

Abstract
Initiation of DNA replication during the mitotic cell cycle requires the activation of a cyclin-dependent protein kinase (CDK). The B-type cyclins Clb5 and Clb6 are the primary activators of the S phase function of the budding yeast CDK Cdc28. However, in mitotically growing cells this role can be fulfilled by the other B-type cyclins Clb1–Clb4. We report here that cells undergoing meiotic development also require Clb dependent CDK activity for DNA replication. Diploid clb5/clb5 clb6/clb6 mutants are unable to perform premeiotic DNA replication. Despite this defect, the mutant cells progress into the meiotic program and undergo lethal segregation of unreplicated DNA suggesting that they fail to activate a checkpoint that restrains meiotic M phase until DNA replication is complete. We have found that a DNA replication checkpoint dependent on the ATM homolog MEC1 operates in wild-type cells during meiosis and can be invoked in response to inhibition of DNA synthesis. Although cells that lack clb5 and clb6 are unable to activate the meiotic DNA replication checkpoint, they do possess an intact DNA damage checkpoint which can restrain chromosome segregation in the face of DNA damage. We conclude that CLB5 and CLB6 are essential for premeiotic DNA replication and, consequently, for activation of a meiotic DNA replication checkpoint.
Keywords: CLB, cyclin, CDK, meiosis, DNA replication, checkpoint
The eukaryotic cell cycle consists of a highly orchestrated series of events that allow the faithful duplication of chromosomes and cellular constituents and promotes their accurate segregation to produce two nearly identical daughter cells (Lew et al. 1997). Maintenance of cell cycle organization is the result of the combined effect of dependent events and cell cycle checkpoints (Hartwell 1974; Hartwell and Weinert 1989). However, our increasing understanding of cell cycle regulation during development is revealing that, despite the stringent requirement for the fidelity of the processes that comprise the cell cycle, the organization of cell cycle events and the duration of cell cycle phases are actually quite malleable (Newport and Kirschner 1984; O’Farrell et al. 1989; Edgar et al. 1994; Orr-Weaver 1994). Of the numerous examples of altered cell cycle organization shown to occur in metazoans during development, those occurring during gametogenesis are among the most dramatic (Orr-Weaver 1994; Su et al. 1998). The process of generating haploid gametes from diploid somatic cells through meiosis involves the dramatic reorganization of cell cycle phases. Diploid cells undergo a round of premeiotic DNA replication followed by two rounds of chromosome segregation without an intervening S phase. Furthermore, substantial alterations in chromatin organization and chromosome dynamics occur during the process of haploidization (McKim and Hawley 1995; Kleckner 1996; Roeder 1997). Although these substantial differences have been recognized for many years, the cell cycle regulatory events that govern them are just beginning to be elucidated. Although the specifics are unclear, it is clear that changes of this magnitude will involve differences both in the implementation of the cell cycle regulatory machinery and in the nature and utilization of cell cycle checkpoints.
Cell cycle organization can be attributed to the pattern of activation of the cell cycle regulatory machinery, the cyclin-dependent protein kinases (CDKs). CDKs, along with their positive regulatory subunits, the cyclins, govern progression through the major transitions of the mitotic cell cycle (Reed 1992; Nasmyth 1993). In the budding yeast, these roles can be attributed to a single CDK, Cdc28. The Cdc28 CDK is activated by at least nine distinct cyclins, three G1 cyclins and six B-type cyclins (Nasmyth 1993). The G1 cyclins, CLN1, CLN2, and CLN3, are essential for cell cycle initiation during G1 phase (Richardson et al. 1989). Two of the B-type cyclins, CLB5 and CLB6, promote the transition from G1 into S phase (Epstein and Cross 1992; Kuhne and Linder 1993; Schwob and Nasmyth 1993), whereas the remaining four, CLB1–CLB4, promote events required for the completion of mitosis (Fitch et al. 1992; Richardson et al. 1992). Substantial functional redundancy exists with both G1 and B-type cyclin subclasses such that elimination of one or more members of a class has only limited effects on the kinetics of cell cycle progression (Richardson et al. 1989, 1992; Fitch et al. 1992). For example, although it is assumed that CLB5 and CLB6 are the physiologically relevant activators of the S phase function of the Cdc28 CDK, their inactivation results in only a modest delay in initiation of DNA replication (Epstein and Cross 1992; Kuhne and Linder 1993; Schwob and Nasmyth 1993). That this results from functional redundancy with CLB1–CLB4 is demonstrated by the fact that inactivation of all six B-type cyclins leads to an absolute block in DNA replication (Schwob et al. 1994).
The importance of CDKs in progression through meiosis is well documented. Some of the earliest studies of cell cycle control in eukaryotes were performed with prophase-arrested amphibian and invertebrate oocytes in which it was found that progression into the first meiotic division was dependent on an activity called maturation promoting factor (MPF) (Masui and Markert 1971). This regulatory factor was subsequently shown to consist of the CDK, Cdc2, and an associated B-type cyclin (Dunphy et al. 1988; Gautier et al. 1988, 1990; Lohka et al. 1988). B-type cyclins are also required for progression through meiotic M phase in both budding yeast and fission yeast (Grandin and Reed 1993; Dahmann and Futcher 1995; Iino et al. 1995). In budding yeast, CLB1, CLB3, and CLB4 are important for progression from pachytene into the first meiotic division (MI) and essential for progression from MI to MII (Dahmann and Futcher 1995). During meiosis, expression of five of the six budding yeast B type cyclins is largely controlled by the meiosis-specific transcription factor, NDT80 (Chu and Herskowitz 1998). CLB2, lacks Ndt80 binding sites and is not expressed significantly during meiosis (Grandin and Reed 1993; Chu and Herskowitz 1998). Consistent with their role in formation and elongation of the spindle during both mitosis and meiosis (Fitch et al. 1992; Dahmann and Futcher 1995), CLB1, CLB3, and CLB4 transcripts, protein, and associated kinase activity all peak about the time of initiation of MI and persist until MII is complete (Grandin and Reed 1993; Chu and Herskowitz 1998). Like those cyclins, which it controls, NDT80 is also required for the pachytene to MI transition (Xu et al. 1995).
Premeiotic S phase, like the meiotic M phases, bears many similarities to its counterpart in the mitotic cell cycle. Many of the same gene products required for DNA replication during the mitotic cycle are also essential for meiosis. These include enzymes involved in the synthesis of DNA precursors (TMP1, RNR1, and CDC8) and in the process of replication itself (CDC2, CDC17, and CDC9) (Simchen et al. 1976; Zamb and Roth 1977; Schild and Byers 1978; Johnson et al. 1982; Budd et al. 1989). In addition, it has been established that the same replication origins are used in both meiotic and mitotic S phases (Collins and Newlon 1994). Thus, it is curious that, in contrast to their essential role in replication during the mitotic cell cycle, experiments using temperature-sensitive mutants of CDC7 and CDC28 suggest that neither is required for premeiotic DNA replication (Schild and Byers 1978; Shuster and Byers 1989). In contrast, inactivation of the Schizosaccharomyces pombe CDK, cdc2, prevents premeiotic S phase (Iino et al. 1995). Despite the fact that S phase of the mitotic cell cycle and meiosis share many features, substantial differences also exist as illustrated by the dependence of premeiotic S phase on a series of gene functions expressed uniquely during meiosis (Kupiec et al. 1997).
In this study, we establish that in meiosis, as in the mitotic cell cycle, CLB5 and CLB6 promote progression into premeiotic S phase. However, unlike their S phase role during the mitotic cycle, their role during premeiotic S phase is essential. We show that premeiotic S phase is inhibited by the Clb/Cdc28-specific CDK inhibitor, Sic1, suggesting that the essential role of Clb5 and Clb6 is executed in conjunction with Cdc28. Despite their failure to replicate DNA, clb5/clb5 clb6/clb6 mutants proceed into the meiotic program and undergo one or more meiotic M phases. Meiotic progression in the clb5/clb5 clb6/clb6 mutants results from the failure to activate a MEC1-dependent DNA replication checkpoint, which we show to be operable in meiotic cells but inoperable when those cells lack CLB5 and CLB6. This study provides the first evidence of an essential role for S-phase cyclins that is distinct from those promoted by CLB1–CLB4. In addition, it provides support for the evolving perception that both the nature and regulation of some checkpoints is conserved between meiosis and mitosis.
Results
The S-phase cyclins, CLB5 and CLB6, are required for meiotic development
Progression from G1 phase into S phase of the mitotic cell cycle requires the activity of three G1 cyclins (CLNs) and two B-type cyclins, CLB5 and CLB6. Whereas deletion of all three CLNs results in a terminal arrest in which cells are unable to form a bud or to enter S phase, inactivation of CLB5 and CLB6 results only in a delay in initiation and slowed progression through S phase (Kuhne and Linder 1993; Schwob and Nasmyth 1993). Surprisingly, we have found that unlike their role in the mitotic cell cycle, G1 cyclins (CLNs) are dispensable for meiosis and sporulation (D. Stuart and C. Wittenberg, unpubl.). Their expression is rapidly repressed when cells are induced to sporulate. In contrast, CLB5 and CLB6 are essential. Inactivation of CLB5 resulted in a dramatic reduction in sporulation efficiency relative to wild-type cells (Table 1), a defect that had been noted previously but not characterized (Epstein and Cross 1992). Although deletion of CLB6 had little effect on sporulation (Table 1), this cyclin can clearly contribute to this process because inactivation of both CLB5 and CLB6 resulted in a more dramatic defect in both tetrad formation and spore viability as compared with clb5/clb5 mutants (Table 1). No full tetrads were observed in the clb5/clb5 clb6/clb6 mutant, but a small number of aberrant triads, dyads, and monads were seen (Table 1). Although clb5/clb5 mutants are severely diminished in their capacity to sporulate and form tetrads, 60% of the spores recovered were viable. In contrast, no viable spores could be recovered from the aberrant asci in clb5/clb5 clb6/clb6 mutant cultures (Table 1). To determine if the severe sporulation defect in clb5/clb5 and clb5/clb5 clb6/clb6 mutants was unique to the SK1 strain background, we examined the effect of these mutations in an independent Saccharomyces cerevisiae strain background, BF264-15Du (Richardson et al. 1989). Despite the inherent difference in sporulation efficiency and kinetics between these two strains, the effect of the clb mutations was comparable (data not shown).
Table 1.
Sporulation frequency and viability of wild-type and CLB mutants
| Sporulation
|
CLB/CLB
|
clb5/clb5
|
clb6/clb6
|
clb5/clb5 clb6/clb6
|
CLB5/clb5 CLB6/clb6
|
HU
|
|---|---|---|---|---|---|---|
| Unsporulated | 9.0 | 94.6 | 10.5 | 96.0 | 10.2 | 100 |
| Monad | 2.9 | 0.9 | 1.0 | 2.0 | 3.0 | 0 |
| Dyad | 5.0 | 2.3 | 9.0 | 1.8 | 10.4 | 0 |
| Triad | 16.9 | 1.6 | 18.0 | 0.2 | 12.8 | 0 |
| Tetrad | 66.2 | 0.6 | 61.5 | 0.0 | 63.6 | 0 |
| Spore viabilitya | 99.2 | 60.0 | 98.6 | 0.0 | 97.8 | N.A.b |
Sporulation frequency is determined as percent cells from a total of 1000 cells counted.
Spore viability was determined by tetrad analysis from at least 25 tetrads for all strains except clb5/clb5 clb6/clb6, which was determined by random spores analysis.
(N.A.) Not applicable; cells in hydroxyurea (HU) failed to sporulate.
clb5 and clb5 clb6 mutants are defective in premeiotic DNA synthesis
The defect in sporulation of clb5/clb5 clb6/clb6 mutants suggested that, like the same mutants in the mitotic cycle, they might exhibit defects in DNA replication. Whereas the precise nature of the DNA replication defect in clb5 clb6 mutants during the mitotic cell cycle is unclear, there is some evidence to suggest that origin firing requires activation of Clb-associated kinase (Schwob et al. 1994; Zou and Stillman 1998). As a consequence, DNA replication is delayed in clb5 clb6 mutants until CLB1–CLB4-associated CDK activity accumulates (Schwob and Nasmyth 1993; Schwob et al. 1994). Therefore, we examined premeiotic DNA synthesis in wild-type, clb5, clb6, or clb5 clb6 homozygous mutants. Analysis of DNA content by flow cytometry demonstrated that CLB5 and CLB6 are required for efficient premeiotic DNA replication, whereas wild-type diploids completed premeiotic S phase within 4 hr subsequent to induction of sporulation (Fig. 1). DNA replication in clb5/clb5 mutants was first detectable at ∼8 hr and appeared to be incomplete in many cells even after 24 hr (Fig. 1). No delay or defect in DNA replication could be detected in clb6/clb6 mutants (data not shown). However, as observed with the defect in sporulation, combining clb5 and clb6 mutations yielded a profound defect in which no premeiotic DNA replication could be detected even after 24 hr (Fig. 1). This flow cytometric profile of DNA content is similar to that observed when wild-type diploids were treated with the DNA synthesis inhibitor hydroxyurea (HU) following induction of sporulation (data not shown). Thus, unlike the modest defect caused by these mutations in S phase of the mitotic cell cycle, inactivation of both CLB5 and CLB6 appears to completely block progression into premeiotic S phase.
Figure 1.

CLB5 and CLB6 are required for efficient premeiotic DNA replication. Synchronous populations of wild-type, clb5/clb5, and clb5/clb5 clb6/clb6 strains were isolated by centrifugal elutriation and induced to sporulate. Samples of each culture were collected every 2 hr and DNA content of the populations was monitored by flow cytometry of propidium iodide-stained cells. The position of 2C and 4C DNA contents is indicated at the bottom of each plot.
The most likely explanation for the DNA replication defect in the Clb5- and Clb6-deficient cells is that they lack the CDK activity required to activate premeiotic DNA replication. However, it was possible that mitotically growing clb5 clb6 mutants have a defect in chromosome metabolism that is subtle during mitotic growth but which carries over from the final division and precludes effective premeiotic DNA replication. To investigate this possibility, a clb5/clb5 clb6/clb6 GAL1–CLB5 strain was grown in the presence of galactose to allow CLB5 expression. Although these cells expressed CLB5 during mitiotic growth, they were unable to sporulate efficiently when the GAL1 promoter was repressed 1 hr prior to inducing sporulation (data not shown). Thus, the meiotic defect of clb5 clb6 mutants results specifically from the absence of Clb5 and Clb6 during meiosis.
Clb5 accumulates during premeiotic S phase and activates the Cdc28 CDK
CLB5 and CLB6 RNA has been shown to accumulate at about the time that cells are undergoing premeiotic S phase and peak during MI and MII (Chu and Herskowitz 1998; data not shown). Consistent with those observations, cells that have been synchronized in G1 phase and then induced to sporulate, begin to accumulate CLB5 RNA prior to the initiation of S phase (Fig. 2A). CLB6 RNA follows the same pattern of accumulation (Chu and Herskowitz 1998). The accumulation of Clb5 protein closely follows the accumulation of mRNA with the protein abundance increasing throughout S phase and reaching a peak at about the time of MI (Fig. 2B). As expected, the histone H1 kinase activity associated with Clb5 also accumulates through S phase, reaching a peak after the completion of DNA replication (Fig. 2C).
Figure 2.
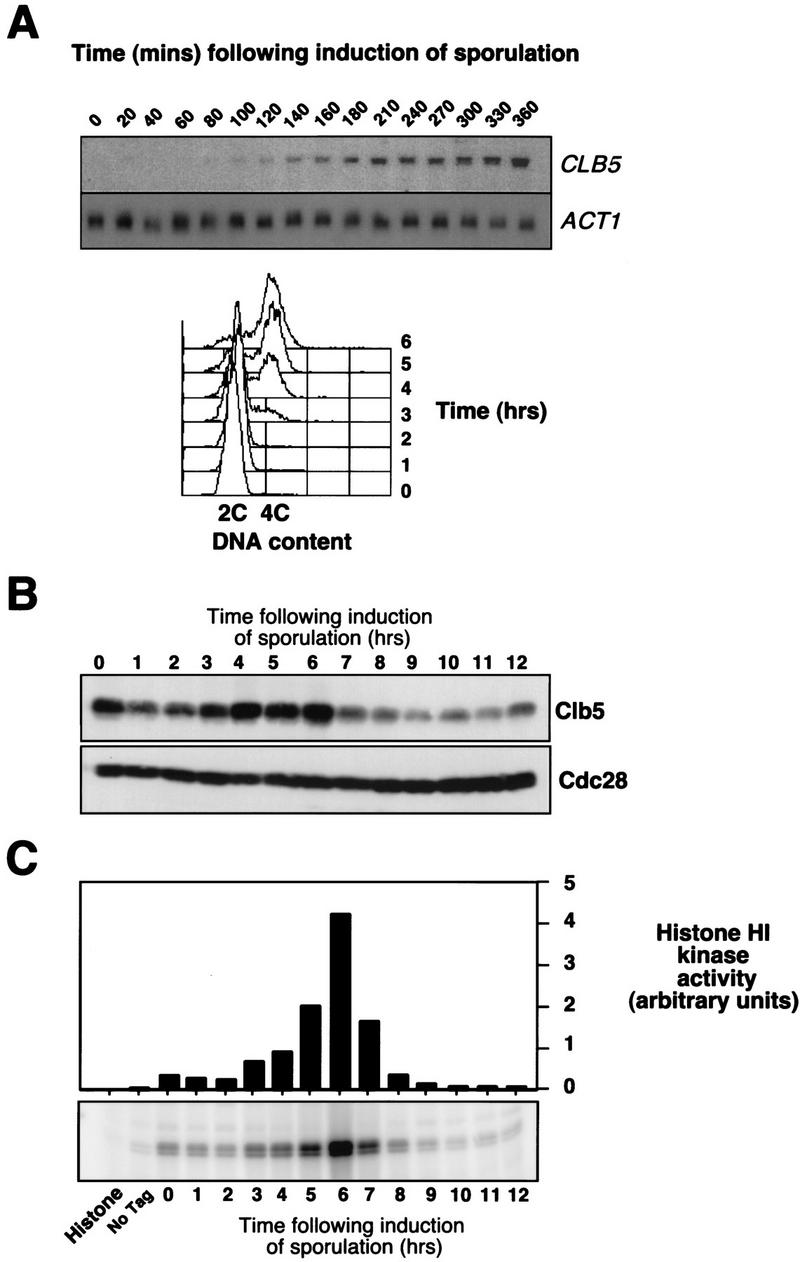
CLB5 RNA, protein, and associated kinase accumulate prior to and throughout meiotic S phase. (A) Synchronous population of G1 cells, isolated by centrifugal elutriation was induced to sporulate and samples collected at the indicated times were analyzed by Northern blotting for the abundance of CLB5 and ACT1 transcripts (top), or for DNA content by FACS (bottom). (B) Western blot analysis of Clb5 protein abundance in a culture grown to late log phase and then induced to sporulate at time 0. The Western blot was probed with 12CA5 anti HA antibody to detect Clb5HA and with anti-Cdc28 antibody as a control for loading. (C) Kinase activity associated with Clb5 was analyzed by immunoprecipitation with anti-HA antibody from samples of CLB5HA/CLB5HA diploids that had been induced to sporulate. Immune complexes were extensively washed and then assayed for kinase activity with histone H1 as a substrate.
Clb5 associates with and activates the histone H1 kinase activity of the Cdc28 CDK during the mitotic cell cycle and is thought to be required for its S phase-promoting activity (Schwob and Nasmyth 1993; Schwob et al. 1994). Clb5 is also detected in association with Cdc28 in extracts from meiotic cells consistent with the notion that the Clb5-associated kinase activity accumulating during meiosis is Cdc28 dependent (Fig. 3A). Furthermore, when Clb5 immune complexes were prepared from cdc28-4 mutants induced to undergo meiosis and sporulation at the permissive temperature, the associated histone H1 kinase activity was severely diminished (Fig. 3B). It has been established previously that those mutants display little or no in vitro kinase activity even when assayed at the permissive temperature (Reed et al. 1985). However, despite the reduced levels of Clb5-associated H1 kinase activity measured in vitro, the cdc28-4 strain expressed Clb5 and underwent premeiotic S phase with similar kinetics to wild-type cells under these conditions (data not shown).
Figure 3.
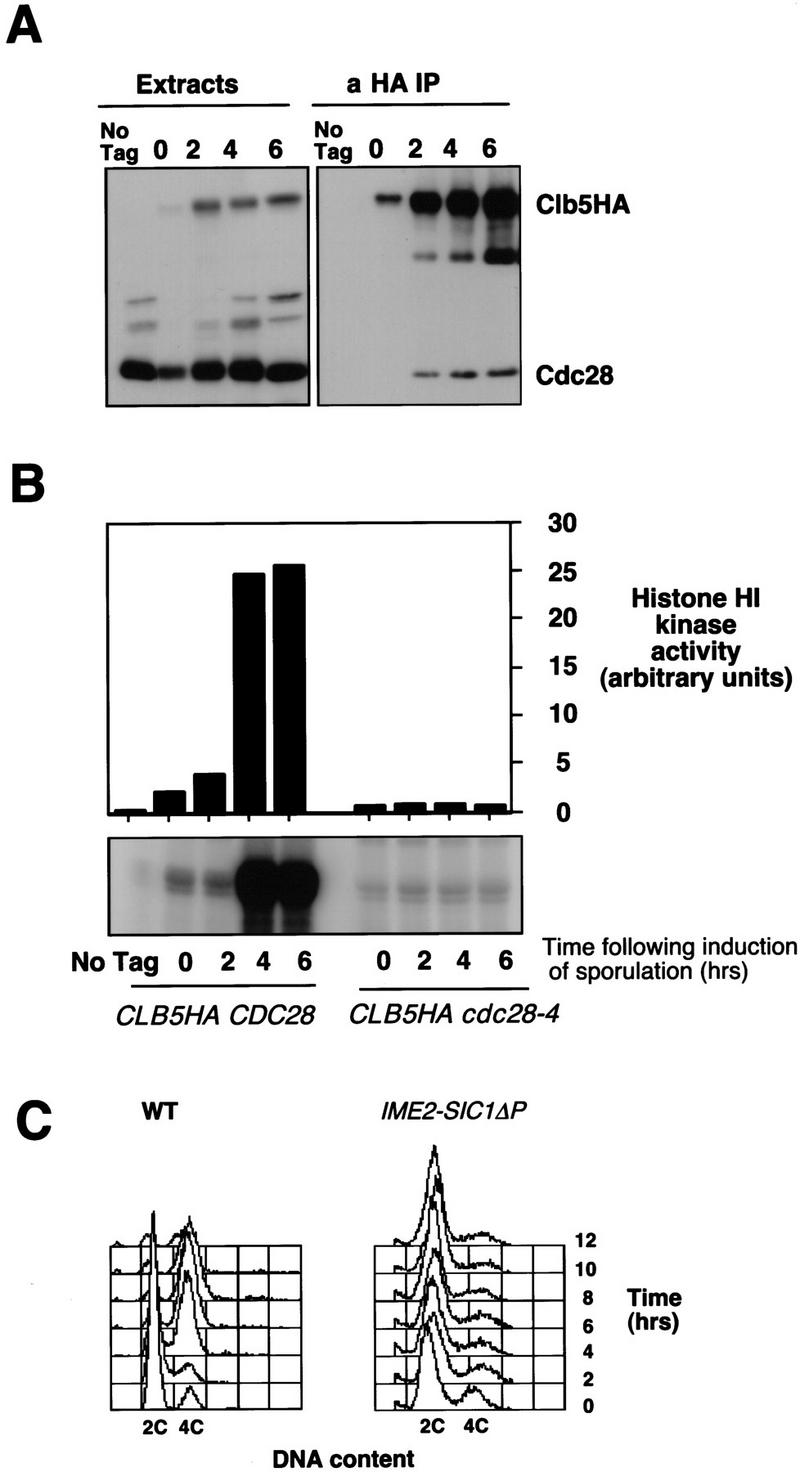
Histone HI kinase activity associated with Clb5 during sporulation is Cdc28 dependent. (A) Whole cell extract (50 μg) or anti-HA immunoprecipitates (from 1 mg of total protein) from either wild-type diploid cells (No Tag) or CLB5HA/CLB5HA diploids were separated by gel electrophoresis and probed for Clb5HA and Cdc28. (B) Histone H1 kinase activity associated with Clb5HA immune complexes prepared from diploid strains expressing wild-type Cdc28 (CLB5HA CDC28), a temperature-sensitive Cdc28-4 (CLB5HA cdc28-4), or a wild-type Cdc28 with untagged CLB5 (No Tag). Strains were grown and induced to sporulate at the permissive temperature of 28°C and kinase activity was assayed at 25°C. (C) DNA content over a time course of sporulation of wild-type diploids and diploids that express a stabilized version of the Clb/Cdc28 CDK inhibitor SIC1 (SIC1ΔP) under the regulation of the meiosis specific IME2 promoter.
Clb-associated CDK activity is essential for premeiotic S phase
The dependence of premeiotic S phase on two B-type cyclins appeared to be at odds with the observation that S phase could be completed in temperature-sensitive cdc28 mutants (Shuster and Byers 1989; Xu et al. 1997). We have independently confirmed these results (data not shown). However, such experiments are inconclusive because they could not be performed at a fully restrictive temperature because of the inherent temperature sensitivity of meiosis. Therefore, to determine whether premeiotic S phase required a Clb-dependent CDK function, we asked whether cells could sporulate in the absence of Clb-associated CDK activity. This was accomplished by expressing a hyperstabilized form of the Clb-specific CDK inhibitor Sic1 (SIC1ΔP) (Verma et al. 1997) in sporulating cells under the control of the meiosis-specific IME2 promoter. Whereas wild-type cells efficiently replicated DNA following the induction of sporulation, diploid cells homozygous for the IME2–SIC1ΔP construct were unable to progress into premeiotic S phase and arrested with a G1 DNA content (Fig. 3C). This was consistent with the effect of inactivating cyclin–CDK activity with stabilized Sic1 in mitotic cells. The most direct interpretation of this series of experiments is that Clb-associated Cdc28 CDK activity is essential for premeiotic S phase, suggesting that the results of experiments with temperature-sensitive cdc28 mutants have been misinterpreted. However, the formal possibility remains that Clb5 activates another Sic1-sensitive CDK that is essential for this process.
clb5 clb6 mutants proceed through the meiotic program in the absence of replicated DNA and undergo a catastrophic M phase
Chemical inhibitors or mutations that block premeiotic DNA replication also block progression into the meiotic program, decrease or prevent meiotic recombination, and inhibit the expression of middle and late sporulation genes (Simchen et al. 1976; Schild and Byers 1978; Mitchell 1994). Thus, the failure to replicate chromosomal DNA would alone be sufficient to explain the inability of clb5/clb5 clb6/clb6 mutants to complete meiosis and undergo sporulation, as indicated by the sporulation defect observed in HU-treated cells (Table 1). However, cells treated with an arresting concentration of HU (100 mm) remain viable and are competent to return to mitotic growth when plated onto rich medium lacking HU (Fig. 4B). In contrast, both clb5/clb5 and clb5/clb5 clb6/clb6 mutants rapidly lose the ability to form colonies on rich growth medium when they are removed from sporulation conditions (Fig. 4A). This suggested that these mutants had an additional defect that caused them to lose viability during sporulation or that simply prevented them from returning to mitotic growth.
Figure 4.

clb5 and clb5 clb6 mutant diploids rapidly lose viability when induced to sporulate. (A) Synchronous population of wild-type (█) clb5/clb5 (▴) or clb5/clb5 clb6/clb6 (•) cells were isolated by centrifugal elutriation and induced to sporulate. The viability of cells during the timecourse was determined by their ability to return to mitotic growth when plated onto rich growth medium at the times indicated. Values represent the average number of colonies derived from two independent samples. (B) Viability of wild-type diploid cells induced to sporulate in the absence of HU (█) or in the presence of 100 mm HU (□). All of the values represent the average number of colonies from two independent samples.
We hypothesized that the mutant cells had become committed to meiotic progression despite having completed little or no DNA replication. The clb5/clb5 (not shown) and clb5/clb5 clb6/clb6 mutant diploids progressed into the meiotic program based on four distinct criteria. First, the mutants executed the normal program of meiotic gene expression as indicated by the transcription of both early (IME1 and IME2) and middle sporulation genes (SPS1, SPS2) (Fig. 5A). Two B-type cyclins, CLB1 and CLB3, were coordinately expressed with SPS1 and SPS2 in both wild-type and mutant cells (Fig. 5A). Surprisingly, despite their continued expression, these cyclins were insufficient to complement the S phase defect of either the clb5/clb5 or clb5/clb5 clb6/clb6 mutant as has been observed in the mitotic cell cycle. Next, fluorescence microscopy revealed the appearance of two discrete DNA masses in a substantial proportion of the clb5/clb5 (not shown) and clb5/clb5 clb6/clb6 mutants (Fig. 5B,C) coincident with execution of the first meiotic M phase in wild-type controls. This was followed by the appearance of three or more chromatin masses in a small proportion of the cells consistent with execution of meiosis II. Finally, the appearance of two or more DNA masses was correlated with the elongation of M phase spindles visualized by green fluorescent protein (GFP)–tubulin fluorescence (Fig. 5D) and with the appearance of separated spindle pole bodies (SPBs) as visualized independently by use of a GFP-tagged SPB component Nuf2 (data not shown). The observations indicate that the clb5/clb5 clb6/clb6 mutants attempted to proceed through meiotic chromosome divisions despite the inability to replicate DNA.
Figure 5.
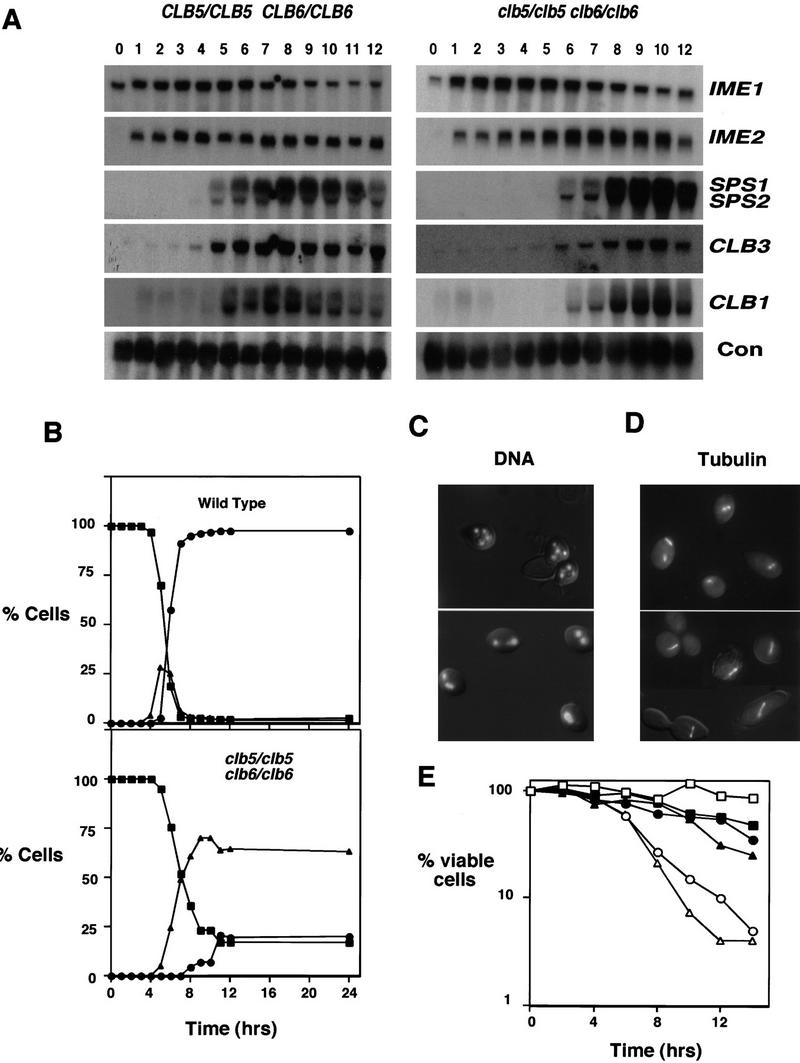
clb5/clb5 clb6/clb6 mutants attempt to progress through meiotic development despite being unable to replicate DNA. (A) Northern blots made with RNA samples from sporulating wild-type diploids (left) or clb5/clb5 clb6/clb6 mutants (right) were sequentially hybridized with probes recognizing RNA transcripts from the early (IME1 and IME2) and middle (SPS1 and SPS2) sporulation genes and the B-type cyclins, CLB1 and CLB3. The constitutively expressed gene C4/2 was used as a loading control (Su and Mitchell 1993). (B) Chromatin segregation in wild-type diploids (top) or clb5/clb5 clb6/clb6 mutants (bottom) following induction of sporulation. The proportion of cells having either one (█), two (▴), or more than two (•) masses of divided chromatin was determined by fluorescence microscopic examination of DAPI-stained cells following induction of sporulation in synchronized populations. (C) Chromatin masses in a representative group of cells from the 8-hr time point of B. Chromatin was visualized by propidium iodide fluorescence and is overlaid on a DIC image of the same cells. The wild-type cells (top) have undergone MI and MII, whereas most of the clb5/clb5 clb6/clb6 mutants (bottom) have apparently undergone a single meiotic division. (D) Meiotic spindles visualized by GFP–tubulin fluorescence in wild-type (top) and clb5/clb5 clb6/clb6 (bottom) 6 hr following the induc-tion of sporulation. The fluorescence image is overlaid on a DIC image of the same cells. (E) Lethal meiosis of clb5/clb5 clb6/clb6 mutants is partially rescued by inhibiting spindle formation. Viability in synchronized populations of wild-type (□,█), clb5/clb5 (○,•) or clb5/clb5 clb6/clb6 (▵,▴) mutant cells treated (solid symbols) with nocodazole (20 μ/ml)/benomyl (30 μg/ml) or left untreated (open symbols) following induction of sporulation.The percent of viable cells was determined by their ability to return to mitotic growth when removed from sporulation conditions and plated onto rich growth medium lacking any inhibitors. Values represent the average number of colonies derived from two independent samples.
The loss of viability in clb5/clb5 clb6/clb6 mutants correlated well with the time at which wild-type cells initiated the segregation of chromosomal DNA at meiosis I (see Fig. 5B, top). Consistent with the view that progression into meiotic M phase in the absence of fully replicated DNA is the cause of lethality in clb5/clb5 and clb5/clb5 clb6/clb6 mutants, the inviability (but not the sporulation defect) of both strains was partially suppressed by treatment with chemical inhibitors of M phase spindle assembly (nocodazole and benomyl) subsequent to the induction of meiosis (Fig. 5E). We assume that the failure of this treatment to more fully suppress the inviability of the mutant cells was caused by the toxicity of the inhibitors because the same treatment reduced the viability of wild-type cells. In fact, it appears that the rescue was very efficient because the proportion of viable cells in the inhibitor-treated wild-type and mutant populations is quite similar. Thus, the inviability of the strains deficient in CLB5 or CLB5 and CLB6 results from their inability to restrict meiotic M phase in the absence of fully replicated chromosomes.
A MEC1-dependent DNA replication checkpoint operates during meiosis
The loss of viability observed in clb5/clb5 clb6/clb6 mutants undergoing meiosis is consistent with the inability of those mutants to activate a checkpoint coupling completion of DNA replication to M phase. Checkpoint controls that monitor DNA damage or imperfect DNA synthesis, recombination, spindle formation, and chromosome segregation have been identified in cells undergoing meiosis (Weber and Byers 1992; Rose and Holm 1993; Thorne 1993; Li and Nicklas 1995; Lydall et al. 1996; Xu et al. 1997). However, it was possible that wild-type cells undergoing meiosis lacked a functional DNA replication checkpoint. Such a checkpoint operating during meiotic development would be expected to prevent the initiation of chromosome divisions when DNA replication is blocked by an arresting dose of the DNA synthesis inhibitor HU and to delay divisions if the completion of DNA replication is slowed by a subarresting dose of HU. To establish the existence of such a checkpoint, cells were induced to sporulate and then treated with 100 mm HU, a concentration sufficient to block DNA replication. HU treatment prevented both sporulation (Table 1) and execution of M phase as indicated by the lack of chromatin segregation (Fig. 6A, bottom). Unlike clb5/clb5 clb6/clb6 mutants, HU-treated wild-type cells maintained high viability on return to growth (Fig. 6A, top). Their ability to maintain viability during this treatment depended on the activity of the MEC1 gene, which is required for both the mitotic DNA replication and DNA damage checkpoints (Weinert et al. 1994; Lydall et al. 1996). As a result, mec1-1/mec1-1 mutants proceeded through meiotic M phase in the presence of HU with the same kinetics as untreated wild-type cells (data not shown) and rapidly lost viability (Fig. 6A, top).
Figure 6.

A DNA replication checkpoint dependent on MEC1 operates during meiotic development. (A) (Top) Viability of wild-type diploid cells (□,█) or mec1-1/mec1-1 mutants (○,•) that were either untreated (solid symbols) or treated with 100 mm HU (open symbols) following induction of sporulation. Viability was assessed by removing samples of each culture at the indicated time and plating onto rich growth medium lacking HU. (Bottom) Percent of wild-type or mec1-1/mec1-1 cells that display one, two, or more than two separated chromatin masses following 12 hr of sporulation in the presence or absence of 100 mm HU. (B) (Top) DNA content of wild-type diploids induced to sporulate in the presence of 2.5 mm HU, a subarresting concentration, at the indicated times following induction of sporulation determined by FACS analysis. (Bottom) Segregation of chromatin, determined by DAPI staining and cell viability as measured by ability to return to growth in wild-type diploids during sporulation in the absence (█) or in the presence (□) of 2.5 mm HU. (C) (Top) DNA content of mec1-1/mec1-1 diploids induced to sporulate in the presence of 2.5 mm HU, a subarresting concentration as determined by FACS analysis. (Bottom) Segregation of chromatin and cell viability of mec1-1/mec1-1 diploid cells during sporulation in the absence (•) or presence (○) of 2.5 mm HU. (D) Percent of wild-type or mec1-1/mec1-1 diploid cells forming asci after being sporulated for 24 hr in the presence or absence of 2.5 mm HU.
A similar experiment was performed by use of an HU concentration that was sufficient to delay, but not to block, progress through DNA replication (Tsui et al. 1997). Wild-type cells treated with 2.5 mm HU proceeded slowly through S phase, but ultimately completed meiosis and sporulated, albeit with somewhat reduced efficiency (Fig. 6B,D). Importantly, chromatin segregation in HU-treated wild-type cells was substantially delayed relative to untreated cells (Fig. 6B, bottom). The HU-induced M-phase delay was abolished in the mec1-1/mec1-1 mutants (Fig. 6C, bottom). Consistent with their failure to delay M phase in the presence of HU, the mec1-1/mec1-1 mutant cells suffered a drastic reduction in viability (Fig. 6C, bottom) and efficiency of sporulation relative to either untreated cells or HU-treated wild-type cells (Fig. 6D). These data provide strong support for the existence of a checkpoint restricting meiotic M phase in the absence of fully replicated DNA.
The meiotic DNA replication checkpoint is inoperable in clb5 clb6 mutants
The failure of clb5/clb5 clb6/clb6 mutant diploids to restrict meiotic M phase in the absence of DNA replication suggests that such cells lack the ability to activate the DNA replication checkpoint. This predicts that treatment of the mutant cells with HU will neither rescue them from inviability, nor restrict the segregation of chromosomal DNA in the absence of DNA replication. Low doses of HU, sufficient to delay MI in wild-type cells (Fig. 7A, top), failed to delay the appearance of divided chromosome masses in clb5/clb5 clb6/clb6 mutants (Fig. 7B, top), or to rescue the mutants from the profound loss of viability observed on return to growth (Fig. 7B, bottom). A high dose of HU provided only a small degree of rescue to the clb5/clb5 clb6/clb6 mutants and was largely unable to prevent the onset of meiosis I (Fig. 7B, top). The degree of viability loss in clb5/clb5 clb6/clb6 mutants is similar to that of mec1/mec1 mutants, suggesting that the small degree of rescue provided by the high dose of HU is independent of the MEC1-mediated checkpoint. The inability of HU to delay or prevent meiotic chromosome segregation demonstrates that the DNA replication checkpoint is inactive and is not able to be activated in clb5/clb5 clb6/clb6 mutants.
Figure 7.

clb5/clb5 clb6/clb6 mutants are unable to delay chromosome segregation in response to HU, but can delay in response to DNA damage. Wild-type (A) or clb5/clb5 clb6/clb6 (B) diploids were induced to sporulate in the absence of HU (█), or with 2.5 mm HU (▴), or 100 mm HU (•). Samples obtained at the indicated intervals were stained with DAPI to determine when chromosome segregation occurred (top, A,B), or were diluted and plated to determine viability (bottom, A,B). (C) Wild-type (□,█) or clb5/clb5 clb6/clb6 (○,•) diploids were induced to sporulate at 30°C and after 2 hr were subjected to either mock irradiation (open symbols) or γ irradiation with 200 gy (solid symbols). Following treatment, cultures were returned to 30°C and samples withdrawn at the indicated times and stained with DAPI to monitor chromosome segregation.
In contrast, clb5/clb5 clb6/clb6 mutants are competent to respond to DNA damage induced during sporulation by delaying meiotic M phase. Treatment with γ irradiation provoked a response that was able to delay the onset of the first meiotic division by ∼2 hr in both wild-type and clb5/clb5 clb6/clb6 mutant cells (Fig. 7C). Similarly, when wild-type or clb5/clb5 clb6/clb6 mutants are treated with UV irradiation, chromosome segregation is delayed (data not shown). These data demonstrate that, despite their defect in the activation of a DNA replication checkpoint, clb5/clb5 clb6/clb6 mutants retain the ability to respond to DNA damage by delaying progression into M phase.
Discussion
CLB5 and CLB6 regulate premeiotic S phase
Clb5 and Clb6 are essential for sporulation because they are required for premeiotic DNA replication. It is surprising that these cyclins are absolutely required during meiotic development because their loss merely delays the initiation and progression through S phase during the mitotic cell cycle (Epstein and Cross 1992; Kuhne and Linder 1993; Schwob and Nasmyth 1993). The ability of CLB1–CLB4 to assume the S-phase function normally performed by CLB5 and CLB6, may explain why these two cyclins are dispensable during mitotic growth, because cells deficient in all six CLBs arrest at the G1/S phase boundary (Schwob et al. 1994). Furthermore, the defect resulting from inactivation of all six CLBs can be suppressed by ectopic expression of CLB1 (S. Haase and S. Reed, pers. comm.). In cells undergoing meiosis, CLB1, CLB3, and CLB4 are expressed even when CLB5 and CLB6 are inactivated. Yet, these B-type cyclins fail to perform the essential CLB-dependent S phase function. That other B-type cyclins are functional and become active in clb5/clb5 clb6/clb6 mutants is demonstrated both by their pattern of expression and their ability to promote Clb-dependent events associated with meiotic M phase. These include separation of SPBs and elongation of meiotic spindles (Fitch et al. 1992; Dahmann and Futcher 1995). Ironically, it is likely the capacity of those B-type cyclins to promote meiotic M phases that results in lethality in Clb5 Clb6-deficient cells. Consistent with that idea, inhibition of all Clb-associated CDK activity by overexpression of Sic1 prevents both DNA replication and chromosome segregation, whereas inactivation of CLB5 and CLB6 prevents DNA replication but not chromosome segregation.
Several lines of evidence suggest that Clb5 and Clb6 perform their essential premeiotic S phase function by activating the Cdc28 CDK. We have shown that Clb5 associates with Cdc28 to form an active protein kinase during meiosis. In addition, expression of Sic1, an inhibitor of the Clb-associated forms of the Cdc28 CDK, is sufficient to block premeiotic S phase as well as subsequent meiotic M phases. This is consistent with the observation that during the mitotic cell cycle inhibition of Clb/Cdc28 kinase, either by the deletion of CLB1–CLB6 or by overexpression of Sic1, prevents DNA replication (Schwob et al. 1994; Verma et al. 1997). The fact that premeiotic S phase is dependent on Cdc2 in S. pombe further supports this contention. However, in agreement with the observations of Shuster and Byers (1989), we have been unable to abrogate meiotic DNA synthesis by use of temperature-sensitive cdc28 mutants. Although this may simply reflect our inability to effectively inactivate Cdc28 under conditions that are permissive for the inherently temperature-sensitive meiotic developmental pathway, we cannot formally exclude the possibility that Clb5 and Clb6 perform a Cdc28-independent function that is required for meiotic DNA replication.
The failure of CLB1, CLB3, and CLB4 to complement a deficiency of CLB5 and CLB6 could be explained if a window of opportunity exists in which Clb-dependent kinase activity can activate DNA replication. Because the expression of CLB5 and CLB6 is rapidly followed by the induction of CLB1–CLB4 during mitotic growth, these four mitotic cyclins can accumulate during the permissive window of time and promote initiation of S phase. However, during meiosis, the interval between the initiation of S phase and CLB1 accumulation is greatly prolonged. As a consequence, CLB1, CLB3, and CLB4 might accumulate too late to adequately replace the S phase function of CLB5 and CLB6 (i.e., subsequent to the window of opportunity) and would simply promote meiotic M phase. A number of scenarios that might lead to the existence of such a window can be envisioned. One reasonable hypothesis is that some aspect of the prereplication complex (pre-RC) is inherently unstable and requires Clb-associated kinases to act prior to decay of the labile state. This hypothesis is not unlike the point of no return hypothesis proposed to explain the requirement for Cdc6 in activating origins of replication (Piatti et al. 1996). That hypothesis holds that if Cdc6 is not provided prior to the activation of Clb-associated kinase activity, it is not able to promote replication, whereas we propose that if Clb-associated kinase is not provided within a specific time frame subsequent to licensing of origins during meiosis, DNA replication will not ensue. Nevertheless, that activity will promote meiotic M phases. Although it is a formal possibility that CLB5 and CLB6 serve some unique function in the activation of premeiotic DNA replication that can not be performed by other forms of Clb–CDK, we think this unlikely because other Clbs can activate DNA replication very effectively during mitotic growth. However, it is also possible that chromosomes become refractory to DNA replication as a consequence of meiotic progression.
The meiotic DNA replication checkpoint is inoperable in clb5 clb6 mutants
The rapid loss of viability that occurs when clb5/clb5 clb6/clb6 mutants enter the meiotic pathway was unexpected because it has been shown that cells can return to mitotic growth if arrested at virtually any stage of meiotic development (Honigberg and Esposito 1994). However, the ultimate demise of the clb5 clb6 mutants stems not from their inability to replicate DNA, but from their inability to restrain chromosome segregation in the absence of DNA replication. This phenotype suggests that the machinery responsible for detecting or responding to a failure in DNA synthesis is defective in these mutants. This defect appears to be meiosis specific because clb5 clb6 mutants display no checkpoint defects during mitotic growth (Li and Cai 1997). Our analysis revealed that a single attempt at chromosome segregation occurs in a relatively large proportion of the clb5/clb5 clb6/clb6 mutants. If chromosomes do not undergo pairing in the absence of replication, the outcome of this attempted division would likely result in unequal chromosome segregation and perhaps in chromosome breakage. DNA damage resulting from that division would likely prevent meiotic progression via the DNA damage checkpoint. This scenario is consistent with our observation that only a small number of the mutant cells ever develop four independent DNA masses.
Although the unreplicated homologous chromosomes may undergo some form of meiotic pairing (Weiner and Kleckner 1994), it is not clear whether such paired chromosomes would behave like those participating in a conventional synapsis (Kleckner 1996; Roeder 1997). One approach to establishing whether chromosomes undergo productive synapsis is to establish the extent to which they recombine. On the basis of an analysis of the rate of recombination of heteroalleles observed on returning cells to growth in rich medium, we conclude that the level of recombination is modestly reduced and delayed in clb5/clb5 mutants relative to wild-type cells. In contrast, clb5/clb5 clb6/clb6 mutants appear not to undergo significant levels of recombination during meiosis (data not shown). Although it might be concluded that the capacity of the later strain to undergo recombination is severely affected, simple interpretations of these results are compromised by the exceedingly small number of viable cells recovered following return to growth as time following induction of meiosis increases (Fig. 4).
Stable arrest in response to inhibitors and mutations that block DNA replication during meiosis is well documented (Simchen et al. 1976; Schild and Byers 1978). This response can now be attributed to the existence of a MEC1-dependent checkpoint that monitors DNA replication and blocks meiotic progression. The same checkpoint is required to delay chromosome segregation when the completion of DNA synthesis is delayed. The target of the DNA replication checkpoint in the mitotic cell cycle is unclear. However, it is known that when replication is blocked with HU during the mitotic cell cycle expression of CLB1–CLB4 persists and a high level of histone H1 kinase activity accumulates (Stueland et al. 1993). When cells undergoing meiosis are similarly treated, they fail to express middle sporulation genes, including CLB1 (Mitchell 1994; data not shown). Although the status of Clb-associated CDK activity in those cells is unclear, they are able to stably arrest without segregating chromosomes and remain viable. It has recently been shown that a similar effect on CLB1 expression occurs in response to DNA damage caused by failure to complete recombination, which then results in a pachytene arrest (Chu and Herskowitz 1998). This restraint mechanism invoked by the DNA replication checkpoint machinery is clearly circumvented in clb5/clb5 clb6/clb6 mutants, which express CLB1 and undergo chromosome segregation.
It is not clear why CLB5 and CLB6 are specifically required for activation of the DNA replication checkpoint. They may be required either to generate a signal concerning the status of DNA replication or to respond to such a signal. The mutants are not generally defective in the generation of checkpoint signals that prevent meiotic M phase because the DNA damage checkpoint is operable in those cells. Several studies suggest that induction of the DNA replication checkpoint during mitotic growth depends on the correct assembly and activation of origins of replication (Li and Deshaies 1993; Piatti et al. 1995; Toyn et al. 1995; Tavormina et al. 1997). In cells arrested by inhibitors of DNA replication, such as HU, at least some pre-RCs are converted to RCs (Diffley et al. 1994) leading to the hypothesis that either the stalled replication forks or some aspect of the activated origin may act as the signal. This is consistent with the observation that mutations which prevent origin activation fail to activate the DNA replication checkpoint (Piatti et al. 1995; Toyn et al. 1995; Tavormina et al. 1997) presumably because no replication signal can be generated. A similar defect may explain the behavior of clb5/clb5 clb6/clb6 mutants during meiosis. Like Cdc6, Clb5 has been shown to interact with origin components, and has been implicated in the activation of replication origins (Epstein and Cross 1992; Kuhne and Linder 1993; Schwob and Nasmyth 1993; Elsasser et al. 1996; Zou and Stillman 1998). That the meiotic DNA replication checkpoint depends on origin activation may not be inconsistent with the ability of clb5/clb5 mutants to undergo some DNA replication, because M phase and concomitant loss of viability precedes detectable DNA replication in those cells. Nevertheless, at least some of those cells appear to progress through meiotic M phase in the presence of replicating DNA, suggesting that Clb5 may also be involved in generating a DNA replication signal. Clb5 need not be the signaling molecule in either of these cases. Instead, Clb5 may be required for correct organization or activation of the replication complex and it may be the state of that complex that is monitored by the checkpoint machinery.
Materials and methods
Strains and growth conditions
All yeast strains used in this study were derived from either BF264-15Du (MATa ade1 bar1Δ his2 leu2 trp1-1 ura3Δ) (Richardson et al. 1989) or SK1 (Kane and Roth 1974) MATa/α ho::LYS2/ho::LYS2 lys2/lys2 ura3/ura3 arg4Bgl/arg4Nsp leu2::hisG/leu2::hisG trp1::hisG/trp1::hisG his4-X/his4-B (Lydall et al. 1996). The relevant genotypes of strains used in this study are shown in Table 2. The phenotypes of clb5, clb6, and clb5 clb6 mutant diploids was comparable in both the BF264-15Du and SK1 strain backgrounds despite the inherent differences in sporulation efficiency of the wild-type parents. Only data obtained from SK1 strains is presented in this report. All strains were constructed by standard genetic methods (Rose et al. 1990). CLB5 was inactivated by replacing a 1.5-kb AflII fragment that contains the entire ORF with either a 1.1-kb URA3 fragment or a kanamycin resistance gene (KANR) from pFA6-KanMX2 (Wach et al. 1994). CLB6 was inactivated by replacing 0.9 kb of the ORF with a 1.1-kb TRP1 fragment. Disrupted alleles were confirmed by PCR analysis. Meiosis-specific expression of a hyperstabilized SIC1 was achieved by fusing the mutant SIC1 ORF (Verma et al. 1997) to the IME2 promoter and integrating the construct at the URA3 locus. cdc28-4 was introduced into the SK1 strain background from BF264-15Du by backcrossing eight times with DSY1030 a MATa derivative of DSY1089. Three copies of an HA epitope were introduced into the carboxy-terminal of CLB5 by site-directed mutagenesis The tagged version of CLB5 was used to replace the wild-type CLB5 in both MATa and MATα strains, which were then mated to produce a homozygous diploid DSY1000. All strains were grown and maintained on rich YEPD medium or synthetic medium at 30°C (Rose et al. 1990). Sporulation experiments were performed essentially as described (Padmore et al. 1991). Strains were initially grown in rich glycerol medium (YEPG) to select for mitochondrial function and then pregrown for 12 hr in rich acetate medium (YEPA), washed once with sporulation medium (SPM) and then inoculated into SPM and incubated at 30°C with vigorous agitation. SPM used in this study is 1% potassium acetate supplemented with 0.0005% arginine, leucine, tryptophan, and uracil. For experiments shown in Figures 1, 4A, and 5, B–D, homogeneous populations of G1 phase cells grown in YEPA were isolated by centrifugual elutriation (Stuart and Wittenberg 1995) and then inoculated into sporulation medium. Cell viability was determined by a return to growth procedure (Esposito and Esposito 1974). Samples of cells from sporulating cultures were withdrawn at the indicated time points, diluted, and plated in duplicate on rich YEPD plates in the absence of any drugs or inhibitors. Colonies were counted after the plates had been incubated for 2 to 3 days at 30°C.
Table 2.
S. cerevisiae strains used in this study
| Strain
|
Relevant genotype
|
Source
|
|---|---|---|
| DSY 1089 | MATa/α | Lydall et al. (1996) |
| DSY 960 | MATa/α clb5::URA3/″ | this study |
| DSY 945 | MATa/α clb6::TRP1/″ | this study |
| DSY 984 | MATa/α clb5::URA3/″clb6::TRP1/″ | this study |
| DSY 1092 | MATa/α clb5::KANR/″clb6::TRP1/″ | this study |
| DSY 1029 | MATa/α CLB5/clb5::URA3 CLB6/clb6::TRP1 | this study |
| DSY 1106 | MATa/α URA3::IME2–SIC1ΔP/″ | this study |
| DSY 1061 | MATa/α TUB1–GFP [LEU2] | this study |
| DSY 1062 | MATa/α clb5::KANR/″clb6::TRP1/″TUB1–GFP [LEU2] | this study |
| DSY 1001 | MATa/α nuf2::NUF2–GFP::KANR/″ | this study |
| DSY 1002 | MATa/α clb5::URA3/″clb6::TRP1/″nuf2::NUF2–GFP::KANR/″ | this study |
| DSY 1057 | MATa/α mecl-1/mecl-1 | Lydall et al. (1996) |
| DSY 1000 | MATa/α CLB5HA–TRP1/″ | this study |
| DSY 1079 | MATa/α cdc28-4/″CLB5HA–TRP1/″ | this study |
γ irradiation
Cultures pregrown to late log phase in YEPA were harvested, washed in SPM, and resuspended in SPM for 2 hr at 30°C. The cultures were then cooled on ice and divided in half. Half of each culture remained on ice while the other half, also on ice, was exposed to γ radiation until a dose of 200gy had been administered. Both irradiated and nonirradiated cells were then resuspended in SPM and samples were withdrawn at the indicated intervals and fixed in 70% ethanol for staining with DAPI, or diluted and plated to determine viability.
Western blotting, immunoprecipitation, and kinase assays
Protein extracts for Western blot analysis were prepared as described (Grandin and Reed 1993). One hundred-microgram samples were electrophoresesd through 10% gels and then transferred to nylon membrane and probed with monoclonal anti HA antibody from BABCO (1:10,000), and polyclonal anti Cdc28 antibody. Assay for Clb5 associated HI kinase activity was performed essentially as described (Grandin and Reed 1993). Clb5HA was immunoprecipitated from 1 mg of extract by addition of anti-HA antibody that had been conjugated to sepharose beads. After 1 hr at 4°C, anti HA beads were collected, washed three times with extraction buffer, then once with low salt buffer, the beads were then divided into two portions. One portion was assayed for HI kinase activity in a 10 μl of reaction as described (Verma et al. 1997) and the other portion was mixed with sample buffer and analyzed by Western blot for the presence of Clb5HA and Cdc28. Histone H1 kinase activity was quantitated by scanning the dried gel in a Molecular Dynamics PhosphorImager.
Analysis of DNA content
Samples of cells taken during sporulation were fixed in 70% ethanol overnight. After rehydration in 50 mm Tris-HCl (pH 7.5), the samples were digested first with RNase A and then briefly with pepsin. Finally the cells were stained overnight in propidium iodide (50 μg/ml). DNA content of the propidium iodide stained cells was then determined by FACS with a Becton Dickinson FACSCAN (Epstein and Cross 1992).
Cytology
Sporulation frequency was determined by counting asci visualized by light microscopy. DNA was stained with propidium iodide or with DAPI and visualized by fluorescence microscopy. For determination of progression through meiosis, cells that contained two or more DAPI staining chromosomal masses were scored as being post-MI. SPBs and meiotic spindles were analyzed by fluorescence microscopy of GFP-Nuf2 (Kahana et al. 1995) or GFP-Tub1 (Straight et al. 1997), respectively.
Analysis of gene expression
RNA was isolated from samples of sporulating cultures as described (Stuart and Wittenberg 1994). Northern blot analysis was performed by use of standard methods (Rose et al. 1990).
Acknowledgments
We thank Aaron Mitchell, Ted Weinert, Rati Verma, and Ray Deshaies for plasmids and strains used in this study. We also offer special thanks to Marisela Guaderrama for technical assistance and to Ted Weinert, Yona Kassir, and members of the laboratories of C. Wittenberg, P. Russell, and S. Reed for helpful discussions. D.S. is a Special Fellow of the Leukemia Society of America. This work was supported by U.S. Public Health Service grant GM46006 and GM43487 to C.W.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL curtw@scripps.edu; FAX (619) 784-2265.
References
- Budd ME, Wittrup KD, Bailey JE, Campbell JL. DNA polymerase I is required for premeiotic DNA replication and sporulation but not for X-ray repair in Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:365–376. doi: 10.1128/mcb.9.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, Herskowitz I. Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol Cell. 1998;1:685–696. doi: 10.1016/s1097-2765(00)80068-4. [DOI] [PubMed] [Google Scholar]
- Collins I, Newlon CS. Chromosomal DNA replication initiates at the same origins in meiosis and mitosis. Mol Cell Biol. 1994;14:3524–3534. doi: 10.1128/mcb.14.5.3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmann C, Futcher B. Specialization of B-type cyclins for mitosis or meiosis in S. cerevisiae. Genetics. 1995;140:957–963. doi: 10.1093/genetics/140.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diffley JFX, Cocker JH, Dowell SJ, Rowley A. Two steps in the assembly of complexes at yeast replication origins in vivo. Cell. 1994;78:303–316. doi: 10.1016/0092-8674(94)90299-2. [DOI] [PubMed] [Google Scholar]
- Dunphy WG, Brizuela L, Beach D, Newport J. The Xenopus cdc2 protein is a component of MPF, a cytoplasmic regulator of mitosis. Cell. 1988;54:423–431. doi: 10.1016/0092-8674(88)90205-x. [DOI] [PubMed] [Google Scholar]
- Edgar BA, Sprenger F, Duronio RJ, Leopold P, O’Farrell P. Distinct molecular mechanisms regulate cell cycle timing at successive stages of Drosophila embryogenesis. Genes & Dev. 1994;8:440–452. doi: 10.1101/gad.8.4.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser S, Lou F, Wang B, Campbell J, Jong A. Interaction between yeast Cdc6 protein and B-type cyclin/Cdc28 kinases. Mol Biol Cell. 1996;7:1723–1735. doi: 10.1091/mbc.7.11.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein CB, Cross FR. CLB5: A novel B cyclin from budding yeast with a role in S phase. Genes & Dev. 1992;6:1695–1706. doi: 10.1101/gad.6.9.1695. [DOI] [PubMed] [Google Scholar]
- Esposito MS, Esposito RE. Genetic recombination and commitment to meiosis in Saccharomyces cerevisiae. Proc Nat Acad Sci. 1974;71:3172–3176. doi: 10.1073/pnas.71.8.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch I, Dahman C, Surana U, Amon A, Nasmyth K, Goetsch L, Byers B, Futcher B. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol Biol Cell. 1992;3:805–818. doi: 10.1091/mbc.3.7.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier J, Norbury C, Lohka M, Nurse P, Maller J. Purified maturation-promoting factor contains the product of a Xenopus homolog of the fission yeast cell cycle control gene cdc2+ Cell. 1988;54:433–439. doi: 10.1016/0092-8674(88)90206-1. [DOI] [PubMed] [Google Scholar]
- Gautier J, Minshull J, Lohka M, Glotzer M, Hunt T, Maller JL. Cyclin is a component of maturation-promoting factor from Xenopus. Cell. 1990;60:487–494. doi: 10.1016/0092-8674(90)90599-a. [DOI] [PubMed] [Google Scholar]
- Grandin N, Reed SI. Differential function and expression of Saccharomyces cerevisiae B-type cyclins in mitosis and meiosis. Mol Cell Biol. 1993;13:2113–2125. doi: 10.1128/mcb.13.4.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH. Saccharomyces cerevisiae cell cycle. Bacteriol Rev. 1974;38:164–198. doi: 10.1128/br.38.2.164-198.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: Controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Honigberg SM, Esposito RE. Reversal of cell determination in yeast meiosis: Post commitment arrest allows return to mitotic growth. Proc Natl Acad Sci. 1994;91:6559–6563. doi: 10.1073/pnas.91.14.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino Y, Hiramine Y, Yamamoto M. The role of cdc2 and other genes in meiosis in Schizosaccharomyces pombe. Genetics. 1995;140:1235–1245. doi: 10.1093/genetics/140.4.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LH, Johnson AL, Game JC. The effect of the cdc9 mutation on premeiotic DNA synthesis in the yeast Saccharomyces cerevisiae. Exp Cell Res. 1982;141:63–69. doi: 10.1016/0014-4827(82)90068-4. [DOI] [PubMed] [Google Scholar]
- Kahana JA, Schnapp BJ, Silver PA. Kinetics of spindle pole body separation in budding yeast. Proc Natl Acad Sci. 1995;92:9707–9711. doi: 10.1073/pnas.92.21.9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane SM, Roth R. Carbohydrate metabolism during ascospore development in yeast. J Bacteriol. 1974;118:8–14. doi: 10.1128/jb.118.1.8-14.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner N. Meiosis: How could it work? Proc Natl Acad Sci. 1996;93:8167–8174. doi: 10.1073/pnas.93.16.8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhne C, Linder P. A pair of B-type cyclins from Saccharomyces cerevisiae that function early in the cell cycle. EMBO J. 1993;12:3437–3447. doi: 10.1002/j.1460-2075.1993.tb06018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupiec M, Byers B, Esposito RE, Mitchell AP. Meiosis and sporulation in Saccharomyces cerevisiae. In: Pringle JR, Broach JR, Jones EW, editors. Molecular and cellular biology of the yeast Saccharomyces. Cell cycle and cell biology. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 889–1036. [Google Scholar]
- Lew DJ, Weinert T, Pringle JR. Cell cycle control in Saccharomyces cerevisiae. In: Pringle JR, Broach JR, Jones EW, editors. The molecular and cellular biology of the yeast Saccharomyces cell cycle and cell biology. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 607–695. [Google Scholar]
- Li JJ, Deshaies RJ. Exercising self restraint: Discouraging illicit acts of S and M in eukaryotes. Cell. 1993;74:223–226. doi: 10.1016/0092-8674(93)90413-k. [DOI] [PubMed] [Google Scholar]
- Li X, Nicklas RB. Mitotic forces control a cell cycle checkpoint. Nature. 1995;373:630–632. doi: 10.1038/373630a0. [DOI] [PubMed] [Google Scholar]
- Li X, Cai M. Inactivation of the cyclin-dependent kinase Cdc28 abrogates cell cycle arrest induced by DNA damage and disassembly of the mitotic spindle in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:2723–2734. doi: 10.1128/mcb.17.5.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohka MJ, Hayes MK, Maller JL. Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc Natl Acad Sci. 1988;85:3009–3013. doi: 10.1073/pnas.85.9.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydall D, Nikolsky Y, Bishop DK, Weinert T. A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature. 1996;383:840–843. doi: 10.1038/383840a0. [DOI] [PubMed] [Google Scholar]
- Masui Y, Markert CL. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J Exp Zool. 1971;177:129–146. doi: 10.1002/jez.1401770202. [DOI] [PubMed] [Google Scholar]
- McKim KS, Hawley RS. Chromosomal control of meiotic cell division. Science. 1995;270:1595–1600. doi: 10.1126/science.270.5242.1595. [DOI] [PubMed] [Google Scholar]
- Mitchell AP. Control of meiotic gene expression in Saccharomyces cerevisiae. Microbiol Rev. 1994;58:56–70. doi: 10.1128/mr.58.1.56-70.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K. Control of the yeast cell cycle by the Cdc28 protein kinase. Curr Opin Cell Biol. 1993;5:166–179. doi: 10.1016/0955-0674(93)90099-c. [DOI] [PubMed] [Google Scholar]
- Newport JW, Kirschner MW. Regulation of the cell cycle during early Xenopus development. Cell. 1984;37:741–742. doi: 10.1016/0092-8674(84)90409-4. [DOI] [PubMed] [Google Scholar]
- O’Farrell PH, Edgar BA, Lakich D, Lehner CF. Directing cell division during development. Science. 1989;246:635–640. doi: 10.1126/science.2683080. [DOI] [PubMed] [Google Scholar]
- Orr-Weaver TL. Developmental modification of the Drosophila cell cycle. Trends Genet. 1994;10:321–327. doi: 10.1016/0168-9525(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Padmore R, Cao L, Kleckner N. Temporal comparison of recombination and synaptonemal complex formation during meiosis in Saccharomyces cerevisiae. Cell. 1991;66:1239–1256. doi: 10.1016/0092-8674(91)90046-2. [DOI] [PubMed] [Google Scholar]
- Piatti S, Lengauer C, Nasmyth K. Cdc6 is an unstable protein whose de novo synthesis in G1 is important for the onset of S phase and for preventing a “reductional” anaphase in the budding yeast Saccharomyces cerevisiae. EMBO J. 1995;14:3788–3799. doi: 10.1002/j.1460-2075.1995.tb00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatti S, Bohm T, Cocker JH, Diffley JFX, Nasmyth K. Activation of S-phase promoting CDKs in late G1 defines a “point of no return” after which Cdc6 synthesis cannot promote DNA replication in yeast. Genes & Dev. 1996;10:1516–1531. doi: 10.1101/gad.10.12.1516. [DOI] [PubMed] [Google Scholar]
- Reed SI. The role of p34 kinases in the G1 to S-phase transition. Annu Rev Cell Biol. 1992;8:529–561. doi: 10.1146/annurev.cb.08.110192.002525. [DOI] [PubMed] [Google Scholar]
- Reed SI, Hadwiger JA, Lorincz AT. Protein kinase activity associated with the product of the yeast cell division cycle gene CDC28. Proc Natl Acad Sci. 1985;82:4055–4059. doi: 10.1073/pnas.82.12.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson HE, Wittenberg C, Cross FR, Reed SI. An essential G1 function for cyclin-like proteins in yeast. Cell. 1989;59:1127–1133. doi: 10.1016/0092-8674(89)90768-x. [DOI] [PubMed] [Google Scholar]
- Richardson HE, Lew DJ, Henze M, Sugimoto K, Reed SI. Cyclin-B homologs in Saccharomyces cerevisiae function in S phase and in G2. Genes & Dev. 1992;6:2021–2034. doi: 10.1101/gad.6.11.2021. [DOI] [PubMed] [Google Scholar]
- Roeder GS. Meiotic chromosomes: It takes two to tango. Genes & Dev. 1997;11:2600–2621. doi: 10.1101/gad.11.20.2600. [DOI] [PubMed] [Google Scholar]
- Rose D, Holm C. Meiosis specific arrest revealed in DNA topoisomerase II mutants. Mol Cell Biol. 1993;13:3445–3455. doi: 10.1128/mcb.13.6.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MD, Winston F, Heiter P. Methods in yeast genetics. A laboratory course manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- Schild D, Byers B. Meiotic effects of DNA-defective cell division cycle mutations of Saccharomyces cerevisiae. Chromosoma. 1978;70:109–130. doi: 10.1007/BF00292220. [DOI] [PubMed] [Google Scholar]
- Schwob E, Nasmyth K. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes & Dev. 1993;7:1160–1175. doi: 10.1101/gad.7.7a.1160. [DOI] [PubMed] [Google Scholar]
- Schwob E, Boehm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1/S transition in Saccharomyces cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Shuster EO, Byers B. Pachytene arrest and other meiotic effects of the start mutations in Saccharomyces cerevisiae. Genetics. 1989;123:29–43. doi: 10.1093/genetics/123.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simchen G, Idar D, Kassir Y. Recombination and hydroxyurea inhibition of DNA synthesis in yeast meiosis. Mol & Gen Genet. 1976;144:21–27. doi: 10.1007/BF00277299. [DOI] [PubMed] [Google Scholar]
- Straight AF, Marshall WF, Sedat JW, Murray AW. Mitosis in living budding yeast: Anaphase A but no metaphase plate. Science. 1997;277:574–578. doi: 10.1126/science.277.5325.574. [DOI] [PubMed] [Google Scholar]
- Stuart D, Wittenberg C. Cell cycle-dependent transcription of CLN2 is conferred by multiple distinct cis-acting regulatory elements. Mol Cell Biol. 1994;14:4788–4801. doi: 10.1128/mcb.14.7.4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— CLN3, not positive feedback, determines the timing of CLN2 transcription in cycling cells. Genes & Dev. 1995;9:2780–2794. doi: 10.1101/gad.9.22.2780. [DOI] [PubMed] [Google Scholar]
- Stueland CS, Lew DJ, Reed SI. Full activation of p34CDC28 histone H1 kinase activity is unable to promote entry into mitosis in checkpoint-arrested cells of the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:3744–3755. doi: 10.1128/mcb.13.6.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su SSY, Mitchell AP. Identification of functionally related genes that stimulate early meiotic gene expression in yeast. Genetics. 1993;133:67–77. doi: 10.1093/genetics/133.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TT, Campbell SD, O’Farrell PH. The cell cycle program in germ cells of the Drosophila embryo. Dev Biol. 1998;196:160–170. doi: 10.1006/dbio.1998.8855. [DOI] [PubMed] [Google Scholar]
- Tavormina PA, Wang Y, Burke DJ. Differential requirements for DNA replication in the activation of mitotic checkpoints in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:3315–3322. doi: 10.1128/mcb.17.6.3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne LW, Byers B. Stage specific effects of X-irradiation on yeast meiosis. Genetics. 1993;134:29–42. doi: 10.1093/genetics/134.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyn JH, Johnson L, Johnston LH. Segregation of unreplicated chromosomes in Saccharomyces cerevisiae reveals a novel G1/M-phase checkpoint. Mol Cell Biol. 1995;15:5312–5321. doi: 10.1128/mcb.15.10.5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui K, Simon L, Norris D. Progression into the first meiotic division is sensitive to histone H2A-H2B dimer concentration in Saccharomyces cerevisiae. Genetics. 1997;145:647–659. doi: 10.1093/genetics/145.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Annan S, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 1997;278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat R, Pohlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Weber L, Byers B. A RAD9-dependent checkpoint blocks meiosis of cdc13 yeast cells. Genetics. 1992;131:55–63. doi: 10.1093/genetics/131.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner BM, Kleckner N. Chromosome pairing via multiple interstitial interactions before and during meiosis in yeast. Cell. 1994;77:977–991. doi: 10.1016/0092-8674(94)90438-3. [DOI] [PubMed] [Google Scholar]
- Weinert T, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes & Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- Xu L, Ajimura M, Padmore R, Klein C, Kleckner N. NDT80, a meiosis specific gene required for exit from pachytene in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:6572–6581. doi: 10.1128/mcb.15.12.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Weiner BM, Kleckner N. Meiotic cells monitor the status of the interhomolog recombination complex. Genes & Dev. 1997;11:106–118. doi: 10.1101/gad.11.1.106. [DOI] [PubMed] [Google Scholar]
- Zamb TJ, Roth R. Role of mitotic replication genes in chromosome duplication during meiosis. Proc Nat Acad Sci. 1977;74:3951–3955. doi: 10.1073/pnas.74.9.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Stillman B. Formation of a preinitiation complex by S-phase cyclin CDK dependent loading of Cdc45p onto chromatin. Science. 1998;280:593–596. doi: 10.1126/science.280.5363.593. [DOI] [PubMed] [Google Scholar]