

Abstract
Genetic studies in worms, flies, and humans have implicated the presenilins in the regulation of the Notch signaling pathway and in the pathogenesis of Alzheimer's Disease. There are two highly homologous presenilin genes in mammals, presenilin 1 (PS1) and presenilin 2 (PS2). In mice, inactivation of PS1 leads to developmental defects that culminate in a perinatal lethality. To test the possibility that the late lethality of PS1-null mice reflects genetic redundancy of the presenilins, we have generated PS2-null mice by gene targeting, and subsequently, PS1/PS2 double-null mice. Mice homozygous for a targeted null mutation in PS2 exhibit no obvious defects; however, loss of PS2 on a PS1-null background leads to embryonic lethality at embryonic day 9.5. Embryos lacking both presenilins, and surprisingly, those carrying only a single copy of PS2 on a PS1-null background, exhibit multiple early patterning defects, including lack of somite segmentation, disorganization of the trunk ventral neural tube, midbrain mesenchyme cell loss, anterior neuropore closure delays, and abnormal heart and second branchial arch development. In addition, Delta like-1 (Dll1) and Hes-5, two genes that lie downstream in the Notch pathway, were misexpressed in presenilin double-null embryos: Hes-5 expression was undetectable in these mice, whereas Dll1 was expressed ectopically in the neural tube and brain of double-null embryos. We conclude that the presenilins play a widespread role in embryogenesis, that there is a functional redundancy between PS1 and PS2, and that both vertebrate presenilins, like their invertebrate homologs, are essential for Notch signaling.
Keywords: Presenilin, notch, somite segmentation, Alzheimer's Disease
The presenilins belong to a family of highly conserved proteins that localize largely to the endoplasmic reticulum and Golgi apparatus (Cook et al. 1996; Kovacs et al. 1996; Walter et al. 1996; De Strooper et al. 1997). The presenilins have also been found in the plasma membrane, in the nuclear envelope, and associated with kinetochores and centrosomes (Dewji and Singer 1997; Li et al. 1997). They are predicted to have between 6 and 8 transmembrane domains and a large hydrophilic loop that, along with the amino and carboxyl termini, is oriented toward the cytoplasm (Doan et al. 1996; Li and Greenwald 1996, 1998). The presenilins exist in a cleaved form as an amino-terminal fragment (NTF) and a carboxy-terminal fragment (CTF) (Thinakaran et al. 1996). They are widely expressed in mammalian embryonic development and in adult tissues, with high expression in the cell bodies and dendrites of neurons in the mammalian central nervous system (CNS) (Cook et al. 1996; Lee et al. 1996). One presenilin gene (Dps) has been identified in Drosophila melanogaster, two genes (Sel-12 and Hop-1) have been isolated in Caenorhabditis elegans, and two homologs, called presenilin-1 (PS1) and presenilin 2 (PS2), have been cloned in mammals.
Insights into the role of the presenilins in normal and pathological processes have emerged from both genetic and biochemical analyses. Biochemical interactions between the presenilins and a variety of proteins have been reported, although the significance of these remains to be determined. For example, several groups have reported independently that the presenilins bind and affect the stabilization of members of the β-catenin family (Zhou et al. 1997; Murayama et al. 1998; Yu et al. 1998; Zhang et al. 1998b; Levesque et al. 1999; Nishimura et al. 1999a). In addition, cytoskeleton- and microtubule-associated proteins, calcium-binding proteins, and G-proteins and GTPases have all been reported to interact with the presenilins (Buxbaum et al. 1998; Smine et al. 1998; Takashima et al. 1998; Zhang et al. 1998a; Dumanchin et al. 1999; Stabler et al. 1999). There have also been studies suggesting that the presenilins directly associate with the amyloid precursor protein (APP) and the Notch receptor (Weidemann et al. 1997; Xia et al. 1997; Ray et al. 1999).
Genetic studies of families with familial Alzheimer's disease (FAD), an autosomal dominant disorder of the CNS have implicated PS1, PS2, and APP in the pathogenesis of this disease (Chartier-Harlin et al. 1991; Murrell et al. 1991; Levy-Lahad et al. 1995; Rogaev et al. 1995; Sherrington et al. 1995). Nearly 50% of FAD patients bear mutations in one of the presenilin genes, whereas <3% of patients bear APP mutations (Blacker and Tanzi 1998; Nishimura et al. 1999b). Genetic studies in C. elegans and D. melanogaster have also demonstrated a role for the presenilins as positive regulators of the Notch pathway (Levitan and Greenwald 1995; Li and Greenwald 1997; Struhl and Greenwald 1999; Ye et al. 1999). Interestingly, FAD patients have elevated Notch-1 expression (Berezovska et al. 1998). Thus, Notch signaling might play a role in this disease.
Analyses of presenilin mutations in mammalian cells and flies have shown that the presenilins function in the processing of membrane-associated proteins such as the Notch-1 receptor and APP (De Strooper et al. 1998, 1999; Struhl and Greenwald 1999; Wolfe et al. 1999; Ye et al. 1999). APP sustains a series of cleavage events, which can culminate in the release of a toxic product, called the Abeta1-42 peptide, that is a major component of the extracellular plaques that are the hallmark of AD. The presenilins appear to control aspects of APP processing, in that cells lacking PS1 make less of the Abeta1-42 peptide, whereas mice bearing FAD-linked PS1 alleles make more (Duff et al. 1996; De Strooper et al. 1998).
The Notch signaling pathway controls embryonic cell-fate decisions in a variety of cell lineages in flies, worms, and mammals (Artavanis-Tsakonas et al. 1999). There is only one Notch gene in flies, whereas there are two and four Notch genes in worms and mammals, respectively. The molecular mechanisms of Notch signaling are conserved in different species and are initiated by the interaction of the Notch receptor with its ligands [Delta-like-1 (Dll1) or -3, or Jagged-1 or -2, in mammals]. This interaction induces a series of proteolytic cleavage events of the Notch receptor. These processing steps culminate in the release of a Notch intracellular fragment that translocates to the nucleus and complexes and activates RBP-Jκ, a transcription factor regulating a number of downstream genes, including HES-5 and Dll1 (Tamura et al. 1995; Lu and Lux 1996; Artavanis-Tsakonas et al. 1999; Ohtsuka et al. 1999). Apparently, the presenilins positively regulate the release of the Notch intracellular fragment (De Strooper et al. 1999; Struhl and Greenwald 1999; Ye et al. 1999).
Consistent with genetic and biochemical studies in flies and worms that indicate a role for the presenilins in regulating the Notch pathway, a targeted inactivation of the PS1 gene in mice results in somite disorganization characteristic of mice harboring single mutations in genes that comprise the Notch pathway (Conlon et al. 1995; Oka et al. 1995; Shen et al. 1997; Wong et al. 1997; Evrard et al. 1998; Zhang and Gridley 1998). In addition to the somite defects, PS1-null mice exhibit rib-cage abnormalities, and hemorrhaging and cell-loss in the forebrain, which result in a perinatal lethality. Interestingly, the developmental abnormalities in mutant PS1-null mice are mild in comparison with those described in Notch-1 null mice (Conlon et al. 1995). Furthermore, the expression of Dll1, normally negatively regulated by the Notch signaling pathway, was reported to be decreased in PS1-null mice (Wong et al. 1997). If Notch signaling is perturbed by PS1-loss, Dll1 expression would be expected to be up-regulated in the PS1-null mice. Together, these observations raise the likely possibility that PS2 is partially compensating for the loss of PS1 in maintaining Notch signaling in these mutant mice. Genetic studies in C. elegans have indicated that there is a genetic interaction between the two worm presenilin genes, Sel-12 and Hop-1 (Li and Greenwald 1997; Westlund et al. 1999). Thus, it is likely that the mammalian presenilins may also have redundant functions, based on their sequence similarity and overlapping patterns of expression (Lee et al. 1996). To test this hypothesis, we have generated PS2-null mice and PS1/PS2 double-null mice. Here, we provide evidence that murine PS1 and PS2 have overlapping, but nonidentical functions and that loss of both known mammalian presenilins leads to profound and widespread developmental defects.
Results
Targeted inactivation of the murine PS2 gene
The PS2 gene was disrupted by homologous recombination in embryonic stem (ES) cells by replacing four amino acids (RCYK) from exon 5, which includes part of the second transmembrane region, with the neo resistance gene (Fig. 1A). ES cell clones that had sustained a homologous recombination event were identified by Southern blot analysis with 3′ external (Fig. 1B) and 5′ external (not shown) probes using HindIII (Fig. 1B) or EcoRI (not shown). Subsequent screening of the resulting mice by PCR confirmed this conclusion, as DNA from PS2-null mice did not give rise to any PCR fragments corresponding to the wildtype locus (Fig. 1C). RT-PCR and sequencing analyses confirmed that PS2−/− cells only expressed PS2 transcripts lacking exon 5 (not shown). Any translation product from these transcripts would be predicted to encode only the first 99 amino acids of the PS2 protein (retaining only one intact transmembrane domain) plus 5 additional nonsense residues, as the deletion of exon 5 should give rise to out-of-frame transcripts. To confirm the absence of PS2 protein in the PS2−/− mice, brain extracts from PS2+/+ and PS2−/− mice were examined by Western blot analysis using three different antibodies directed against the hydrophilic loop and amino terminus. As expected, neither the PS2 CTF (Fig. 1D) nor the PS2 NTF (not shown) were detected in PS2−/− mice but were observed in PS2+/+ (Fig. 1D) and PS2+/− mice (not shown). We conclude that the PS2−/− mice are PS2-null.
Figure 1.

Disruption of the murine PS2 locus by homologous recombination in ES cells. (A) Map of the murine PS2 locus showing the major start of translation in exon 3. Southern analysis using genomic DNA digested with HindIII followed by hybridization with 3′ external or 5′ external probes (█), would yield a 12-kb fragment from the untargeted locus and a 5.6-kb from the targeted locus. Primers (arrows) designed to amplify both the unmodified and the modified loci were used for genotyping by PCR. (NEO) PGK–Neomycin resistance gene; loxP sites (▸) flank the NEO gene; (TK) thymidine kinase gene; (E) EcoRI, (H) HindIII, (X) XbaI, (C) ClaI, (S) StuI, (B) BamHI, (K) KpnI. (B) Southern analysis of HindIII-digested genomic DNA from targeted ES cell clones probed with a PS2 3′ external probe, showing the expected sized fragment for the correct integration of the PS2 targeting vector into the murine PS2 locus in two independent cell lines designated by +/−. (C) PCR analysis on tail DNA from weaning-age mice from PS2 +/− intercrosses using primers indicated in A. Note the absence of the PCR fragment corresponding to the wild-type locus in −/− lanes and the strong band from the doubly targeted PS2 loci. Band sizes are indicated at right. (D) Western blot analysis of brain extracts from PS2+/+ and PS2−/− mice blotted with a PS2 antibody. (CTF) Carboxy-terminal fragment); (C) control extract from human HEK 293t cells transfected with an expression construct driving a mutated (VG) PS2 cDNA. (S) standard lane (NEB). The size of the standard band included in the panel is indicated at right.
Generation of mice lacking both PS1 and PS2
Crosses between the heterozygous PS2+/− mice, maintained on several genetic backgrounds (CD1, 129, and 129/C57Bl6/J F1) gave rise to expected Mendelian ratios of phenotypically normal PS2-null mice (not shown). To date, we have not observed any defects in adult mice that are PS2-null, suggesting that PS1 is compensating for the loss of PS2. To examine this further, we generated doubly heterozygous PS1+/−; PS2+/− mice (on a C57Bl6/129 F1 hybrid background) by crossing PS2+/− mice (maintained on a 129 background) described here with 129/C57B6j F1 PS1+/− mice described previously (Wong et al. 1997). The doubly heterozygous animals exhibited no apparent phenotype. Analysis of >290 progeny [at embryonic day 8(E8) to E9.5] from intercrosses of these doubly heterozygous or doubly heterozygous crossed to PS1+/−; PS2−/− mice demonstrated that loss of both or even one copy of PS2 on a PS1−/− background resulted in an early embryonic lethality prior to E13.5 (Table 1). However, loss of one copy of PS1 on a PS2-null background had no consequence (Table 1). Thus, vertebrate presenilins, like their homologs in C. elegans (Li and Greenwald 1997; Westlund et al. 1999), genetically interact and are functionally redundant, and PS2 is haploinsufficient in the absence of PS1. We also conclude that the absence of any discernible phenotype in the PS2−/− mice is due to the presence of PS1.
Table 1.
Presenilin mutant embryos
| Embryonic stage
|
Total no. of pups
|
PS1−/− PS2−/−
|
PS1−/− PS2+/−
|
PS1−/− PS2+/+
|
PS1+/+ PS2−/−
|
PS1+/− PS2−/−
|
PS1+/− PS2+/−
|
All other genotypesa
|
|
|---|---|---|---|---|---|---|---|---|---|
| Cross:
|
PS1+/−;PS2+/− × PS1+/−;PS2+/−
|
||||||||
| E8–E8.75 | 93 | 8b | 15b | 6 | 5 | 7 | 25 | 27 | |
| E9–E9.5 | 79 | 5c | 6b | 10 | 8 | 11 | 17 | 22 | |
| E13–E15.5 | 44 | 0 | 0 | 4 | 2 | 8 | 16 | 14 | |
| Cross: | PS1+/−;PS2−/− × PS1+/−;PS2+/− | ||||||||
| E8–E8.75 | 121 | 16b | 12b | 0 | 14 | 29 | 36 | 14 | |
| E9–E9.5 | 19 | 3c | 4b | 0 | 1 | 3 | 4 | 4 | |
Embryos with genotypes not detailed appeared wild type.
Embryos grossly abnormal (see text).
Embryos dead upon dissection.
Notch signaling defects in embryos lacking all presenilins
At E8.5–E9, double-null embryos failed to show somite segmentation and exhibited a kinked neural tube (n = 16) (Fig. 2I,L), whereas PS1−/−; PS2+/− embryos showed a more variable phenotype, ranging from no somite segmentation (n = 15) to the formation of highly disorganized somites (n = 5) (Fig. 2H,K). Mild somite disorganization defects as have been described previously (Shen et al. 1997; Wong et al. 1997) were observed in PS1-null, and none were observed in PS2-null embryos at E8.5 (n = 6, n = 15, respectively; data not shown) and at E13.5 (n = 12, n = 14, respectively; data not shown). To determine whether the somite abnormalities in the double-null embryos were attributable to a paraxial mesoderm defect, we tested the expression of Dll1 (Bettenhausen et al. 1995) in presomitic embryos. The level and spatial pattern of Dll1 expression in the paraxial mesoderm of E8 double-null embryos was similar to that in PS1+/−; PS2+/− embryos (n = 3) (Fig. 3A,B). However, at E8.5, whereas Dll1 expression remained high in the paraxial mesoderm of double-null embryos, Dll1 expression was undetectable in the segmental plate (n = 4) (Fig. 3D). Instead, ectopic expression of Dll1 was observed in the neural tube, hindbrain, and forebrain of double-mutant embryos (n = 4) (Fig. 3D). Ectopic neural tube Dll1 expression was also observed in some PS1−/−; PS2+/− (n = 4) and PS1-null embryos (n = 3) (Fig. 2F,H), whereas others of these genotypes showed the expected somitic or segmental plate expression at E9 at levels comparable to those in PS1+/−; PS2+/− embryos (Fig. 2K,L). These observations should be contrasted with a previous report of decreased Dll1 expression in PS1-null mice (Wong et al. 1997).
Figure 2.
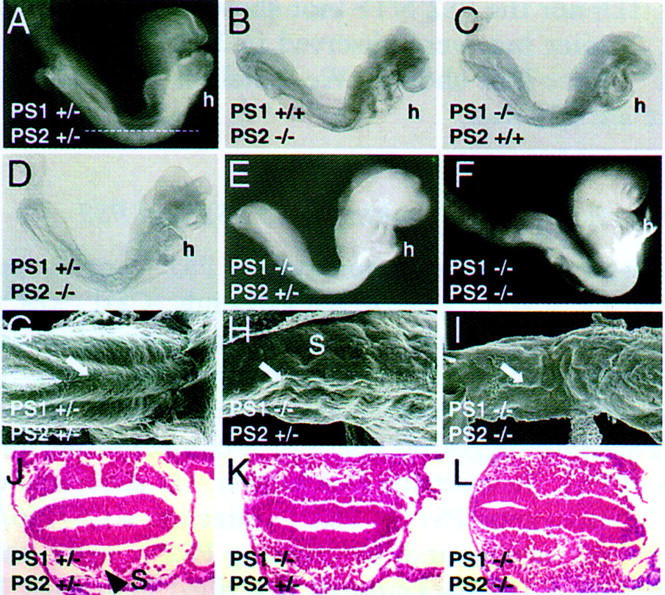
Phenotype of presenilin-mutant embryos. E8.5 PS1+/−; PS2+/−(A), PS1+/+; PS2−/−, (B) PS1−/−; PS2+/+(C), and PS1+/−; PS2−/− (D) embryos, which look normal. Note the normal development of the heart (h). The line in A shows the approximate position of the sections shown in J–L. E8.5 PS1−/−; PS2+/− (E) and PS1−/−; PS2−/− (F) embryos, which show no somite segmentation (see K,L), an underdeveloped heart (h, see Fig. 5) and an open anterior neuropore (see Fig. 4). (G–I) SEM of E9 PS1+/−; PS2+/− (G), PS1−/−; PS2+/−(H), and PS1−/−; PS2−/− (I) embryos, showing the neural tube (arrow). Tail bud is on the left. (S) Somites. Transverse sections through E8.5 PS1+/−; PS2+/− (J), PS1−/−; PS2+/−, and PS1−/−; PS2−/− embryos, showing disorganization of the trunk paraxial mesoderm and a kinked neural tube in the presenilin mutants, whereas segmented somites (S, arrowhead) are evident in the PS1+/−; PS2+/− embryo (J).
Figure 3.
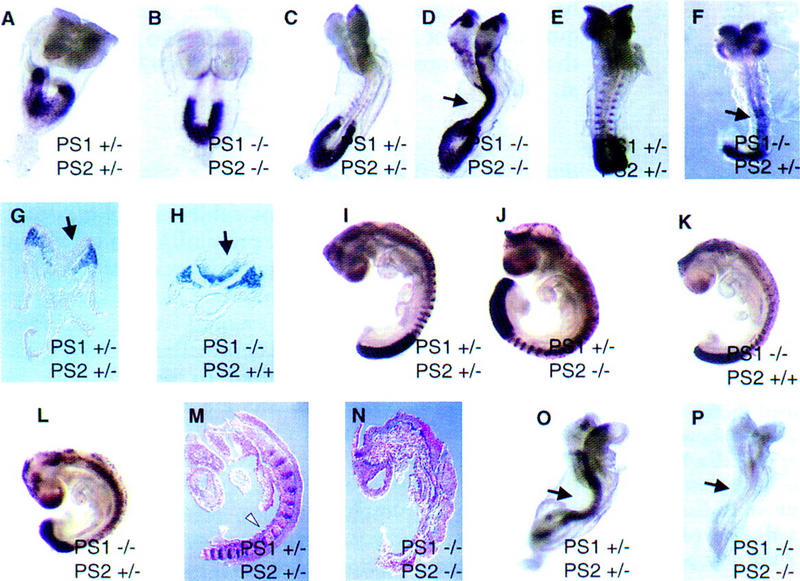
Notch-associated defects of presenilin mutant embryos. Whole-mount Dll1 expression in E8 (A,B) and E8.5 (C,D) PS1+/−; PS2+/−, and PS1−/−; PS2−/− embryos showing normal expression in the paraxial mesoderm, but ectopic upregulation in the neural tube (arrow) and head of E8.5 double-null embryos. Note the absence of somite expression in the double-null embryo at E8.5. (E–F) Dll1 expression in E8.5 PS1+/−; PS2+/−, and PS1−/−; PS2+/− embryos showing ectopic expression in the neural tube (arrow) of the PS1−/−; PS2+/− embryo (F). (G–H) Transverse sections taken from the tail region of E8.5 double heterozygous and PS1-null embryos hybridized with Dll1 showing expression in the paraxial mesoderm and the neural tube (arrow) of the PS1-null mutant. (I–L) Dll1 expression in E9 presenilin mutant embryos showing PS1−/−; PS2+/− and PS1-null embryos that did not show ectopic expression in the neural tube (K,L). Note the irregularity of Dll1 signal in the presumptive segmentalplate of the PS1−/−; PS2+/− embryo (L).(M–N) UncX4.1 expression in presenilin-mutant embryos. E9 embryos were purposely overstained to ensure detection of any signal on sections from double-null embryos. Saggital sections show UncX4.1 expression in the posterior somitic compartments (white arrowhead) of the normal embryo (M) and no expression in the presenilin double-null embryo (N). (O) Hes-5 expression in the neural tube (arrow) of a normal E8.5 embryo. (P) Absence of Hes-5 expression in an E8.5 presenilin double-null embryo (neural tube is denoted with arrow).
To confirm the absence of somites in the double-null embryos, we analyzed expression of UncX4.1, a marker for posterior somitic compartments (Mansouri et al. 1997). E9 embryos were purposely overstained to ensure detection of any UncX4.1 expression in double-null embryos (Fig. 3M,N). Expression of UncX4.1 was undetectable in the segmental plate of double-null embryos (n = 3) (Fig. 3N), concurrent with a lack of somitic identity in the mutants. Loss of somite identity was confirmed by the lack of Dll1 expression in a metameric somitic pattern in presenilin double-null embryos (Fig. 2D). In addition, the expression of murine Twist, a mesodermal marker, was also absent from presomitic mesoderm adjacent to the neural tube in double-null embryos (data not shown).
Because other Notch pathway mutant mice show ectopic Dll1 expression in the neural tube (de la Pompa et al. 1997), it is likely that Notch signaling was ablated by the loss of both presenilin genes. To test this hypothesis further, we analyzed expression of HES-5, a murine homolog of Enhancer of split, a gene that lies in the Notch pathway in D. melanogaster, and is down-regulated in Notch pathway mutant mice (de la Pompa et al. 1997). HES-5 expression was reduced in PS1−/− and PS1−/−; PS2+/− embryos (data not shown), whereas it was completely abolished in the presenilin double-null embryos (n = 3) (Fig. 3, cf. O and P), consistent with lack of Notch signaling caused by presenilin loss.
Pleiotropic defects of presenilin double-null embryos
The somite segmentation defects of presenilin double-null embryos were anticipated in light of the fact that mice carrying mutations in genes that comprise the Notch pathway also exhibit similar abnormalities (Conlon et al. 1995; Oka et al. 1995; Evrard et al. 1998; Zhang and Gridley 1998). However, presenilin double-null embryos also exhibited a wide range of additional phenotypes. For example, there was a delay in the closure of the anterior neuropore at E8.5 (n = 16) (Fig. 4A) and E9 (n = 3) (Fig. 5C), as well as cell loss in the mesenchyme of the presumptive midbrain at E8.5 (n = 4) (Fig. 4, cf. B and C). Furthermore, presenilin double-null embryos had underdeveloped second branchial arches (Fig. 5C) and unlooped hearts at E8.5 (n = 16) and E9 (n =4) (Fig. 5C,F). Some PS1−/−; PS2+/− exhibited heart looping delays at E8.5 and E9 (n = 12 and 5, respectively) (Fig. 5B,E), whereas others exhibited partial heart looping at E9 (n = 2) (not shown), and no cardiac defects were observed in PS1-null (Shen et al. 1997; Wong et al. 1997) or PS2-null embryos (Fig. 5A). Cardiac looping defects generally lead to cardiovascular failure and probably contribute to the lethality in the double-null embryos at E9–E9.5. In addition, we observed chorioallantoic fusion defects in double-null (n = 10), but not PS1−/−; PS2+/− embryos (n = 14) (not shown), which would also lead to the demise of the double-null embryos. Our data do not exclude the possibility that the heart looping, branchial arch and neuropore closure defects are due to a developmental delay arising from a primary defect of mesoderm patterning.
Figure 4.
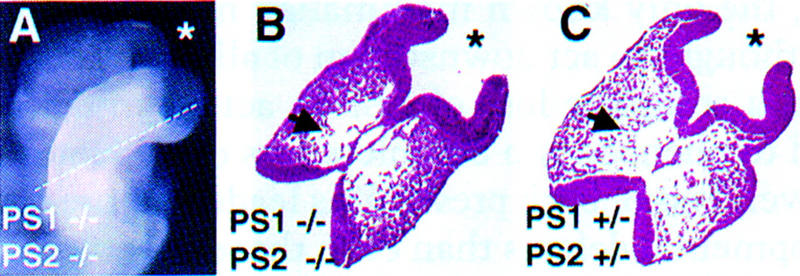
Brain abnormalities in presenilin double-null embryos. (A) Close-up of the head region of an E8.5 double-null embryo showing an open anterior neuropore and an apparent expanded forebrain (*). The line demarcates the approximate position of the section shown in B. (B) Section through the head of a PS1−/−; PS2−/− embryo, which shows loss of mesenchyme cells (arrow). (*) Forebrain. (C) Corresponding section taken from a normal embryo.
Figure 5.

Heart and second branchial arch defects of presenilin-mutant embryos. (A) SEM of an E9 PS1+/−; PS2−/− embryo that looks normal. Note the looped heart (solid arrow), normal second branchial arch (open arrow), and closed anterior neural folds (arrowhead). (B,C) S.E.M of E9 PS1−/−; PS2+/− (B) and PS1−/−; PS2−/− (C) embryos exhibiting small, unlooped hearts (solid arrows), absence of second branchial arch development (open arrows), and open neural folds (arrowheads). SEM were photographed at 130× magnification. (D–F) Transverse sections through E8.5 presenilin mutants showing small unlooped hearts (arrows) in the double-null (F) and PS1−/−; PS2+/− (E) embryos.
Finally, E8.5 double-null embryos exhibit a severe disorganization of the trunk ventral neural tube (n = 4) (Fig. 6C,D). In some instances, the notochord was completely surrounded by disordered ventral neural tube cells (n = 3) (Fig. 6E). To address whether notochord defects might be the primary cause of the ventral neural tube disorganization in the presenilin double-null embryos, we analyzed expression of Sonic Hedgehog (Shh). At E9, Shh is expressed in the notochord and the ventral midline of the neural tube (i.e., floor plate) in both the head and trunk (Echelard et al. 1993). Expression of SHH in the notochord of E9 double-null embryos was comparable to that of PS1+/−; PS2+/− embryos (n = 3) (Fig. 6G–J), suggesting that the ventral neural tube defect observed in those mutants is not secondary to notochord abnormalities. However, ventral neural tube expression of Shh in the double-null embryos was restricted to the head region (Fig. 2H) and was completely absent in the trunk (Fig. 6J) (n = 3). The ventral neural tube in the trunk of presenilin double-null embryos also failed to express Nkx2.2 (not shown), a gene whose expression pattern in wild-type embryos defines a population of presumptive neurons in the midline of the ventral neural tube. These observations support the hypothesis that distinct mechanisms govern the patterning of the neural tube along the rostrocaudal axis (Dale et al. 1997; Ensini et al. 1998) and suggests that the consequences of presenilin loss is restricted to ventral neural tube patterning in the trunk region.
Figure 6.
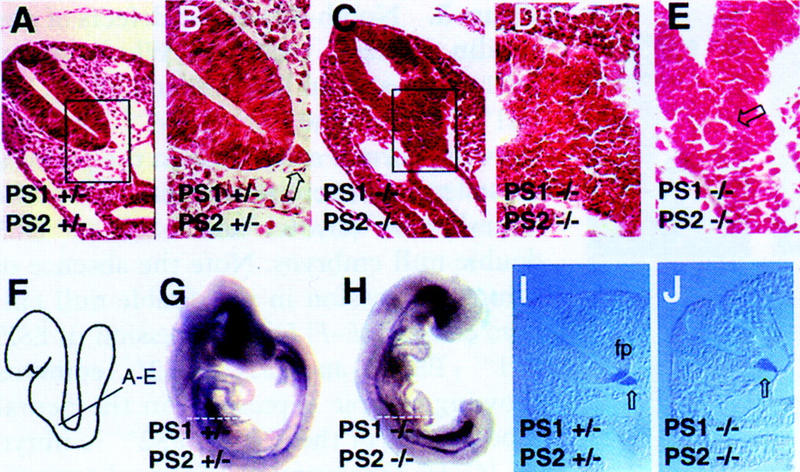
Neural tube defects of presenilin mutant embryos. (A) Transverse section through a neural tube of a PS1+/−; PS2+/− embryo. The box demarcates the area magnified in the next panel. (B) The boxed region of the dorsal neural tube shown in A at 100× showing ordered columnar morphology of cells. Note the notochord (open arrow) in proximity of the floor plate. (C) The ventral neural tube of a PS1−/−; PS2−/− embryo is disorganized. No notochord is visible, but other sections showed notochord (see E). The boxed region is magnified in the next panel. D Section shown in C is shown at 100×. (E) Transverse section through a PS1−/−; PS2−/− embryo, showing the presence of a notochord (open black arrow), which is surrounded by disorganized cells. (F) Diagram of the approximate position from which the sections shown in (A–E) were derived. E9 PS1+/−; PS2+/− (G) and PS1−/−; PS2−/− (H) embryos hybridized with a probe to Shh. Note the strong expression of Shh in the brain of the double-null embryo (H). The broken lines indicate the position of the sections shown in I–J. (I) Shh expression in the notochord (open arrow) and floor plate (fp) in the trunk region of a normal embryo. (J) Shh expression is detected only in the notochord, and not in the floor plate, in the trunk region of a presenilin double-null embryo.
Discussion
The mammalian presenilins have overlapping but not identical functions
Mice lacking PS1 die at birth with a truncated skeleton, rib-cage defects, and forebrain hemorrhaging and cell loss, whereas mice lacking PS2, and even those lacking PS2 and one copy of PS1, are viable and phenotypically normal. However, loss of even one copy of PS2 in the absence of PS1 greatly exacerbates the PS1-null phenotype, in that those embryos die between E9.5 and E13.5 of pleiotropic defects. Loss of both presenilins results in a severe, complex phenotype, and lethality before E9.5. These results show that PS1 and PS2 have different, but partially overlapping, activities. Studies in C. elegans have also demonstrated overlapping, but distinct requirements for Sel-12 and Hop-1, the two presenilins in the worm genome (Li and Greenwald 1997; Westlund et al. 1999).
Both PS1 and PS2 positively regulate Notch signaling
Presenilin double-null embryos exhibit a complex phenotype demonstrating that the presenilins are widely required in embryogenesis. Certain aspects of the double-null phenotype, such as the complete lack of somite organization, an undulating neural tube, anterior neuropore closure defects, and chorioallantoic fusion abnormalities, could be attributed to perturbations in Notch signaling. Similar defects are exhibited by mice mutated in RBP–Jk, a gene that encodes a transcription factor downstream in the Notch pathway (Oka et al. 1995; de la Pompa et al. 1997). Furthermore, two genes downstream of Notch signaling, HES-5 and Dll1, are misexpressed in both presenilin double-null and RBP–Jk-null embryos, demonstrating that presenilin loss in mammals directly affects Notch signaling. Because the RBP–Jk-null-like phenotypes were only observed when both PS1 and PS2 were mutated, we conclude that both presenilins play a role in the Notch pathway.
The complexity of the presenilin-null phenotype
Embryos lacking both PS1 and PS2 exhibit a variety of defects not described previously in mice with single mutations in Notch pathway genes, including defects in heart looping, branchial arch development, midbrain mesenchyme cell loss, and trunk ventral neural tube disorganization. This pleiotropy may indicate that the presenilins affect the processing of all four mammalian Notch proteins, and therefore, that the presenilin-null phenotype reflects a complete absence of all mammalian Notch signaling activity. To date, only mice carrying a mutation in the Notch-1 gene have been described (Conlon et al. 1995), and these mice exhibit a less complex set of phenotypes than those described here in presenilin-null embryos. It is possible that mice with a combination of targeted mutations in all four Notch genes would exhibit phenotypes identical to those of the presenilin double-null embryos. However, the RBP–Jk transcription factor, the only known mammalian member of its family, is thought to act downstream of all four Notch genes. Hence, a complete loss of RBP-Jk activity might be expected to phenocopy a complete loss of all Notch genes. However, loss of both presenilins leads to more profound developmental defects than even those observed in RBP–Jk−/− embryos, raising the possibility that mammalian presenilins might function within and outside the Notch pathway. For example, the presenilins can associate with multiple proteins (see above) and may have a role in apoptosis, the regulation of calcium homeostasis, or the regulation of G proteins. In addition, the presenilins can associate with proteins that belong to the Armadillo family and can affect β-catenin stabilization and trafficking, suggesting that they may play a role in the Wnt/Wingless pathway (Zhou et al. 1997; Murayama et al. 1998; Yu et al. 1998; Zhang et al. 1998b; Nishimura et al. 1999a). Alternatively, the more pleiotropic effects observed in presenilin double-null mice, in comparison to those observed in RPB–Jk-null mice, may indicate that Notch receptor signaling may funnel into the nucleus to other transcription factors besides RBP-Jk. Consistent with this hypothesis, studies in D. melanogaster have indicated that Notch can also function independently of Suppressor of Hairless, the RBP-Jk homolog (Matsuno et al. 1997).
The complex phenotypes observed in the presenilin-null embryos may be attributable to a global mesoderm-patterning defect. For example, disorganization of the paraxial mesoderm could affect both somite segmentation and the patterning of the trunk ventral neural tube. Furthermore, the disorganization of head mesenchyme, as evidenced by cell loss in the presumptive midbrain region of presenilin double-null embryos, could contribute to the anterior neuropore closure defects, as proposed by genetic analysis of the murine Twist gene (Chen and Behringer 1995). The branchial arch and heart looping defects may also be due to the mispatterning of cephalic and lateral plate mesoderm, respectively. Interestingly, we have not observed any obvious morphological differences or changes in gene expression in primitive streak mesoderm in E6.5–E7 presenilin double-null embryos (not shown).
Presenilin double-null embryos and Alzheimer's disease
It is interesting that mutant mice that completely lack PS2 have no discernible phenotype. In contrast, humans with a point mutation in only one copy of PS2 develop FAD. There are several possible explanations for these contrasting effects of PS2 mutations in mice and humans. First, it is possible that the mutant alleles in FAD, which are point mutations, are actually gain-of-function alleles that confer a novel activity onto the PS2 protein. Second, it is also possible that the nervous system in humans is significantly more sensitive to presenilin gene dosage than in mice, such that hypomorphic loss-of-function mutations in even one allele of PS2 is sufficient to result in severe pathology. If this is the case, a more severe reduction in murine presenilin gene dosage, such as in the viable PS1+/−; PS2−/− mice generated in this study, might result in AD-like pathologies. A third possibility is that mutant FAD alleles may be acting in a dominant-negative fashion interfering with the activity of the remaining three wild-type alleles. Complementation studies involving the introduction of FAD alleles of PS1 into mice and worms argues against this latter possibility (Levitan et al. 1996; Baumeister et al. 1997; Davis et al. 1998; Qian et al. 1998).
Regardless of whether the FAD-linked mutations cause a gain or a loss of function, the availability of cells and tissues derived from embryos lacking both presenilins will significantly facilitate functional studies aimed at understanding the role of these proteins in Notch signaling and in normal and disease cellular processes.
Materials and methods
Generation of PS2-targeted mice
P1 clones containing the murine PS2 gene were isolated from a 129 genomic library (Genome Systems) by PCR with primers from exon 4 of the published murine PS2 sequence (5′-TCCTCCACTGGGCAGTG, 5′-GGAAGAGGTGTGTGATGAGC). A 12-kb HindIII fragment containing PS2 exons 4–6 was cloned into the pZERO vector (Invitrogen) and mapped by Southern analysis and sequencing. A StuI–EcoRI 2.2-kb fragment from intron 5 was blunt-cloned into the BamHI and KpnI sites of pPNT–loxP–Neo vector, followed by the insertion of the XbaI–ClaI 2.4-kb fragment containing exon 4 and most of exon 5 into the corresponding sites in the vector. This vector was electroporated into R1 ES cells using standard techniques (Pirity et al. 1998). Following positive/negative selection, homologous recombination events were detected at a 1:250 frequency, as determined by Southern analyses with probes generated by PCR using primers designed to exons 3 (5′-ATGTCAGCCGAGAGCCCCACATC, 5′-GGCTGGACAGATGGCTCAGCAGT) and 6 (5′-CATGGCTGGCTGATCATGTCCT, 5′-GGACAGCATACAGAGTCTACTC). PCR genotyping of mice and embryos was performed with primers for the Neo gene (5′-GCCTGAAGAACGAGATCAGCA), PS2 exon 5 (5′-AAGTATCGATGCTACAAGGTGAGG), and intron 5 (5′-CCCACATGATAAAAGGAGAGC). Two independent targeted lines were used in aggregation with CD1 morulae (Pirity et al. 1998). Progeny from backcrosses of founder mice (which transmitted the 129 ES-cell derived allele of PS2 through the germ line) to 129svcp mice were pure 129.
Western blot analysis
Brains from PS2+/+, PS2+/−, and PS2(−/− adult mice were dissected and homogenized (Dounce) five times in lysis buffer A (Anafi et al. 1997), followed by sonication on ice 3 times for 20 sec. The homogenate was spun for 15 min, and the supernatant was quantitated using the BCA reagent assay (Bio-Rad), according to manufacturer's protocols. Total protein (75 μg) was loaded in each lane of a 10% polyacrylamide gel, followed by transfer onto nitrocellulose and blocking in 5% nonfat milk in TBS overnight. Two different antibodies directed against the hydrophilic loop of human PS2, antiPS2L1 (Fig. 1D) (Oyama et al. 1998) and PS2 Ab-1 (Oncogene) (not shown), were diluted in milk at 1:500 and 1:100, respectively, and incubated with the blot for 1 hr, followed by 4 washes (10 min) with TBS and 2 washes (10 min) with TBS-T. This was followed by an incubation for 45 min with HRP-conjugated goat-anti-rabbit antibody (Bio-Rad) diluted 1:20,000 in TBS-T. After 4 washes (10 min) in TBS-T, detection of immunocomplexes was revealed using ECL reagents (Amersham). Both anti-PS2 antibodies gave consistent results in multiple experiments with independent mice and primary cells derived from these mice.
Analysis of mutant mice
Mice were kept on a 12-hr light/dark cycle. Noon on plug date was designated day 0.5, although the embryos were staged according to standard morphological landmarks. Embryos were dissected in HEPES-buffered Dulbecco's modified Eagle medium (DMEM) containing 10% serum, their yolk sacs were removed directly into PCR lysis buffer, whereas the embryos were fixed in 4% paraformaldehyde for histology or in situ analysis. Embryos were embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Unless indicated otherwise, all sections shown were transverse. In situ analysis was performed as described (Conlon and Rossant 1992), using probes for Dll1 (Bettenhausen et al. 1995), UncX4.1 (Mansouri et al. 1997), Hes-5 (Akanawa et al. 1992), and Shh (Echelard et al. 1993). Scanning electron microscopy (SEM) was performed as described (Hayat 1974).
Acknowledgments
We thank P. Hunter and S. Vesely for assistance with in situ and Western analysis; S. Tondat for ES cell aggregations; K. Harpal for sectioning of embryos; D. Holmyard for SEM; E. Fan for RNA analysis; P. Cheung for Southern blot analyses; F. Oyama, Y. Ihara, and T. Iwatsubo for PS2 antibodies; J. Rossant for in situ probes, helpful discussions, and comments on the manuscript; R. Conlon for in situ probes and helpful discussions; J. Cross, S. Cordes, and C.C. Hui for help with the analysis of the histological sections; S. Egan and G. Boulianne for critical reading of the manuscript; and S. Osadchuk for assistance with the genotyping. This work was supported by grants from the Medical Research Council of Canada to A.B. and P.H.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL donoviel@mshri.on.ca; FAX (416) 586-8857.
References
- Akanawa C, Sasai Y, Nakanishi S, Kageyama R. Molecular characterization of a rat negative regulator with a basic helix-loop-helix structure predominantly expressed in the developing nervous system. J Biol Chem. 1992;267:21879–21885. [PubMed] [Google Scholar]
- Anafi M, Kiefer F, Gish GD, Mbamalu G, Iscove NN, Pawson T. SH2/SH3 adaptor proteins can link tyrosine kinases to a ste20-related protein kinase, HPK1. J Biol Chem. 1997;272:27804–27811. doi: 10.1074/jbc.272.44.27804. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: Cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Baumeister R, Leimer U, Zweckbronner I, Jakubek C, Grunberg J, Haass C. Human presenilin-1, but not familial Alzheimer's disease (FAD) mutants, facilitate Caenorhabditis elegans Notch signalling independently of proteolytic processing. Genes Funct. 1997;1:149–159. doi: 10.1046/j.1365-4624.1997.00012.x. [DOI] [PubMed] [Google Scholar]
- Berezovska O, Xia MQ, Hyman BT. Notch is expressed in adult brain, is coexpressed with presenilin-1, and is altered in Alzheimer disease. J Neuropathol Exp Neurol. 1998;57:738–745. doi: 10.1097/00005072-199808000-00003. [DOI] [PubMed] [Google Scholar]
- Bettenhausen B, Hrabe de Angelis M, Simon D, Guenet JL, Gossler A. Transient and restricted expression during mouse embryogenesis of Dll1, a murine gene closely related to Drosophila Delta. Development. 1995;121:2407–2418. doi: 10.1242/dev.121.8.2407. [DOI] [PubMed] [Google Scholar]
- Blacker D, Tanzi RE. The genetics of Alzheimer disease: Current status and future prospects. Arch Neurol. 1998;55:294–296. doi: 10.1001/archneur.55.3.294. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Choi EK, Luo Y, Lilliehook C, Crowley AC, Merriam DE, Wasco W. Calsenilin: A calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat Med. 1998;4:1177–1181. doi: 10.1038/2673. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, et al. Early-onset Alzheimer's disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- Chen ZF, Behringer RR. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes & Dev. 1995;9:686–699. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- Conlon RA, Rossant J. Exogenous retinoic acid rapidly induces anterior ectopic expression of murine Hox-2 genes in vivo. Development. 1992;116:357–368. doi: 10.1242/dev.116.2.357. [DOI] [PubMed] [Google Scholar]
- Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–1545. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- Cook DG, Sung JC, Golde TE, Felsenstein KM, Wojczyk BS, Tanzi RE, Trojanowski JQ, Lee VM, Doms RW. Expression and analysis of presenilin 1 in a human neuronal system: Localization in cell bodies and dendrites. Proc Natl Acad Sci. 1996;93:9223–9228. doi: 10.1073/pnas.93.17.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale JK, Vesque C, Lints TJ, Sampath TK, Furley A, Dodd J, Placzek M. Cooperation of BMP7 and SHH in the induction of forebrain ventral midline cells by prechordal mesoderm. Cell. 1997;90:257–269. doi: 10.1016/s0092-8674(00)80334-7. [DOI] [PubMed] [Google Scholar]
- Davis JA, Naruse S, Chen H, Eckman C, Younkin S, Price DL, Borchelt DR, Sisodia SS, Wong PC. An Alzheimer's disease-linked PS1 variant rescues the developmental abnormalities of PS1-deficient embryos. Neuron. 1998;20:603–609. doi: 10.1016/s0896-6273(00)80998-8. [DOI] [PubMed] [Google Scholar]
- de la Pompa JL, Wakeham A, Correia KM, Samper E, Brown S, Aguilera RJ, Nakano T, Honjo T, Mak TW, Rossant J, Conlon RA. Conservation of the Notch signalling pathway in mammalian neurogenesis. Development. 1997;124:1139–1148. doi: 10.1242/dev.124.6.1139. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, Moechars D, Bollen M, Fraser P, St George Hyslop P, Van Leuven F. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer's disease-associated presenilins. J Biol Chem. 1997;272:3590–3598. doi: 10.1074/jbc.272.6.3590. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Dewji NN, Singer SJ. Cell surface expression of the Alzheimer disease-related presenilin proteins. Proc Natl Acad Sci. 1997;94:9926–9931. doi: 10.1073/pnas.94.18.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan A, Thinakaran G, Borchelt DR, Slunt HH, Ratovitsky T, Podlisny M, Selkoe DJ, Seeger M, Gandy SE, Price DL, Sisodia SS. Protein topology of presenilin 1. Neuron. 1996;17:1023–1030. doi: 10.1016/s0896-6273(00)80232-9. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Dumanchin C, Czech C, Campion D, Cuif MH, Poyot T, Martin C, Charbonnier F, Goud B, Pradier L, Frebourg T. Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum Mol Genet. 1999;8:1263–1269. doi: 10.1093/hmg/8.7.1263. [DOI] [PubMed] [Google Scholar]
- Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- Ensini M, Tsuchida TN, Belting HG, Jessell TM. The control of rostrocaudal pattern in the developing spinal cord: Specification of motor neuron subtype identity is initiated by signals from paraxial mesoderm. Development. 1998;125:969–982. doi: 10.1242/dev.125.6.969. [DOI] [PubMed] [Google Scholar]
- Evrard YA, Lun Y, Aulehla A, Gan L, Johnson RL. lunatic fringe is an essential mediator of somite segmentation and patterning. Nature. 1998;394:377–381. doi: 10.1038/28632. [DOI] [PubMed] [Google Scholar]
- Hayat M. Principles and techniques of scanning electron microscopy: Biological applications. New York, NY: Van Nostrand Rienhold; 1974. [Google Scholar]
- Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, et al. Alzheimer-associated presenilins 1 and 2: Neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- Lee MK, Slunt HH, Martin LJ, Thinakaran G, Kim G, Gandy SE, Seeger M, Koo E, Price DL, Sisodia SS. Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J Neurosci. 1996;16:7513–7525. doi: 10.1523/JNEUROSCI.16-23-07513.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque G, Yu G, Nishimura M, Zhang DM, Levesque L, Yu H, Xu D, Liang Y, Rogaeva E, Ikeda M, et al. Presenilins interact with armadillo proteins including neural-specific plakophilin-related protein and beta-catenin. J Neurochem. 1999;72:999–1008. doi: 10.1046/j.1471-4159.1999.0720999.x. [DOI] [PubMed] [Google Scholar]
- Levitan D, Greenwald I. Facilitation of lin-12-mediated signalling by sel12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature. 1995;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, Sisodia SS, Greenwald I. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci. 1996;93:14940–14944. doi: 10.1073/pnas.93.25.14940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Li J, Xu M, Zhou H, Ma J, Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90:917–927. doi: 10.1016/s0092-8674(00)80356-6. [DOI] [PubMed] [Google Scholar]
- Li X, Greenwald I. Membrane topology of the C. elegans SEL-12 presenilin. Neuron. 1996;17:1015–1021. doi: 10.1016/s0896-6273(00)80231-7. [DOI] [PubMed] [Google Scholar]
- ————— HOP-1, a Caenorhabditis elegans presenilin, appears to be functionally redundant with SEL-12 presenilin and to facilitate LIN-12 and GLP-1 signaling. Proc Natl Acad Sci. 1997;94:12204–12209. doi: 10.1073/pnas.94.22.12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Additional evidence for an eight-transmembrane-domain topology for Caenorhabditis elegans and human presenilins. Proc Natl Acad Sci. 1998;95:7109–7114. doi: 10.1073/pnas.95.12.7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu FM, Lux SE. Constitutively active human Notch1 binds to the transcription factor CBF1 and stimulates transcription through a promoter containing a CBF1- responsive element. Proc Natl Acad Sci. 1996;93:5663–5667. doi: 10.1073/pnas.93.11.5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri A, Yokota Y, Wehr R, Copeland NG, Jenkins NA, Gruss P. Paired-related murine homeobox gene expressed in the developing sclerotome, kidney, and nervous system. Dev Dyn. 1997;210:53–65. doi: 10.1002/(SICI)1097-0177(199709)210:1<53::AID-AJA6>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Go MJ, Sun X, Eastman DS, Artavanis-Tsakonas S. Suppressor of Hairless-independent events in Notch signaling imply novel pathway elements. Development. 1997;124:4265–4273. doi: 10.1242/dev.124.21.4265. [DOI] [PubMed] [Google Scholar]
- Murayama M, Tanaka S, Palacino J, Murayama O, Honda T, Sun X, Yasutake K, Nihonmatsu N, Wolozin B, Takashima A. Direct association of presenilin-1 with beta-catenin. FEBS Lett. 1998;433:73–77. doi: 10.1016/s0014-5793(98)00886-2. [DOI] [PubMed] [Google Scholar]
- Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Yu G, Levesque G, Zhang DM, Ruel L, Chen F, Milman P, Holmes E, Liang Y, Kawarai T, et al. Presenilin mutations associated with Alzheimer disease cause defective intracellular trafficking of beta-catenin, a component of the presenilin protein complex. Nat Med. 1999a;5:164–169. doi: 10.1038/5526. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Yu G, St George-Hyslop PH. Biology of presenilins as causative molecules for Alzheimer disease. Clin Genet. 1999b;55:219–225. doi: 10.1034/j.1399-0004.1999.550401.x. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, Kageyama R. Hes1 and Hes5 as Notch effectors in mammalian neuronal differentiation. EMBO J. 1999;18:2196–2207. doi: 10.1093/emboj/18.8.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka C, Nakano T, Wakeham A, de la Pompa JL, Mori C, Sakai T, Okazaki S, Kawaichi M, Shiota K, Mak TW, Honjo T. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 1995;121:3291–3301. doi: 10.1242/dev.121.10.3291. [DOI] [PubMed] [Google Scholar]
- Oyama F, Sawamura N, Kobayashi K, Morishima-Kawashima M, Kuramochi T, Ito M, Tomita T, Maruyama K, Saido TC, Iwatsubo T, et al. Mutant presenilin 2 transgenic mouse: Effect on an age-dependent increase of amyloid beta-protein 42 in the brain. J Neurochem. 1998;71:313–322. doi: 10.1046/j.1471-4159.1998.71010313.x. [DOI] [PubMed] [Google Scholar]
- Pirity M, Hadjantonakis AK, Nagy A. Embryonic stem cells, creating transgenic animals. Methods Cell Biol. 1998;57:279–293. doi: 10.1016/s0091-679x(08)61585-x. [DOI] [PubMed] [Google Scholar]
- Qian S, Jiang P, Guan XM, Singh G, Trumbauer ME, Yu H, Chen HY, Van de Ploeg LH, Zheng H. Mutant human presenilin 1 protects presenilin 1 null mouse against embryonic lethality and elevates Abeta1-42/43 expression. Neuron. 1998;20:611–617. doi: 10.1016/s0896-6273(00)80999-x. [DOI] [PubMed] [Google Scholar]
- Ray WJ, Yao M, Nowotny P, Mumm J, Zhang W, Wu JY, Kopan R, Goate AM. Evidence for a physical interaction between presenilin and Notch. Proc Natl Acad Sci. 1999;96:3263–3268. doi: 10.1073/pnas.96.6.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Smine A, Xu X, Nishiyama K, Katada T, Gambetti P, Yadav SP, Wu X, Shi YC, Yasuhara S, Homburger V, Okamoto T. Regulation of brain G-protein go by Alzheimer's disease gene presenilin-1. J Biol Chem. 1998;273:16281–16288. doi: 10.1074/jbc.273.26.16281. [DOI] [PubMed] [Google Scholar]
- Stabler SM, Ostrowski LL, Janicki SM, Monteiro MJ. A myristoylated calcium-binding protein that preferentially interacts with the Alzheimer's disease presenilin 2 protein. J Cell Biol. 1999;145:1277–1292. doi: 10.1083/jcb.145.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc Natl Acad Sci. 1998;95:9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, Honjo T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H) Curr Biol. 1995;5:1416–1423. doi: 10.1016/s0960-9822(95)00279-x. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Walter J, Capell A, Grunberg J, Pesold B, Schindzielorz A, Prior R, Podlisny MB, Fraser P, Hyslop PS, Selkoe DJ, Haass C. The Alzheimer's disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol Med. 1996;2:673–691. [PMC free article] [PubMed] [Google Scholar]
- Weidemann A, Paliga K, Durrwang U, Czech C, Evin G, Masters CL, Beyreuther K. Formation of stable complexes between two Alzheimer's disease gene products: presenilin-2 and beta-amyloid precursor protein. Nat Med. 1997;3:328–332. doi: 10.1038/nm0397-328. [DOI] [PubMed] [Google Scholar]
- Westlund B, Parry D, Clover R, Basson M, Johnson CD. Reverse genetic analysis of Caenorhabditis elegans presenilins reveals redundant but unequal roles for sel-12 and hop-1 in Notch-pathway signaling. Proc Natl Acad Sci. 1999;96:2497–2502. doi: 10.1073/pnas.96.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Wong PC, Zheng H, Chen H, Becher MW, Sirinath-singhji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- Xia W, Zhang J, Perez R, Koo EH, Selkoe DJ. Interaction between amyloid precursor protein and presenilins in mammalian cells: Implications for the pathogenesis of Alzheimer disease. Proc Natl Acad Sci. 1997;94:8208–8213. doi: 10.1073/pnas.94.15.8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Lukinova N, Fortini ME. Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature. 1999;398:525–529. doi: 10.1038/19096. [DOI] [PubMed] [Google Scholar]
- Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, St George-Hyslop PH, Fraser PE. The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains beta-catenin. J Biol Chem. 1998;273:16470–16475. doi: 10.1074/jbc.273.26.16470. [DOI] [PubMed] [Google Scholar]
- Zhang N, Gridley T. Defects in somite formation in lunatic fringe-deficient mice. Nature. 1998;394:374–377. doi: 10.1038/28625. [DOI] [PubMed] [Google Scholar]
- Zhang W, Han SW, McKeel DW, Goate A, Wu JY. Interaction of presenilins with the filamin family of actin-binding proteins. J Neurosci. 1998a;18:914–922. doi: 10.1523/JNEUROSCI.18-03-00914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998b;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]
- Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M, Kosik KS. Presenilin 1 interaction in the brain with a novel member of the Armadillo family. Neuroreport. 1997;8:2085–2090. doi: 10.1097/00001756-199705260-00054. [DOI] [PubMed] [Google Scholar]