

Abstract
The mitogen-activated protein kinases (MAPKs) play critical roles in many signal transduction processes. Several MAPKs have been found in Saccharomyces cerevisiae, including Fus3 in the mating pathway and Hog1 in the osmotic-stress response pathway. Cells lacking Fus3 or Hog1 activity are deficient in mating or adaptation to osmotic shock, respectively. However, constitutive activation of either Fus3 or Hog1 is lethal. Therefore, yeast cells have to tightly regulate both the activation and inactivation of Fus3 and Hog1 MAPKs, which are controlled mainly by phosphorylation and dephosphorylation. Previous studies have shown that Fus3 activity is negatively regulated by protein tyrosine phosphatase Ptp3. In contrast, the Hog1 MAPK is mainly dephosphorylated by Ptp2 even though the two phosphatases share a high degree of sequence similarity. To understand the mechanisms of MAPK regulation, we examined the molecular basis underlying the in vivo substrate specificity between phosphatases and MAPKs. We observed that the amino-terminal noncatalytic domain of Ptp3 directly interacts with Fus3 via CH2 (Cdc25 homology) domain conserved among yeast PTPases and mammalian MAP kinase phosphatases and is responsible for the in vivo substrate selectivity of the phosphatase. Interaction between Ptp3 and Fus3 is required for dephosphorylation and inactivation of Fus3 under physiological conditions. Mutations in either Ptp3 or Fus3 that abolish this interaction cause a dysregulation of the Fus3 MAPK. Our data demonstrate that the specificity of MAP kinase inactivation in vivo by phosphatases is determined by specific protein–protein interactions outside of the phosphatase catalytic domain.
Keywords: Fus3, MAP kinase, Ptp3, protein tyrosine phosphatase, mating response
Mitogen-activated protein kinases (MAPKs), also known as extracellular signal-regulated kinases (ERKs), are involved in a wide range of eukaryotic signaling processes (Lewis et al. 1998). A typical MAPK signaling pathway contains an evolutionarily conserved kinase cascade consisting of Raf or MAPK kinase kinase (MEKK), MAPK kinase (MEK), and MAPK. In response to a variety of extracellular signals, MAPK activity is regulated through phosphorylation on the conserved TXY motif by MEKs (Cobb and Goldsmith 1995). Each individual MAPK is activated by distinct extracellular signals, and activation of MAPKs results in specific physiological responses including mitogenesis, adaptation to environmental stress, differentiation and development in high eukaryotes (Lewis et al. 1998; Tan and Kim 1999). In Saccharomyces cerevisiae, five MAPK signaling pathways have been identified that regulate mating, osmotic-stress response, cell wall integrity, filamentous growth, and spore wall formation (Herskowitz 1995; Gustin et al. 1998).
The S. cerevisiae mating pheromone response represents a well-characterized MAPK signaling pathway that controls the formation of diploid cells from haploid cells (Bardwell et al. 1994). Treatment of a or α haploid yeast cells with mating pheromone (α- or a-factor), respectively, triggers activation of a MAPK cascade that is composed of Ste11 MEKK, Ste7 MEK, and Fus3 MAPK. Activation of this MAPK pathway results in alteration of cellular morphology, induction of pheromone-inducible genes, and G1 cell cycle arrest in preparation for zygote formation (Gustin et al. 1998). Hog1, another well-characterized yeast homolog of mammalian P38/JNK stress-activated MAPKs, is involved in cellular adaptation to osmotic stress. High osmolarity causes activation of the Hog1 MAPK that leads to accumulation of intracellular glycerol to counteract the extracellular hyperosmolarity (Gustin et al. 1998; Millar 1999).
Although Fus3 activation is required for mating pheromone response, constitutive activation of the mating pathway can cause lethality possibly as a result of irreversible cell cycle arrest (Miyajima et al. 1987; Nomoto et al. 1990; Stevenson et al. 1992). Similarly, activation of Hog1 is crucial for cell viability under hyperosmotic conditions, yet constitutive activation of the Hog1 MAPK is lethal (Brewster et al. 1993; Maeda et al. 1993, 1994). Therefore, the activities of both Fus3 and Hog1 MAP kinases must be tightly regulated in response to specific environmental conditions because either lack of activity or overstimulated activity can be detrimental to yeast cells. MAPKs are mainly activated by Tyr and Thr phosphorylation catalyzed by MEKs. In addition, MAPKs including Fus3 can also undergo autophosphorylation, leading to autoactivation at lower efficiency (Errede et al. 1993). It is critical for cells to repress the basal activity of Fus3 in the absence of pheromone to avoid unelicited mating responses. It is equally critical for yeast cells to deactivate Fus3 to resume normal cell growth after pheromone stimulation. In contrast to the well characterized activation mechanism that requires MEKs, much less is known about the equally crucial mechanism for repressing basal activity and deactivating MAPK following stimulation.
In our previous study we demonstrated that Fus3 is coordinately regulated by two phosphatases (Zhan et al. 1997). Constitutively expressed tyrosine-specific Ptp3 prevents the autoactivation by regulating basal tyrosine phosphorylation. Pheromone stimulation induces a dual-specificity phosphatase, Msg5 (Doi et al. 1994), and Msg5 together with Ptp3 dephosphorylate and inactivate Fus3, allowing cells to recover following mating response. Ptp3 shares extensive sequence homology to another yeast tyrosine phosphatase, Ptp2. In the absence of Ptp3, Ptp2 can partially substitute for Ptp3 (Zhan et al. 1997). Genetic data from other groups suggest that Ptp2 is more critical than Ptp3 in the dephosphorylation and inactivation of Hog1 MAPK, whereas Ptp3 plays a minor role (Jacoby et al. 1997; Wurgler-Murphy et al. 1997). Therefore, Ptp3 and Ptp2 display relative substrate selectivity towards Fus3 MAPK in pheromone response and Hog1 in osmotic stress response, respectively.
In this study we investigated the molecular mechanisms responsible for the determination of Ptp3 substrate preference in vivo. Analysis of chimeric molecules in which the functional domains of Ptp2 and Ptp3 were swapped demonstrates that substrate selectivity is determined by the amino-terminal noncatalytic domains. In addition, Ptp3 is targeted to Fus3 MAPK in vivo through specific and direct protein–protein interaction mediated by the Ptp3 amino-terminal domain instead of the phosphatase domain. Despite the limited overall sequence homology between tyrosine phosphatase Ptp3 and mammalian dual-specificity MAPK phosphatases (MKPs), we identified a stretch of residues conserved among Ptp3 and MKPs outside of the catalytic domain that is required for Ptp3 and Fus3 interaction. We further demonstrate that such interaction is essential for Fus3 MAPK regulation, as mutant forms of Ptp3 that have lost Fus3 binding are unable to dephosphorylate Fus3 in vivo. On the other hand, Fus3 mutants, which abrogate the Ptp3 interaction, become constitutively phosphorylated on Tyr and render cells carrying these mutant alleles more sensitive to pheromone. These results demonstrate that the in vivo targeting of protein tyrosine phosphatase Ptp3 to its substrate Fus3 MAPK is required in the MAPK regulation and in the determination of substrate selectivity of phosphatases.
Results
The amino-terminal noncatalytic domains determine Ptp2 and Ptp3 substrate specificity in vivo
To understand the molecular mechanism responsible for substrate selectivity between Ptp2 and Ptp3, we first set out to test the differential regulation of Fus3 or Hog1 by PTPases biochemically by examining kinase activities in vivo in PTPase gene disruption strains. The Fus3 kinase was immunoprecipitated from wild type, ptp2Δ, ptp3Δ, or ptp2Δptp3Δ double disruption cells treated with pheromone. As previously observed (Zhan et al. 1997), ptp2Δ single disruption had no effect on the kinase activity and Tyr phosphorylation of HA–Fus3 (Fig. 1A, top and middle, lanes 5,6) compared with wild type (lanes 3,4). In contrast, ptp3Δ single disruption enhanced both kinase activity (twofold increase) and Tyr phosphorylation (lanes 7,8). Double disruption of ptp2Δptp3Δ further increased the kinase activity (up to threefold) and Tyr phosphorylation (top and middle, lanes 9,10). Interestingly, when Hog1 activity and Tyr phosphorylation were determined in cells challenged with high osmolarity (0.4 m NaCl), ptp3Δ disruption had little effect (Fig. 1B, top and middle, lanes 7,8) compared with wild-type cells (lanes 3,4), whereas ptp2Δ single disruption increased both the basal, as well as the stimulated, activity (twofold increase, top, lanes 5,6) and Tyr phosphorylation (middle, lanes 5,6). Double disruption of ptp2Δptp3Δ resulted in further enhancement (up to fourfold in kinase activity, top and middle, lanes 9,10). In all the immunoprecipitation experiments, a similar amount of HA–Fus3 or HA–Hog1 was detected (Fig. 1A,B, bottom). Therefore, these data are consistent with previous observations that in vivo Fus3 and Hog1 MAPKs are differentially regulated by Ptp3 and Ptp2, respectively (Jacoby et al. 1997; Wurgler-Murphy et al. 1997; Zhan et al. 1997).
Figure 1.

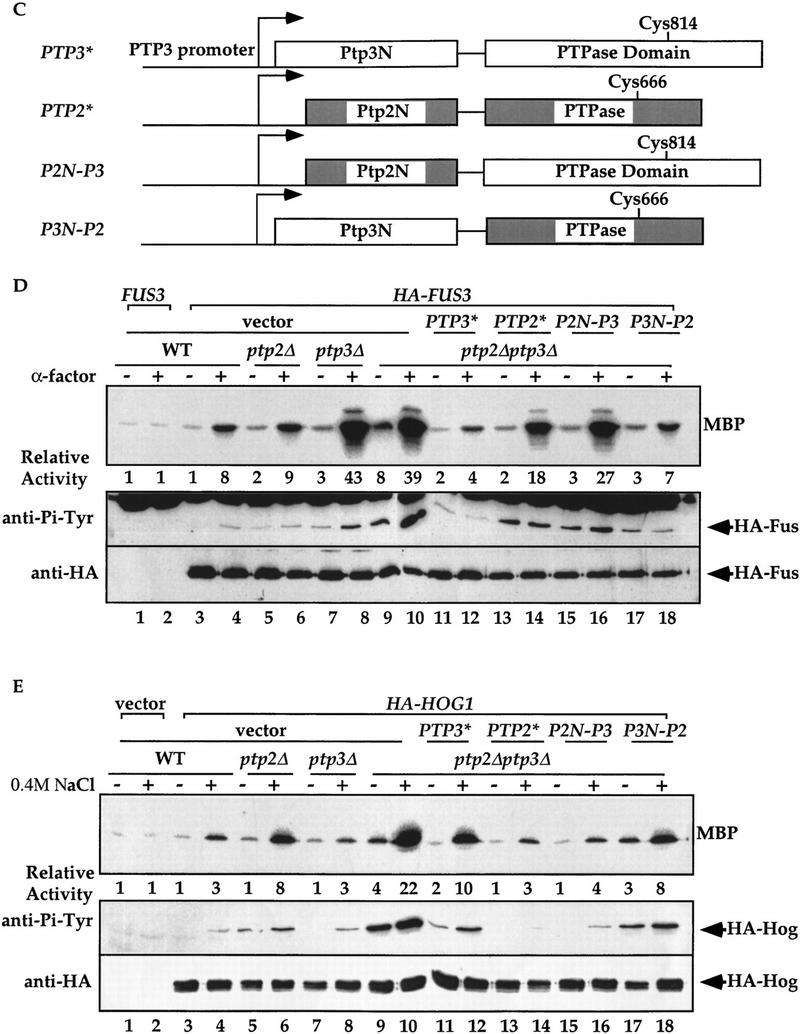
The amino-terminal noncatalytic domain determines Ptp2 and Ptp3 substrate specificities in vivo. (A) Fus3 activity and Tyr–Pi are elevated with ptp3Δ disruption. HA–Fus3 (pXZ147) was expressed in wild type (Y568), ptp2Δ (Y572), ptp3Δ (Y567), or ptp2Δptp3Δ double disruption cells (Y578). Logarithmically growing cells were stimulated with 2.5 μm α-factor at 30°C for 30 min. HA–Fus3 was immunoprecipitated, and kinase activities were determined by in vitro MBP kinase assays (top). The MBP phosphorylation levels were quantitated with a PhosphorImager (Molecular Dynamics) and normalized to the activity in unstimulated wild-type cells expressing untagged FUS3 (pXZ148; lane 1). In parallel, anti-Pi–Tyr and anti-HA immunoblotting were performed on the same immunoprecipitated samples to determine Tyr phosphorylation (middle), and HA–Fus3 protein levels (bottom). *IgG(H) denotes the immunoglobulin heavy chain. (B) Hog1 activity and Tyr–Pi are elevated with ptp2Δ disruption. Wild type (Y568), ptp2Δ (Y572), ptp3Δ (Y567), or ptp2Δptp3Δ double disruption cells (Y578) expressing a functional HA-tagged HOG1 (pXZ331) was subjected to 0.4 m NaCl stimulation at 30°C for 10 min. The kinase activity, Tyr–Pi, and protein levels of HA–Hog1 were determined by performing immunoprecipitation, in vitro kinase assay using MBP as substrate (top), and immunoblotting with anti-Pi–Tyr (middle) and anti-HA (bottom) mAbs as described in the Materials and Methods. The kinase activity is normalized to the level in unstimulated wild-type cells expressing vector (top, lane 1). (C) Schematic representation of PTP2/PTP3 chimeric constructs. Open boxes and shaded boxes depict PTP2 and PTP3, respectively. The active site Cys residues are indicated. PTP3* and PTP2* represent synthetic genes that contain PTP3 or PTP2 ORF subcloned at the 3′ end of the PTP3 promoter. All plasmids use the PTP3 promoter to drive the expression of native or chimera proteins. (D) The amino-terminal domain of Ptp3 dictates the phosphatase specificity towards Fus3. Centromeric plasmids that express PTP3 (pXZ273), PTP2 (pXZ315), P2N-P3 (pXZ316), and P3N-P2 (pXZ317) from the endogenous PTP3 promoter were introduced into ptp2Δptp3Δ double disruption cells carrying HA–FUS3 (pXZ147). These transformants or wild-type (Y568), ptp2Δ (Y572), ptp3Δ (Y567), and ptp2Δptp3Δ cells (Y578) carrying HA–FUS3 (pXZ147) + vector were stimulated with 2.5 μm α-factor. Immunoprecipitation, in vitro kinase assay, and immunoblotting were performed to measure the kinase activity (top), Tyr–Pi (middle), and HA–Fus3 (bottom) protein levels as described for A. (E) The amino-terminal domain of Ptp2 determines its selectivity towards Hog1. Centromeric plasmids that express PTP3 (pXZ273), PTP2 (pXZ315), P2N-P3 (pXZ316), and P3N-P2 (pXZ317) were introduced into ptp2Δptp3Δ double disruption cells carrying HA–HOG1 (pXZ331). Wild type (Y568), ptp2Δ cells (Y572), ptp3Δ cells (Y567), or ptp2Δptp3Δ cells (Y578) carrying the indicated plasmids were treated with 0.4 m NaCl for 5 min at 30°C. HA–Hog1 kinase activity (top), Tyr–Pi levels (middle), and protein levels (bottom) were determined as described for B. All experiments were repeated with similar results.
PTP2 and PTP3 encode proteins that share the highest degree of homology in the carboxy-terminal phosphatase domains. The amino-terminal noncatalytic domains are much less conserved than the phosphatase domains. To investigate which domain contributes to the in vivo substrate selectivity, the amino-terminal domains of Ptp2 and Ptp3 were swapped to create P2N-P3 or P3N-P2 chimeric molecules (Fig. 1C). These chimeric molecules were expressed in ptp2Δptp3Δ double disruption cells from centromeric plasmids under the control of the PTP3 promoter. Kinase activity and Tyr phosphorylation of Fus3 (Fig. 1D) or Hog1 (Fig. 1E) were measured. Introduction of PTP3 into ptp2Δptp3Δ cells reduced Fus3 kinase activity (Fig. 1D, top, lanes 9–12) and Tyr phosphorylation (middle, lanes 9–12) to the level comparable in wild type (top and middle, lanes 3,4) or ptp2Δ disruption cells (top and middle, lanes 5,6). Expression of PTP2 or P2N-P3, which encodes a chimeric protein with Ptp2 amino-terminal and Ptp3 phosphatase domain, failed to reduce Fus3 kinase activity (top, lanes 13–16) and Tyr phosphorylation (middle, lanes 13–16) in ptp2Δptp3Δ cells to the levels of wild-type cells (lanes 3,4). In fact, the activity and Tyr phosphorylation remained at levels comparable with ptp3Δ disruption cells (lanes 7,8). Interestingly, when P3N-P2, which encodes Ptp3 amino-terminal fused to Ptp2 phosphatase domain, was expressed in ptp2Δptp3Δ double disruption cells, both Fus3 kinase activity (top, lanes 17,18) and Tyr phosphorylation (middle, lanes 17,18) were reduced to the levels comparable with ptp2Δ single disruption cells (lanes 5,6).
In parallel, the kinase activity and Tyr phosphorylation of Hog1 in cells expressing these chimeric molecules were examined. Expression of PTP2 (lanes 13,14) or P2N-P3 chimera (lanes 15,16) in ptp2Δptp3Δ cells decreased the Hog1 activity and Tyr phosphorylation to the levels comparable with ptp3Δ single disruption (lanes 7,8) or wild-type cells (lanes 4,5). In contrast, expression of PTP3 (lanes 11,12) or P3N-P2 (lanes 17,18) failed to reduce the Hog1 activity and Tyr phosphorylation to the levels of wild-type (lanes 3,4) or ptp3Δ cells (lanes 7,8).
These data demonstrate that P2N-P3 functionally complements defects of Hog1 regulation in ptp2Δ cells, whereas the P3N-P2 chimera complements defective Fus3 regulation in ptp3Δ cells. Therefore, the functions of Ptp2/Ptp3 chimeras in vivo are dictated by the amino-terminal noncatalytic domains instead of the carboxy-terminal phosphatase domains. Although we were unable to assess the expression level and stability of these chimeric molecules, the observation that P2N-P3 or P3N-P2 could functionally complement ptp2Δ or ptp3Δ, respectively, argues against the possibility that the expression of these chimeric molecules is abnormal. Taken together, our data strongly suggest that the amino-terminal noncatalytic domains of Ptp2 and Ptp3 determine their substrate specificity in vivo.
Specific interaction between Fus3 and Ptp3 in vivo
To investigate how the selectivity of Ptp3 towards Fus3 is achieved, we test the interaction between Fus3 and three yeast tyrosine phosphatases, Ptp1, Ptp2, or Ptp3, by in vivo copurification. A functional HA epitope-tagged Fus3 was coexpressed with GST phosphatase fusion proteins in yeast cells. The GST fusion proteins and their associated proteins were purified from the total cellular extract using glutathione (GSH)–agarose, eluted from the affinity resin with free GSH and subsequently analyzed by immunoblotting using anti-HA or anti-GST antibody. HA–Fus3 was found to copurify with GST–Ptp3 (Fig. 2A). In contrast, very little HA–Fus3 copurified with GST–Ptp2 and no HA–Fus3 was associated with GST–Ptp1 or GST (top). Differences in binding affinities were unlikely to be due to lack of expression of GST, GST–Ptp1, or Ptp2 because these proteins are readily detected in either lysates or GSH eluates (Fig. 2A, bottom). As controls, neither HA-tagged yeast Cdc28 kinase nor phosphoglycerate kinase Pgk3, an abundant yeast protein, copurified with GST–Ptp3 in similar assays (Fig. 2B). The interaction between Fus3 and Ptp3 was further examined in a yeast two hybrid system (Gyuris et al. 1993). PTP3 was expressed as a lexA DNA-binding domain fusion (BD fusion). FUS3 or its upstream activator STE7 MEK was fused to the bacterial B42 transactivation domain fusion (AD fusion). The expression of AD fusion proteins was controlled by a galactose inducible promoter. The lexA operator-driven LEU2 (lexAop–LEU2) reporter gene was induced when BD–PTP3 and AD–FUS3 were coexpressed on medium containing galactose, suggesting a positive interaction (Fig. 2C). Although AD–STE7 was able to interact with BD–FUS3, no interaction was detected between BD–PTP3 and AD–STE7. Based on results from these two independent approaches, we conclude that Ptp3 specifically interacts with Fus3.
Figure 2.

Specific interaction between Fus3 and Ptp3. (A) Fus3 interacts with GST–Ptp3 but not GST–Ptp1 or GST–Ptp2 in vivo. Total cellular lysates were prepared from wild-type cells (Y568) co-expressing HA–FUS3 (pXZ147) with either vector, GST (pXZ134), GST–PTP1 (pXZ110), GST–PTP2 (pXZ113), or GST–PTP3 (pXZ123). GST fusion proteins were purified on GSH–agarose and eluted with free GSH. Western blotting analysis was performed on total lysates or GSH eluates with anti-GST or anti-HA antibody to detect GST fusion proteins or HA–Fus3. Lysates prepared from cells transformed with untagged FUS3 (pXZ148) and GST–PTP3 (pXZ123) were used as a negative control. (*) A protein in lysate cross-reactive with anti-HA 12CA5 mAb. Representative data from at least two independent experiments are shown. (B) GST–Ptp3 does not interact with HA–Cdc28 or Pgk3. HA–CDC28 (pSF19) and either vector, GST (pXZ134), GST–PTP3 (pXZ123), or GST–ptp3C814G (pXZ136) were introduced into wild-type cells (Y568) as a control. After GSH–agarose affinity purification of the GST fusion proteins, Western blotting analysis was performed on total lysates or GSH eluates with anti-HA (12CA5), anti-Pgk3 (yeast phosphoglycerate kinase 3), or anti-GST antibody. (*) A protein in lysate cross-reactive with anti-HA 12CA5 mAb. (C) PTP3 interacts with FUS3 but not STE7 in yeast two-hybrid assay. A two-hybrid host strain (EGY48) was transformed with lexA DNA binding domain fusion BD–PTP3 (pXZ152) with either vector or B42 transactivation domain fusion AD–FUS3 (pXZ302) or AD–STE7 (pXZ292). Alternatively, the host strain was transformed with BD–FUS3 (pXZ322) with either vector or AD–STE7 (pXZ292). At least two transformants were picked at random and streaked on SC-HWU/Dextrose to maintain the plasmids or on SC-HWUL/Galactose/Rafinose to induce the GAL1,10 promoter-driven AD fusion proteins and to test the expression of LEU2 reporter. The dependence of LEU2 reporter expression on the AD fusion was tested by streaking transformants on SC-HWUL/Dextrose plates on which the expression of AD fusion genes were repressed.
The Fus3 and Ptp3 interaction is direct
The interaction between Ptp3 and Fus3 can be direct or, alternatively, indirectly mediated by other components in the Fus3 MAPK pathway. Ste5 has been shown to function as a scaffold that organizes multiple components of pheromone signaling pathway into a protein complex through its interactions with Ste11 MEKK, Ste7 MEK, and Fus3, etc. (Choi et al. 1994; Marcus et al. 1994; Printen and Sprague 1994). In addition, Fus3 also forms a complex with its upstream activating kinase, the Ste7 MEK (Bardwell et al. 1996). To exclude the possibility that Ptp3 and Fus3 interaction is mediated by Ste5 or Ste7, in vivo copurification was performed to determine the Ptp3/Fus3 interaction in ste5Δ, ste7Δ, or ste5Δste7Δ disruption cells (Fig. 3A). The interaction was not significantly affected in any disruption strains compared with wild-type cells, suggesting that this interaction is independent of the Ste5 scaffold protein and the Ste7 MEK.
Figure 3.


The Fus3 and Ptp3 interaction is direct. (A) The Fus3 and Ptp3 interaction is independent of Ste5 scaffold or St7 MEK. HA–FUS3 (pXZ147) and GST–PTP3 (pXZ123) expression plasmids were transformed into wild type (Y568), ste5Δ disruption cells (Y305), ste7Δ disruption cells (Y306), or ste5Δste7Δ double disruption cells (Y313). In vivo copurification assay and immunoblotting were conducted as described in the legend to Fig. 2A. (B) Direct interaction of Fus3/Ptp3 detected by in vitro binding assay. Full-length Ptp3 labeled with [35S]Met was synthesized in reticulocyte-coupled transcription/translation system. The labeled Ptp3 was partially purified and subjected to binding assay with recombinant GST or GST fused to Elk1, Cdk6, Erk1, and Fus3 purified from E. coli. as described in Materials and Methods. Recombinant proteins were visualized by Coomassie blue staining, and Ptp3 was detected by autoradiography. (C) The Fus3 and Ptp3 interaction is independent of the Tyr phosphorylation or pheromone stimulation. Wild-type cells (Y568) harboring HA–FUS3 (pXZ147) or HA–fus3Y182F (pXZ148) and with either vector, GST (pXZ134), GST–PTP3 (pXZ123), or GST–ptp3C814G (pXZ136) were stimulated with 2.5 μm α-factor for 30 min. In vivo copurification assay and immunoblotting were performed as described in the legend to Fig. 2A.
To test whether Ptp3 and Fus3 can directly interact with each other, an in vitro binding assay was performed using recombinant GST fusion protein purified from Escherichia coli and Ptp3 synthesized by in vitro coupled transcription/translation. The [35S]Met-labeled Ptp3 was partially purified after in vitro synthesis and incubated with immobilized GST or GST fused to transcription factor Elk1, mammalian cyclin-dependent kinase Cdk6, mammalian MAPK Erk1, or Fus3. The proteins retained on GSH–agarose affinity resin were analyzed by SDS-PAGE, Coomassie blue staining and autoradiography. Ptp3 associated only with GST–Fus3 (Fig. 3B). Based on the concentration of Ptp3 used in the binding assay and the amount of Ptp3 bound, we estimated the dissociation constant (Kd) is ∼200 nm for Fus3 and Ptp3, indicating a high-affinity association. Although it is formally possible that Ste5 or Ste7 may modulate the association of Fus3 and Ptp3 in vivo, these results support a model of direct interaction.
Pheromone stimulation induces phosphorylation at residue Tyr-182 in the activation loop of Fus3 kinase, and this Tyr phosphorylation is essential for Fus3 activity (Gartner et al. 1992). We have shown that Ptp3 regulates the phosphorylation of Tyr-182. To test whether the interaction between the phosphatase Ptp3 and its substrate Fus3 is affected by Tyr phosphorylation or pheromone stimulation, we performed in vivo copurification to determine the association of a Fus3Y182F mutant with Ptp3 or a previously described catalytically inactive C814G mutant (Zhan et al. 1997) in unstimulated or pheromone-stimulated cells (Fig. 3C). The association between Fus3 and Ptp3 was detected in unstimulated cells and was not significantly affected following stimulation (lanes 9,10). The interaction was also not affected by the Y182F mutation (lanes 11,12), which abolished the Tyr phosphorylation site on Fus3, suggesting that interaction is independent of Fus3 Tyr phosphorylation. It has been demonstrated in several Tyr phosphatases that mutations of active site will stabilize the phosphatase–substrate complex, presumably as a result of the inability of the phosphatases to dephosphorylate and subsequently release substrates (Sun et al. 1993; Garton et al. 1996). Interestingly, the C814G mutation at the essential Cys in Ptp3 abolished the Tyr phosphatase activity (data not shown) yet had no significant effect on Fus3 binding (Fig. 3C, lanes 13–16). Therefore, the catalytic domain of Ptp3 may not be responsible for Fus3 interaction, consistent with the functional assays in Figure 1.
The amino-terminal noncatalytic domain of Ptp3 mediates the interaction with Fus3 and is essential for Ptp function
To map the Fus3 interaction site, we constructed a series of truncated Ptp3 molecules fused to GST (Fig. 4A) and examined their abilities to interact with HA–Fus3 by in vivo copurification assay. Immunoblotting with anti-GST mAb detected the expression of truncated Ptp3 molecules (Fig. 4B, bottom). HA–Fus3 associated with full-length, wild-type Ptp3 or the C814G mutant (Fig. 4B, middle, lanes 4,5). HA–Fus3 also copurified with GST–ΔC3 (amino acid residues 1–171), GST–ΔC2 (residues 1–350), or GST–ΔC1 (residues 1–686) (middle, lanes 6,7,9), suggesting that the amino-terminal domain is sufficient for Fus3 association. In contrast, when the amino-terminal noncatalytic domain was deleted, the interaction between Fus3 and Ptp3 was abolished (middle, lane 8). In addition, in the yeast two hybrid assay, AD–FUS3 could associate with full-length BD–PTP3, BD–ptp3ΔC(1–350) but not with BD–ptp3ΔN(380–931) (data not shown). From these results we conclude that the amino-terminal noncatalytic domain (residues 1–171) of Ptp3 is necessary and sufficient for Fus3 interaction.
Figure 4.

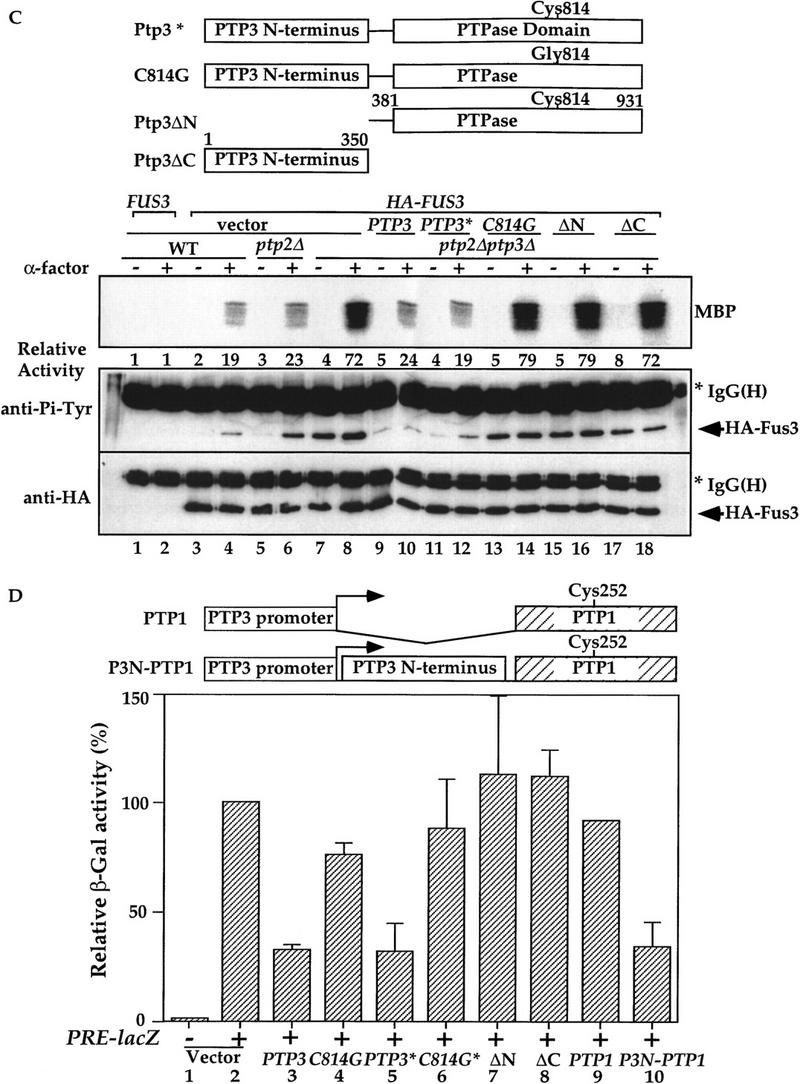
The amino-terminal noncatalytic domain of Ptp3 is necessary and sufficient for Fus3 interaction and is essential for Ptp3 function. (A) Wild type and various truncated Ptp3 molecules are depicted schematically. The numbers represent the number of amino acid residues in Ptp3. (+) A positive interaction with Fus3; (-) no interaction. (B) The amino-terminal domain is necessary and sufficient for Fus3 binding. Wild-type cells carrying HA–FUS3 (pXZ147) was transformed with either GST–PTP3 (pXZ123), GST–ptp3C814G (pXZ136), GST–ΔC3 (pXZ125), GST–ΔC2 (pXZ126), GST–ΔN (pXZ207), or GST–ΔC1 (pXZ204). In vivo copurification assay and immunoblotting were performed as described in the legend to Fig. 2A. (C) The amino-terminal domain is required for Ptp3 function in vivo. Wild-type PTP3 (pXZ273) (lanes i11,12), ptp3C814G (pXZ274) (lanes 13,14), ptp3ΔN (pXZ275) (lanes 15,16), or ptp3ΔC (pXZ276) (lanes 17,18) driven by endogenous PTP3 promoter from single copy plasmids were expressed in ptp2Δptp3Δ double disruption cells (Y578) bearing HA–FUS3 (pXZ147). A single copy genomic clone of PTP3 (lanes 9,10) was used as a control. Wild type (Y568), ptp2Δ single disruption (Y572), or ptp2Δptp3Δ double disruption cells (Y578) carrying the indicated plasmids were stimulated with 2.5 μm α-factor for 30 min at 30°C. Anti-HA immunoprecipitation, in vitro MBP kinase assays, anti-Pi–Tyr, and anti-HA immunoblotting were performed to determine kinase activity (top), Tyr phosphorylation (middle), and HA–Fus3 protein levels (bottom). Representative results from at lease two independent experiments are shown. (D) Suppression of STE11-4 induced PRE–lacZ by multicopy PTP3 wild type, C814G, ptp3ΔN, and ptp3ΔC. Ptp1 and chimeric molecule in which Ptp1 is fused to Ptp3 amino-terminal domain were depicted schematically. SY1866 (STE11-4) carrying PRE–lacZ reporter plasmid (pUZ4) was transformed with vector, multicopy genomic PTP3 (pXZ213), ptp3C814G (pXZ214), PTP3* (pXZ77), C814G* (pXZ278), ΔN (pXZ279), ΔC (pXZ280), PTP1 (pXZ324), or P3N-P1 (pXZ325). β-Galactosidase activity in the absence of α-factor was measured, and the control activity of SY1866 carrying only pUZ4 reporter plasmid and vector was normalized to 100%. Each value is the mean activity measured in duplicate from two independent transformants.
To investigate the physiological function of the Fus3 binding domain of Ptp3, we examined the effects of deleting this domain on PTP3 function by two assays: complementation of chromosomal ptp3Δ disruption and suppression of STE11-4. In ptp3Δ cells, the basal and stimulated Tyr phosphorylation as well as kinase activity of Fus3 are elevated. We tested the ability of wild-type PTP3, ptp3C814G, ptp3ΔN(381–931), or ptp3ΔC(1–350) to reverse the effects of a chromosomal ptp3Δ disruption when these molecules were expressed from the endogenous promoter on centromeric plasmids (Fig. 4C). The HA–Fus3 proteins were analyzed for Tyr phosphorylation and kinase activity. HA–Fus3 displayed similar levels of Tyr phosphorylation and activity in wild type (Fig. 4C, lanes 3,4) or ptp2Δ single disruption cells (lanes 5,6). Basal Tyr phosphorylation of Fus3, as well as the pheromone-stimulated phosphorylation and kinase activity are elevated in ptp2Δptp3Δ double disruption cells (lanes 7,8). Elevated Tyr phosphorylation and kinase activity were reduced when a genomic PTP3 clone (lanes 9,10) or the single copy PTP3 gene driven by the endogenous PTP3 promoter (PTP3*; lanes 11,12) was expressed in ptp2Δptp3Δ. In contrast, expression of neither the catalytically deficient C814G mutant (lanes 13,14), ptp3ΔN (residues 381–931, lanes 15,16), nor ptp3ΔC (residues 1–350, lanes 17,18) was able to decrease the Fus3 Tyr phosphorylation and pheromone-stimulated kinase activity to the levels in wild-type or ptp2Δ cells. Because of the single copy number and low expression of the PTP3 gene from its endogenous promoter, we were unable to assess the protein levels of PTP3, ptp3C814G, ptp3ΔN, and ptp3ΔC with immunoblotting. However, when these molecules were fused to GST and expressed from a strong yeast ADH1 promoter, all were expressed well (Fig. 4B; data not shown). In addition, the lack of complementation by ptp3ΔN is unlikely due to lack of phosphatase activity because the purified ptp3ΔN has been shown to possess enzymatic activity (Fig. 5D, bar 5; Zhan et al. 1997).
Figure 5.



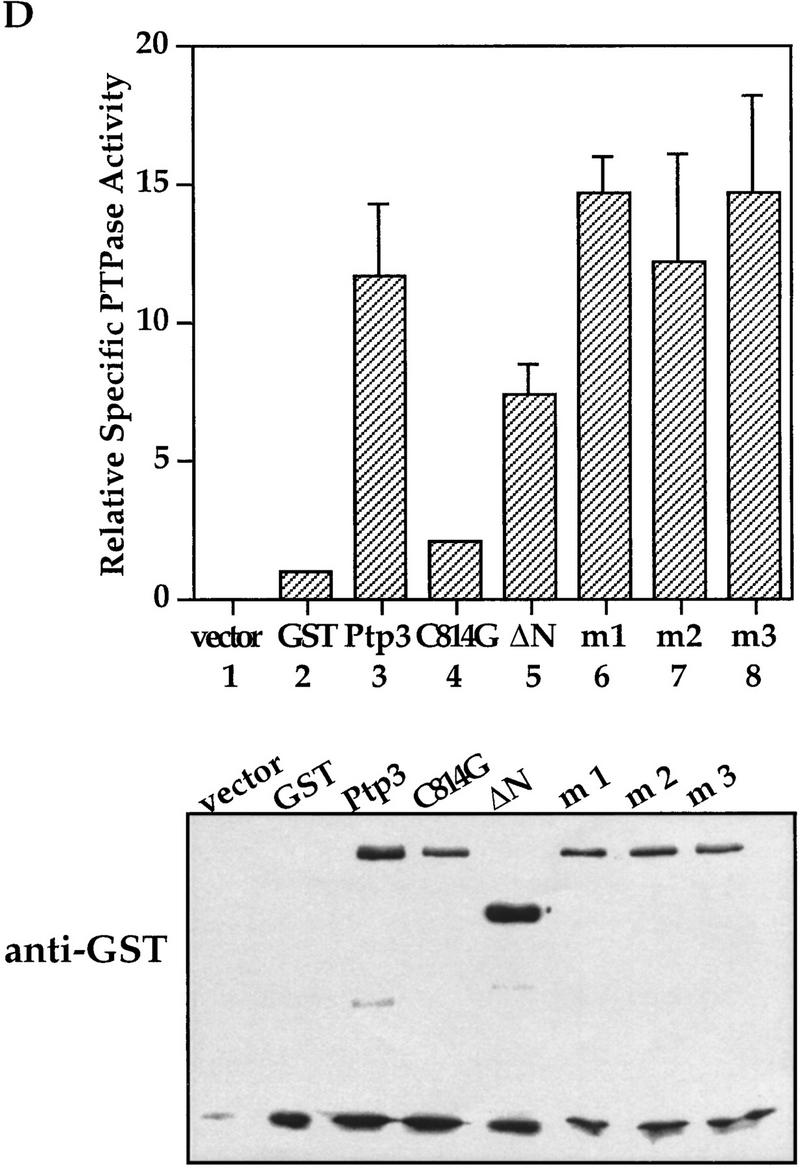
Mutations in the CH2 domain of Ptp3 abrogate Fus3 binding and Pp3 function in vivo. (A) The amino-terminal region of Ptp3 contains conserved amino acid residues present in yeast tyrosine phosphatases and mammalian MKPs. Multiple amino acid sequence alignment of S. cerevisiae Ptp3 and Ptp2, S. pombe pyp1 and pyp1, and mammalian MKPs were obtained with ClustalW (MacVector) program. The CH2A motif is underlined. The amino acid substitutions of mutants Ptp3 (m1, m2, or m3) generated in this study are indicated. The accession numbers for aligned sequences are as follows: human MKP1 (X68277), MKP2 (U21108), and PAC1 (L11329); Rat MKP3 (Q64346); S. pombe SpPYP1 (P27574) and SpPYP2 (P32586); and S. cerevisiae ScPTP2 (P29461) and ScPTP3 (P40048). (B) Mutations in the CH2 domain affect the interaction with Fus3. Copurification assays were performed using lysates prepared from wild-type cells expressing HA–Fus3 (pXZ147) together with either GST (pXZ134), GST–Ptp3 (pXZ123), GST–mutant 1 (m1; pXZ339), GST–mutant 2 (m2; pXZ340), or GST–mutant 4 (m4; pXZ341). GST fusion proteins or HA–Fus3 were detected by anti-HA or anti-GST immunoblotting, respectively. (C) The CH2 domain is required for PTP3 function in vivo. Wild-type PTP3 (pXZ273), m1 (pXZ342), m2 (pXZ343), or m3 (pXZ344) driven by endogenous PTP3 promoter from single copy plasmids were expressed in ptp2Δptp3Δ double disruption cells (Y578) bearing HA–FUS3 (pXZ147). The complementation of ptp3Δ by PTP3, m1, m2, or m3 was tested by determining the Fus3 kinase activity and Tyr–Pi as described in the legend for Fig. 1D. (D) Mutations in the CH2 domain have no effect on Ptp3 Tyr phosphatase activity. GST, GST–Ptp3 wild-type, m1, m2, or m3 protein was purified from wild-type yeast cells expressing the respective proteins. The Tyr phosphatase activity (top) was determined by using MBP as a substrate, which was labeled in vitro by v-Src kinase on Tyr residues. Phosphatase activities were normalized to the amount of purified proteins quantified by anti-GST immunoblotting (bottom) and densitometery scanning. Data points are the mean of duplicated measurement.
The significance of the amino-terminal Fus3 binding domain in Ptp3 function was further tested by examining the ability of multicopy PTP3 to suppress STE11-4-induced pheromone-responsive element (PRE)–lacZ reporter expression (Fig. 4D). In cells bearing a chromosomal STE11-4 allele that encodes a mutant MEKK with constitutive activity, the PRE–lacZ reporter gene expression is elevated in the absence of stimulation (Fig. 4D, bar 2). This high-level reporter gene expression can be suppressed when PTP3 is expressed from a multicopy plasmid (bars 3 and 5), and the suppression is significantly diminished by multicopy catalytically deficient C814G mutant (bars 4 and 6). When the carboxy-terminal phosphatase domain was deleted, the truncated ptp3ΔC failed to reduce PRE–lacZ expression (bar 8). Interestingly, the PRE–lacZ expression in STE11-4 cells was not suppressed by multicopy ptp3ΔN that lacks the amino-terminal Fus3 binding domain but retains the intact phosphatase domain (bar 7). The lack of suppression by ptp3ΔN was not due to lack of expression because the multicopy HA-tagged ptp3ΔN used in this experiment can be readily detected in the cell extract (data not shown). Our data suggest that the amino-terminal noncatalytic domain is required for Ptp3 function in vivo, possibly for targeting Ptp3 to Fus3 kinase thereby allowing dephosphorylation by the phosphatase domain.
One prediction from this hypothesis is that if the Ptp3 amino-terminal domain is fused to a different phosphatase that is normally not involved in Fus3 regulation, by providing a Fus3 binding site, then this chimeric molecule may mimic the function of Ptp3. To test this possibility, the PTP1 gene or a chimeric P3N-P1 molecule created by fusing a Ptp3 amino-terminal domain to Ptp1 was expressed in STE11-4 cells. Multicopy PTP1 was unable to decrease PRE–lacZ expression (Fig. 4D, bar 9) (Zhan et al. 1997). In contrast, multicopy P3N-P1 reduced β-galactosidase activity (bar 10) to a level similar to PTP3 (bar 3 or 5). Taken together, these data strongly suggest that the amino-terminal noncatalytic domain of Ptp3 is essential for PTP3 function in vivo. Our data also indicate that the Ptp3 amino-terminal domain targets the phosphatase to its physiological substrate Fus3, thus allowing the phosphatase domain to dephosphorylate and inactivate Fus3. This hypothesis is further tested in the following experiments using Ptp3 with point mutations.
Point mutations in the amino-terminal CH2 motif severely compromise Fus3 interaction and Ptp3 function
Close inspection of the Ptp3 amino-terminal amino acid sequence (residues 1–180) revealed intriguing sequence motifs that are also found in mammalian MKPs, designated as CH2A and CH2B (Cdc25 homology region) (Keyse and Ginsburg 1993; Kwak et al. 1994; Muda et al. 1996a, 1997). In Ptp3 the homology is the highest within the CH2A motif (Fig. 5A). The CH2A and CH2B regions of Cdc25 flank the active site and function to maintain the structure as revealed by crystallographic study (Fauman et al. 1998). In mammalian MKPs, the CH2 motifs are located amino-terminal to the dual-specificity phosphatase domain. The presence of amino-terminal CH2 motifs is a unique feature shared by all MKPs, yet the significance of these motifs is not fully understood (Keyse 1995). Interestingly, in S. cerevisiae or Schizosaccharomyces pombe these two motifs are present at the amino-terminal region of tyrosine phosphatases instead of dual-specificity phosphatases. Additionally, we also identified a stretch of amino acids (residues 142–160 in Ptp3; Fig. 5A) that are highly conserved between Ptp3 and mammalian MKPs but are absent in the Cdc25 phosphatase. Furthermore, the sequence similarity within the CH2A motif among Ptp3 (residues 114–137) and mammalian MKPs is higher than the similarity between yeast Ptp3 and Ptp2 (Fig. 5A). The observations that Ptp3 and MKPs all possess these CH2 motifs in the amino terminus raises a tantalizing hypothesis: Do the CH2 motifs of Ptp3 serve as a Fus3-binding site? We generated three clusters of mutations in the conserved motifs (Fig. 5A) and tested the abilities of these mutant proteins to interact with Fus3 in vivo. All three mutants were soluble and were expressed at similar levels as wild-type Ptp3 (Fig. 5B, bottom), suggesting that the mutations did not cause deleterious changes on global protein structure. However, the ability to interact with Fus3 was significantly reduced in mutant #2 (m2) and abolished in mutant #1 (m1) or mutant #3 (m3) (Fig. 5B, middle). Therefore, these residues conserved among yeast PTPases and mammalian MKPs are critical for Fus3 and Ptp3 interaction.
We assessed the abilities of PTP3 mutants to complement chromosomal ptp3Δ deletion when these mutant genes were expressed from the endogenous PTP3 promoter on single copy plasmids. Introducing the wild-type PTP3 gene into ptp2Δptp3Δ cells (Fig. 5C, lanes 9,10) effectively reduced the elevated Fus3 kinase activity (Fig. 5C, top) and Tyr–Pi (Fig. 5C, middle) to the levels in wild-type (lanes 3,4) or ptp2Δ cells (lanes 5,6). The ptp3 m2 mutant (lanes 13,14) reduced the kinase activity and Tyr–Pi to a level between wild-type cells and (lanes 3,4) ptp2Δptp3Δ double disruption cells (lanes 7,8), suggesting partial complementation. In contrast, when ptp3 m1 (lanes 11,12) or m3 (lanes 15,16) was expressed, Fus3 kinase activity and Tyr–Pi remained as high as the levels in the ptp2Δptp3Δ double disruption cells (lanes 7,8; three- to fourfold higher than activity in wild type), indicating a lack of complementation.
To determine whether these mutations affect Ptp3 phosphatase activity, the Ptp3 mutants were purified as GST fusion protein from yeast cell lysates and tyrosine phosphatase activities were measured. In vitro phosphatase activity was assayed using myelin basic protein (MBP) phosphorylated on Tyr residue by v-Src Tyr kinase as a substrate (Fig. 4D). All three mutant proteins (bars 6–8) displayed phosphatase activity similar to wild-type Ptp3 (bar 3). Therefore, the lack of complementation by mutant Ptp3 is not attributable to lack of phosphatase activity. Our data demonstrate that the CH2 motifs of Ptp3 are required for interaction with Fus3 and the physiological function of Ptp3.
Mutations at residue Asp-317 of FUS3 increase sensitivity of cells to pheromone, elevate Fus3 Tyr phosphorylation and kinase activity
If the Fus3 and Ptp3 interaction is critical for the physiological regulation of Fus3 MAPK, then a mutant kinase unable to interact with Ptp3 should no longer be regulated by Ptp3 and show elevated Tyr phosphorylation even in the presence of wild-type Ptp3. Furthermore, the FUS3 mutant should behave as a dominant gain-of-function allele. One previously identified gain-of-function allele is a Leu substitution at the Ile-161 residue that is not highly conserved among MAPKs. The Fus3I161L mutant has elevated autophosphorylation activity, thereby increasing pheromone sensitivity (Brill et al. 1994). Another mutation, substitution of Asp-317 to Gly, results in insensitivity to the repression of mating pathway by the Hog1 MAPK pathway under osmotic-shock condition (Hall et al. 1996). Interestingly, Asp-317 is evolutionarily conserved among all members of the MAPK family. An analogous mutation termed Sevenmaker (rlsevenmaker), which contains a single amino acid substitution of Asn-334 for Asp (D334N) in the Drosophila MAPK, displays a dominant gain of function and activates several developmental pathways (Brunner et al. 1994). Similarly, the mammalian ERK2 D319N mutant is also more sensitive to activation by stimuli and appears to be resistant to inactivation by dual-specificity phosphatases in transfected cells (Bott et al. 1994; Chu et al. 1996; Camps et al. 1998). However, the molecular mechanism underlying this gain-of-function mutation is unknown.
We examined the effects of FUS3D317N and FUS3D317G on the pheromone sensitivity of wild-type yeast cells (Y568). Pheromone-induced cell cycle arrest was determined by spotting serially diluted culture from cells expressing vector, single copy FUS3 wild type, D317N, or D317G on medium containing various concentrations of α-factor. D317N and D317G had no adverse effect on the growth of wild-type cells in the absence of pheromone (Fig. 6A, left). In contrast, both mutants rendered the cells more sensitive towards pheromone-induced cell cycle arrest than vector or wild-type FUS3 alone (Fig. 6A, right). The enhanced sensitivity was also confirmed by the halo assay (data not shown). Then, the effect on pheromone-inducible PRE–lacZ reporter gene expression was determined. Expression of single copy D317N or D317G in wild-type cells (Y568) resulted in more than twofold increase in basal (Fig. 6B, left) as well as pheromone-stimulated (Fig. 6B, right) β-galactosidase activity compared with cells carrying vector or wild-type FUS3. Similar expression levels of Fus3, D317N, and D317G were detected in wild-type cells by immunoblotting (data not shown). Therefore, these results demonstrate that cells carrying D317N or D317G mutant display enhanced sensitivity towards pheromone similar to the previously characterized I161L mutant (Brill et al. 1994).
Figure 6.
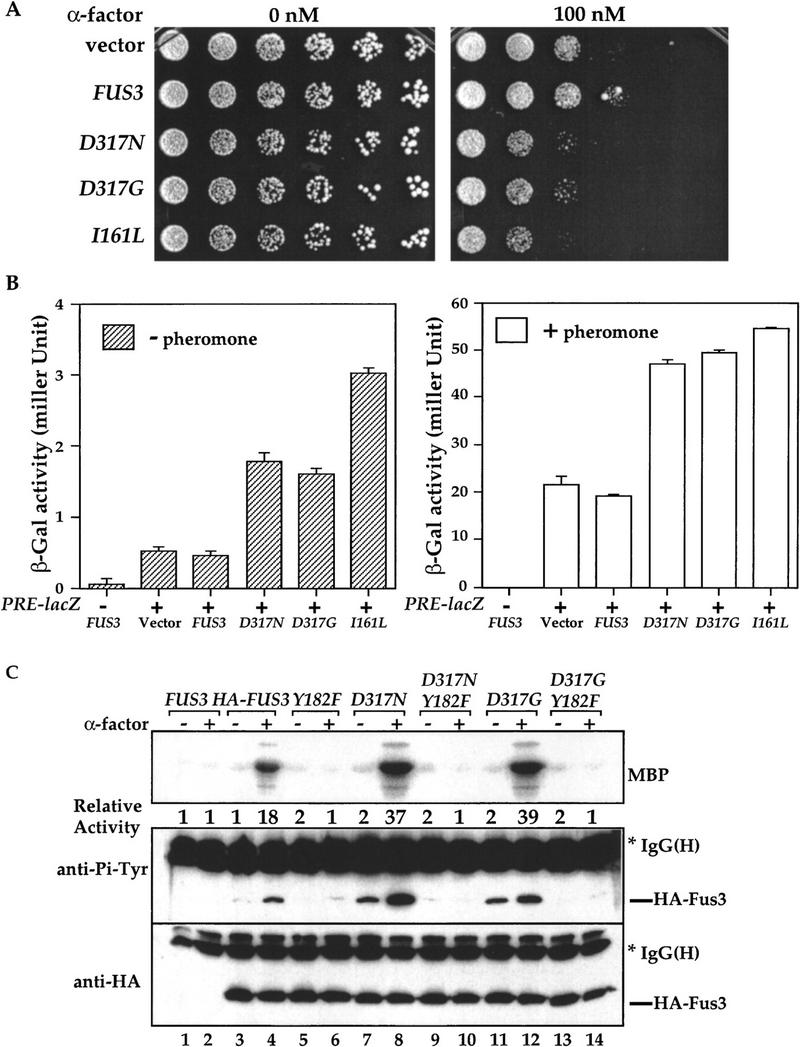

Mutations at residue Asp-317 of FUS3 increase Tyr phosphorylation, kinase activity, and sensitivity to pheromone. (A) Sensitivities of cells carrying FUS3, FUS3D317N, FUS3D317G, or FUS3I161L to pheromone-induced growth arrest. Centromeric plasmids containing either vector, wild-type FUS3 (pXZ287), FUS3D317N (pXZ289), FUS3D317G (pXZ290), or FUS3I161L (pXZ291) expressed from endogenous FUS3 promoter were transformed into wild-type cells (Y568). Aliquots of the indicated strains were serially diluted and spotted onto selective agar media in the absence (left) or presence (right) of α-factor. Plates were incubated for 3 days at 30°C and photographed. Representative results from at least three experiments using independent transformants are shown. (B) Fus3 mutants increase expression of lacZ reporter gene driven by pheromone-responsive promoter. β-Galactosidase activities in wild-type cells (Y568) carrying pUZ4 reporter with either empty vector, single copy FUS3 (pXZ287), FUS3D317N (pXZ289), FUS3D317G (pXZ290), or FUS3I161L (pXZ291) were determined in the absence of α-factor (left) or presence of 2.5 μm α-factor for 2 hr (right). Y568 cells carrying FUS3 (pXZ287) without the reporter was included as a control. The activities in Miller unit are the mean values from duplicated assays with two independent transformants. (C) Enhanced kinase activity and Tyr–Pi of Fus3 D317N and D317G. Wild-type cells (Y568) expressing the HA-tagged FUS3 (pXZ147), Y182F (pXZ150), D317N (pXZ281), D317N/Y182F (pXZ283), D317G (pXZ282), or D317N/Y182F (pXZ284) were treated with 2.5 μm α-factor for 30 min. The kinase activity (top) and Tyr–Pi (middle), and protein levels (bottom) were determined as described in the legend to Fig. 1A. (D) D317N and D317G mutations have no effect on Fus3 autophosphorylation activity. Fus3 (not tagged; pXZ148), HA-tagged Fus3 (pXZ147), Y182F (pXZ150), D317N (pXZ281), D317N/Y182F (pXZ283), D317G (pXZ282), or D317G/Y182F (pXZ284) were immunoprecipitated from wild-type cells (Y568) without pheromone treatment. The autophosphorylation activities of Fus3 were determined by adding [γ-32P]ATP to the immunoprecipitants and incubating at 30°C for 45 min. Proteins were resolved on SDS-PAGE and analyzed by autoradiography (top). Tyr phosphorylation (middle) and protein levels (bottom) were determined by anti-HA and anti-Pi–Tyr immunoblotting, respectively. (E) The elevated Tyr phosphorylation of Fus3 mutants is independent of STE7. HA– FUS3 (pXZ147), FUS3D317N (pXZ281), or D317G (pXZ282) was expressed in either wild-type (Y568) cells (lanes 3,4,7,8,11,12) or ste7Δ disruption (Y306) cells (lane 5,6,9,10,13,14). After 30-min stimulation with 2.5 μm α-factor, the kinase activities and Tyr–Pi of Fus3 proteins in these cells were determined. (F) The elevated basal Tyr phosphorylation of Fus3 D317N mutant requires its kinase activity. HA–FUS3 (pXZ147), catalytically deficient K42R (pXZ345), D317N (pXZ281), or K42R/D317N (pXZ347) was expressed in wild-type (Y568) cells. After 30-min stimulation with 2.5 μm α-factor, the kinase activities and Tyr–Pi of Fus3 proteins were determined.
To elucidate the biochemical mechanisms of Fus3 mutations at residue Asp-317, we measured kinase activity and Tyr phosphorylation of HA-tagged Fus3, D317N, or D317G mutants (Fig. 6C). Upon pheromone stimulation, the activity (Fig. 6C, top) of HA–D317N (lane 8) or HA–D317G (lane 12) was reproducibly twofold higher than Fus3 wild-type activity (lane 4). When the Tyr phosphorylation status was monitored by anti-Pi–Tyr immunoblotting (Fig. 6C, middle), the Tyr phosphorylation of wild-type Fus3 is only detected when cells were stimulated with pheromone (lanes 3,4). To the contrary, a dramatic increase in basal Tyr phosphorylation (in the absence of pheromone) was consistently observed on both D317N (lane 7) and D317G (lane 11) mutants compared with wild-type Fus3 (lane 3). The Tyr phosphorylation was further increased upon pheromone stimulation (lanes 8,12). Fus3, like all MAPK, requires phosphorylation on the Tyr residue in the conserved TXY motif (Tyr-182) for activation and signaling. When Tyr-182 was replaced by Phe (Y182F) alone or in combination with D317N or D317G, no kinase activity was detected (Fig. 6C, top, lanes 5,6,9,10,13,14). Furthermore, Tyr phosphorylation of wild-type Fus3, D317N, or D317G mutant was eliminated by the Y182F substitution, suggesting that Y182 is the site that becomes constitutively phosphorylated. The differences in Tyr–Pi and kinase activity were not attributable to variations in protein expression levels because similar amounts of HA-tagged proteins were present as shown by anti-HA immunoblotting. Taken together, mutations at residue Asp-317 of Fus3 result in an increased sensitivity to pheromone, elevated Tyr phosphorylation, and higher kinase activity.
Several possible mechanisms can be envisioned to explain the above results. First, the Fus3 mutants may have augmented autophosphorylation activity towards the Tyr-182 residue as in the case of Fus3I161L mutant (Brill et al. 1994). To test whether mutations at Asp-317 enhance autophosphorylation activity, HA-tagged Fus3, D317N, and D317G were immunoprecipitated, [γ-32P]ATP was added to the immunoprecipitants, and autophosphorylation activity was measured (Fig. 6D). No significant difference was observed on the autophosphorylation activity of Fus3 wild type (top panel, lane 2), D317N (lane 4), and D317G (lane 6). The second possibility is that Fus3, D317N or D317G mutants are better substrates for Ste7 MEK than wild type, resulting in more efficiently phosphorylation catalyzed by Ste7. If this is correct, then it can be predicted that in ste7Δ disruption cells, the constitutive Tyr phosphorylation of D317N or D317G should be eliminated. To test this alternative mechanism, the kinase activity and Tyr phosphorylation of wild-type Fus3, D317N, and D317G mutants were compared in wild-type or ste7Δ disruption cells. No pheromone-inducible kinase activities of wild-type Fus3, D317N, and D317G mutants were detected in ste7Δ cells (Fig. 6E, top, lanes 5,6,9,10,13,14). Importantly, the basal Tyr phosphorylation of D317N and D317G mutants in ste7Δ cells remained at similar levels as in wild-type cells (lanes 7,9,11,12), suggesting that the elevated basal Tyr phosphorylation is independent of Ste7. However, the induction of Tyr phosphorylation of wild type, D317N, and D317G by pheromone was eliminated by ste7Δ disruption (lanes 6,8,10,12,14), indicating that the induction still requires Ste7.
If the basal Tyr phosphorylation of Fus3 mutants is attributable to autophosphorylation, then the constitutive phosphorylation in the absence of pheromone should be abolished by eliminating Fus3 catalytic activity. To test this hypothesis, we mutated a conserved Lys-42 residue that is required for kinase activity (Errede et al. 1993) in D317N mutant (Fig. 6F). The kinase deficient K42R mutant did not display any kinase activity (top, lanes 5,6), but its Tyr phosphorylation was still induced by pheromone (middle, lanes 5,6), due to the action of Ste7 (Gartner et al. 1992). The K42R mutation in D317N abolished high basal Tyr phosphorylation without affecting the pheromone-inducible Tyr phosphorylation (middle, lanes 7–10), indicating that autophosphorylation is responsible for the elevated basal Tyr phosphorylation levels in Fus3 D317N and D317G mutants.
Together, the above data demonstrate that the D317N or D317G mutation does not enhance the autophosphorylation activity of Fus3 on its Tyr residue. The elevated basal Tyr phosphorylation in the mutants is independent of Ste7 but requires the autophosphorylation activity of Fus3. The balance of protein phosphorylation is maintained by the opposing reactions of phosphorylation and dephosphorylation. Because the phosphorylation aspect of Fus3 D317N and D317G mutant is not altered and Fus3 basal Tyr phosphorylation is controlled by Ptp3 (Zhan et al. 1997), our data suggest a possibility that these mutants are defective to be regulated by Ptp3.
D317N and D317G mutants are unable to interact with Ptp3 and are more resistant to Ptp3 dephosphorylation
Because the interaction between Fus3 and Ptp3 is essential for Ptp3 function (Figs. 4 and 5), lack of interaction with Ptp3 will render Fus3 mutants resistant to Ptp3 dephosphorylation, consequently resulting in constitutive Tyr–Pi. To directly test the interaction between Ptp3 and D317N or D317G mutant, their ability to copurify with Ptp3 in vivo was determined. Although HA-tagged Fus3, D317N, and D317G proteins were expressed to similar levels in wild-type cells (Fig. 7A, immunoblotting on total lysate), the amount of HA-tagged mutant proteins copurified with GST–Ptp3 was dramatically diminished compared with HA–Fus3 wild type (Fig. 7A, immunoblotting on GSH eluates). The interactions between Ptp3 and D317N or D317G were further examined by the yeast two-hybrid assay. Neither D317N nor D317G interacted with Ptp3, whereas Fus3 showed a positive interaction with Ptp3 (Fig. 7B). We also tested the interaction between Fus3 mutants with the Ptp3 amino-terminal (Ptp3ΔC) or the phosphatase domain (Ptp3ΔN) in the two-hybrid system. No interaction was detected between Ptp3ΔC or Ptp3ΔN and D317N or D317G (data not shown).
Figure 7.

Fus3 D317N and D317G mutants are unable to interact with Ptp3 and are more resistant to Ptp3 dephosphorylation. (A) Fus3 D317N and D317G mutants are not copurified with GST–Ptp3. HA–FUS3 (pXZ147), FUS3D317N (pXZ281), or D317G (pXZ282) was coexpressed with GST–PTP3 (pXZ123) in wild-type cells (Y568). In vivo copurification assay was performed as described in the legend to Fig. 2A to determine the interaction of Ptp3 with Fus3 wild type or mutants. Cells expressing untagged FUS3 (pXZ148) with GST–PTP3 (pXZ123), or HA–FUS3 (pXZ147) with either vector or GST (pXZ134) were used as controls. Proteins of interests in lysates or GSH eluates were detected by immunoblotting with anti-GST or anti-HA mAbs. (B) Lack of interaction between Ptp3 and Fus3 mutants in the yeast two-hybrid assay. The two-hybrid host strain (EGY48) was transformed with lexA BD–PTP3 (pXZ152) together with either empty vector or B42 transactivation domain fusion AD–FUS3 (pXZ302), AD–FUS3D317N (pXZ285), AD–FUS3D317G (pXZ286), or AD–STE7 (pXZ292). Two transformants were picked at random and streaked out on SC-HWUL/galactose/rafinose or SC-HWUL/dextrose plates to test the interaction. (C) Fus3 D317N and D317G mutants are resistant to inactivation by Ptp3. Multicopy plasmids containing wild-type PTP3 gene (pXZ213) (lanes 5,6,11,12,15,16) or ptp3C814G (pXZ214), were introduced into wild-type cells (Y568) carrying either HA-tagged FUS3 (pXZ147), D317N (pXZ281), or D317G (pXZ282). The effects of multicopy PTP3 on Fus3 kinase activity and Tyr phosphorylation were determined by immunoprecipitation followed by in vitro kinase assay (top) and immunoblotting (middle and bottom).
To further test the hypothesis that the mutant Fus3 is resistant to dephosphorylation by Ptp3, the effects of multicopy PTP3 on kinase activity and Tyr phosphorylation of Fus3, D317N, or D317G were examined. Compared with the vector control (Fig. 7C, lanes 3,4), the kinase activity (Fig. 7C, top, threefold reduction, lanes 5,6) and Tyr phosphorylation (middle, lanes 5,6) of wild-type Fus3 was decreased when PTP3 was present in multiple copies. Reduction of activity and Tyr phosphorylation was dependent on phosphatase activity because the catalytically deficient C814G mutant had no effect (lanes 7,8). In contrast, expressing multiple-copy PTP3 had no effect on D317N activity and Tyr phosphorylation (lanes 9–12). Tyr phosphorylation of D317G was slightly reduced in cells overexpressing PTP3 (lanes 13–16) but still higher than the Tyr phosphorylation of wild-type Fus3 in cells carrying only empty vector (lanes 3,4). These data demonstrate that D317N and D317G mutations in Fus3 significantly decrease the ability of Fus3 to interact with Ptp3, resulting in resistance to dephosphorylation and inactivation by Ptp3.
Discussion
MAPK activity must be tightly regulated because dysregulation of MAPK can be detrimental. For instance, although ERK activity is required for normal cell growth, constitutive activation of the mammalian ERK1/2 pathway by v-Ras can result in cellular transformation (Joneson and Bar-Sagi 1997). In S. cerevisiae the activation of Fus3 MAPK is required for the mating pheromone response (Elion et al. 1990), but unregulated activation will cause growth defects or even lethality (Sprague and Thorner 1992; Stevenson et al. 1992; Sprague 1998). Similarly, the activation of Hog1 stress-activated MAPK in S. cerevisiae is essential to maintain the balance of intra- and extracellular osmolarity (Gustin et al. 1998; Millar 1999). The viability of cells is affected by either constitutive Hog1 activation or lack of Hog1 activity when cells are challenged with high osmolarity (Brewster et al. 1993; Maeda et al. 1993, 1994). Therefore, yeast cells have to specifically modulate different MAPK activities in response to distinct extracellular stimuli. One mechanism to achieve this is via selective activation of upstream activating kinase MEKs (Cobb and Goldsmith 1995). However, phosphatases must also be responsible for the highly specific inactivation of MAPKs (Tonks and Neel 1996; Keyse 1998; Van Vactor et al. 1998).
The Fus3 MAPK is negatively regulated by the actions of a tyrosine-specific Ptp3 and a dual-specific phosphatase Msg5 (Doi et al. 1994; Zhan et al. 1997). In parallel, Hog1 is regulated by a related tyrosine phosphatase Ptp2 (Jacoby et al. 1997; Wurgler-Murphy et al. 1997). Despite the extensive similarity between Ptp3 and Ptp2 and high degree of sequence identity between respective substrates Fus3 and Hog1, Ptp3 shows a distinctive specificity towards Fus3, whereas Ptp2 is selective to Hog1. What molecular mechanism determines the in vivo substrate specificity of different phosphatases? In this study we have demonstrated that the amino-terminal noncatalytic domains of Ptp2 and Ptp3 dictate the substrate specificity. By swapping the amino-terminal noncatalytic domain between Ptp2 and Ptp3, the substrate specificity can be switched. We have also established that Ptp3 is targeted to its physiological substrate Fus3 MAPK via specific and direct protein–protein interaction mediated by the amino-terminal noncatalytic domain of Ptp3. We have further demonstrated that the interaction of Ptp3 and Fus3 is required for proper Fus3 MAPK regulation: When the interaction with Fus3 is abolished by either deletion or point mutations in the CH2 motif, the mutant Ptp3 proteins can no longer dephosphorylate and deactivate Fus3 in vivo even though these mutants retain phosphatase activity. Consistently, cells carrying Fus3 mutants that have lost the ability to interact with Ptp3 exhibit increased sensitivity to pheromone. In addition, the mutant Fus3 displays elevated Tyr phosphorylation and is more resistant to dephosphorylation by Ptp3. Our study provides direct evidence that targeting of tyrosine phosphatase Ptp3 to Fus3 MAPK is essential for the in vivo regulation of MAPK and is a key mechanism to ensure the highly specific regulation of MAPKs by phosphatases.
Fus3, similar to other MAPKs, is able to autophosphorylate on Tyr. Genetic study in yeast has pointed to the physiological significance of the Fus3 autophosphorylation in setting the sensitivity of MAPK activation to extracellular stimuli (Brill et al. 1994). In the absence of stimulation, basal Tyr phosphorylation of Fus3 is governed by the balance between autophosphorylation by the kinase and dephosphorylation by Ptp3 phosphatase. The elevated Tyr phosphorylation of Fus3 D317N and D317G was not due to an increase in autophosphorylation. It is more likely attributable to the lack of dephosphorylation by Ptp3 because the D317N and D317G mutants can no longer interact with Ptp3. In contrast, the other gain-of-function mutant, Fus3I161L, displays enhanced autophosphorylation; as a result, these cells are more sensitive to pheromone (Brill et al. 1994).
The residue D317 of Fus3 is conserved in all MAPK family members from yeast to human. Genetic analysis on an analogous mutation termed Sevenmaker (rlsevenmaker) that contains Asp-334 to Asn (D334N) substitution in Drosophila MAPK rolled suggests that this mutant displays dominant gain-of-function phenotypes (Brunner et al. 1994). The analogous mutation in mammalian ERK2 (ERK2 D319N mutant) appears to be resistant to inactivation by dual-specificity phosphatases in transfected cells (Bott et al. 1994; Chu et al. 1996). In vitro biochemical analysis has shown that recombinant ERK2 but not the D319N mutant can bind to the dual-specificity phosphatase (Camps et al. 1998). Therefore, the interaction between MAPKs and phosphatases represents a mechanism for MAPK regulation conserved through evolution.
An emerging family of dual-specificity phosphatase MKP has been implicated in MAPK inactivation. This family of proteins shares a conserved dual-specificity protein phosphatase domain and an amino-terminal domain that has two short-sequence motifs (CH2A and CH2B) (Keyse and Ginsburg 1993; Kwak et al. 1994; Muda et al. 1996a, 1997), also present in the catalytic domain of the Cdc25 phosphatase (Fauman et al. 1998), which are involved in dephosphorylation and activation of Cdc2 kinase. The significance of these motifs in MKP functions is unclear. Interestingly, the amino-terminal portion of MKP-3 may contribute to its substrate specificity towards ERK1/2 MAPKs (Muda et al. 1996b, 1998). Sequences similar to CH2 motifs are also identified in the amino-terminal domain of Ptp3. Mutational analysis has demonstrated that the conserved LLLDxR (residues 117–122 in Ptp3) and KK(R/K) (residues 145–147) residues in Ptp3 are required for Fus3 interaction. These Ptp3 mutants are severely compromised in their function in Fus3 regulation. In contrast, the Ptp3 m2 mutant, encoding substitutions in the less conserved residues, only partially decreased the interaction with Fus3 (Fig. 6). Interestingly, ptp3 m2 also showed a partial loss of function in the complementation test. Our data provide the first evidence that CH2 motifs may serve as MAPK binding sites and determine the in vivo substrate selectivity of the Ptp3 phosphatase. We propose that the CH2 domain in other phosphatases also serve as a MAPK binding motif to determine substrate specificity. Consistent with this hypothesis, CH2 motifs are present in the S. pombe pyp1 and pyp2 that also interact with and regulate spc1/sty1 stress-activated MAPK (Millar et al. 1995; Shiozaki and Russell 1995). We speculate that sequence divergence at the CH2 motif between Ptp2 and Ptp3 might determine phosphatase specificity towards Hog1 and Fus3, respectively.
Although the dual specificity Msg5 phosphatase is also involved in Fus3 regulation, it does not possess CH2 motifs. A growing body of evidence points to a complex network of MAPK regulation by both tyrosine phosphatases and dual-specificity phosphatases as first demonstrated in S. cerevisiae. Recent studies from mammalian tissue culture system and Drosophila genetics have demonstrated the involvement of tyrosine phosphatases PTP-ER (Karim and Rubin 1999) or PTP-SL/STEP (Pulido et al. 1998; Zuniga et al. 1999) in MAPK regulation. However, protein–protein interaction motifs termined KIM (kinase interaction motif), present in several proteins known to interact with MAPK (Bardwell et al. 1996; Pulido et al. 1998; Karim and Rubin 1999; Zuniga et al. 1999), are used to mediate the interaction between PTP-SL/STEP and MAPK. The KIM motif, which contains a 16-amino-acid element (G/N)L(L/F)xRRGSN(H/Q)S(I/L)TL(N/K) and is unrelated to CH2 domain, is not found in yeast Ptp2 and Ptp3. A second type of MAPK interaction motif, the so-called docking sites for ERK, FxFP motif (DEF motif), represents another evolutionarily conserved MAPK binding site and is often found in close proximity to ERK phosphorylation sites on MAPK substrates (Jacobs et al. 1999). We did identify a single FQFP motif (amino acid residues 372–375) in Ptp3, but it is located outside of the domain (amino acid residues 1–180) necessary and sufficient for Fus3 binding, and there is no ERK phosphorylation consensus site in the close proximity of this motif. It is becoming an emerging paradigm that multiple MAPK pathways must be specifically activated/deactivated in response to distinct extracellular stimuli. To achieve high specificity in vivo, the MKPs and yeast tyrosine phosphatases use two functional domains, catalytic phosphatase domains, and CH2 domains as a substrate targeting domain. It is likely that the CH2 domain of each MKP specifically interacts with one or a set of MAPKs, thus determining the specificity of MAPK dephosphorylation in vivo.
Materials and methods
Strains and plasmids
S. cerevisiae strains used in this study are listed in Table 1. Standard methods were used for yeast genetic manipulations (Rose et al. 1990; Guthrie and Fink 1991). Gene replacements were performed to disrupt STE7 or STE5 gene with HIS3 or Kanr, respectively. Disruptions were confirmed by genomic PCR analysis with specific primers and mating assay. Plasmids are listed in Table 2. Details of plasmid constructions are available upon request. All DNA fragments generated by PCR were verified by sequencing analysis.
Table 1.
S. cerevisiae strains used in this study
| Straina
|
Genotype
|
Reference of source
|
|---|---|---|
| Y568 | MATa trp1 his3 leu2 ura3 | lab stock |
| Y572 | MATa trp1 his3 leu2 ura3 ptp2Δ::LEU2 | Guan et al. (1992) |
| Y567 | MATa trp1 his3 leu2 ura3 ptp3Δ::URA3 | Zhan et al. (1997) |
| Y578 | MATa trp1 his3 leu2 ura3 ptp2Δ::LEU2 ptp3Δ::URA3 | Zhan et al. (1997) |
| Y305 | MATa trp1 his3 leu2 ura3 ste5Δ::Kanr | this study |
| Y306 | MATa trp1 his3 leu2 ura3 ste7Δ::HIS3 | this study |
| Y313 | MATa trp1 his3 leu2 ura3 ste5Δ::Kanr ste7Δ::HIS3 | this study |
| EGY48 | MATα trp1 his3 ura3-52 leu2::pLEU2-LexAop6 | Gyuris et al. (1993) |
| SY1866 | MATα STE11-4 fus1::FUS1::HIS3 leu2 ura3 trp1 his3Δ200::ura3 pep4Δ::ura3 can1 | Stevenson et al. (1992) |
All strains are isogenic except for EGY48 and SY1866.
Table 2.
Plasmids used in this study
| Plasmid
|
Markera
|
Reference of source
|
|---|---|---|
| phosphatase | plasmid | |
| pXZ128 | AmpR 6xHis tagged PTP3 ORF | this study |
| pXZ134 | URA3 2μ PADH1–GST | this study |
| pXZ110 | URA3 2μ PADH–GST–PTP1 | this study |
| pXZ113 | URA3 2μ PADH–GST–PTP2 | this study |
| pXZ123 | URA3 2μ PADH–GST–PTP3 | this study |
| pXZ136 | URA3 2μ PADH–GST–ptp3C814G | this study |
| pXZ204 | URA3 2μ PADH–GST–ptp3ΔC1(1–686) | this study |
| pXZ126 | URA3 2μ PADH–GST–ptp3ΔC(1–350) | this study |
| pXZ125 | URA3 2μ PADH–GST–ptp3ΔC3(1–171) | this study |
| pXZ207 | URA3 2μ PADH–GST–ptp3ΔN(381–931) | this study |
| pXZ339 | URA3 2μ PADH–GST–ptp3m1(L117A/L118A/D119N/R121M) | this study |
| pXZ340 | URA3 2μ PADH–GST–ptp3m2(P122A/F123A/T124A/E125A) | this study |
| pXZ341 | URA3 2μ PADH–GST–ptp3m3(R145M/R146A/K147m) | this study |
| pXZ213 | LEU2 2μ PTP3 | Zhan et al. (1997) |
| pXZ214 | LEU2 2μ ptp3C814G | Zhan et al. (1997) |
| pXZ277 | LEU2 2μ PPTP3–HA–PTP3 | this study |
| pXZ278 | LEU2 2μ PPTP3–HA–ptp3C814G | this study |
| pXZ279 | LEU2 2μ PPTP3–HA–ptp3ΔN(381–931) | this study |
| pXZ280 | LEU2 2μ PPTP3–HA–ptp3ΔC(1–350) | this study |
| pXZ324 | LEU2 2μ PPTP3–HA–PTP13 | this study |
| pXZ325 | LEU2 2μ PPTP3–HA–ptp3ΔC(1–350)-PTP1 | this study |
| pXZ209 | HIS3 CEN genomic PTP3 | this study |
| pXZ273 | HIS3 CEN PPTP3–PTP3 | this study |
| pXZ274 | HIS3 CEN PPTP3–ptp3C814G | this study |
| pXZ275 | HIS3 CEN PPTP3–ptp3ΔN(381–931) | this study |
| pXZ276 | HIS3 CEN PPTP3–ptp3ΔC(1–350) | this study |
| pXZ315 | HIS3 CEN PPTP3–PTP2 | this study |
| pXZ316 | HIS3 CEN PPTP3–ptp2N(1–350)–ptp3C(381–931) | this study |
| pXZ317 | HIS3 CEN PPTP3–ptp3N(1–380)–ptp2C(351–835) | this study |
| pXZ342 | HIS3 CEN PPTP3–ptp3m1(L117A/L118A/D119N/R121M) | this study |
| pXZ343 | HIS3 CEN PPTP3–ptp3m2(P122A/F123A/T124A/E125A) | this study |
| pXZ344 | HIS3 CEN PPTP3–ptp3m3(R145M/R146A/K147M) | this study |
| kinase | plasmid | |
| pSF19 | TRP1 2μ PADH1–HA–CDC28 | Sorger and Murray (1992) |
| pXZ147 | TRP1 CEN PADH1–HA–FUS3 | Zhan et al. (1997) |
| pXZ148 | TRP1 CEN PADH1–FUS3 | Zhan et al. (1997) |
| pXZ150 | TRP1 CEN PADH1–HA–fus3Y182F | Zhan et al. (1997) |
| pXZ281 | TRP1 CEN PADH1–HA–FUS3D317N | this study |
| pXZ282 | TRP1 CEN PADH1–HA–FUS3D317G | this study |
| pXZ283 | TRP1 CEN PADH1–HA–fus3D317N, Y182F | this study |
| pXZ284 | TRP1 CEN PADH1–HA–fus3D317G, Y182F | this study |
| pXZ345 | TRP1 CEN PADH1–HA–fus3K42R | this study |
| pXZ347 | TRP1 CEN PADH1–HA–fus3 D317N/K42R | this study |
| pXZ287 | URA3 CEN PFUS3–HA–FUS3 | this study |
| pXZ289 | URA3 CEN PFUS3–HA–FUS3D317N | this study |
| pXZ290 | URA3 CEN PFUS3–HA–FUS3D317G | this study |
| pXZ291 | URA3 CEN PFUS3–HA–FUS3I161L | this study |
| pXZ331 | TRP1 2μ PGPD1–HOG1–HA | this study |
| 2-hybrid | plasmid | |
| pXZ152 | HIS3 2μ PADH1–lexA BD–PTP3 | this study |
| pXZ321 | HIS3 2μ PADH1–lexA BD–STE7 | this study |
| pXZ302 | TRP1 2μ PGAL1–B42 AD–FUS3 | this study |
| pXZ292 | TRP1 2μ PGAL1–B42 AD–STE7 | this study |
| pXZ285 | TRP1 2μ PGAL1–B42 AD–FUS3D317N | this study |
| pXZ286 | TRP1 2μ PGAL1–B42 AD–FUS3D317G | this study |
PADH1, PPTP3, PFUS3, PGPD1, and PGAL1 are ADH1, PTP3, FUS3, GPD1, and GAL1 promoters, respectively.
Preparation of yeast extract, in vivo copurification, in vitro tyrosine phosphatase assay, and immunoblotting
Yeast cells harboring expression plasmids were grown to an OD600 of ∼1.0 and harvested. Whole cell extracts were prepared by vortexing with glass beads in lysis buffer (25 mm Tris-HCl at pH 7.4, 250 mm NaCl, 15 mm EGTA, 15 mm MgCl2, 1 mm DTT, 1.0%Triton X-100) with protease inhibitor cocktail (Boehringer Mannheim). Protein concentration was determined by Bradford assay (Bio-Rad). Cellular extracts containing 1 mg of total proteins were incubated with 25 μl of 50%suspension of prewashed GSH–agarose (Sigma) at 4°C for 1 hr. The resin was washed with 1 ml of lysis buffer for three times. GST fusion proteins and its associated complexes were eluted from GSH–agarose with lysis buffer containing 15 mm free GSH. Proteins of interests in extracts or GSH eluates were detected by immunoblotting using Enhanced Chemiluminescence detection system (Amersham). Mouse monoclonal antibody against HA epitope (12CA5), GST, or yeast phosphoglycerate kinase (Pgk3) was obtained from Boehringer Mannheim, Zymed, or Molecular Probes, respectively. The in vitro tyrosine phosphatase activities were measured as described (Guan and Dixon 1990).
Two-hybrid analysis
A two-hybrid system using lexA DNA binding domain fusion (BD fusion), B42 trans-activation domain fusion (AD fusion), and lexA-operator driven LEU2 or lacZ reporter genes was used to detect protein–protein interactions as descibed (Gyuris et al. 1993).
In vitro coupled transcription/translation and binding assay
pXZ128 plasmid DNA was linearized with EcoRI and used as template in T7 TNT quick coupled rabbit reticulocyte transcription/translation system (Promega) to synthesize full-length 6 His-tagged Ptp3 labeled with l-[35S]Met (NEN). The reaction mix was buffered with HEPES (pH 7.4) to 20 mm final concentration and loaded on a DEAE Sephacryl mini column (0.5-ml bed volume). The column was washed with 20 mm HEPES (pH 7.4), and bound proteins were eluted in buffer containing 20 mm HEPES (pH 7.4) and 0.5 m NaCl. Ptp3 was purified from the eluates on a mini Ni–NTA column (0.3-ml bed volume). The amount of Ptp3 was determined by scintillation counting. The in vitro synthesized Ptp3 was mixed with recombinant GST fusion proteins purified from E. coli, and GSH–agarose in the binding buffer (20 mm Tris-HCl at pH 7.2, 150 mm NaCl, and 0.1%Tween 20); after 1-hr incubation at 4°C, the resin was washed three times with 0.5 ml of buffer, and proteins were resolved by 12.5%SDS-PAGE. The gel was stained with Coomassie brilliant blue and dried, followed by autoradiography.
Pheromone-induced G1 arrest and β-galactosidase reporter assay
Cells carrying vector or different FUS3 alleles (pXZ287, pXZ289, pXZ290, or pXZ291) were grown in SC-Ura. Sensitivity to pheromone-induced G1 arrest was assessed by serially diluting cultures from an initial concentration of ∼2 × 106 cells/ml by a factor of 5 in 200 μl volume in a sterile 96-well plate. A 48-pin multipronged replicator was used to transfer ∼3 μl of cell suspension to SC-Ura plates containing various concentration of α-factor (from 0 nm to 100 nm). Cells bearing pUZ4 that encodes a lacZ reporter gene driven by pheromone-responsive element were grown to OD600 of ∼0.7–1.0, then incubated with or without 2.0 μm α-factor for 2 hr. β-Galactosidase reporter activity was assayed as described by Zhan et al. (1997). The activity was calculated as Miller unit [OD420 × 1000]/[OD600 × time (min) × volume of culture (ml)].
Immunoprecipitation and in vitro kinase assay
For Fus3 kinase assay, yeast cells were grown to an OD600 of 0.7–0.8 in SC media lacking the appropriate nutrient to maintain plasmids and stimulated with 2.5 μm α-factor for 30 min. Immunoprecipitation of HA–Fus3 and in vitro kinase assay using MBP as substrate were performed as described (Zhan et al. 1997). For Fus3 autophosphorylation assay, HA–Fus3 immunocomplex precipitated from extracts containing 100 μg of total proteins was mixed with 15 μl of mixture containing 6 μm ATP, 80 μCi [γ-32P]ATP (7000 Ci/mmole; ICN) in kinase buffer and incubated at 30°C for 45 min. The reaction was terminated by adding 5 μl of 4× SDS-PAGE sample buffer, and samples were analyzed by 10%SDS-PAGE and autoradiography.
To determine Hog1 kinase activity, yeast cells were grown to an OD600 of 0.7–0.8 and stimulated with NaCl at 0.4 m final concentration for 10 min. Whole cell extracts were prepared, and immunoprecipitation with anti-HA mAb 12CA5 (Boehringer Mannheim) was performed as described for Fus3 kinase assay except that lysates containing 1 mg of total cellular proteins were used. The immunocomplex was washed three times with 1 ml of lysis buffer and three times with 1 ml of RIPA buffer (50 mm Tris-HCl at pH 7.5, 150 mm NaCl, 50 mm NaF, 1 mm EDTA, 1 mm EGTA, 0.1%SDS, 1%Triton X-100, 0.5%sodium deoxycholic acid, 1 mm DTT, 0.25 mm sodium orthovanadate). Kinase assay was performed as described for Fus3 kinase assay. Phosphorylation of substrate was quantified by PhosphorImaging scanning (Molecular Dynamics).
HA–Fus3 protein in immunocomplex and its Tyr phosphorylation were detected by immunoblotting as described (Zhan et al. 1997). Because HA–Hog1 comigrates with mouse IgG heavy chain, a biotin-conjugated anti-HA mAb 12CA5 (Boehringer Mannheim) or a rabbit polyclonal anti-Pi–Tyr antibody (Zymed) was used to detect HA–Hog1 or Tyr phosphorylation levels, respectively.
Acknowledgments
We are grateful to the following for generous gifts. Drs. R. Brent, E.A. Elion, H.D. Madhani, D.A. Mitchell, and I. Ota. We thank Drs. J.E. Dixon, X.-D. Liu, and M. Muda for helpful discussion and Drs. D.J. Thiele, R.S. Fuller, K. Morano, and P.C. Liu for critical reading of the manuscript. This work is supported by a grant from the National Institutes of Health (GM55642) to K.-L.G.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kunliang@umich.edu; FAX (734) 763-4581.
References
- Bardwell L, Cook JG, Inouye CJ, Thorner J. Signal propagation and regulation in the mating pheromone response pathway of the yeast Saccharomyces cerevisiae. Dev Biol. 1994;166:363–379. doi: 10.1006/dbio.1994.1323. [DOI] [PubMed] [Google Scholar]
- Bardwell L, Cook JG, Chang EC, Cairns BR, Thorner J. Signaling in the yeast pheromone response pathway: Specific and high-affinity interaction of the mitogen-activated protein (MAP) kinases Kss1 and Fus3 with the upstream MAP kinase kinase Ste7. Mol Cell Biol. 1996;16:3637–3650. doi: 10.1128/mcb.16.7.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott CM, Thorneycroft SG, Marshall CJ. The sevenmaker gain-of-function mutation in p42 MAP kinase leads to enhanced signalling and reduced sensitivity to dual specificity phosphatase action. FEBS Lett. 1994;352:201–205. doi: 10.1016/0014-5793(94)00958-9. [DOI] [PubMed] [Google Scholar]
- Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. An osmosensing signal transduction pathway in yeast. Science. 1993;259:1760–1763. doi: 10.1126/science.7681220. [DOI] [PubMed] [Google Scholar]
- Brill JA, Elion EA, Fink GR. A role for autophosphorylation revealed by activated alleles of FUS3, the yeast MAP kinase homolog. Mol Biol Cell. 1994;5:297–312. doi: 10.1091/mbc.5.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner D, Oellers N, Szabad J, Biggs WH, III, Zipursky SL, Hafen E. A gain-of-function mutation in Drosophila MAP kinase activates multiple receptor tyrosine kinase signaling pathways. Cell. 1994;76:875–888. doi: 10.1016/0092-8674(94)90362-x. [DOI] [PubMed] [Google Scholar]
- Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C, Boschert U, Arkinstall S. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase [see comments] Science. 1998;280:1262–1265. doi: 10.1126/science.280.5367.1262. [DOI] [PubMed] [Google Scholar]
- Choi KY, Satterberg B, Lyons DM, Elion EA. Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S. cerevisiae. Cell. 1994;78:499–512. doi: 10.1016/0092-8674(94)90427-8. [DOI] [PubMed] [Google Scholar]
- Chu Y, Solski PA, Khosravi-Far R, Der CJ, Kelly K. The mitogen-activated protein kinase phosphatases PAC1, MKP-1, and MKP-2 have unique substrate specificities and reduced activity in vivo toward the ERK2 sevenmaker mutation. J Biol Chem. 1996;271:6497–6501. doi: 10.1074/jbc.271.11.6497. [DOI] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- Doi K, Gartner A, Ammerer G, Errede B, Shinkawa H, Sugimoto K, Matsumoto K. MSG5, a novel protein phosphatase promotes adaptation to pheromone response in S. cerevisiae. EMBO J. 1994;13:61–70. doi: 10.1002/j.1460-2075.1994.tb06235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion EA, Grisafi PL, Fink GR. FUS3 encodes a cdc2+/CDC28-related kinase required for the transition from mitosis into conjugation. Cell. 1990;60:649–664. doi: 10.1016/0092-8674(90)90668-5. [DOI] [PubMed] [Google Scholar]
- Errede B, Gartner A, Zhou Z, Nasmyth K, Ammerer G. MAP kinase-related FUS3 from S. cerevisiae is activated by STE7 in vitro. Nature. 1993;362:261–264. doi: 10.1038/362261a0. [DOI] [PubMed] [Google Scholar]
- Fauman EB, Cogswell JP, Lovejoy B, Rocque WJ, Holmes W, Montana VG, Piwnica-Worms H, Rink MJ, Saper MA. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell. 1998;93:617–625. doi: 10.1016/s0092-8674(00)81190-3. [DOI] [PubMed] [Google Scholar]
- Gartner A, Nasmyth K, Ammerer G. Signal transduction in Saccharomyces cerevisiae requires tyrosine and threonine phosphorylation of FUS3 and KSS1. Genes & Dev. 1992;6:1280–1292. doi: 10.1101/gad.6.7.1280. [DOI] [PubMed] [Google Scholar]
- Garton AJ, Flint AJ, Tonks NK. Identification of p130(cas) as a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol Cell Biol. 1996;16:6408–6418. doi: 10.1128/mcb.16.11.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science. 1990;249:553–556. doi: 10.1126/science.2166336. [DOI] [PubMed] [Google Scholar]
- Guan KL, Deschenes RJ, Dixon JE. Isolation of a second protein tyrosine phosphatase gene, PTP2 from saccharomyces cerevisiae. J Biol Chem. 1992;267:10024–10030. [PubMed] [Google Scholar]
- Gustin MC, Albertyn J, Alexander M, Davenport K. MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1998;62:1264–1300. doi: 10.1128/mmbr.62.4.1264-1300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie, C. and R.G. Fink. 1991. Guide to yeast genetics and molecular biology. Methods Enzymol. 194. [PubMed]
- Gyuris J, Golemis E, Chertkov H, Brent R. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- Hall JP, Cherkasova V, Elion E, Gustin MC, Winter E. The osmoregulatory pathway represses mating pathway activity in Saccharomyces cerevisiae: Isolation of a FUS3 mutant that is insensitive to the repression mechanism. Mol Cell Biol. 1996;16:6715–6723. doi: 10.1128/mcb.16.12.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz I. MAP kinase pathways in yeast: For mating and more. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes & Dev. 1999;13:163–175. [PMC free article] [PubMed] [Google Scholar]
- Jacoby T, Flanagan H, Faykin A, Seto AG, Mattison C, Ota I. Two protein-tyrosine phosphatases inactivate the osmotic stress response pathway in yeast by targeting the mitogen-activated protein kinase, Hog1. J Biol Chem. 1997;272:17749–17755. doi: 10.1074/jbc.272.28.17749. [DOI] [PubMed] [Google Scholar]
- Joneson T, Bar-Sagi D. Ras effectors and their role in mitogenesis and oncogenesis. J Mol Med. 1997;75:587–593. doi: 10.1007/s001090050143. [DOI] [PubMed] [Google Scholar]
- Karim FD, Rubin GM. PTP-ER, a novel tyrosine phosphatase, functions downstream of Ras1 to downregulate MAP kinase during Drosophila eye development. Mol Cell. 1999;3:741–750. doi: 10.1016/s1097-2765(01)80006-x. [DOI] [PubMed] [Google Scholar]
- Keyse SM. An emerging family of dual specificity MAP kinase phosphatases. Biochim Biophys Acta. 1995;1265:152–160. doi: 10.1016/0167-4889(94)00211-v. [DOI] [PubMed] [Google Scholar]
- ————— Protein phosphatases and the regulation of MAP kinase activity. Semin Cell Dev Biol. 1998;9:143–152. doi: 10.1006/scdb.1997.0219. [DOI] [PubMed] [Google Scholar]
- Keyse SM, Ginsburg M. Amino acid sequence similarity between CL100, a dual-specificity MAP kinase phosphatase and cdc25. Trends Biochem Sci. 1993;18:377–378. doi: 10.1016/0968-0004(93)90092-2. [DOI] [PubMed] [Google Scholar]
- Kwak SP, Hakes DJ, Martell KJ, Dixon JE. Isolation and characterization of a human dual specificity protein-tyrosine phosphatase gene. J Biol Chem. 1994;269:3596–3604. [PubMed] [Google Scholar]
- Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- Maeda T, Tsai AY, Saito H. Mutations in a protein tyrosine phosphatase gene (PTP2) and a protein serine/threonine phosphatase gene (PTC1) cause a synthetic growth defect in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:5408–5417. doi: 10.1128/mcb.13.9.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Wurgler-Murphy SM, Saito H. A two-component system that regulates an osmosensing MAP kinase cascade in yeast [see comments] Nature. 1994;369:242–245. doi: 10.1038/369242a0. [DOI] [PubMed] [Google Scholar]
- Marcus S, Polverino A, Barr M, Wigler M. Complexes between STE5 and components of the pheromone-responsive mitogen-activated protein kinase module. Proc Natl Acad Sci. 1994;91:7762–7766. doi: 10.1073/pnas.91.16.7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JB. Stress-activated MAP kinase (mitogen-activated protein kinase) pathways of budding and fission yeasts. Biochem Soc Symp. 1999;64:49–62. [PubMed] [Google Scholar]
- Millar JB, Buck V, Wilkinson MG. Pyp1 and Pyp2 PTPases dephosphorylate an osmosensing MAP kinase controlling cell size at division in fission yeast. Genes & Dev. 1995;9:2117–2130. doi: 10.1101/gad.9.17.2117. [DOI] [PubMed] [Google Scholar]
- Miyajima I, Nakafuku M, Nakayama N, Brenner C, Miyajima A, Kaibuchi K, Arai K, Kaziro Y, Matsumoto K. GPA1, a haploid-specific essential gene, encodes a yeast homolog of mammalian G protein which may be involved in mating factor signal transduction. Cell. 1987;50:1011–1019. doi: 10.1016/0092-8674(87)90167-x. [DOI] [PubMed] [Google Scholar]
- Muda M, Boschert U, Dickinson R, Martinou JC, Martinou I, Camps M, Schlegel W, Arkinstall S. MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J Biol Chem. 1996a;271:4319–4326. doi: 10.1074/jbc.271.8.4319. [DOI] [PubMed] [Google Scholar]
- Muda M, Theodosiou A, Rodrigues N, Boschert U, Camps M, Gillieron C, Davies K, Ashworth A, Arkinstall S. The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J Biol Chem. 1996b;271:27205–27208. doi: 10.1074/jbc.271.44.27205. [DOI] [PubMed] [Google Scholar]
- Muda M, Boschert U, Smith A, Antonsson B, Gillieron C, Chabert C, Camps M, Martinou I, Ashworth A, Arkinstall S. Molecular cloning and functional characterization of a novel mitogen-activated protein kinase phosphatase, MKP-4. J Biol Chem. 1997;272:5141–5151. doi: 10.1074/jbc.272.8.5141. [DOI] [PubMed] [Google Scholar]
- Muda M, Theodosiou A, Gillieron C, Smith A, Chabert C, Camps M, Boschert U, Rodrigues N, Davies K, Ashworth A, Arkinstall S. The mitogen-activated protein kinase phosphatase-3 N-terminal noncatalytic region is responsible for tight substrate binding and enzymatic specificity. J Biol Chem. 1998;273:9323–9329. doi: 10.1074/jbc.273.15.9323. [DOI] [PubMed] [Google Scholar]
- Nomoto S, Nakayama N, Arai K, Matsumoto K. Regulation of the yeast pheromone response pathway by G protein subunits. EMBO J. 1990;9:691–696. doi: 10.1002/j.1460-2075.1990.tb08161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printen JA, Sprague GF., Jr Protein-protein interactions in the yeast pheromone response pathway: Ste5p interacts with all members of the MAP kinase cascade. Genetics. 1994;138:609–619. doi: 10.1093/genetics/138.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido R, Zuniga A, Ullrich A. PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 1998;17:7337–7350. doi: 10.1093/emboj/17.24.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MD, Winston F, Hieter P. Methods in yeast genetics: A laboratory course manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- Shiozaki K, Russell P. Cell-cycle control linked to extracellular environment by MAP kinase pathway in fission yeast. Nature. 1995;378:739–743. doi: 10.1038/378739a0. [DOI] [PubMed] [Google Scholar]
- Sorger PK, Murray AW. S-phase feedback control in budding yeast independent of tyrosine phosphorylation of p34cdc28. Nature. 1992;355:365–368. doi: 10.1038/355365a0. [DOI] [PubMed] [Google Scholar]
- Sprague GF., Jr Control of MAP kinase signaling specificity or how not to go HOG wild. Genes & Dev. 1998;12:2817–2820. doi: 10.1101/gad.12.18.2817. [DOI] [PubMed] [Google Scholar]
- Sprague GF, Jr, Thorner JW. Pheromone response and signal transduction during the mating process of Saccharomyces cerevisiae. In: Jones EW, Pringle JR, Broach JR, editors. The molecular and cellular biology of the yeast Saccharomyces. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. pp. 657–744. [Google Scholar]
- Stevenson BJ, Rhodes N, Errede B, Sprague GF., Jr Constitutive mutants of the protein kinase STE11 activate the yeast pheromone response pathway in the absence of the G protein. Genes & Dev. 1992;6:1293–1304. doi: 10.1101/gad.6.7.1293. [DOI] [PubMed] [Google Scholar]
- Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- Tan PB, Kim SK. Signaling specificity: The RTK/RAS/MAP kinase pathway in metazoans. Trends Genet. 1999;15:145–149. doi: 10.1016/s0168-9525(99)01694-7. [DOI] [PubMed] [Google Scholar]
- Tonks NK, Neel BG. From form to function: Signaling by protein tyrosine phosphatases [see comments] Cell. 1996;87:365–368. doi: 10.1016/s0092-8674(00)81357-4. [DOI] [PubMed] [Google Scholar]
- Van Vactor D, O'Reilly AM, Neel BG. Genetic analysis of protein tyrosine phosphatases. Curr Opin Genet Dev. 1998;8:112–126. doi: 10.1016/s0959-437x(98)80070-1. [DOI] [PubMed] [Google Scholar]
- Wurgler-Murphy SM, Maeda T, Witten EA, Saito H. Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by the PTP2 and PTP3 protein tyrosine phosphatases. Mol Cell Biol. 1997;17:1289–1297. doi: 10.1128/mcb.17.3.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan XL, Deschenes RJ, Guan KL. Differential regulation of FUS3 MAP kinase by tyrosine-specific phosphatases PTP2/PTP3 and dual-specificity phosphatase MSG5 in Saccharomyces cerevisiae. Genes & Dev. 1997;11:1690–1702. doi: 10.1101/gad.11.13.1690. [DOI] [PubMed] [Google Scholar]
- Zuniga A, Torres J, Ubeda J, Pulido R. Interaction of mitogen-activated protein kinases with the kinase interaction motif of the tyrosine phosphatase PTP-SL provides substrate specificity and retains ERK2 in the cytoplasm. J Biol Chem. 1999;274:21900–21907. doi: 10.1074/jbc.274.31.21900. [DOI] [PubMed] [Google Scholar]