

Abstract
The Hin DNA invertase becomes catalytically activated when assembled in an invertasome complex containing two Fis dimers bound to an enhancer segment. The region of Fis responsible for transactivation of Hin contains a mobile β-hairpin arm that extends from each dimer subunit. We show here that whereas both Fis dimers must be capable of activating Hin, Fis heterodimers that have only one functional activating β-arm are sufficient to form catalytically competent invertasomes. Analysis of homodimer and heterodimer mixes of different Hin mutants suggests that Fis must activate each subunit of the two Hin dimers that participate in catalysis. These experiments also indicate that all four Hin subunits must be coordinately activated prior to initiation of the first chemical step of the reaction and that the process of activation is independent of the catalytic steps of recombination. We propose a molecular model for the invertasome structure that is consistent with current information on protein–DNA structures and the topology of the DNA strands within the recombination complex. In this model, a single Fis activation arm could contact amino acids from both Hin subunits at the dimer interface to induce a conformational change that coordinately positions the active sites close to the scissile phosphodiester bonds.
Keywords: Site-specific DNA recombination, DNA invertase, Fis, enhancer, invertasome
Precise control of specialized recombination is necessary because of the potentially severe consequences of aberrant chromosome rearrangements. In the Hin-catalyzed site-specific DNA inversion reaction, the Fis regulatory protein bound to a remote enhancer segment controls the directionality and rate of the reaction (Johnson 1991; van de Putte and Goosen 1992). Fis activates the Hin recombinase only when the protein-bound enhancer and two specific recombination sites have assembled into a topologically correct structure for generating inversion. Many types of nucleic acid transactions, including transcription, replication, and RNA splicing, are regulated from distant sites. The relative simplicity of the Hin recombination reaction combined with the current structural knowledge about the participating proteins make it an attractive system for deciphering the biochemical mechanisms involved in the regulation of enzymatic activity from a remote enhancer.
Hin is one of a number of homologous DNA invertases that mediate the alternate expression of different bacterial or viral genes in various contexts (Glasgow et al. 1989b). The Hin-catalyzed DNA inversion reaction enables Salmonella sp. to escape a primary immune response by switching the orientation of a promoter located near one end of a 1-kb segment of the chromosome. In one orientation, this promoter transcribes one of the two flagellin genes, whose product is a major cell-surface antigen, as well as a repressor of the other flagellin gene. When the promoter is inverted into the opposite orientation, the unlinked flagellin gene is transcribed because of the absence of its repressor. Two recombination sites, hixL and hixR, are located at the boundaries of the invertible segment (see Fig. 1A). Hin dimers bound to these sites can interact to form an inactive ‘paired-hix’ synaptic complex (Heichman and Johnson 1990). For recombination to occur, the synaptic complex must assemble at a 65-bp recombination enhancer located on the same DNA molecule, forming an ‘invertasome’ that is capable of initiating catalysis. DNA supercoiling is required to juxtapose correctly the three sites at the base of a plectonemic DNA branch and provide the energy to overcome otherwise weak Fis–Hin interactions (Kanaar et al. 1988; Heichman and Johnson 1990; Kanaar and Cozzarelli 1992). The hix sites within the active invertasome complex are oriented in the appropriate configuration to generate inversion of the intervening DNA upon exchange of DNA strands (Kanaar et al. 1990; Moskowitz et al. 1991). The requirement for a recombinational enhancer to be in the general vicinity of hixL and hixR also provides an additional level of specificity by decreasing the probability of undesired DNA rearrangements between psuedo recombination sites.
Figure 1.
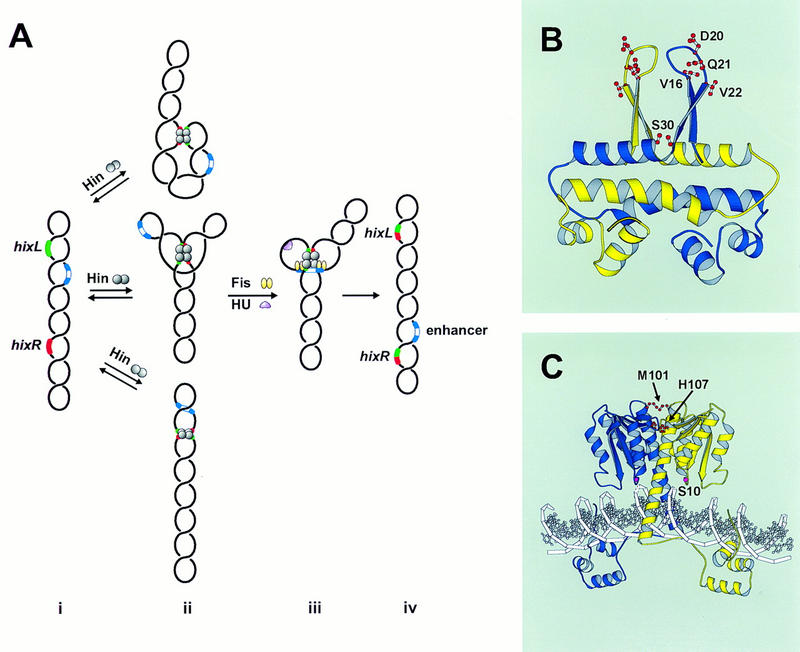
Hin-catalyzed site-specific DNA inversion. (A) Pathway of invertasome assembly. (i) The supercoiled plasmid substrate contains two recombination sites hixL and hixR and a recombinational enhancer containing two Fis-binding sites. (ii) Hin binds to hixL and hixR and can assemble these sites into a relatively unstable paired-hix complex by random collision. Several types of structures that can form are depicted schematically, as shown by topological studies on the Gin M114V and Hin H107Y mutants that can catalyze recombination in the absence of Fis (Crisona et al. 1994; S.K. Merickel and R.C. Johnson, unpubl.). (iii) Assembly of an invertasome containing hixL and hixR associated with the enhancer segment bound by two Fis dimers in a topologically defined structure at the base of a plectonemic branch on DNA. The DNA-bending activity of HU protein facilitates this assembly when the enhancer is located close to one of the recombination sites. Fis activates Hin to initiate double-strand cleavage at hixL and hixR followed by exchange of DNA strands and ligation. (iv) Resolution of the invertasome after completion of strand exchange results in inversion of the orientation of the DNA segment between hixL and hixR. (B) Structure of the Fis dimer showing the locations of Val-16 (V16), Asp-20 (D20), Gln-21 (Q21), Val-22 (V22), and Ser-30 (S30) as revealed in the crystal structure of K36E (Safo et al. 1997). Val-16, Asp-20, Gln-21, and Val-22 are within the β-hairpin activating motifs, whereas the two Ser-30 residues are directly across from each other on the A/A′ helices. (C) Model of the Hin dimer bound to DNA (Haykinson et al. 1996) showing the locations of Met-101 (M101), His-107 (H107), and the active site residue Ser-10 (S10).
The recombinational enhancer consists of two essential binding sites for the Fis protein whose positions relative to each other are critical for enhancer function (Johnson and Simon 1985; Kahmann et al. 1985; Johnson et al. 1987). Fis is a homodimer containing two 98-amino acid subunits (Johnson et al. 1988; Koch et al. 1988). X-ray crystallography has shown that each subunit contains four α-helices that associate in the dimer to generate a compact ellipsoid core with the carboxy-terminal helices forming a helix–turn–helix DNA-binding motif (Kostrewa et al. 1991; Yuan et al. 1991). A recent crystal structure of a mutant Fis has revealed that previously unresolved amino acids 12–26 near the amino terminus of each subunit form a β-hairpin arm that protrudes over 20 Å from the α-helical core (Fig. 1B; Safo et al. 1997). Whereas the positions of the two β-arms in the dimer are fixed by crystal–lattice interactions, disulfide cross-linking between cysteines introduced at different positions within the β-arms demonstrate that the arms are mobile in solution. Extensive mutagenesis of this region has shown that three amino acids, Val-16, Asp-20, and Val-22, near the tip of the β-arm are critical for Fis activation of Hin inversion (Fig. 1B; Safo et al. 1997). Certain solvent-exposed amino acids within the A α-helix may also contact Hin within the invertasome structure.
Hin is a member of an extended family of resolvases and DNA invertases that share extensive amino acid homology, particularly over the catalytic and dimerization domains that constitute the amino-terminal three-fourths of the protein (Sherratt 1989). The molecular structure of Hin bound to hix has been modeled from crystal structures of the Hin DNA-binding domain and γδ resolvase complexes with DNA (Fig. 1C; Feng et al. 1994b; Rice and Steitz 1994b; Yang and Steitz 1995; Haykinson et al. 1996). Crystal structures of the γδ resolvase dimer show that the active site serines within the catalytic domain of each subunit are not positioned appropriately to initiate site-specific cleavage of DNA (Rice and Steitz 1994b; Yang and Steitz 1995). This implies that a conformational change that repositions the active site close to the scissile phosphodiester bonds must precede DNA cleavage. Several pieces of evidence suggest that a quaternary change in the dimer interface accompanies catalytic activation. The dimeric contacts are primarily between the long α-helix (helix E) in each subunit that also traverses the DNA to connect to the DNA binding domain (Yang and Steitz 1995). When disulfide crosslinks are formed between cysteines introduced into the amino-terminal end of helix E, thereby restricting movement between subunits, catalytic activity is abolished, though resolvase or Hin binding to DNA is unaffected (Hughes et al. 1993; Lim 1994; Haykinson et al. 1996). Recombination by helix E cysteine mutants is no longer sensitive to oxidation once Fis–Hin interactions occur in an invertasome complex (Haykinson et al. 1996). These data imply that activation by Fis induces a conformational change directly or indirectly within the Hin dimer interface. Further support for the regulatory importance of the dimer interface comes from the observation that certain detergents that impair Hin-dimer interactions enhance Hin-mediated DNA cleavage and that certain amino acid substitutions within the region lead to hyperactive DNA invertase mutants, which can promote recombination in the absence of Fis or a recombinational enhancer (Haffter and Bickle 1988; Klippel et al. 1988a; Haykinson et al. 1996). Most substitutions within this region, however, inhibit catalysis even though they are well separated from the active site pocket (Lim 1994; Haykinson et al. 1996).
DNA inversion proceeds in two experimentally separable chemical steps once the invertasome has been assembled. In the first step, the four Hin subunits bound to the synapsed hix sites mediate a coordinated DNA attack to create a 2-bp staggered double-strand break at the center of each site (Johnson and Bruist 1989). In this process, invertase subunits become covalently joined to each 5′ end of the broken strands via an ester linkage at serine 10 (Klippel et al. 1988b). In the second step, DNA strand exchange, the cleaved DNA strands are positioned into the recombinant configuration and the DNA is religated through reversal of the protein–DNA linkage. Whereas the cleavage and strand-exchange steps are normally highly concerted, reactions performed in the absence of magnesium and presence of ethylene glycol stall at the cleaved intermediate stage (Johnson and Bruist 1989). The enhancer remains associated with the Hin invertasome throughout the course of the reaction, though it can be released prior to ligation under certain conditions for Hin and readily for the Gin DNA invertase (Kanaar et al. 1990; Heichman et al. 1991; Crisona et al. 1994). Thus, whereas the Fis-bound enhancer sequence is required to initiate catalysis, it is not required for the final chemical step.
In this paper we address how Fis and Hin communicate to initiate the coordinated attack of the DNA strands. Specifically, we examine whether one or both of the activating β-hairpin arms in a Fis dimer are required to activate Hin and whether all or a subset of the Hin subunits must be activated directly by Fis. To investigate these issues, we employ homodimer and heterodimer mixes of different Fis and Hin mutants. Surprisingly, we find that only one functional β-hairpin activating arm is required in each Fis dimer to activate Hin. In contrast, the data indicate that each of the Hin subunits must be activated prior to initiation of any DNA chemistry, though they need not be proficient in catalysis. We also present a molecular model for the invertasome that is consistent with past and present information on its structure.
Results
Two activating Fis dimers are required to initiate recombination by Hin
We have established previously that Fis dimers must be associated with both binding sites within the hin enhancer to form activated invertasomes. This was demonstrated by (1) mutagenesis studies showing that removal of either binding site or changes in the spacing between binding sites essentially abolish Fis-activation of inversion; (2) electron microscopy showing the absence of invertasome structures on substrates containing only one Fis binding domain; and (3) stoichiometry measurements indicating two Fis dimers per activated invertasome (Johnson and Simon 1985; Bruist et al. 1987; Johnson et al. 1987; Heichman and Johnson 1990). It has not been determined, however, if both Fis dimers are required to activate Hin protomers to catalyze inversion in addition to performing structural roles in organizing or stabilizing the invertasome complex. For example, both dimers may be required to assemble an invertasome with the correct DNA topology, but the activating amino-terminal region of only one dimer may be sufficient to initiate catalysis.
To determine whether the amino-terminal activating β-arms are required on both Fis dimers to activate Hin in an invertasome complex, mixing experiments were performed with active and inactive Fis mutants, whose salient properties are listed in Table 1. Fis S30C, which forms a disulfide linkage between the two subunits within the A α-helices when oxidized (S30Cox), was used for the active protein (Fig. 1B; Safo et al. 1997). The Cys-30 covalent linkage does not affect any activities of Fis but prevents the exchange of subunits between dimers, which readily occurs in solution (S.K. Merickel and R.C. Johnson, unpubl.). The conditionally active Fis mutant Q21C displays near wild-type activity when reduced (Q21Cred). However, it is inactive for stimulating Hin inversion when oxidized (Q21Cox) because of distortion of the β-hairpin loops by the Cys-21 disulfide bond (Fig. 1B and 2, lanes 4 and 5; Safo et al. 1997). DNA binding and transcriptional activation are not affected by the disulfide linkage.
Table 1.
Fis and Hin mutants used in this work
| Mutant
|
Relevant properties
|
Source
|
|---|---|---|
| Fis | ||
| Q21C | conditionally defective in Hin activation: inactive when oxidized; active when reduced | Safo et al. (1997) |
| D20K | defective in Hin activation | Safo et al. (1997) |
| S30C | active for Hin activation when reduced or oxidized | Safo et al. (1997) |
| Hin | ||
| S10G | catalytic active site mutant | M. Lainé and R. Johnson |
| M101C | conditionally defective in Hin activity: inactive when oxidized; active when reduced | Haykinson et al. (1996) |
| H107Y | Fis-independent recombination | Haykinson et al. (1996); this paper |
Figure 2.

Activation of Hin by homodimer mixes of active and inactive Fis mutants. In vitro cleavage reactions were incubated for 15 min in the absence of DTT. The units of Fis homodimer mutants used in each reaction are indicated above the lanes. One unit of Fis is defined as the limiting amount of Fis (in its active form) required to stimulate ∼30% cleavage in a 15-min cleavage reaction, which reflects final yields. An equivalent amount (4 ng) of Fis D20K was added in lane 7. The percent of cleavage activity is indicated below each lane with the no Fis basal activity (14%, lane 8) subtracted. The locations of the primary Hin cleavage products, the vector, and invertible segment bands result from double-strand cleavage at both hix sites, are denoted along with the supercoiled plasmid substrate. Normal Hin-catalyzed DNA cleavage reactions generate a small amount of double-strand cleavage at a single hix site generating linear plasmids and no increase over the starting level of single-strand cleaved open circle DNA. The lack of any increase in cleavage products in lanes 3 and 7 over lane 1 indicates that enhancers containing a Fis homodimer with mutated β-arm regions (Q21Cox or D20K) plus a fully active Fis dimer (S30C) are not capable of assembling active invertasomes. This and subsequent ethidium bromide-stained gels are printed in reverse contrast.
Oxidized or reduced Q21C was added to Hin cleavage reactions containing limiting amounts of S30Cox (Fig. 2). If the activation domains of only one of the two Fis dimers in an invertasome complex are sufficient to catalytically activate Hin, the addition of Q21Cox to reactions containing limiting S30Cox would result in an increase in cleaved invertasomes over that observed from S30Cox alone. This increase in activity would be derived from invertasome complexes that contain one active and one inactive Fis dimer. However, if functional amino-terminal arms are required in both Fis dimers to form an activated invertasome complex, the amount of Hin-cleavage products would not increase with the addition of Q21Cox. When limiting amounts of S30Cox (Fig. 2, lane 1) were supplemented with an equivalent amount of S30Cox (lane 2) or Q21Cred (lane 6), a similar increase in activity was observed, reflecting the expected increase in the number of activated invertasome complexes formed. In contrast, when the limiting activity of S30C is supplemented with an equivalent amount of Q21Cox (lane 3), the yield of Hin-cleavage products decreased slightly. The amount of single hix site cleavage, which could have reflected catalytic activation of one Hin dimer by one Fis dimer, did not increase under these conditions. The same result is observed when the limiting activity of S30Cox is supplemented with D20K (lane 7) or D20G (data not shown), which contain inactivating amino acid substitutions at the critical aspartic acid in the β-hairpin arms (Fig. 1B; Safo et al. 1997). These experiments indicate that both Fis dimers must contain a functional β-arm region to form a catalytically active invertasome complex.
Only one functional β-hairpin arm within a Fis dimer is required to activate Hin
To ascertain whether the β-hairpin arms of both subunits within each Fis dimer are required to activate Hin inversion, heterodimers composed of one subunit with an active β-arm and one subunit with an inactive β-arm were constructed. Purified preparations of S30C and D20K/S30C/His, a carboxy-terminally His-tagged Fis containing the S30C mutation and the inversion-inactivating mutation D20K, were mixed, denatured under reducing conditions, and renatured to form a mixture of heterodimers and the two homodimeric forms (see Fig. 3A). The resulting dimers were stabilized by covalently linking the subunits through a disulfide bond at Cys-30 under oxidizing conditions. The S30C:D20K/S30C/Hisox heterodimers, along with the inactive D20K/S30C/Hisox homodimers, were then separated from the active S30Cox homodimers by nickel chromatography. Nonreducing SDS-PAGE demonstrated that S30C:D20K/S30C/Hisox heterodimers constituted ∼75% of the protein in the imidazole-eluted fraction with no detectable S30Cox homodimers. Figure 3, B and C, shows the results of Hin cleavage and inversion reactions performed with the renatured mix before and after the nickel chromatography step. The purified S30C:D20K/S30C/Hisox heterodimer fraction efficiently activated Hin in both reactions (Fig. 3B,C; lane 1), demonstrating that only one active β-hairpin arm in each Fis dimer is sufficient to activate a Hin dimer. Detectable amounts of nicked products that could have arisen from single-strand cleavages at one or both hix sites were not generated from these reactions. The same result was obtained in experiments employing heterodimers in which one of the subunits was deleted for the entire β-hairpin loop [Fis Δ(17–21)] (data not shown).
Figure 3.

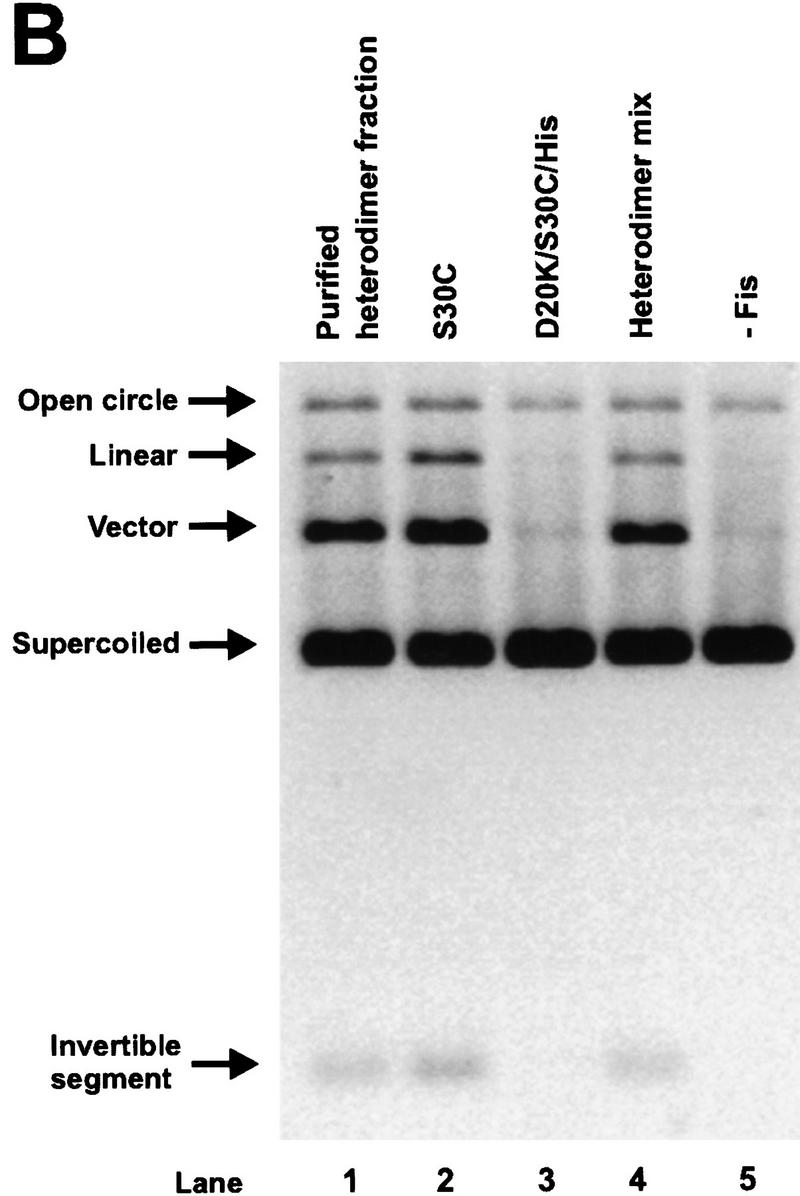
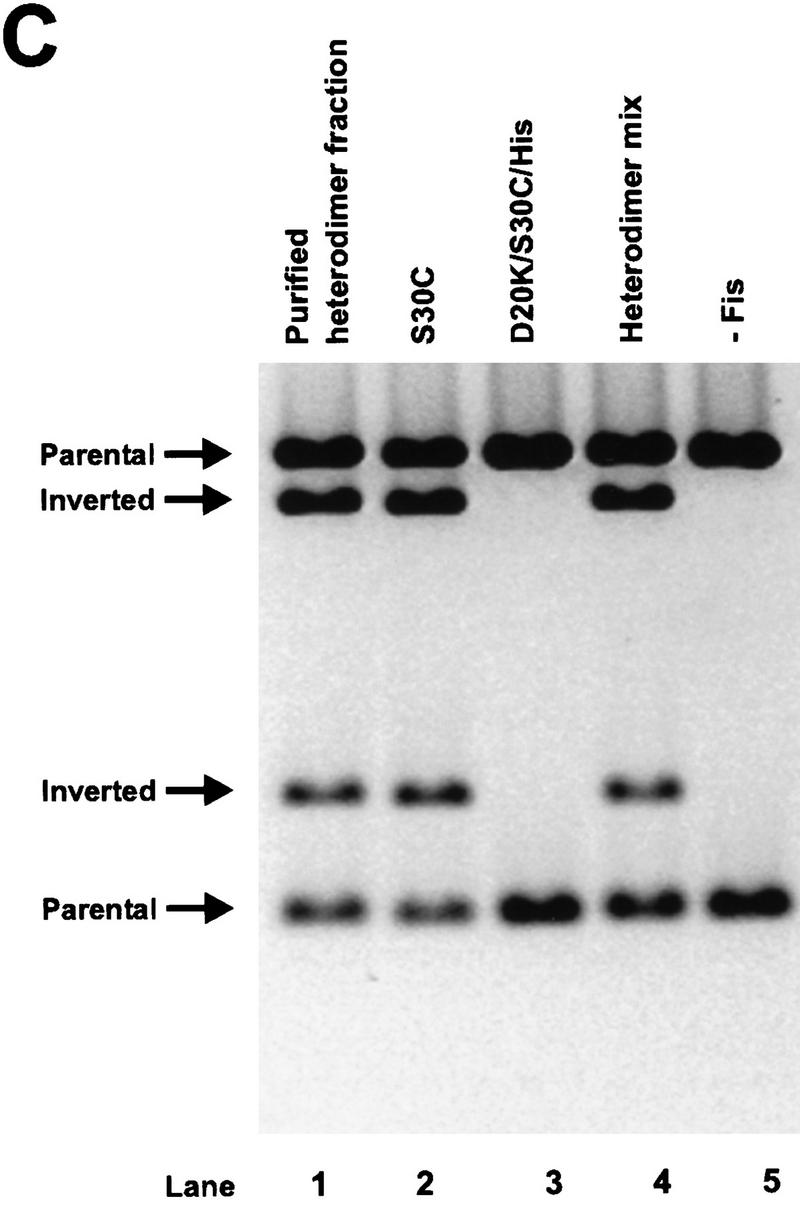
Activation of Hin by Fis heterodimers containing one active β-arm and one inactive β-arm. (A) Schematic diagram of the protocol used to form covalently linked Fis heterodimers and remove active homodimers from the population. (B) In vitro cleavage reactions using refolded S30C (lane 2), D20K/S30C/His (lane 3), the heterodimer mix (S30C and D20K/S30C/His homodimers and heterodimers, lane 4), the purified heterodimer fraction from the nickel column (heterodimers and inactive D20K/S30C/His homodimers, lane 1), and a no Fis control (lane 5). Reactions were incubated for 2 min in the absence of DTT. Product and substrate bands are labeled as in Fig. 2. The low amounts of open circle and linear DNA indicate that the Fis heterodimers are stimulating normal concerted double-strand cleavage of the DNA at both hix sites. (C) Two-min inversion assays performed in the absence of DTT. After the inversion assay, the DNA was digested with PstI and HindIII to determine the orientation of the invertible segment. The DNA fragments labeled ‘inverted’ are the inversion products, whereas fragments labeled ‘parental’ result from DNA in the parental orientation. The cleavage and inversion products present in lane 1 of panels A and B, respectively, indicate that the Fis heterodimers can efficiently activate Hin.
Properties of Hin mutants used to investigate Hin activation
To investigate the mechanism of Hin activation, several Hin mutants that either block the reaction at particular steps or catalyze recombination without activation by Fis were examined. Their relevant properties are summarized in Table 1. Hin S10G is mutated at the serine nucleophile that attacks the DNA phosphodiester during the first chemical step of the recombination reaction (Fig. 1C) and consequently displays no DNA cleavage activity (e.g., Fig. 5B, lane 3, below). Unlike other substitutions at this position (Adams et al. 1997), S10G has no detrimental effect on hix binding (M. Lainé and R.C. Johnson, unpubl.) and synapsis (Fig. 4, lane 4), which is assayed by the formation of paired-hix complexes after glutaraldehyde cross-linking and gel electrophoresis (Heichman and Johnson 1990). Additionally, we provide evidence below that Fis-activation within an invertasome remains unaffected in this mutant. Thus, S10G is blocked specifically at the step of DNA cleavage.
Figure 5.

Activity of homodimer mixes of wild-type and inactive Hin mutants in the presence of Fis. (A) Reactions employing M101C and Hin–WT. The units of Hin homodimers used in each 15-min in vitro cleavage reaction are indicated above the lanes. One unit of Hin was empirically determined as the limiting amount of Hin (in its active form) required to produce ∼30% cleavage in a 15-min cleavage reaction. To prevent potential subunit exchange in solution, Hin homodimers were added sequentially. The activity of M101Cred was assayed by prebinding M101Cox to the DNA, reducing with 5 mm DTT for 1 min at 37°C, and starting the reaction with Fis. The percentage of cleavage activity is indicated below each lane. The lack of any increase in cleavage products in lane 4 as compared to lane 1, particularly single hix-cleaved linear plasmids, indicates that activated invertasomes containing Hin–WT and M101Cox homodimers are not formed. (B) Reactions employing S10G and Hin–WT homodimers. For S10G, one unit was taken as the equivalent amount of Hin (20 ng) required to generate 1 unit of Hin–WT activity, because the catalytic activity of S10G could not be measured.This may result in slightly more functional S10G than Hin–WT, as S10G appears to have a slightly greater specific activity with respect to hixL binding than Hin–WT. The presence of enhanced levels of single hix-cleaved linear plasmids in lane 2 demonstrates that activated invertasomes containing Hin–WT and S10G homodimers are forming.
Figure 4.

Formation of synaptic complexes by Hin mutants. In vitro paired-hix assays were performed on the Hin proteins employed in the mixing experiments. Using pMS634, which contains two hixL sites (hixL-1 and hixL-2), Hin cleavage reactions were cross-linked with glutaraldehyde, digested with PstI, and electrophoresed in an agarose gel. In lane 1 glutaraldehyde was not added to the reaction, and in lane 3 SDS was added to a final concentration of 0.7% immediately prior to loading on the gel. The Hin proteins used in each assay are indicated above the lanes. The location of the paired-hix complex as well as the unpaired hixL DNA fragments are indicated. Hin binding to the smaller PstI fragment containing hixL-2 results in a slower migrating complex, but Hin binding to the larger PstI fragment containing hixL-1 cannot be resolved as a separate complex in this gel. M101Cox was generated by long-term storage in <1 mm DTT and contained 90% disulfide-linked dimers.
Methionine 101 is predicted to be located at the amino-terminal end of the E α-helix within the dimer interface (Fig. 1C). A cysteine at this position readily forms a disulfide linkage between the two dimer subunits (M101Cox) (Lim 1994; Haykinson et al. 1996). M101C is conditionally defective for basal and Fis-activated recombination. Under oxidizing conditions, hix cleavage and subsequent DNA exchange is blocked, but hix binding (Lim 1994; Haykinson et al. 1996) and synapsis (Fig. 4, lane 7) by M101Cox remain efficient. Under reducing conditions, M101C displays near-normal Fis-dependent inversion (M101Cred). Covalent linkage of the Hin dimer probably prevents a quaternary change involving the dimer interface that is required for catalytic activation (Hughes et al. 1993; Lim 1994; Haykinson et al. 1996). Experiments measuring the kinetics of hix cleavage and disulfide formation upon a shift of M101C to oxidizing conditions indicate that oxidative modifications that do not result in a disulfide linkage also effectively block catalysis (data not shown; Fig. 6, below).
Figure 6.
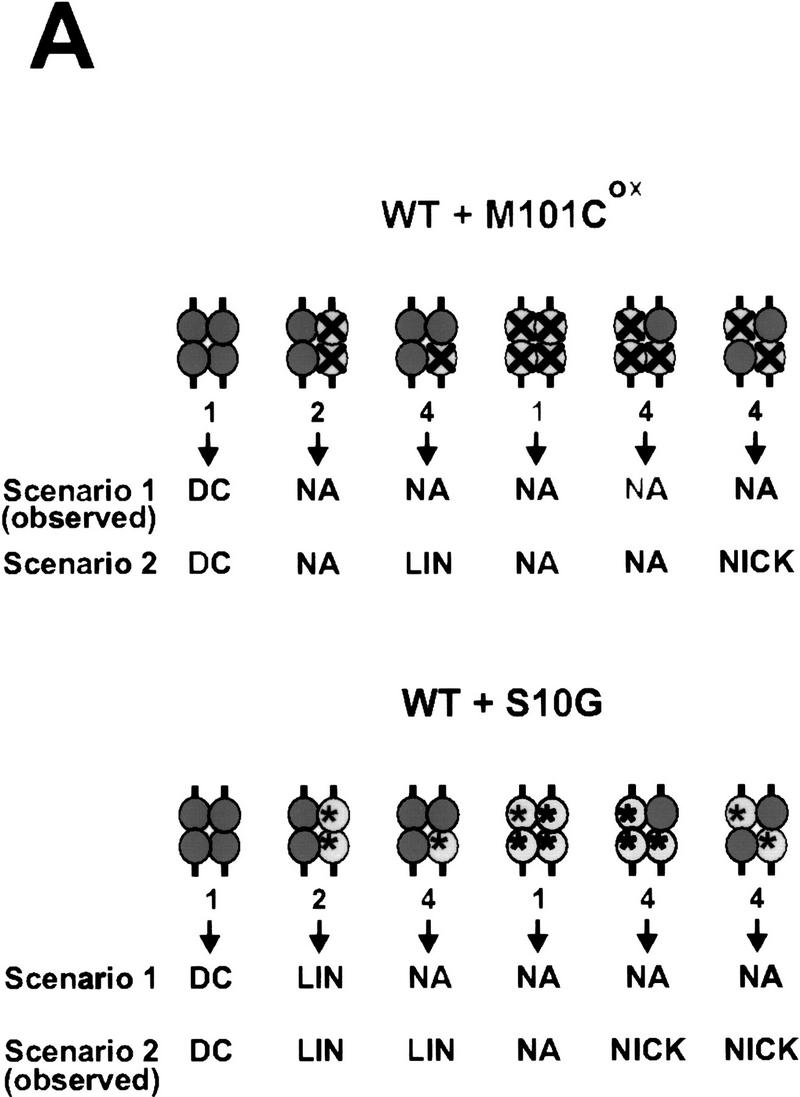

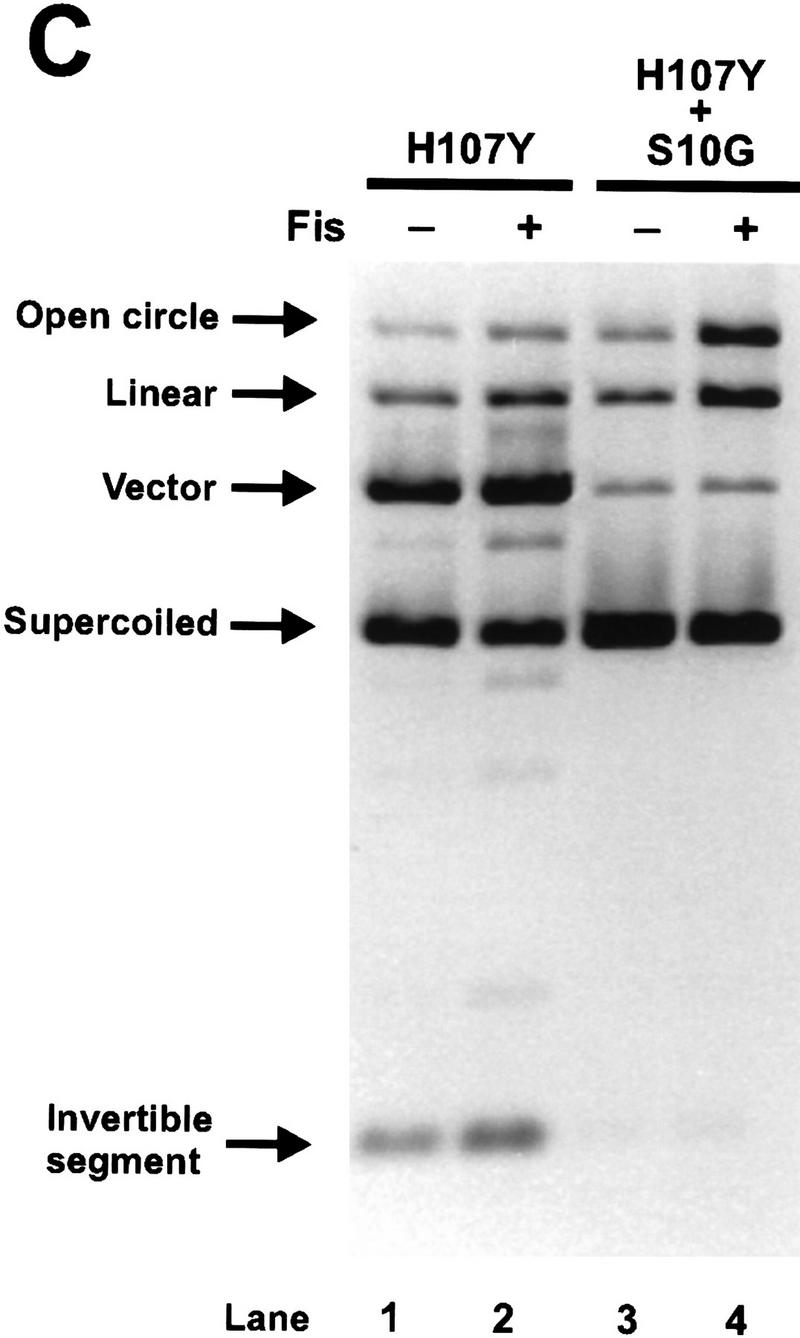

Activity of Hin heterodimers. (A) Schematic diagrams illustrating the combinations of Hin dimers found in invertasome complexes formed in the Hin heterodimer mix assays. The Fis-bound enhancer has been omitted for clarity. The relative amounts of each type of complex predicted to form in the heterodimer mix assays are indicated below the complex. (Top) Predicted results from M101Cox + Hin–WT heterodimer mixing assays. (Dark, solid circles) Hin–WT subunits; (light circles containing an X) inactive M101Cox subunits. Scenario 1 depicts the predicted results if a Hin–WT subunit in a M101Cox:Hin–WT heterodimer is unable to become activated and cleave the DNA. Scenario 2 describes the predicted products from each complex if a Hin–WT subunit in a M101Cox:Hin–WT heterodimer becomes activated and cleaves the DNA. The predicted activities of complexes containing an inactive M101Cox homodimer are based on the results from the M101Cox homodimer mixing assays (Fig. 5A). The predicted activities shown below the arrows include (DC) double-strand cleavage at both hix sites; (NA) no activity; (LIN) linear DNA resulting from double-strand cleavage at only one hix site; and (NICK) nicked DNA resulting from single-strand cleavage at one or both hix sites. Scenario 1 is consistent with the results observed in the M101Cox:Hin–WT heterodimer mixing assays (Fig. 6B). (Bottom) Predicted results from S10G + Hin–WT heterodimer mixing assays. (Dark, solid circles) Hin–WT subunits; (light circles containing an asterisk) catalytically inactive S10G subunits. Scenario 1 indicates the predicted results if a Hin–WT subunit in a S10G:Hin–WT heterodimer cannot become activated and cleave the DNA. Scenario 2 shows the predicted activities if a Hin–WT subunit in a S10G:Hin–WT heterodimer becomes activated and cleaves the DNA. The predicted activities of complexes containing an inactive S10G homodimer are based on the result from the S10G homodimer mixing assays (Fig. 5B). Scenario 2 is consistent with the results observed in the S10G:Hin–WT heterodimer mixing assays (Fig. 6B). (B) Two-min in vitro cleavage reactions employing denatured and refolded Hin–WT (lanes 1,2,7), M101C + Hin–WT heterodimer mix (M101C and Hin–WT homodimers and heterodimers; lanes 3,4), M101C (lanes 5,6), and S10G + Hin–WT heterodimer mix (S10G and Hin–WT homodimers and heterodimers; lane 8). All Hin mutants were allowed to bind to the DNA for 5 min at 37°C and the reactions were started with the addition of Fis. Hin–WT, M101C, and the M101C + Hin–WT heterodimer mix were bound to the substrate and assayed in reducing (Red) conditions containing 5 mm DTT or oxidizing (Ox) conditions containing 10 mm oxidized glutathione as indicated. The low amount of cleavage products generated by the M101Cox + Hin–WT preparation (lane 4) implies that invertasomes containing M101Cox:Hin–WT heterodimers are not catalytically active. The substantial amounts of single-strand hix cleavage (open circle) and double-strand cleavage at a single hix site (linear) produced by the S10G + Hin–WT preparation (lane 8) are indicative of invertasomes containing S10G:Hin–WT heterodimers and S10G + Hin–WT homodimers, respectively. (C) Two-min in vitro cleavage assays of refolded H107Y homodimers and a H107Y + S10G heterodimer mix (H107Y and S10G homodimers and heterodimers) performed in the presence or absence of Fis as indicated above the lanes. The formation of single-strand hix cleavage (open circle) and double-strand cleavage at a single hix site (linear) produced by the H107Y + S10G preparation in the presence of Fis (lane 4) is indicative of synaptic complexes containing H107Y:S10G heterodimers and H107Y + S10G homodimers, respectively. The low amount of cleavage products generated in the absence of Fis (lane 3) implies that most catalytic activity, including single-strand cleavage from H107Y:S10G heterodimers, is dependent upon Fis. (D) Two-min in vitro cleavage reactions of refolded H107Y, Hin–WT, and H107Y + Hin–WT heterodimer mixes in which H107Y and Hin–WT were mixed at 1:1 (lanes 5,6) and 1:2 (lanes 7,8) molar ratios before refolding. Because H107Y is more active than Hin–WT, these ratios overestimate the functional amount of H107Y. These preparations were assayed in the presence or absence of Fis as indicated above the lanes. Note the strong stimulation by Fis with Hin–WT (lanes 3,4), but efficient cleavage by H107Y regardless of the presence of Fis (lanes 1,2) under these relatively long incubation times. Complexes assembled from the mixed populations appear to be stimulated strongly by Fis for catalytic activity, implying that H107Y:Hin–WT heterodimers are not active even for single-strand cleavage without Fis.
Gain-of-function mutants that support recombination in the absence of Fis have been isolated and characterized in the Gin, Cin, and Hin DNA invertases (Haffter and Bickle 1988; Klippel et al. 1993; Haykinson et al. 1996). H107Y is the strongest Hin mutant isolated to date that can promote in vivo and in vitro recombination without Fis. H107Y catalyzes efficient hix cleavage in the absence of Fis, under standard conditions (e.g., Fig 6, C and D, lanes 1 and 2, below). Like the Fis-activated wild-type reaction, DNA cleavage by H107Y without Fis activation appears concerted between hix sites and primarily double-strand cuts are generated at each site. Thus, hix cleavage by H107Y occurs within the context of paired-hix complexes, which form efficiently and are very stable (Fig. 4, lane 5). H107Y remains further activatable by Fis because cleavage and inversion rates are increased when Fis is present (Fig. 6C, lanes 1 and 2, below; data not shown), indicating that His-107 is probably not directly contacted by Fis. Histidine 107 is located in the E α-helix region, but unlike Met-101, its imidazole ring is predicted to be oriented towards residues 92–93 of the partner subunit within the dimer (Fig. 1C). We postulate that His-107 may be making an important regulatory contact within the dimer that modulates Hin activity in response to Fis.
Both Hin dimers must be activated to initiate hix cleavage
To determine whether both Hin dimers bound to the synapsed hix sites within an invertasome must be activated for any catalysis to occur, we performed reactions containing different combinations of Hin proteins. The Hin proteins were added sequentially to minimize potential subunit mixing prior to hix site binding. Previous studies have shown that Hin forms a very stable dimer once bound to hix (Glasgow et al. 1989a). In addition, control experiments were performed in which (1) Hin binding to a labeled fragment was followed by gel-mobility shift assays after competition with excess Hin recombination substrate DNA, and (2) Hin recombination was measured in reactions in which a second substrate was added after prebinding to the first substrate. These experiments indicated that once bound, Hin does not exchange between hix sites during the time frame of the Hin reactions performed below (data not shown).
Mixtures of Hin–WT and M101Cox, were assayed to determine whether both Hin dimers in an invertasome must be activatable to observe any cleavage activity. In Figure 5A, a limiting amount of disulfide-linked M101Cox was bound to the substrate DNA, resulting in substrates containing two hix-bound Hin dimers, one hix-bound Hin dimer, or no hix-bound Hin dimers. The cleavage reaction was then supplemented with an equivalent amount of Hin–WT and initiated by the addition of Fis. The activity of M101Cred was assayed by reducing the DNA-bound M101Cox into an activatable form with 5 mm DTT. Prebinding of covalently linked M101Cox dimers helped to ensure that subunit mixing between Hin dimers did not occur. As expected, the yield of cleavage products from M101Cred plus Hin–WT (Fig. 5A, lane 3) was increased over that obtained with Hin–WT or M101Cred (lanes 1, 5) and similar to the 2× Hin–WT (lane 2) controls. However, no increase in the reaction was observed with M101Cox plus Hin–WT (lane 4). Significantly, this reaction did not produce an increase in single hix cut (double stranded or nicked) products that could have been produced by invertasomes containing one active Hin–WT and one inactive M101Cox dimer. These results imply that both Hin dimers that participate directly in the reaction must be activatable for any hix cleavage to occur.
Mixing experiments were also performed with the active site mutant S10G and Hin–WT. Limiting amounts of S10G were bound to the substrate followed by the addition of Hin–WT, and the nature of the Hin cleavage products in the presence of Fis was determined. If S10G is competent for activation, but defective in catalysis, invertasomes containing one Hin–WT dimer and one S10G dimer would be expected to be catalytically active but cleave only one of the hix sites in the substrate. As shown in Figure 5B, lane 2, single hix cut plasmids accumulated in the S10G plus Hin–WT reaction. This result demonstrates that activation and cleavage can be mechanistically separated. Although both Hin dimers bound to the hix sites must be activatable, DNA cleavage by both dimers is not necessary to observe catalytic activity.
Both subunits within the Hin dimer bound to hix must be activated to initiate catalysis
Hin heterodimers were used to determine whether both subunits in a Hin dimer must be activatable to initiate catalysis. Equal amounts of M101C and Hin–WT were denatured and refolded together under dilute conditions in the presence of high concentrations of CHAPS to generate heterodimers. The heterodimer mix was assayed for cleavage activity after a brief incubation with oxidized glutathione. Under these oxidation conditions, M101C subunits can be inactivated by oxidative modifications at individual Cys-101 residues, but Hin–WT activity is not affected (Fig. 6B, lane 2). If Hin–WT subunits in M101Cox:Hin–WT heterodimers are unable to become activated and cleave DNA, we would expect to observe only a small amount of double-strand cleavage at both hix sites as depicted in scenario 1 of Figure 6A (top). However, if Hin–WT subunits are able to cleave the DNA even when associated with nonactivatable M101Cox subunits in heterodimers, we would predict that double-strand cleavage at one hix site and nicked products would also be generated as shown in scenario 2 of Figure 6A (top). We find that Fis-activated reactions performed with M101Cox:Hin–WT heterodimer mixes only generate a trace of double-strand cleavage (Fig. 6B, lane 4), consistent with scenario 1 of Figure 6A (top). We interpret the small amount of activity present in the M101Cox:Hin–WT heterodimer mix reaction to have originated from invertasomes containing two wild-type Hin dimers. The oxidation-sensitive population contains at least one M101Cox:Hin–WT heterodimer or M101Cox homodimer (Fig. 6A, top, scenario 1). The absence of an increase in nicked products in these reactions indicates that Hin–WT subunits within complexes containing M101Cox:Hin–WT heterodimers do not catalyze single-strand cleavage at hix at a detectable level (Fig. 6A, top, scenario 1).
Reactions performed with S10G:Hin–WT heterodimer mixes confirm that functional heterodimers are formed in the renaturing protocol and that a Hin dimer can be induced into an activated state without both subunits being catalytically competent. Scenario 1 of Figure 6A (bottom) depicts the expected results if the Hin–WT subunit of an S10G:Hin–WT heterodimer is not able to become activated and cleave the DNA. Scenario 2 of Figure 6A (bottom) illustrates the predicted results if S10G:Hin–WT heterodimers are able to become activated and the Hin–WT subunit is able to cleave the DNA even though the S10G subunit is catalytically deficient. Fis-activated S10G:Hin–WT heterodimer mix reactions generate a significant number of both nicked and linear DNA products (Fig. 6B, lane 8), representing single-strand cleavage at one or both hix sites and double-strand cleavage at only one hix site, respectively. These results are consistent with the predicted types of products illustrated in scenario 2 of Figure 6A (bottom). The nicked DNA products would presumably be generated from invertasomes containing two S10G:Hin–WT heterodimers or one S10G:Hin–WT heterodimer and one S10G homodimer. This is the first experimental condition in which Hin has been found to generate single-strand cleavages. Invertasomes generating linear products would contain one catalytically competent wild-type homodimer and one dimer containing at least one subunit of S10G. The presence of the nicked products indicates that only one subunit within a Hin dimer needs to be catalytically competent to observe cleavage products. The inactivity of the M101Cox:Hin–WT heterodimer, combined with the nicking activity of the S10G:Hin–WT heterodimers suggests that both subunits within the dimer must be activatable by Fis but not necessarily catalytically competent, to observe cleavage activity.
Reactions were also performed on heterodimer mixes of the Fis-independent mutant H107Y and either S10G or Hin–WT. Because H107Y is more active than wild type, even in the presence of Fis, molar equivalent mixtures of H107Y and Hin–WT or S10G probably functionally over-represent H107Y activity. As shown in Figure 6C, reactions employing a heterodimer mix of H107Y and S10G generated substantial amounts of nicked and linear products in the presence of Fis (lane 4), but little reaction was observed in the absence of Fis (lane 3). As discussed above, the presence of nicked DNA products confirms that heterodimers were present in these reactions. The Fis dependence of the H107Y:S10G heterodimer nicking activity demonstrates that both subunits in a Hin dimer must be either activated by Fis or contain a mutation conferring Fis independence to initiate catalysis. Thus, alteration of only one of the two dimer contacts involving residue 107 is not sufficient to induce Fis independence in the Hin dimer. Reactions employing H107Y:Hin–WT heterodimer mixes also displayed little activity without Fis, particularly in the 1:2 mixing reaction, but were activated by Fis to generate double-strand cleavages at both hix sites (Fig. 6D, lanes 5–8). The absence of single-strand nicking in the reactions without Fis indicates that a H107Y subunit within a heterodimer is not capable of catalyzing cleavage unless the other subunit is activated by Fis.
Discussion
The initiation of the chemical steps of Hin recombination by the concerted nucleophilic attack of serine 10 in all four Hin subunits bound to the hix sites is regulated by two different protein–protein interactions within the invertasome: synaptic interactions between Hin dimers and Fis–Hin interactions. The present work focuses on the role of Fis–Hin interactions in the regulation of coordinated catalytic activity of the Hin protomers.
Communication between Fis and Hin dimers in the coordinate activation of the invertasome
We have shown previously that Fis dimers must be bound to both enhancer domains to activate DNA cleavage by Hin (Johnson and Simon 1985; Bruist et al. 1987). We show here that enhancers containing activating and nonactivating Fis dimers are incapable of assembling catalytically active invertasomes. Likewise, our experiments suggest that both Hin dimers bound to the synapsed hix sites must be activatable to initiate the chemical steps of recombination. Invertasomes containing one activatable and one nonactivatable dimer do not promote any DNA cleavage. The lack of single hix site cleavage in these reactions strengthens the argument that the Hin dimers must communicate to coordinate their catalytic activities. Although we have not directly proven that invertasomes containing activatable and nonactivatable Hin dimers are assembling, we note that nonactivatable M101Cox Hin dimers readily form paired-hix synaptic complexes. Moreover, addition of M101Cox strongly inhibits reactions containing saturating amounts of Hin–WT (data not shown). Taken together, the Fis and Hin dimer mixing experiments suggest that Fis must activate both of the synapsed Hin dimers to initiate any catalysis.
The Hin homodimer mixing experiments also demonstrate that activation and catalysis are biochemically separable events. Invertasomes assembled from wild-type Hin and a catalytic site mutant (Hin S10G) are catalytically active but generate double-strand cleavages at only one hix site. Given the requirement discussed previously that both Hin dimers must be activatable to initiate catalysis, we interpret the single-site cleavage to imply that invertasomes containing mixtures of wild-type and S10G dimers can be activated even though only one dimer is catalytically competent. Experiments with γδ resolvase have also shown that synaptic complexes containing a catalytic site mutant dimer and a wild-type dimer at the crossover sites generate double-strand cleavages at one res site (Boocock et al. 1995).
Communication between Fis and Hin subunits in the coordinate activation of the invertasome
Because the activation region of the Fis dimer contains two structurally independent and mobile β-hairpin arm motifs that protrude from the core, it is tempting to consider a model in which each β-arm contacts one of the four Hin subunits that are bound to a hix site in the invertasome to initiate recombination. However, we found that only one β-arm in each Fis dimer was sufficient to stimulate both Hin DNA cleavage and strand exchange. Reactions employing Fis heterodimers with only one active β-arm appeared to be as efficient as active homodimers in stimulating Hin inversion. The activity of the Fis heterodimers is entirely consistent with an earlier surprising observation that Fis dimers containing the two mobile β-arms linked together by a disulfide bridge at one of several positions within the β-strands or β-hairpin loops (e.g., between amino acids 15, 18, and 19) were able to activate Hin efficiently (Safo et al. 1997). The covalent linkage would prevent the two β-arms from interacting simultaneously with two separate regions of the Hin dimer.
The requirement for only one of the two regulatory regions on a dimeric protein has also been described for transcriptional activation by the CRP protein, in which heterodimers composed of activating and nonactivating CRP subunits were found to potentiate transcription when the activating region was oriented appropriately with respect to the target RNA polymerase (Zhou et al. 1993, 1994). Whereas the very efficient activation by Fis heterodimers implies that they may function when bound in either orientation at each enhancer domain, this conclusion is tempered by the fact that catalytic activation, not invertasome assembly, is likely be the slow step in the Hin inversion reaction (Haykinson et al. 1996). Therefore, different binding arrangements of the Fis heterodimers could be sampled in the time frame required to initiate cleavage. Directing the orientation of a Fis heterodimer using altered binding specificity mutations, as was done with CRP, is probably not possible because of the degenerate DNA-recognition sequence.
The finding that only one of the Fis dimer β-arms is required to activate a Hin dimer led us to investigate whether activation of only one Hin subunit within a hix-bound dimer is sufficient to initiate recombination. Using newly established methods for effectively refolding denatured Hin, a series of different Hin mutant heterodimers were assayed for their ability to initiate recombination in the presence or absence of Fis. The results of these experiments are consistent with the conclusion that both Hin subunits within the dimer must be activated by Fis to initiate recombination. For example, invertasomes assembled using heterodimer mixes containing wild-type and the activation-defective M101Cox Hin subunits were not catalytically active, even for single-strand cleavage at hix. On the other hand, Fis-activated reactions employing heterodimers containing the activatable catalytic site mutant S10G plus wild-type Hin generated substantial levels of nicked (as well as double-strand cleavages at one hix site) products. Thus, the catalytic activity of both subunits within the Hin dimer is coordinately activated by Fis prior to DNA cleavage.
Heterodimer mixes containing Fis-independent (H107Y) and Fis-dependent (S10G or wild-type) subunits were assayed to determine whether a single Fis-independent subunit could confer Fis-independence to a Hin dimer. These experiments suggested that complexes containing H107Y:S10G or wild-type heterodimers were only active in the presence of Fis. As was the case with the Fis-activatable:Fis-nonactivatable Hin heterodimers, nicking by the hyperactive H107Y subunit dimerized with a wild-type subunit was not observed. These results provide further support for the conclusion that both subunits within the Hin dimer must be in an activated state to initiate hix cleavage.
Potential mechanisms of Hin dimer activation by a single β-hairpin arm on Fis
There are several possible mechanisms by which a single Fis β-arm could activate both subunits of a Hin dimer within the invertasome. In one model, a Fis β-arm could interact sequentially with a common site in each Hin subunit. Once one Hin subunit has been activated, the mobile β-arm would move to contact the second subunit. Such a movement, however, would seem unlikely within the context of an assembled invertasome (see model below), as the β-arm would need to swing around to the opposite side of the Hin dimer to contact the analogous position within the symmetrically oriented subunit. Moreover, reactions employing Fis heterodimers do not generate detectable levels of nicked DNA intermediates, which could arise from activation of the first Hin subunit prior to the second. A second model predicts that one Fis β-arm interacts with only one Hin dimer subunit, perhaps triggering a conformational change, which in turn, induces a similar change in the second dimer subunit to result in a coordinate attack of the hix DNA. Whereas this type of sequential activation mechanism also remains possible, we do not favor it because of the data implying that both Hin subunits within the dimer must be either activated by Fis or contain the Fis-independent activating mutation to initiate catalysis. In a third model, a single Fis β-arm contacts Hin within or adjacent to the dimer interface in such a way as to trigger a simultaneous conformational change in both subunits. This quaternary change would result in the repositioning of the active sites of both Hin subunits to enable a coordinate attack of both DNA strands at the center of the hix sites (Feng et al. 1994a; Rice and Steitz 1994b). In this regard, the consequence of Fis–Hin interactions may resemble the situation found with the single amino acid substitutions within the dimer interface (such as H107Y) that allow the DNA invertases to promote recombination without Fis.
Structural model of the Hin invertasome
We have constructed a molecular model for the Fis–Hin invertasome complex that provides a framework for further elucidating the mechanism of Fis–Hin and Hin–Hin communication during the process of DNA inversion. Previous studies place a number of constraints on the structure of the invertasome that must be satisfied. The spacing of the two Fis-binding sites within the enhancer, 48 bp between their centers, is critical for enhancer function. Whereas addition of one complete helical turn of DNA is partially tolerated, addition of more than one helical turn, deletion of one turn, or addition of a partial helical turn essentially destroys enhancer activity (Johnson et al. 1987; R.C. Johnson, unpubl.). Fis-induced DNA bending angles and the path of DNA when bound at the hixL proximal and distal binding sites in the enhancer have been determined by gel mobility shift assays and from mapping the locations and efficiencies of scission sites by different Fis-1,10 phenanthroline chimeras (Pan et al. 1996; Perkins-Balding et al. 1997). For both binding sites, Fis-1,10 phenanthroline copper cleavage data have shown that the DNA flanking the core Fis-binding sites that is oriented towards the center of the enhancer is more severely bent than the outer flanking DNA segments. This information, along with the determination of the helical repeat of supercoiled DNA (11.2 bp/turn) around the hixL-enhancer region (Haykinson and Johnson 1993), allows the structure of the Fis-bound enhancer to be modeled (see Fig. 7). The activation domains of the two Fis dimers are separated by about 120 Å, as measured from the base of the flexible β-arms, and are rotated ∼115° with respect to each other.
Figure 7.
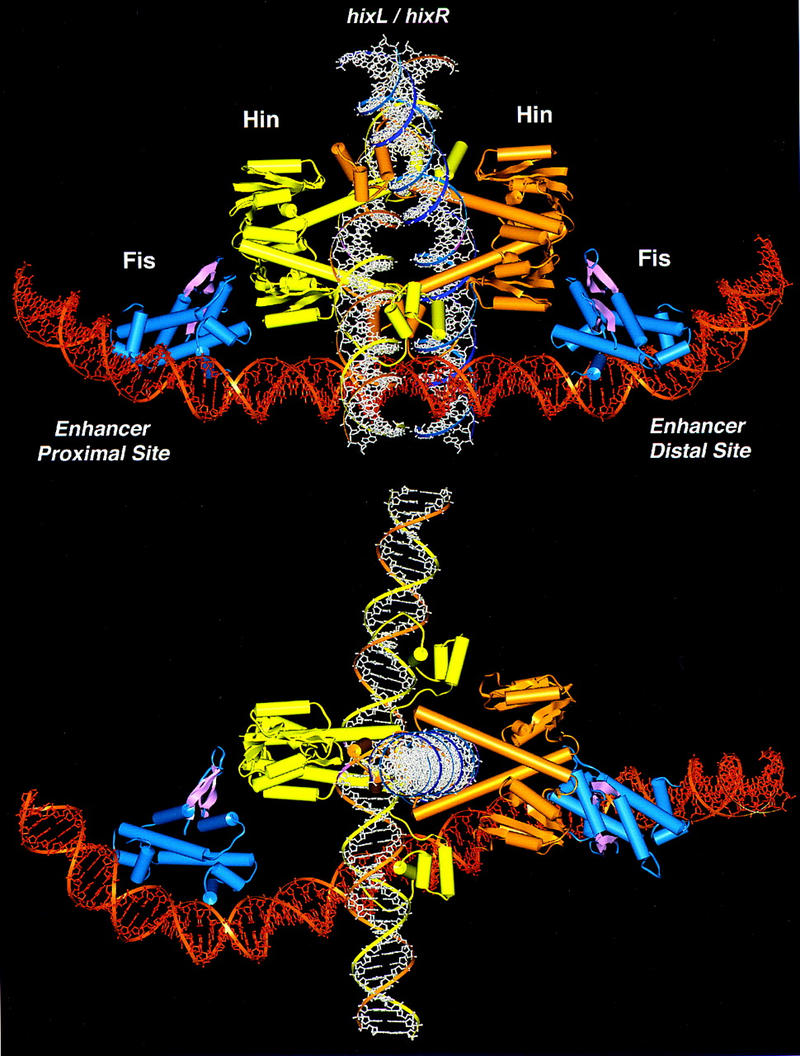
Model of the invertasome complex. Two Fis dimers, depicted in blue with the β-arm activation domains highlighted in pink, are bound to an enhancer segment of DNA shown in dark orange. Yellow and light orange ribbon diagrams of Hin dimers are complexed with recombination sites on DNA fragments shown in white. (Bottom) The model has been rotated 90° about the x-axis.
A complex consisting of two hix recombination sites each bound by at least a dimer of Hin must fit within the spatial constraints of the enhancer. Measurements of the diameters of crosslinked paired-hix synaptic complexes by dark-field diffraction electron microscopy are consistent with a tetramer of Hin (S. Ryazantsev, M.J. Haykinson, and R.C. Johnson, unpubl.). Topological studies of the Hin- and Gin-promoted inversion reactions have provided strong evidence that two negative DNA nodes are trapped in the invertasome complex such that the two hix DNA segments are located on opposite sides of the enhancer DNA (Kanaar et al. 1988; Heichman et al. 1991). Of the many possibilities considered, the most plausible configuration of two Hin bound hix sites that fits the spatial arrangement of Fis dimers and the geometric arrangement of DNA strands positions the catalytic domains on the outside of the Hin tetramer adjacent to the activating regions of Fis (Fig. 7). Within this model, the hix DNA segments are located in the center of the Hin tetramer and oriented at ∼90° with respect to each other. This arrangement of the Hin tetramer is similar to one of the models proposed for site I association in the resolvase synaptosome and is consistent with high-resolution electron microscopy of paired-hix complexes (Rice and Steitz 1994a; Yang and Steitz 1995; S. Ryazantsev, M.J. Haykinson, and R.C. Johnson, unpubl.). Whereas the Fis dimers are closer to one of the subunits in each Hin dimer, one of the Fis β-arms is directed towards and can easily contact the Hin dimer interface. Thus, this structural model of the invertasome is consistent with the proposed mechanism of Hin activation by Fis. Although this model is compatible with current information on the Hin inversion reaction, the predicted protein interfaces remain to be determined experimentally. We note that the core nucleotides that are exchanged upon recombination are located close to each other such that a relatively modest movement could position the cleaved DNA ends into the recombinant configuration. A mechanism by which multiple DNA exchanges could occur within the complex, leading to the complex knotted DNA products observed from processive recombination, is not apparent (Kanaar et al. 1990; Heichman et al. 1991).
Comparison with the Mu transposon enhancer
The enhancer elements in both phage Mu transposition and site-specific DNA inversion are believed to be assembled into topologically similar structures and function to increase reaction fidelity, though the precise mechanisms by which the two regulatory elements activate recombination differ somewhat. Like the paired-hix complex by Hin, the MuA transposase can readily assemble a paired-end complex, which is not active for DNA cleavage (Watson and Chaconas 1996). The Mu enhancer facilitates the assembly of a catalytically active MuA transposase tetramer by the formation of transient bivalent interactions between binding sites within the enhancer and the Mu ends (Lavoie and Chaconas 1995). As a functional catalytic unit in the Mu transpososome is assembled from multiple MuA subunits (Aldaz et al. 1996; Savilahti and Mizuuchi 1996; Yang et al. 1996), a catalytically competent complex is formed in conjunction with enhancer-assisted tetramer assembly. For the resolvase-invertase family, the active site pocket is exclusively contained within one protomer, though amino acids from the partner subunit in the dimer may influence its catalytic activity (Boocock et al. 1995; Yang and Steitz 1995). We propose that the hin enhancer, acting via the bound Fis dimers, functions to induce a conformational change in the Hin dimer to reposition the individual active sites close to the four strands of DNA for a concerted attack. Thus, somewhat different regulatory controls have evolved to lock the Mu transposase and the Hin DNA invertase in a catalytically inactive state even in a paired-end or paired-hix complex. The cis-acting enhancer elements unlock these inhibitory controls and at the same time ensure that only the topologically correct genetic rearrangement occurs. The Hin mutations causing Fis independence have released these controls and lead to indiscriminate rearrangements. Deletions that remove a substantial portion of the MuA amino terminus and lead to partial enhancer-independence also release these controls for Mu transposition (Yang et al. 1995).
Materials and methods
Fis mutants
The Fis mutants used in this study were described in Safo et al. (1997) and listed in Table 1. Fis D20K and S30C were combined by standard recombinant DNA methods, and a carboxy-terminal 6× His tag was added to D20K/S30C using PCR creating D20K/S30C/His. Fis mutant proteins were purified from cells containing derivatives of pRJ1077, in which fis was expressed from the T7 promoter as described (Pan et al. 1996).
Fis heterodimers were formed by denaturing and renaturing equivalent amounts of S30C and D20K/S30C/His. The proteins were denatured in 6 m guanidine chloride, 20 mm HEPES (pH 7.5), 0.2 m NaCl, 10 mm DTT, 20% glycerol, 1 mm CHAPS, and 0.1 mm EDTA at a final protein concentration of 100 μg/ml. Equal amounts of the denatured proteins were mixed in a dialysis bag and renatured by dialyzing into the same buffer with decreasing guanidine chloride concentrations over the course of two days. Prior to nickel chromatography, the refolded Fis dimers were dialyzed into 20 mm sodium phosphate (pH 7.4), 0.2 m NaCl, and 20% glycerol and then incubated with 10 mm oxidized glutathione at 23°C for 3.5 hr to form disulfide-linked dimers. Fis dimers containing at least one 6× His-tagged subunit were purified with the Qiagen Ni-NTA Spin Kit as specified by the manufacturer except that 0.2 m NaCl was present throughout. The purification was monitored by electrophoresis of the sample on a SDS–polyacrylamide gel in the absence of reducing agent and staining with Coomassie blue. No S30C homodimer, which lacked the His tag, was detected in the 250 mm imidazole elution.
Hin mutants
The properties of Hin M101C were described in Haykinson et al. (1996). The hin mutation H107Y, which was initially isolated as an inversion-proficient suppressor of the inactive F104C mutant (Haykinson et al. 1996), was introduced into an otherwise wild-type hin gene using a two-step PCR-mediated site-directed mutagenesis protocol (Landt et al. 1990). Hin S10G was generated by a single-step PCR using the corresponding mutant primer that overlapped the unique ClaI site in the hin gene (M. Lainé and R.C. Johnson, unpubl.). For purification, the mutant hin genes were cloned into pRJ1518, a derivative of pET11a (Novagen) containing the wild-type hin gene under the control of the T7 promoter. Two liters of Luria broth cultures of RJ1852 [BL21(DE3) fis–767] containing derivatives of pRJ1518 were grown to an OD600 = 0.6 and induced with 0.1 mm IPTG at 10°C for 10 hr. Cells were lysed and Hin purified essentially as described previously (Haykinson et al. 1996).
Hin heterodimers were formed by denaturing and mixing the two proteins in the desired ratio in 6 m guanidine chloride, 20 mm HEPES (pH 7.5), 0.1 m NaCl, 10 mm DTT, 20% glycerol, 50 mm CHAPS, and 0.1 mm EDTA at a final protein concentration of 100 μg/ml. The denatured proteins were placed in dialysis bags in 100 ml of denaturing buffer and renatured over the course of two days by pumping renaturation buffer (same as above except that no guanidine and 1 m NaCl was included) into the dialysis buffer while pumping out the dialysis buffer at the same rate (5 ml/hr). After an additional 6 hr dialysis in 100% renaturing buffer, the proteins were transferred to Hin storage buffer containing 20 mm HEPES (pH 7.5), 1 m NaCl, 50% glycerol, 4 mm CHAPS, and 0.1 mm EDTA. Purification of heterodimers by a method similar to that used for Fis is not feasible for Hin because Hin activity is abolished by amino-terminal tags and reduced significantly by carboxy-terminal tags.
In vitro recombination assays
The in vitro cleavage and inversion assays were performed and quantitated as described previously in Haykinson et al. (1996) except that reactions employing oxidized mutants were performed in the absence of DTT. The Hin proteins were titrated to determine the minimum amounts required for maximum activity. Unless otherwise noted, these saturating amounts of protein were used in the in vitro reactions. Disulfide-linked preparations of Fis Q21C and S30C as well as Hin M101C were obtained after long-term storage in buffer containing ≤1 mm DTT. Oxidized Hin M101Cox in wild-type:M101C and M101Cox renatured preparations was generated by incubation with 10 mm oxidized glutathione for 5 min in the reaction cocktail prior to initiating the reaction with Fis. Paired-hix complexes formed on pMS634 (Johnson and Simon 1985) were crosslinked with 0.15% glutaraldehyde for 40 min at 23°C. Forty millimolar lysine was added to inactivate remaining cross-linker, and the products were digested with PstI prior to electrophoresis on a 1% agarose gel.
Molecular modeling
Modeling of the invertasome was performed on an SGI Indigo 2 workstation using the Insight II software package (Molecular Simulations). Hin bound to hixL on a 50-bp DNA segment was modeled as described in Haykinson et al. (1996). The K36E Fis structure (Safo et al. 1997) was docked onto the two binding sites within an 83-bp hin enhancer segment such that the overall DNA bending angles at the proximal and distal domains were 65° and 74°, respectively, and the DNA curvature at each flank conformed with the Fis · 1,10-phenanthroline-copper scission data as described in Pan et al. (1996). The helical repeat of the enhancer DNA was set at 11.2 bp, corresponding to twist angles of −32.14° (Haykinson and Johnson 1993).
Acknowledgments
We thank Leah Corselli for isolation and purification of the Fis mutants, Muriel Lainé for isolation and purification of Hin S10G, Hanna Yuan for valuable discussions, and Samson Chow for critical reading of the manuscript. We also thank Yi-Meng Yen for help with figure preparation. This work was supported by grant GM38509 from the National Institutes of Health and by an American Cancer Society Faculty Research Award to R.C.J. S.K.M. was supported in part by U.S. Public Health Service National Research Service Award GM07185.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rcjohnson@mednet.ucla.edu; FAX (310) 206-5272.
References
- Adams CW, Nanassy O, Johnson RC, Hughes KT. Role of arginine-43 and arginine-69 of the Hin recombinase catalytic domain in the binding of Hin to the hix DNA recombination sites. Mol Microbiol. 1997;24:1235–1247. doi: 10.1046/j.1365-2958.1997.4141789.x. [DOI] [PubMed] [Google Scholar]
- Aldaz H, Schuster E, Baker TA. The interwoven architecture of the Mu transposase couples DNA synapsis to catalysis. Cell. 1996;85:257–269. doi: 10.1016/s0092-8674(00)81102-2. [DOI] [PubMed] [Google Scholar]
- Boocock MR, Zhu X, Grindley NDF. Catalytic residues of gamma delta resolvase act in cis. EMBO J. 1995;14:5129–5140. doi: 10.1002/j.1460-2075.1995.tb00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruist MF, Glasgow AC, Johnson RC, Simon MI. Fis binding to the recombinational enhancer of the Hin DNA inversion system. Genes & Dev. 1987;1:762–772. doi: 10.1101/gad.1.8.762. [DOI] [PubMed] [Google Scholar]
- Crisona NJ, Kanaar R, Gonzalez TN, Zechiedrich EL, Klippel A, Cozzarelli NR. Processive recombination by wild-type Gin and an enhancer-independent mutant. Insight into the mechanisms of recombination selectivity and strand exchange. J Mol Biol. 1994;243:437–457. doi: 10.1006/jmbi.1994.1671. [DOI] [PubMed] [Google Scholar]
- Feng J-A, Dickerson RE, Johnson RC. Proteins which promote DNA inversion and deletion. Curr Opin Struct Biol. 1994a;4:60–66. [Google Scholar]
- Feng JA, Johnson RC, Dickerson RE. Hin recombinase bound to DNA: The origin of specificity in major and minor groove interactions. Science. 1994b;263:348–355. doi: 10.1126/science.8278807. [DOI] [PubMed] [Google Scholar]
- Glasgow AC, Bruist MF, Simon MI. DNA-binding properties of the Hin recombinase. J Biol Chem. 1989a;264:10072–10082. [PubMed] [Google Scholar]
- Glasgow AC, Hughes KT, Simon MI. Bacterial DNA inversion systems. In: Berg DE, Howe MM, editors. Mobile DNA. Washington, D.C.: American Society for Microbiology; 1989b. pp. 637–659. [Google Scholar]
- Haffter P, Bickle TA. Enhancer-independent mutants of the Cin recombinase have a relaxed topological specificity. EMBO J. 1988;7:3991–3996. doi: 10.1002/j.1460-2075.1988.tb03287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haykinson MJ, Johnson RC. DNA looping and the helical repeat in vitro and in vivo: Effect of HU protein and enhancer location on Hin invertasome assembly. EMBO J. 1993;12:2503–2512. doi: 10.1002/j.1460-2075.1993.tb05905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haykinson MJ, Johnson LM, Soong J, Johnson RC. The Hin dimer interface is critical for Fis-mediated activation of the catalytic steps of site-specific DNA inversion. Curr Biol. 1996;6:163–177. doi: 10.1016/s0960-9822(02)00449-9. [DOI] [PubMed] [Google Scholar]
- Heichman KA, Johnson RC. The Hin invertasome: Protein-mediated joining of distant recombination sites at the enhancer. Science. 1990;249:511–517. doi: 10.1126/science.2166334. [DOI] [PubMed] [Google Scholar]
- Heichman KA, Moskowitz IPG, Johnson RC. Configuration of DNA strands and mechanism of strand exchange in the Hin invertasome as revealed by analysis of recombinant knots. Genes & Dev. 1991;5:1622–1634. doi: 10.1101/gad.5.9.1622. [DOI] [PubMed] [Google Scholar]
- Hughes RE, Rice PA, Steitz TA, Grindley ND. Protein-protein interactions directing resolvase site-specific recombination: A structure-function analysis. EMBO J. 1993;12:1447–1458. doi: 10.1002/j.1460-2075.1993.tb05788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RC. Mechanism of site-specific DNA inversion in bacteria. Curr Opin Genet Dev. 1991;1:404–411. doi: 10.1016/s0959-437x(05)80307-7. [DOI] [PubMed] [Google Scholar]
- Johnson RC. Site-specific recombinases and their interactions with DNA. In: Lilley D, editor. Frontiers in molecular biology: DNA–protein: Structural interactions. Oxford, UK: IRL Press; 1995. pp. 141–176. [Google Scholar]
- Johnson RC, Simon MI. Hin-mediated site-specific recombination requires two 26 bp recombination sites and a 60 bp recombinational enhancer. Cell. 1985;41:781–791. doi: 10.1016/s0092-8674(85)80059-3. [DOI] [PubMed] [Google Scholar]
- Johnson RC, Bruist MF. Intermediates in Hin-mediated DNA inversion: A role for Fis and the recombinational enhancer in the strand exchange reaction. EMBO J. 1989;8:1581–1590. doi: 10.1002/j.1460-2075.1989.tb03542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RC, Glasgow AC, Simon MI. Spatial relationship of the Fis binding sites for Hin recombinational enhancer activity. Nature. 1987;329:462–465. doi: 10.1038/329462a0. [DOI] [PubMed] [Google Scholar]
- Johnson RC, Ball CA, Pfeffer D, Simon MI. Isolation of the gene encoding the Hin recombinational enhancer binding protein. Proc Natl Acad Sci. 1988;85:3484–3488. doi: 10.1073/pnas.85.10.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahmann R, Rudt F, Koch C, Mertens G. G inversion in bacteriophage Mu DNA is stimulated by a site within the invertase gene and a host factor. Cell. 1985;41:771–780. doi: 10.1016/s0092-8674(85)80058-1. [DOI] [PubMed] [Google Scholar]
- Kanaar R, Cozzarelli NR. Roles of supercoiled DNA structure in DNA transactions. Curr Opin Struct Biol. 1992;2:369–379. [Google Scholar]
- Kanaar R, Klippel A, Shekhtman E, Dungan JM, Kahmann R, Cozzarelli NR. Processive recombination by the phage Mu Gin system: Implications for the mechanisms of DNA strand exchange, DNA site alignment, and enhancer action. Cell. 1990;62:353–366. doi: 10.1016/0092-8674(90)90372-l. [DOI] [PubMed] [Google Scholar]
- Kanaar R, van de Putte P, Cozzarelli NR. Gin-mediated DNA inversion: Product structure and the mechanism of strand exchange. Proc Natl Acad Sci. 1988;85:752–756. doi: 10.1073/pnas.85.3.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A, Cloppenborg K, Kahmann R. Isolation and characterization of unusual Gin mutants. EMBO J. 1988a;7:3983–3989. doi: 10.1002/j.1460-2075.1988.tb03286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A, Kanaar R, Kahmann R, Cozzarelli NR. Analysis of strand exchange and DNA binding of enhancer-independent Gin recombinase mutants. EMBO J. 1993;12:1047–1057. doi: 10.1002/j.1460-2075.1993.tb05746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A, Mertens G, Patschinsky T, Kahmann R. The DNA invertase Gin of phage Mu: Formation of a covalent complex with DNA via a phosphoserine at amino acid position 9. EMBO J. 1988b;7:1229–1237. doi: 10.1002/j.1460-2075.1988.tb02935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch C, Vandekerckhove J, Kahmann R. Escherichia coli host factor for site-specific DNA inversion: Cloning and characterization of the fis gene. Proc Natl Acad Sci. 1988;85:4237–4241. doi: 10.1073/pnas.85.12.4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrewa D, Granzin J, Koch C, Choe HW, Raghunathan S, Wolf W, Labahn J, Kahmann R, Saenger W. Three-dimensional structure of the E. coli DNA-binding protein FIS. Nature. 1991;349:178–180. doi: 10.1038/349178a0. [DOI] [PubMed] [Google Scholar]
- Landt O, Grunert HP, Hahn U. A general method for rapid site-directed mutagenesis using the polymerase chain reaction. Gene. 1990;26:125–128. doi: 10.1016/0378-1119(90)90351-q. [DOI] [PubMed] [Google Scholar]
- Lavoie BD, Chaconas G. Transposition of phage Mu DNA. In: Saedler H, Gierl A, editors. Current topics in microbiology and immunology. New York, NY: Springer-Verlag; 1995. pp. 83–102. [DOI] [PubMed] [Google Scholar]
- Lim HM. Analysis of subunit interaction by introducing disulfide bonds at the dimerization domain of Hin recombinase. J Biol Chem. 1994;269:31134–31142. [PubMed] [Google Scholar]
- Moskowitz IP, Heichman KA, Johnson RC. Alignment of recombination sites in Hin-mediated site-specific DNA recombination. Genes & Dev. 1991;5:1635–1645. doi: 10.1101/gad.5.9.1635. [DOI] [PubMed] [Google Scholar]
- Pan CQ, Finkel SE, Cramton SE, Feng JA, Sigman DS, Johnson RC. Variable structures of Fis-DNA complexes determined by flanking DNA-protein contacts. J Mol Biol. 1996;264:675–695. doi: 10.1006/jmbi.1996.0669. [DOI] [PubMed] [Google Scholar]
- Perkins-Balding D, Dias DP, Glasgow AC. Location, degree, and direction of DNA bending associated with the Hin recombinational enhancer sequence and Fis-enhancer complex. J Bacteriol. 1997;179:4747–4753. doi: 10.1128/jb.179.15.4747-4753.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice PA, Steitz TA. Model for a DNA-mediated synaptic complex suggested by crystal packing of gamma delta resolvase subunits. EMBO J. 1994a;13:1514–1524. doi: 10.2210/pdb1gdr/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice PA, Steitz TA. Refinement of gamma delta resolvase reveals a strikingly flexible molecule. Structure. 1994b;2:371–384. doi: 10.1016/s0969-2126(00)00039-3. [DOI] [PubMed] [Google Scholar]
- Safo MK, Yang WZ, Corselli L, Cramton SE, Yuan HS, Johnson RC. The transactivation region of the Fis protein that controls site-specific DNA inversion contains extended mobile β-hairpin arms. EMBO J. 1997;16:6860–6873. doi: 10.1093/emboj/16.22.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savilahti H, Mizuuchi K. Mu transpositional recombination: Donor DNA cleavage and strand transfer in trans by the Mu transposase. Cell. 1996;85:271–280. doi: 10.1016/s0092-8674(00)81103-4. [DOI] [PubMed] [Google Scholar]
- Sherratt DJ. Tn3 and related tansposable elements: Site-specific recombination and transposition. In: Berg DE, Howe MM, editors. Mobile DNA. Washington, D.C.: American Society for Microbiology; 1989. pp. 163–184. [Google Scholar]
- van de Putte P, Goosen N. DNA inversions in phages and bacteria. Trends Genet. 1992;8:457–462. doi: 10.1016/0168-9525(92)90331-w. [DOI] [PubMed] [Google Scholar]
- Watson MA, Chaconas G. Three-site synapsis during Mu DNA transposition: A critical intermediate preceding engagement of the active site. Cell. 1996;85:435–445. doi: 10.1016/s0092-8674(00)81121-6. [DOI] [PubMed] [Google Scholar]
- Yang JY, Jayaram M, Harshey RM. Enhancer-independent variants of phage Mu transposase: Enhancer-specific stimulation of catalytic activity by a partner transposase. Genes & Dev. 1995;9:2545–2555. doi: 10.1101/gad.9.20.2545. [DOI] [PubMed] [Google Scholar]
- Yang JY, Jayaram M, Harshey RM. Positional information within the Mu transposase tetramer: Catalytic contributions of individual monomers. Cell. 1996;85:447–455. doi: 10.1016/s0092-8674(00)81122-8. [DOI] [PubMed] [Google Scholar]
- Yang W, Steitz TA. Crystal structure of the site-specific recombinase gamma delta resolvase complexed with a 34 bp cleavage site. Cell. 1995;82:193–207. doi: 10.1016/0092-8674(95)90307-0. [DOI] [PubMed] [Google Scholar]
- Yuan HS, Finkel SE, Feng JA, Kaczor-Grzeskowiak M, Johnson RC, Dickerson RE. The molecular structure of wild-type and a mutant Fis protein: Relationship between mutational changes and recombinational enhancer function or DNA binding. Proc Natl Acad Sci. 1991;88:9558–9562. doi: 10.1073/pnas.88.21.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Busby S, Ebright RH. Identification of the functional subunit of a dimeric transcription activator protein by use of oriented heterodimers. Cell. 1993;73:375–379. doi: 10.1016/0092-8674(93)90236-j. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Pendergrast PS, Bell A, Williams R, Busby S, Ebright RH. The functional subunit of a dimeric transcription activator protein depends on promoter architecture. EMBO J. 1994;13:4549–4557. doi: 10.1002/j.1460-2075.1994.tb06776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]