

Abstract
In this study we elucidated the role of nonactive JNK in regulating p53 stability. The amount of p53–JNK complex was inversely correlated with p53 level. A peptide corresponding to the JNK binding site on p53 efficiently blocked ubiquitination of p53. Similarly, p53 lacking the JNK binding site exhibits a longer half-life than p53wt. Outcompeting JNK association with p53 increased the level of p53, whereas overexpression of a phosphorylation mutant form of JNK inhibited p53 accumulation. JNK–p53 and Mdm2–p53 complexes were preferentially found in G0/G1 and S/G2M phases of the cell cycle, respectively. Altogether, these data indicate that JNK is an Mdm2-independent regulator of p53 stability in nonstressed cells.
Keywords: JNK, p53, ubiquitination, degradation, Mdm2
The p53 tumor suppressor protein is a potent transcription factor (Kern et al. 1991; Zambetti et al. 1992; Friedlander et al. 1996) that is activated in response to various DNA-damaging agents (Fritsche et al. 1993; Hall et al. 1993; Zhan et al. 1993), leading to cell cycle arrest and/or apoptosis (Canman et al. 1995; Polyak et al. 1996). Disruption of this pathway occurs in a wide range of human cancers and is highly correlated with the tumorigenic phenotype (Harris 1996; Levine 1997). The key to the magnitude and duration of p53 activities lies in its stability (Maki et al. 1996; Brown and Pagano 1997). In normally growing cells, p53 half-life is limited to minutes, whereas cellular stress or exposure to DNA-damaging agents prolongs it to hours (Maltzman and Czyzyk 1984). Proteins known to alter p53 stability include HPV16–E6 (Huibregtse et al. 1991), WT-1 (Maheswaran et al. 1995), E1B/E4orf6 (Querido et al. 1997), SV40 T-antigen (Reihsaus et al. 1990; Tiemann et al. 1995), and Mdm2 (Haupt et al. 1997; Kubbutat et al. 1997). Whereas association of SV40 T antigen, WT1, or E1B/E4orf6 with p53 increases its stability, the binding of E6 or Mdm2 with p53 accelerates its degradation. To date, Mdm2 is the only cellular protein whose direct association with p53 results in its ubiquitination and subsequent degradation (Haupt et al. 1997; Honda et al. 1997; Kubbutat et al. 1997; Fuchs et al. 1998a). The regulation of p53 stability has been associated with post-translational modifications, including phosphorylation on amino-terminal residues (Shieh et al. 1997; Siliciano et al. 1997).
In previous studies we found that Jun-N (amino)-terminal kinase (JNK) targets the ubiquitination and stability of its associated proteins, c-Jun (Fuchs et al. 1996), JunB, and ATF2 (Fuchs et al. 1997). JNK targeting for ubiquitination occurs in a phosphorylation-dependent manner as phosphorylated forms of c-Jun and ATF2 were found to be protected against JNK-targeted ubiquitination. Essential for JNK’s ability to target the ubiquitination of ATF2, c-Jun, and JunB is its association with each of these proteins (Fuchs et al. 1996, 1997). Recent evidence for JNK association with p53 (Adler et al. 1997) provided the foundation for our hypothesis that JNK also has a role in the regulation of p53 stability.
Results and Discussion
The association between JNK and p53 in vivo was first demonstrated via coimmunoprecipitations (Fig. 1a; Adler et al. 1997). JNK–p53 complex was preferentially found in nonstressed cells; after UV irradiation its concentration decreased immensely (Fig. 1a). Whereas >30% of p53 is in complex with JNK 0.5 hr after UV irradiation, <2% of p53 is bound to JNK after 4–8 hr. The extent of JNK association with p53 is inversely correlated with p53 expression levels, suggesting that JNK could affect p53 stability in nonstressed cells.
Figure 1.





(a) In vivo association of JNK with p53 before and after UV irradiation. Mouse fibroblasts (BALB/3T3/12.1) were subjected to sham (C) or UV treatment, and proteins (4 mg) prepared at the time points indicated were immunoprecipitated with antibodies to JNK. Immunoprecipitated material (IP) was subjected to immunoblot (IB) using p53 antibodies (top) or antibodies to JNK (middle). (Bottom) The amount of p53 in whole-cell extracts. (b) JNK binding to p53 is inhibited by p7. Binding of bacterially expressed his-tagged human p53 to a purified form of JNK (lane −) was assessed in the presence of p7 (A), c7 peptide (B) at 1, 2, 4, or 8 μg, or when p7 was added after incubation of JNK with p53 (C). (Top) An immunoblot with JNK antibodies; (middle) a Ponceau S-stained membrane; (bottom) an autoradiograph of p53 phosphorylation by JNK in the presence of p7, c7, or no peptide (C). (c) Effect of p7 on JNK–p53 association in vivo. (Right) BALB/3T3/12.1 cells were transfected with cDNA of either p7 or c7 (cloned in-frame with penetratin–HA sequence into pcDNA3). (Top) The level of p53 in whole-cell extracts. p53–JNK association was monitored via immunoprecipitation with antibodies to JNK before immunoblot with antibodies to p53 (middle). The same immunoblot was reprobed with antibodies to JNK (bottom). (Left) p53 null cells (10.1) were transfected with either p53wt or p53Δp7 constructs. Protein extracts prepared 36 hr after transfection and 8 hr after treatment with lactacystin (5 μm) were processed as indicated above. (d) In vivo ubiquitination of p53 in BALB/3T3/12.1 cells. cDNAs of hisp53, ubiquitin–HA, and either c7, p7, or c-Jun were cotransfected into BALB/3T3/12.1 cells. p53 ubiquitination was monitored in immunblots performed on nickel resin-purified proteins. The upper part of the membranes (above 55 kD) was probed with antibodies to HA to identify any p53-associated tagged ubiquitin–HA; the lower part was analyzed using antibodies to p53. (e) In vivo stability of p53Δp7. p53wt or p53Δp7 constructs were transfected into 10.1 p53 null cells that were metabolically labeled with 0.5 mCi/ml [35S]methionine for 10 min and chased with medium supplemented with 2 mm of unlabeled methionine. Proteins prepared at the indicated time points were immunopurified using PAb 421 antibodies and analyzed on SDS–polyacrylamide gels via autoradiography.
Previous studies showed that JNK association with p53 requires amino acids 97–155 within the p53 central domain (Adler et al. 1997). A 20-amino acid peptide spanning amino acids 97–116 (designated p7) was found capable of altering p53 phosphorylation (Adler et al. 1997). To test the effect of p7 on JNK association with p53, increasing concentrations of the p7 peptide, or its control peptide (c7), were added in vitro to purified forms of JNK and p53. As shown in Figure 1b, p7, but not c7, caused a dose-dependent inhibition of JNK association with p53 (Fig. 1b). Although able to inhibit the formation of the JNK–p53 complex, p7 would not dissociate the preformed JNK–p53 complex (Fig. 1b). When added to a solid-phase kinase reaction, p7 inhibited p53 phosphorylation by JNK (Fig. 1b, bottom).
To test the effect of JNK–p53 association on p53 ubiquitination in vivo, we utilized BALB/3T3/12.1 cells (Harvey and Levine 1991); these cells express normal levels of Mdm2, which exhibited weak association with p53wt (not shown). Transfection of p7 or c7 cDNA to BALB/3T3/12.1 revealed that p7 (but not c7) inhibited the association of endogenous p53 with JNK (Fig. 1c). To determine the relationship between p53–JNK association and p53 ubiquitination, BALB/3T3/12.1 cells were cotransfected with hisp53 and HA-tagged ubiquitin. This approach allows one to follow the amount of the polyubiquitin chains formed on a substrate in vivo (Treier et al. 1994). This assay revealed that p7 (but not c7) transfection markedly decreased p53 ubiquitination (Fig. 1d).
Further support for JNK’s role in regulating p53 stability comes from the use of a p53 construct whose JNK-binding domain was deleted (p53Δp7). This mutant was not found in the complex with JNK as assayed by coimmunoprecipitation (Fig. 1c). Lack of JNK association with p53Δp7 in 10.1 p53 null cells coincided with prolonged half-life of p53Δp7 as compared with the p53wt (Fig. 1e). The importance of the 100–150 amino acid region for p53 stability was demonstrated previously as its fusion with a long-lived protein, ornithine decarboxylase, reduced stability of the chimeric protein (Li and Coffino 1996).
Because JNK association with its substrate is a prerequisite for targeting ubiquitination of c-Jun, JunB, and ATF2 (Fuchs et al. 1996, 1997), we determined whether outcompeting JNK with another substrate, c-Jun, would affect p53 stability. Transfecting increasing amounts of c-Jun led to a dose-dependent increase in p53 level (Fig. 2a, I). These changes were not observed in cells treated with the proteasome inhibitor lactacystin, suggesting that c-Jun does affect p53 stability (Fig. 2a, II). Importantly, transfection of amino-terminal Jun1–110 (which lacks DNA-binding capacity) or amino-terminal JNK1–202 increased p53 level (Fig. 2a, III) and decreased the amount of p53 that could be coimmunoprecipitated with JNK (Fig. 2a, III). Similar to its effect in mouse fibroblasts, JNK1–202 expression led to accumulation of endogenous p53wt in human melanoma and breast cancer cells (Fig. 2b). Whereas cotransfection of c-Jun inhibited in vivo ubiquitination of p53 (Fig. 1d), pulse-chase labeling with [35S]methionine revealed that the p53 half-life in cells that express high levels of c-Jun was extended to >8 hr (Fig. 2a, IV). Cotransfection of c-Jun with p53Δp7 did not elevate the expression of this p53 form (Fig. 3a), which lacks JNK-binding sites, suggesting that overexpression of c-Jun stabilizes p53 through a JNK-dependent mechanism. Together, these data suggest that c-Jun affects p53 stability by squelching JNK.
Figure 2.

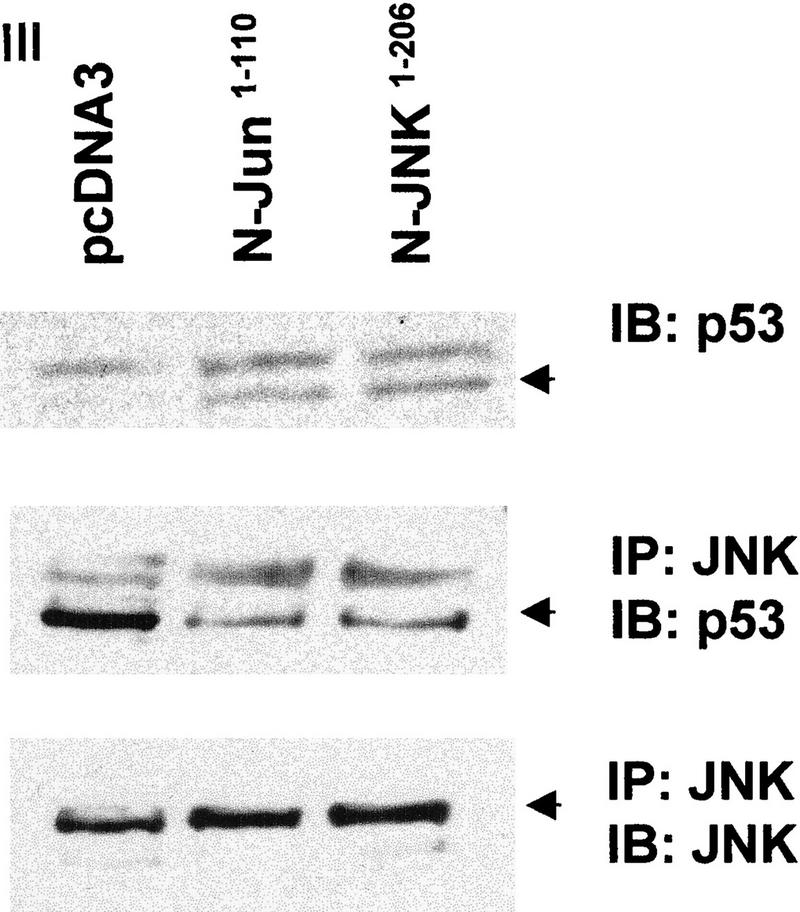





(a) Overexpression of c-Jun stabilizes expression of p53. (I) BALB/3T3/12.1 cells were transfected with increasing amounts (μg) of HA-tagged c-Jun (Treier et al. 1994) as indicated. The total amount of DNA in all transfections was equalized. Whole-cell extracts prepared 24 hr after transfection were analyzed via immunoblot using antibodies to p53 (top) and antibodies to HA (bottom, upper band). (II) BALB/3T3/12.1 cells were transfected with HA-tagged c-Jun (8 μg) and cells treated with 5 μm lactacystin (LC) for 8 hr as indicated. Extracts prepared 24 hr after transfection were analyzed as in I. (III) Amino-terminal Jun (N-Jun1–110) or amino-terminal JNK (N-JNK1–202) was transfected into BALB/3T3/12.1 cells, and proteins prepared from cells 24 hr after transfection were analyzed via immunoblot to determine p53 level (top). The complex between JNK and p53 was measured via immunoblots with antibodies to p53 on material that was first immunoprecipitated (from 800 μg of the same extracts) with antibodies to JNK (middle). (Bottom) Expression level of full-length JNK. Transfection with pcDNA3 was used as control. (IV) Half-life of p53 increases in c-Jun-overexpressing cells. pcDNA3 or c-Jun–HA was transfected into BALB/3T3/12.1 cells that were metabolically labeled with [35S]methionine for 10 min and chased with 2 mm unlabeled methionine for the indicated time periods. Endogenous p53 was immunopurified from the extracts prepared at the indicated time points of methionine chase. Immunoprecipitates were separated by SDS-PAGE and analyzed via autoradiography. (b) Amino-terminal JNK (N-JNK1–206) was transfected (3.5 or 7 μg , lanes 2 and 3, respectively) into breast cancer (MCF7)- or human melanoma (Lu-1205)-derived cell lines. p53 level was determined via immunoblot with antibodies pAb 421. (Bottom) Expression of HA-tagged N-JNK1–206, which was identified with the aid of HA antibodies. (c) Rat p53, human Flag-tagged JNK2183,185, and Jun1–110 were expressed in BALB/3T3/12.1 cells as indicated. p53 was immunoprecipitated with pAb 421 and analyzed by immunoblotting with anti-p53 polyclonal antibody. (d) MCF7 cells were transfected with Flag-tagged JNK2183,185 (left) or empty pcDNA3 vector (right) and 24 hr later treated with taxol (1 μm). Protein extracts, prepared 4, 8, and 24 hr after taxol addition, were analyzed by immunoblotting with anti-p53 polyclonal antibody (top) or M2 anti-Flag monoclonal antibody (IBI Kodak; bottom).
Figure 3.


(a) Effect of Mdm2 and c-Jun on levels of p5322,23 and p53Δp7. p53 (of the forms indicated) was cotransfected into p53 null (10.1) cells together with pCMV–Mdm2, pCMV–Jun–HA, or pCMV–Jun1–110 as indicated. p53 proteins were immunopurified using pAb 421 and analyzed via immunoblot probed with the p53 polyclonal antibody. (Bottom) Immunoblots controlling for the amount of Jun–HA expressed. (b) Analysis of JNK and Mdm2 association with p53 at different phases of the cell cycle. (Top) Swiss 3T3 cells were synchronized at the G0 phase. Cells were then fed 10% serum, and samples were collected after 0, 8, 20, and 24 hr representing the majority of cells at the G0, G1, S, and G2/M phases, respectively. (Bottom) Swiss 3T3 cells maintained under the same conditions also served as a source for analysis of p53 association with JNK or Mdm2.
The effect of Mdm2 on p53 degradation was demonstrated previously via overexpression of Mdm2 (Haupt et al. 1997; Kubbutat et al. 1997). Unlike Mdm2 expression, JNK is constitutively expressed at high levels, because of which further increase in JNK expression is expected to have a limited effect on the half-life of JNK-associated proteins. To overcome this problem, we have tested the effect of inactive mutant JNK2 construct (T183A, Y185F; Galcheva-Gargova et al. 1994) on elevated levels of p53. The mutant form of JNK2183,185 has attenuated the increase in p53 level mediated by p53 overexpression with or without Jun1–110 in BALB/3T3/12.1 mouse fibroblasts (Fig. 2c). Moreover, expression of mutant JNK2183,185 prevented taxol-induced accumulation of p53 in MCF7 breast cancer cells (Fig. 2d). These data suggest that phosphorylation-deficient JNK is capable of targeting p53 degradation. The kinase activity of JNK was also found to be dispensable for targeting ubiquitination of c-Jun (Fuchs et al. 1996).
To investigate the relationship between Mdm2 and JNK targeting of p53 degradation, the level of p53 mutants that cannot associate with Mdm2 (p5322,23; Lin et al. 1994) or JNK (p53Δp7; Fig. 1c) was monitored in cells that had been transfected with either c-Jun (to squelch JNK) or Mdm2. Although cotransfection of Mdm2 did not affect the level of p5322,23 (Fig. 3a; Kubbutat et al. 1997), cotransfection of c-Jun increased the level of the p5322,23 form (Fig. 3a), indicating that the stability of p53 that no longer responds to Mdm2 can still be affected by JNK. C-Jun had similar effects on the expression level of p53wt. Cotransfection of Mdm2 with p53Δp7 decreased the level of this p53 mutant, which is not affected by c-Jun overexpression. Both c-Jun and Mdm2 affected the level of cotransfected p53wt (Fig. 3a). These observations confirm that Mdm2 and JNK independently regulate p53 stability.
Because Mdm2 was found to associate with p53 and mediate its degradation, we explored the possible interplay between Mdm2 and JNK. We monitored Mdm2–p53 and JNK–p53 complexes at different phases of the cell cycle in Swiss 3T3 cells that were synchronized by serum starvation. Analysis at 0, 8, 20, and 24 hr after growth release (representing G0, G1, S, and G2/M phases of the cell cycle, respectively) revealed that JNK–p53 complexes were preferentially found in G0/G1, whereas Mdm2–p53 complexes were primarily found in S and G2/M phases of the cell cycle (Fig. 3b).
Further support for the role of JNK in targeting p53 ubiquitination was obtained through the use of a solid-phase in vitro ubiquitination assay. In this assay, beads-bound human hisp53 was incubated with targeting proteins, followed by extensive washing and subsequent ubiquitination using reticulocyte lysates that were immunodepleted of JNK and Mdm2. In this system, adding exogenously purified JNK as the targeting molecule resulted in increased ubiquitination of p53 as indicated by the smear at the top of the gel (Fig. 4). JNK’s ability to target p53 ubiquitination was inhibited when p7, but not c7, was added at the targeting step of our in vitro ubiquitination assay. P7’s ability to block JNK targeting of p53 ubiquitination suggests that its effects in vivo (Fig. 1c,d) are primarily mediated through interference with JNK interaction with the endogenous p7 domain on p53. Wild-type Mdm2, added as exogenously purified protein, efficiently targeted p53 ubiquitination, whereas mutant Mdm2, which cannot bind p53, failed to mediate this targeting (Fig. 4b). In contrast to its inhibitory effects on JNK, p7 did not inhibit Mdm2 targeting of p53 ubiquitination. Efficient targeting of p53 ubiquitination was also achieved using protein extracts from Mdm2/p53 null cells (Jones et al. 1996). However, targeting p53 ubiquitination by Mdm2/p53 null cell proteins could be inhibited by adding p7. Similarly, immunodepleting JNK from Mdm2/p53 null cell extracts significantly reduced p53-targeted ubiquitination. That JNK immunodepletion did not completely abolish p53 ubiquitination suggests that other p53 targeting molecules may exist in these JNK-depleted Mdm2 null extracts. Supplementing JNK-immunodepleted extracts with purified JNK restored targeting p53 for ubiquitination.
Figure 4.

JNK and extracts from Mdm2/p53 null cells target p53 ubiquitination in vitro. Targeting ubiquitination of hisp53 in vitro was performed using either purified components (JNK; Mdm2) or whole-cell extracts (WCE) from Mdm2/p53 null cells (Jones et al. 1996). Where indicated, whole cell extracts were immunodepleted with antibodies to JNK or with naive rabbit serum (NRS). Peptides (p7 or c7) were added (40 μm per reaction) in parallel to addition of the respective targeting protein(s), as indicated. In all cases hisp53-associated proteins were subjected to extensive washes before proceeding to the ubiquitination step, which utilizes reticulocyte lysates in the presence of Ub–HA (Fuchs et al. 1997). (I) Immunoblot using antibodies to HA. Polyubiquitinated p53 is marked on the left. (II) A Ponceau S stain of the same membrane to verify that equal amounts of p53 were used throughout the reaction.
To determine the degree of p53 ubiquitination mediated by JNK, we used in vitro-translated [35S]methionine-labeled p53 as the substrate. Quantifying the polyubiquitinated form of p53 revealed that within 30 min, ∼15% of p53 was targeted for ubiquitination by JNK (data not shown). Targeting of p53 ubiquitination can also be mediated by in vitro-translated wild-type or phosphorylation mutant (JNK2183,185) forms (data not shown), suggesting that JNK does not require its kinase activities to target p53 ubiquitination, as was observed previously with ATF2 and c-Jun (Fuchs et al. 1996, 1997). Similar targeting occurred when baculovirus-produced human or murine p53 was used as the substrate for these reactions (not shown). These data suggest that via its association, JNK directly targets p53 ubiquitination.
In sum, this study demonstrates that in nonstressed normally growing cells, p53 ubiquitination and degradation are also mediated by JNK. In both Mdm2/p53 null cells and BALB/3T3/12.1 cells, JNK appears to be the principal regulator of p53 ubiquitination. Our data also suggest that Mdm2 and JNK represent two independent pathways for targeting p53 stability. The fact that Mdm2–p53 complexes were found at S and G2/M phases of the cell cycle, but JNK–p53 complexes were in G0/G1 phases, suggests that through their targeting of p53 stability in nonstressed cells, Mdm2 and JNK may regulate different cellular functions of p53 during normal cell growth. Our data do not preclude the existence of other targeting molecules, as immunodepletion of JNK from Mdm2 null cell lysates did not completely abolish p53 targeting for in vitro ubiquitination.
In line with our previous studies, stress-mediated JNK activation inversely correlates with targeting of its associated proteins, as shown here for p53. JNK activation via MEKK1 results in p53 phosphorylation, inhibition of Mdm2 association, and p53 ubiquitination as reflected by a prolonged p53 half-life (Fuchs et al. 1998b). Yet because JNK from UV-treated cells can still associate with recombinant p53 in vitro, it is possible that in vivo p53 phosphorylation in response to stress requires multiple stress kinases, which mediate sufficient changes to p53 conformation to result in p53 dissociation from its targeting molecules.
The emerging model supported by our current data suggests that p53 stability is affected by JNK independently of Mdm2 in a cell cycle-dependent manner. In this model, prolonged half-life, which is characteristic of mutant forms of p53, could be attributed to lack of p53 association with one or both targeting molecules. JNK is likely to be among the growing number of adapter molecules that participate in the formation of the E3 ubiquitin/ligase complex. Such adapters were shown to have key roles in substrate recognition and targeting ubiquitination of yeast CDK inhibitor protein Sic1 (Feldman et al. 1997; Skowyra et al. 1997). A similar mechanism was described for HPV-mediated p53 degradation through the recruiting of E6–AP ubiquitin ligase with the aid of the E6 viral protein (Huibregtse et al. 1991). As a targeting molecule that mediates the stability of the oncogene, c-Jun, and the tumor suppressor p53, JNK emerges as a key regulator of cell growth in normally growing cells.
Materials and methods
Preparation of JNK and Mdm2
JNK was purified from 600 mg of protein extract prepared from UV-irradiated (60 J/m2) BALB/3T3 cells as described (Adler et al. 1995). A purified form of JNK (54 kD, as revealed by immunoblotting) was used for in vitro ubiquitination assays (∼1 μg/assay). Mdm2 was prepared from Sf9 cells infected with baculovirus-expressing human Mdm2 cDNA in either wild-type or mutant (Δ1–150) forms and purified as described previously (Chen et al. 1996). The purity of Mdm2 and JNK was confirmed as single bands seen on silver-stained gels.
Immunoprecipitations/immunodepletions
JNK was immunodepleted from reticulocyte lysates and protein extracts of Mdm2/p53 null cells by incubating 700 μg of proteins with 1 μg of antibody to JNK (C-17; Santa Cruz) for 16 hr at 4°C. Protein A/G beads were added to this mixture for 2 hr at 4°C, followed by quick centrifugation. Supernatants were used as JNK-depleted proteins. Monoclonal antibodies were used for immunoprecipitation (clone 333, PharMingen) and for immunodepletion of JNK (clone 666) as described (Fuchs et al. 1997).
Construction of expression vectors
To express peptides in vivo, oligonucleotides bearing the sequence of p7 (VPSQKTYHGSYGFRLGFLHSG) or c7 control peptide (SPPVVPSQSKSTSYGQGYRF) were cloned, respectively, in-frame into pcDNA3 that carries the penetratin sequence (RQIKIWFQNRRMKWKK), followed by the sequence encoding the HA tag (YPYDVPDYASL). Using antibodies to HA enables detection of these fusion peptides (not shown). To generate a p53 expression vector that is histidine-tagged, cDNA of rat p53 was cloned into a pcDNA3 vector by PCR using 5′ primer that encodes amino-terminal 6xhis, thus generating the hisp53 fusion protein. To generate p53 whose p7 sequence has been deleted, the rat cDNA of p53 (pCMV–hisp53) was deleted from amino acids 95–114 (corresponding to amino acids 97–116 of human p53, which constitute the p7 domain) using site-directed mutagenesis (Quick Change, Stratagene), resulting in the p53Δp7 construct. Amino-terminal JNK (amino acids 1–202) and amino-terminal c-Jun (amino acids 1–110) constructs were cloned by PCR into pcDNA3. The integrity and expression of all constructs were verified on the basis of sequencing and immunoblots.
In vitro ubiquitination assay and transfection
The in vitro ubiquitination assay was performed as described (Fuchs et al. 1997). Transfection was performed via lipofection (DOTAP) into subconfluent 60-mm plates. In all cases, equal amounts of DNA were transfected (by adjusting the concentrations of respective constructs with empty mammalian expression vector).
Acknowledgments
We thank Bin Xie for technical assistance, Fred Friedman for peptide synthesis, Z.Q. Pan for Sf-9 cells, T. Soussi for p53 construct, D. Bohmann for ubiquitin–HA and Jun–HA vectors, Roger Davis and Michael Karin for JNK constructs, James Manfredi for the Taxol reagent, Craig Monnel of PharMingen for the JNK antibodies, and Arnold Levine for Mdm2 reagents. We also thank Stuart Aaronson, Victor Fried, and Ed Johnson for critically reading the manuscript. These studies were supported in part by National Cancer Institute grants CA59908 and CA78419 to Z.R.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL zeev_ronai@smtplink.mssm.edu; FAX (212) 849-2446.
References
- Adler V, Pincus MR, Brandt-Rauf PW, Ronai Z. Complexes of p21RAS with JUN N-terminal kinase and JUN proteins. Proc Natl Acad Sci. 1995;92:10585–10589. doi: 10.1073/pnas.92.23.10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler V, Pincus MR, Minamoto T, Fuchs S, Bluth MJ, Brandt-Rauf PW, Friedman FK, Robinson RC, Chen JM, Wang XW, Harris CC, Ronai Z. Conformation-dependent phosphorylation of p53. Proc Natl Acad Sci. 1997;94:1686–1691. doi: 10.1073/pnas.94.5.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JP, Pagano M. p53 stability. Biochem Biophys Acta. 1997;1332:1–6. doi: 10.1016/s0304-419x(96)00048-0. [DOI] [PubMed] [Google Scholar]
- Canman CE, Gilmer TM, Coutts SB, Kastan MB. Growth factor modulation of p53-mediated growth arrest versus apoptosis. Genes & Dev. 1995;9:600–611. doi: 10.1101/gad.9.5.600. [DOI] [PubMed] [Google Scholar]
- Chen J, Wu X, Lin J, Levine AJ. Mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol Cell Biol. 1996;16:2445–2452. doi: 10.1128/mcb.16.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman RMR, Correl CC, Kaplan KB, Deshaies RJ. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Friedlander P, Legros Y, Soussi T, Prives C. Regulation of mutant p53 temperature-sensitive DNA binding. J Biol Chem. 1996;271:25468–25478. doi: 10.1074/jbc.271.41.25468. [DOI] [PubMed] [Google Scholar]
- Fritsche M, Haessler C, Brandner G. Induction of cellular p53 accumulation of the tumor-suppressor protein by DNA-damaging agents. Oncogene. 1993;8:307–318. [PubMed] [Google Scholar]
- Fuchs SY, Dolan L, Davis RJ, Ronai Z. Phosphorylation-dependent targeting of c-Jun ubiquitination by Jun N-kinase. Oncogene. 1996;13:1531–1535. [PubMed] [Google Scholar]
- Fuchs SY, Xie B, Adler V, Fried VA, Davis RJ, Ronai Z. c-Jun NH2-terminal kinases target the ubiquitination of their associated transcription factors. J Biol Chem. 1997;272:32163–32168. doi: 10.1074/jbc.272.51.32163. [DOI] [PubMed] [Google Scholar]
- Fuchs, S.Y., V. Adler, T. Buschmann, X. Wu, and Z. Ronai. 1998a. Mdm2 association with p53 targets its ubiquitination. Oncogene (in press). [DOI] [PubMed]
- Fuchs, S.Y., V. Adler, M.R. Pincus, and Z. Ronai. 1998b. MEKK1/JNK signaling stabilizes and activates p53. Proc. Natl. Acad. Sci. (in press). [DOI] [PMC free article] [PubMed]
- Galcheva-Gargova Z, Derijard B, Wu I-H, Davis RJ. An osmosensing signal transduction pathway in mammalian cells. Science. 1994;265:806–808. doi: 10.1126/science.8047888. [DOI] [PubMed] [Google Scholar]
- Hall PA, McKee PH, Menage HP, Dover R, Lane DP. High levels of p53 protein in UV-irradiated normal human skin. Oncogene. 1993;8:203–207. [PubMed] [Google Scholar]
- Harris CC. Structure and function of the p53 tumor suppressor gene: Clues for rational cancer therapeutic strategies. J Natl Cancer Inst. 1996;88:1442–1455. doi: 10.1093/jnci/88.20.1442. [DOI] [PubMed] [Google Scholar]
- Harvey DM, Levine AJ. P53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes & Dev. 1991;5:2375–2385. doi: 10.1101/gad.5.12b.2375. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–298. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10:4129–4135. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SN, Sands AT, Hancock AR, Vogel H, Douchouer LA, Linke SP, Wahl GM, Bradley A. The tumorigenic potential and cell growth characteristics of p53-deficient cells are equivalent in the presence or absence of Mdm2. Proc Natl Acad Sci. 1996;93:14106–14111. doi: 10.1073/pnas.93.24.14106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. Identification of p53 as sequence-specific DNA-binding protein. Science. 1991;252:1708–1711. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- Kubbutat MHG, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–302. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Levine A. P53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Li Z, Coffino P. Identification of a region of p53 that confers liability. J Biol Chem. 1996;271:4447–4451. doi: 10.1074/jbc.271.8.4447. [DOI] [PubMed] [Google Scholar]
- Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids in the p53 amino terminal domain are required for transcriptional activation, binding to mdm2 and the adenovirus 5 E1B 55kd protein. Genes & Dev. 1994;8:1235–1246. doi: 10.1101/gad.8.10.1235. [DOI] [PubMed] [Google Scholar]
- Maheswaran S, Englert C, Bennett P, Heinrich G, Haber DA. The WT1 product stabilizes p53 and inhibits p53-mediated apoptosis. Genes & Dev. 1995;9:2143–2156. doi: 10.1101/gad.9.17.2143. [DOI] [PubMed] [Google Scholar]
- Maki CG, Huibregtse JM, Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res. 1996;56:2649–2654. [PubMed] [Google Scholar]
- Maltzman W, Czyzyk L. UV irradiation stimulate levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol. 1984;4:1689–1694. doi: 10.1128/mcb.4.9.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Waldman T, He C, Kinzler KW, Vogelstein B. Genetic determinants of p53-induced apoptosis and growth arrest. Genes & Dev. 1996;10:1945–1952. doi: 10.1101/gad.10.15.1945. [DOI] [PubMed] [Google Scholar]
- Querido E, Marellus RC, Lai A, Charbonneau R, Teodoro JG, Ketner G, Branton PE. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J Virol. 1997;71:3788–3798. doi: 10.1128/jvi.71.5.3788-3798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reihsaus E, Kohler M, Kraiss S, Oren M, Montenarh M. Regulation of the level of the oncoprotein p53 in non-transformed and transformed cells. Oncogene. 1990;5:137–145. [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damaged induced phosphorylation of p53 alleviates inhibition by Mdm2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes & Dev. 1997;11:3471–3481. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Tiemann F, Zerrah J, Deppert W. Cooperation of simian virus 40 large and small T antigens in metabolic stabilization of tumor suppressor p53 during cellular transformation. J Virol. 1995;69:6115–6121. doi: 10.1128/jvi.69.10.6115-6121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treier M, Staszewski L, Bohmann D. Ubiquitin-dependent c-jun degradation in vivo is mediated by the δ domain. Cell. 1994;78:787–798. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- Zambetti GP, Bargonetti J, Walker K, Prives C, Levine AJ. Wild-type p53 mediates positive regulation of gene expression through a specific DNA sequence element. Genes & Dev. 1992;6:1143–1152. doi: 10.1101/gad.6.7.1143. [DOI] [PubMed] [Google Scholar]
- Zhan Q, Carrier F, Fornace AJ. Induction of cellular p53 activity by DNA-damaging agents and growth arrest. Mol Cell Biol. 1993;13:4242–4250. doi: 10.1128/mcb.13.7.4242. [DOI] [PMC free article] [PubMed] [Google Scholar]