate could proceed around the entire λ chromosome (circle, Fig. 7A) and the replisome then might push the junction rightward (Morgan and Severini 1990). For λ, the junction need only move past the next packaging origin encountered (cos, Fig. 7A) to produce a packageable replicated recombinant. Although other explanations are also possible, this one and variations on the theme in Figure 7 (see Bosco and Haber 1998) are simple in that they do not require any special properties of the replication associated with recombination that are not seen for replication in general. These models also make testable predictions. Further work will be required to address the possibilities raised by findings reported here.
Materials and methods
Bacterial and phage strains
Bacterial strains are E. coli K12 derivatives and are listed in Table 2. New genotypes were constructed using standard phage P1-mediated transduction (Miller 1992). The presence of recA, recG, ruvA, ruvB, and ruvC alleles was confirmed by the increased UV light sensitivity phenotypes. For ruv recG double mutants, extreme UV sensitivity (Lloyd 1991) was verified. SMR650 was constructed from SMR632 by transduction of ruvC53 eda-51::Tn10 (Lloyd 1991) from CS85, then recG258::Tn10minikan from RDK2655 (Lloyd and Buckman 1991, obtained from R. Kolodner). SMR3124 was constructed similarly [RDK2641 donated ruvA59::Tn10 (Shurvinton et al. 1984)]. SMR632 transduced with P1 from SMR540 (lab collection, allele from R. Maurer, Case Western Reserve University, Cleveland, OH) yielded SMR4594. SMR4600 is SMR4594 with ruvC53 eda-51::Tn10 (Lloyd and Buckman 1991) and recG258::Tn10minikan (Lloyd and Buckman 1991). SMR4601 was made by from SMR4594 with P1 grown on SMR624 (Harris et al. 1994).
Table 2.
Bacterial strains
| Strain
|
Relevant properties
|
Source or reference
|
|---|---|---|
| 594 | Su−rec+ | (Weigle 1966) |
| C600 | SuII+rec+ | (Appleyard 1954) |
| AFT196 | C600 Δ(srlR–recA)306∷Tn10 | Lab collection |
| KR3a | SuIII+ recA | Lab collection |
| RDK2641 | ruvA59∷Tn10 | R. Kolodner |
| CS85 | ruvC53 eda-51∷Tn10 | R.G. Lloyd, via R. Kolodner |
| RDK2655 | recG258∷Tn10minikan | R. Kolodner |
| RM5258 | eda-57∷Tn10∷cam | (Foster, et al. 1996) |
| SMR423 | C600 Sull III recD1903∷Tet hsdrK− mK+ | Lab collection |
| SMR632 | 594 hsdrK− mK+ | Lab collection |
| SMR650 | SMR632 ruvC53 eda-51∷Tn10 recG258∷Tn10minikan | This work |
| SMR3124 | SMR632 ruvA59∷Tn10 recG258∷Tn10minikan | This work |
| SMR4594 | SMR632 dnaEts486 zae∷Tn10d-Cam | This work |
| SMR4600 | SMR632 dnaEts486 zae∷Tn10d-Cam ruvC53 eda-51∷Tn10 recG258∷Tn10minikan | This work |
| SMR4601 | SMR632 dnaEts486 zae∷Tn10d-Cam Δ(srlR-recA)306∷Tn10 | This work |
| SMR3731 | SMR632 grpD55 malF∷Tn10∷kan (λ Jts15 red3 gam210 Δnin5 Sam7) | This work |
| SMR3732 | SMR632 grpD55 malF∷Tn10∷kan recG162 zib-636∷Tn10 ruvC53 eda-57∷Tn10∷cam (λ Jts15 red3 gam210 Δnin5 Sam7) | This work |
| JAS36 | C600 (λ Jts15 red3 gam210 imm434 Δnin5 Sam7) | (Sawitzke and Stahl 1997) |
| JAS38 | Δ(srlR-recA)306∷Tn10 recD1009 (λ Jts15 red3 gam210 imm434 Δnin5 Sam7) | (Sawitzke and Stahl 1997) |
| FC40 | ara Δ(lac-pro)XIIIthi RifR [F‘ lacI33 ΩlacZ proAB] | (Cairns and Foster 1991) |
| RSH45 | FC40 ruvC53 eda-51∷Tn10 recG258∷Tn10minikan | (Harris et al. 1996) |
| RSH160 | FC40 ruvA59∷Tn10 recG258∷Tn10minikan | (Harris et al. 1996) |
SMR3731 was made by lysogenizing SMR632 with λJts15 red3 gam210 Δnin5 Sam7 [λSR459 (Sawitzke and Stahl 1997)], followed by transduction to kanamycin resistance with P1 grown on a grpD55 malF::Tn10::kan strain (Bull and Hayes 1996). SMR3732 was made from SMR3731 by transducing ruvC53 eda57::Tn10::cam (obtained from a transductant of CS85 × P1 RM5258) and recG162 zib-636::Tn10 (Storm et al. 1971).
λ phages are either from the λSR collection or were gifts from F.W. Stahl (University of Oregon, Eugene) or S. Hayes (University of Saskatchewan, Saskatoon, Canada). Phage genotypes used in crosses to measure the frequency of recombinants (Figs. 2 and 3) are from Razavy et al. (1996) and λΔb2 red3 gam210 cI857 Sam7 (nin+); λbio1 (nin+). The phages used in Figure 4 were λSR27, bio1 Δnin5, and in Figure 6 were MMS1816, λJts15 int4 red3 gam210 cI857 Δnin5; MMS1817, λint4 red3 gam210 Δnin5 Sam7, and homoimmune prophage MMS2076, λJts15 red3 gam210 Δnin5 Sam7, with helper packaging functions provided by MMS2084, λJts15 int4 red3 gam210 imm434 Δnin5 Sam7 (Sawitzke and Stahl 1997).
Growth of phage stocks and E. coli cultures
dnaEts strains were grown at 28°C. ruv recG double mutants are slow growing and form small colonies, such that cultures are prone to accumulation of faster-growing and larger mutant colonies carrying suppressor mutations as well as true reversions (Lloyd and Buckman 1991; Harris et al. 1996). ruv recG double mutant strains were grown at 32°C to avoid the accumulation of suppressors normally associated with growing these strains at higher temperatures (Harris et al. 1996). The UV and drug-sensitivity phenotypes of all strains were confirmed for cultures used in each experiment (and/or for ∼30 colonies from a given culture). Cultures were also routinely monitored for possible accumulation of suppressors or revertants as described previously (Harris et al. 1996).
λ phage stocks (carrying light isotopes) were grown and plaque assays were performed according to standard procedures (Murray 1983). Stocks of λ phage density labeled with 13C and 15N were grown according to procedures of Stahl et al. (1972) on prototrophic bacteria for 12–14 hr at 32°C.
Recombination assays
Experiments in Figures 2 and 3 were performed as previously (Razavy et al. 1996), except that cultures for mixed infections were grown at 32°C, inoculated from 10–30 μl of the frozen bacterial cultures.
λ recombination assay in the absence of DNA replication
Prewarmed, density-labeled λ red gam nin (λSR27) were infected into prewarmed SMR4594, 4600, and 4601 (at 43.5°C, m.o.i. = 10) that had been grown at 28°C to 2 × 108 cell/ml in tryptone broth + 1% yeast extract, 0.2% maltose, and 0.01 mg/ml of vitamin B1 (+25 μg/ml of kanamycin for ruv recG strains, to avoid accumulation of recG revertants formed by transposon excision). Infected cells were bubbled for 30 min, diluted with 4 ml of prewarmed broth (above), bubbled for 35 min at 43.5°C, then washed with cold TM buffer (10 mm, Tris Mg), resuspended in chilled broth (above), lysed with lysozyme and chloroform, and pelleted, and the supernatants were collected. A density gradient prepared for each lysate (McMilin and Russo 1972) was collected as two-drop fractions into 1 ml of TB and titered on SMR423.
Assay for central and end λ recombinants formed under partial replication block
Partial replication block was achieved by homoimmune repression and heteroimmune helper phage infection, as described by Sawitzke and Stahl (1997), except that our E. coli strains also carried the grpD55 mutation. grpD55 encodes a DnaB helicase that does not interact with the λ replication proteins (Bull and Hayes 1996). In the absence of this allele, λ replication could not be blocked sufficiently in ruv recG strains to allow resolution of any unreplicated phage (HH peaks). Recombinants were assayed on strains JAS36 and JAS38 as described (Sawitzke and Stahl 1997).
Acknowledgments
This paper is dedicated to the memory of our cherished colleague and friend, Dr. A. Richard Morgan. We thank S. Hayes, R. Kolodner, R.G. Lloyd, R. Maurer, S. Sandler, J. Sawitzke, F.W. Stahl, and the E. coli Genetic Stock Center for generous gifts of strains. We are grateful to H.J. Bull, J. Haber, R.S. Harris, P.J. Hastings, I. Kobayashi, M.-J. Lombardo, A.R. Morgan, G. Mosig, D.B. Roth, and M. Russell for helpful discussions and comments on the manuscript, J. van der Velde for help with experiments, and M.-J. Lombardo for work on the manuscript. This work was supported in part by grants from the Medical Research Council of Canada (MRC), by grants R01 GM53158 and R01 AI43917 from the U.S. National Institutes of Health, and by an MRC Scientist Award and an Alberta Heritage Foundation for Medical Research Senior Scholarship to S.M.R.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL smr@bcm.tmc.edu; FAX (713) 798-8704.
References
- Anderson DG, Kowalczykowski SC. The recombination hot spot chi is a regulatory element that switches the polarity of DNA degradation by the RecBCD enzyme. Genes & Dev. 1997;11:571–581. doi: 10.1101/gad.11.5.571. [DOI] [PubMed] [Google Scholar]
- Appleyard RK. Segregation of new lysogenic types during growth of a doubly lysogenic strain derived from Escherichia coli K12. Genetics. 1954;39:440–452. doi: 10.1093/genetics/39.4.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco G, Haber JE. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics. 1998;150:1037–1047. doi: 10.1093/genetics/150.3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull H, Hayes S. The grpD55 locus of Escherichia coli appears to be an allele of dnaB. Mol Gen Genet. 1996;252:755–760. doi: 10.1007/BF02173984. [DOI] [PubMed] [Google Scholar]
- Cairns J, Foster PL. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SK, Low KB, Yuan A, Radding CM. Resolution of an early RecA-recombination intermediate by a junction-specific endonuclease. Proc Natl Acad Sci. 1997;94:6079–6083. doi: 10.1073/pnas.94.12.6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AJ, Sandler SJ. Homologous genetic recombination: The pieces begin to fall into place. Critical Rev Microbiol. 1994;20:125–142. doi: 10.3109/10408419409113552. [DOI] [PubMed] [Google Scholar]
- Connolly B, Parsons CA, Benson FE, Dunderdale HJ, Sharples GJ, Lloyd RG, West SC. Resolution of Holliday junctions in vitro requires the Escherichia coli ruvC gene product. Proc Natl Acad Sci. 1991;88:6063–6067. doi: 10.1073/pnas.88.14.6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelle J, Carswell-Crumpton C, Hanawalt PC. RecF and RecR are required for resumption of replication at DNA replication forks in Escherichia coli. Proc Natl Acad Sci. 1997;94:3714–3719. doi: 10.1073/pnas.94.8.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DL, Schroeder JL, Szybalski W, Sanger F, Coulson AR, Hong GF, Hill DF, Petersen GB, Blattner FR. Appendix II. Complete annotated lambda sequence. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA, editors. Lambda II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1983. pp. 519–676. [Google Scholar]
- Dannenberg R, Mosig G. Semiconservative DNA replication is initiated at a single site in recombination-deficient gene32 mutants of bacteriophage T4. J Virol. 1981;40:890–900. doi: 10.1128/jvi.40.3.890-900.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Early intermediates in bacteriophage T4 DNA replication and recombination. J Virol. 1983;45:813–831. doi: 10.1128/jvi.45.2.813-831.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutreix M, Rao BJ, Radding CM. The effects on strand exchange of 5′ versus 3′ ends of single-stranded DNA in RecA nucleoprotein filaments. J Mol Biol. 1991;219:645–654. doi: 10.1016/0022-2836(91)90661-o. [DOI] [PubMed] [Google Scholar]
- Eggleston AK, West SC. Exchanging partners: Recombination in E. coli. Trends Genet. 1996;12:20–26. doi: 10.1016/0168-9525(96)81384-9. [DOI] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye T-Z, Straughen J, Lennon DL, Ciocci S, Proytcheva M, German J. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- Feiss M, Becker A. DNA packaging and cutting. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA, editors. Lambda II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1983. pp. 305–330. [Google Scholar]
- Formosa T, Alberts B. Purification and characterization of the T4 bacteriophage uvsX protein. J Biol Chem. 1986;261:6107–6118. [PubMed] [Google Scholar]
- Foster PL, Gudmundsson G, Trimarchi JM, Cai H, Goodman MF. Proofreading-defective DNA polymerase II increases adaptive mutation in Escherichia coli. Proc Natl Acad Sci. 1995;92:7951–7955. doi: 10.1073/pnas.92.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM, Maurer RA. Two enzymes, both of which process recombination intermediates, have opposite effects on adaptive mutation in Escherichia coli. Genetics. 1996;142:25–37. doi: 10.1093/genetics/142.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman-Ohana R, Cohen A. Heteroduplex joint formation in Escherichia coli recombination is initiated by pairing of a 3′-ending strand. Proc Natl Acad Sci. 1998;95:6909–6914. doi: 10.1073/pnas.95.12.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber JE. Meiosis. Searching for a partner. Science. 1998;279:823–824. doi: 10.1126/science.279.5352.823. [DOI] [PubMed] [Google Scholar]
- ————— DNA repair: Gatekeepers of recombination. Nature. 1999;398:665–666. doi: 10.1038/19423. [DOI] [PubMed] [Google Scholar]
- Hagemann AT, Rosenberg SM. Chain bias in Chi-stimulated heteroduplex patches in the λ ren gene is determined by the orientation of λ cos. Genetics. 1991;129:611–621. doi: 10.1093/genetics/129.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Longerich S, Rosenberg SM. Recombination in adaptive mutation. Science. 1994;264:258–260. doi: 10.1126/science.8146657. [DOI] [PubMed] [Google Scholar]
- Harris RS, Ross KJ, Rosenberg SM. Opposing roles of the Holliday junction processing systems of Escherichia coli in recombination-dependent adaptive mutation. Genetics. 1996;142:681–691. doi: 10.1093/genetics/142.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Bull HJ, Rosenberg SM. A direct role for DNA polymerase III in adaptive reversion of a frameshift mutation in Escherichia coli. Mutat Res. 1997;375:19–24. doi: 10.1016/s0027-5107(96)00244-8. [DOI] [PubMed] [Google Scholar]
- Harris RS, Kong Q, Maizels N. Somatic hypermutation and the three R's: Repair, replication and recombination. Rev Mutat Res. 1999;436:157–178. doi: 10.1016/s1383-5742(99)00003-4. [DOI] [PubMed] [Google Scholar]
- Henderson D, Weil J. Recombination-deficient deletions in bacteriophage λ and their interaction with chi mutations. Genetics. 1975;79:143–174. doi: 10.1093/genetics/79.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollifield WC, Kaplan EN, Huang HV. Efficient RecABC-dependent, homologous recombination between coliphage lambda and plasmids requires a phage ninR region gene. Mol Gen Genet. 1987;210:248–255. doi: 10.1007/BF00325690. [DOI] [PubMed] [Google Scholar]
- Holmes A, Haber JE. Double-strand break-repair in yeast requires both leading and lagging strand DNA polymerases. Cell. 1999;96:415–424. doi: 10.1016/s0092-8674(00)80554-1. [DOI] [PubMed] [Google Scholar]
- Kanaar R, Hoeijmakers JH. Recombination and joining: Different means to the same ends. Genes Funct. 1998;1:165–174. doi: 10.1046/j.1365-4624.1997.00016.x. [DOI] [PubMed] [Google Scholar]
- Kemper B, Jensch F, von Depka-Prondzynski M, Fritz HJ, Borgmeyer U, Mizuuchi K. Resolution of Holliday structures by endonuclease VII as observed in interactions with cruciform DNA. Cold Spring Harb Symp Quant Biol. 1984;49:815–825. doi: 10.1101/sqb.1984.049.01.092. [DOI] [PubMed] [Google Scholar]
- Kobayashi I, Murialdo H, Crasemann JM, Stahl MM, Stahl FW. Orientation of cohesive end site cos determines the active orientation of χ sequence in stimulating recA·RecB-mediated recombination in phage λ lytic infections. Proc Nat Acad Sci. 1982;79:5981–5985. doi: 10.1073/pnas.79.19.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi I, Stahl MM, Leach D, Stahl FW. The interaction of cos with Chi is separable from DNA packaging and recA-recBC-mediated recombination of bacteriophage lambda. Genetics. 1983;104:549–570. doi: 10.1093/genetics/104.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi I, Stahl MM, Fairfield FR, Stahl FW. Coupling with packaging explains apparent nonreciprocality of Chi-stimulated recombination of bacteriophage lambda by RecA and RecBC functions. Genetics. 1984;108:773–794. doi: 10.1093/genetics/108.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T. Stable DNA replication: Interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev. 1997;61:212–238. doi: 10.1128/mmbr.61.2.212-238.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T, Cadwell GW, Barnard KG, Asai T. Requirement of the DNA replication priming protein, PriA, for homologous recombination and double-strand break repair. J Bacteriol. 1996;178:1258–1264. doi: 10.1128/jb.178.5.1258-1264.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol. 1995;16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- Kuzminov A, Stahl FW. Double-strand end repair via the RecBC pathway in Escherichia coli primes DNA replication. Genes & Dev. 1999;13:345–356. doi: 10.1101/gad.13.3.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam ST, Stahl MM, McMilin KD, Stahl FW. Rec-mediated hot spot activity in bacteriophage lambda. II. A mutation which causes hot spot activity. Genetics. 1974;77:425–433. doi: 10.1093/genetics/77.3.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xu L, Sandler SJ, Marians KJ. Replication fork assembly at recombination intermediates is required for bacterial growth. Proc Natl Acad Sci. 1999;96:3552–3555. doi: 10.1073/pnas.96.7.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG. Conjugational recombination in resolvase-deficient ruvC mutants of Escherichia coli depends on recG. J Bacteriol. 1991;173:5414–5418. doi: 10.1128/jb.173.17.5414-5418.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG, Buckman C. Genetic analysis of the recG locus of Escherichia coli K-12 and its role in recombination and DNA repair. J Bacteriol. 1991;173:1004–1011. doi: 10.1128/jb.173.3.1004-1011.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG, Low KB. Homologous Recombination. In: Neidhardt FC, et al., editors. Escherichia coli and Salmonella cellular and molecular biology. Washington, D.C.: ASM Press; 1996. pp. 2236–2255. [Google Scholar]
- Lombardo M-J, Rosenberg SM. Hypermutation in stationary-phase E. coli: Tales from the lac operon. J Genet. 1999;78:13–21. [Google Scholar]
- Luder A, Mosig G. Two alternative mechanisms for initiation of DNA replication forks in bacteriophage T4: Priming by RNA polymerase and by recombination. Proc Natl Acad Sci. 1982;79:1101–1105. doi: 10.1073/pnas.79.4.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMilin KD, Russo VEA. Maturation and recombination of bacteriophage lambda DNA molecules in the absence of DNA duplication. J Mol Biol. 1972;68:49–55. doi: 10.1016/0022-2836(72)90261-6. [DOI] [PubMed] [Google Scholar]
- McMilin KD, Stahl MM, Stahl FW. Rec-mediated hot spot activity in bacteriophage lambda. I. Hot spot activity associated with spi deletions and bio substitutions. Genetics. 1974;77:409–423. doi: 10.1093/genetics/77.3.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meselson M. On the mechanism of genetic recombination between DNA molecules. J Mol Biol. 1964;9:734–745. doi: 10.1016/s0022-2836(64)80178-9. [DOI] [PubMed] [Google Scholar]
- Meselson M, Weigle J. Chromosome breakage accompanying recombination in bacteriophage. Proc Natl Acad Sci. 1961;47:857–868. doi: 10.1073/pnas.47.6.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesel L, Roth JR. Evidence that functions of the “RecF pathway” contribute to RecBCD-dependent transductional recombination. J Bacteriol. 1996;178:3146–3155. doi: 10.1128/jb.178.11.3146-3155.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. A short course in bacterial genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- Morgan AR, Severini A. Interconversion of replication and recombination structures: Implications for terminal repeats and concatemers. J Theor Biol. 1990;144:195–202. doi: 10.1016/s0022-5193(05)80318-2. [DOI] [PubMed] [Google Scholar]
- Morrow DM, Connelly C, Hieter P. “Break-copy” duplication: A model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics. 1997;147:371–382. doi: 10.1093/genetics/147.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosig G. Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu Rev Genet. 1998;32:379–413. doi: 10.1146/annurev.genet.32.1.379. [DOI] [PubMed] [Google Scholar]
- Murray NE. Phage lambda and molecular cloning. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RW, editors. Lambda II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1983. pp. 395–432. [Google Scholar]
- Myers RS, Stahl FW. χ and RecBCD enzyme of Escherichia coli. Annu Rev Genet. 1994;28:49–70. doi: 10.1146/annurev.ge.28.120194.000405. [DOI] [PubMed] [Google Scholar]
- Nugent CI, Bosco G, Ross LO, Evans SK, Salinger AP, Moore JK, Haber JE, Lundblad V. Telomere maintenance is dependent on activities required for end repair of double-strand breaks. Curr Biol. 1998;8:657–660. doi: 10.1016/s0960-9822(98)70253-2. [DOI] [PubMed] [Google Scholar]
- Razavy H. “Single-strand DNA ends in recombination in E. coli.” M.Sc. thesis. Edmonton, Canada: University of Alberta; 1997. [Google Scholar]
- Razavy H, Szigety SK, Rosenberg SM. Evidence for both 3′ and 5′ single-strand DNA ends in intermediates in Chi stimulated recombination in vivo. Genetics. 1996;142:333–339. doi: 10.1093/genetics/142.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SM. Mutation for survival. Curr Opin Genet Devel. 1997;7:829–834. doi: 10.1016/s0959-437x(97)80047-0. [DOI] [PubMed] [Google Scholar]
- Rosenberg SM, Hastings PJ. The split-end model for homologous recombination at double-strand breaks and at Chi. Biochimie. 1991;73:385–397. doi: 10.1016/0300-9084(91)90105-a. [DOI] [PubMed] [Google Scholar]
- Rosenberg SM, Motamedi MR. Homologous recombination during bacterial conjugation. In: Atlas RM, et al., editors. Encyclopedia of life Sciences. London, UK: Macmillan Press, Limited; 1999. . (In press). [Google Scholar]
- Rosenberg SM, Stahl MM, Kobayashi I, Stahl FW. Improved in vitro packaging of coliphage lambda DNA: A one-strain system free from endogenous phage. Gene. 1985;38:165–175. doi: 10.1016/0378-1119(85)90215-x. [DOI] [PubMed] [Google Scholar]
- Sawitzke JA, Stahl FW. Roles for lambda Orf and Escherichia coli RecO, RecR and RecF in lambda recombination. Genetics. 1997;147:357–369. doi: 10.1093/genetics/147.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- Shan Q, Bork JM, Webb BL, Inman RB, Cox MM. RecA protein filaments: End-dependent disassociation from ssDNA and stabilization by RecO and RecR proteins. J Mol Biol. 1997;265:519–540. doi: 10.1006/jmbi.1996.0748. [DOI] [PubMed] [Google Scholar]
- Sharples GJ, Corbett LM, Graham IR. λ Rap protein is a structure-specific endonuclease involved in phage recombination. Proc Natl Acad Sci. 1998;95:13507–13512. doi: 10.1073/pnas.95.23.13507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurvinton CE, Lloyd RG, Benson FE, Attfield PV. Genetic analysis and molecular cloning of the Escherichia coli ruv gene. Mol Gen Genet. 1984;194:322–329. doi: 10.1007/BF00383535. [DOI] [PubMed] [Google Scholar]
- Siegel J. Extent and location of DNA synthesis associated with a class of Rec-mediated recombinants of bacteriophage lambda. J Mol Biol. 1974;88:619–628. doi: 10.1016/0022-2836(74)90413-6. [DOI] [PubMed] [Google Scholar]
- Skalka A. A replicator's view of recombination (and repair) In: Grell RR, editor. Mechanisms in recombination. New York, NY: Plenum Press; 1974. pp. 421–432. [Google Scholar]
- Smith GR. Conjugational recombination in E. coli: Myths and mechanisms. Cell. 1991;64:19–27. doi: 10.1016/0092-8674(91)90205-d. [DOI] [PubMed] [Google Scholar]
- Smith GR, Stahl FW. Homologous recombination promoted by Chi sites and RecBC enzyme of Escherichia coli. BioEssays. 1985;2:244–249. [Google Scholar]
- Smith KN, Nicolas A. Recombination at work for meiosis. Curr Opin Genet Devel. 1998;8:200–211. doi: 10.1016/s0959-437x(98)80142-1. [DOI] [PubMed] [Google Scholar]
- Stahl MM, Stahl FW. DNA synthesis associated with recombination I. Recombination in a DNA-negative host. In: Hershey AD, editor. The bacteriophage lambda. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1971. pp. 431–442. [Google Scholar]
- Stahl FW, McMilin KD, Stahl MM, Malone RE, Nozu Y, Russo VEA. A role for recombination in the production of “free-loader” lambda bacteriophage particles. J Mol Biol. 1972;68:57–67. doi: 10.1016/0022-2836(72)90262-8. [DOI] [PubMed] [Google Scholar]
- Stahl FW, Shurvinton CE, Thomason LC, Hill S, Stahl MM. On the clustered exchanges of the RecBCD pathway operating on phage λ. Genetics. 1995;139:1107–1121. doi: 10.1093/genetics/139.3.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm PK, Hoekstra WPM, De Haan PG, Verhoef C. Genetic recombination in Escherichia coli. IV. Isolation and characterization of recombination-deficient mutants of Escherichia coli K12. Mutat Res. 1971;13:9–17. doi: 10.1016/0027-5107(71)90121-7. [DOI] [PubMed] [Google Scholar]
- Strathern JN, Shafer BK, McGill CB. DNA synthesis errors associated with double-strand-break repair. Genetics. 1995;140:965–972. doi: 10.1093/genetics/140.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AF, Smith GR. Strand-specificity of nicking of DNA at Chi sites by RecBCD enzyme: Modulation by ATP and magnesium levels. J Biol Chem. 1995;270:24459–24467. doi: 10.1074/jbc.270.41.24459. [DOI] [PubMed] [Google Scholar]
- Thaler DS, Stahl FW. DNA double-chain breaks in recombination of phage λ and yeast. Annu Rev Genet. 1988;22:169–197. doi: 10.1146/annurev.ge.22.120188.001125. [DOI] [PubMed] [Google Scholar]
- Thaler DS, Sampson E, Siddiqi I, Rosenberg SM, Thomason LC, Stahl FW, Stahl MM. Recombination of bacteriophage λ in recD mutants of Escherichia coli. Genome. 1989;31:53–67. doi: 10.1139/g89-013. [DOI] [PubMed] [Google Scholar]
- Thomason LC, Thaler DS, Stahl MM, Stahl FW. In vivo packaging of bacteriophage λ monomeric chromosomes. J Mol Biol. 1997;267:75–87. doi: 10.1006/jmbi.1996.0870. [DOI] [PubMed] [Google Scholar]
- Tsukamoto Y, Ikeda H. Double-strand break repair mediated by DNA end-joining. Genes Cells. 1998;3:135–144. doi: 10.1046/j.1365-2443.1998.00180.x. [DOI] [PubMed] [Google Scholar]
- Walker GC. The SOS response of Escherichia coli. In: Neidhardt FC, et al., editors. Escherichia coli and Salmonella cellular and molecular biology. Washington, D.C.: ASM Press; 1996. pp. 1400–1416. [Google Scholar]
- Weigle J. Assembly of phage lambda in vitro. Proc Natl Acad Sci. 1966;55:1462–1466. doi: 10.1073/pnas.55.6.1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SC. Enzymes and molecular mechanisms of genetic recombination. Annu Rev Biochem. 1992;61:603–640. doi: 10.1146/annurev.bi.61.070192.003131. [DOI] [PubMed] [Google Scholar]
- Yu C-E, Oshima J, Fu Y-H, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]