

Abstract
The Escherichia coli RNA degradosome is the prototype of a recently discovered family of multiprotein machines involved in the processing and degradation of RNA. The interactions between the various protein components of the RNA degradosome were investigated by Far Western blotting, the yeast two-hybrid assay, and coimmunopurification experiments. Our results demonstrate that the carboxy-terminal half (CTH) of ribonuclease E (RNase E) contains the binding sites for the three other major degradosomal components, the DEAD-box RNA helicase RhlB, enolase, and polynucleotide phosphorylase (PNPase). The CTH of RNase E acts as the scaffold of the complex upon which the other degradosomal components are assembled. Regions for oligomerization were detected in the amino-terminal and central regions of RNase E. Furthermore, polypeptides derived from the highly charged region of RNase E, containing the RhlB binding site, stimulate RhlB activity at least 15-fold, saturating at one polypeptide per RhlB molecule. A model for the regulation of the RhlB RNA helicase activity is presented. The description of RNase E now emerging is that of a remarkably complex multidomain protein containing an amino-terminal catalytic domain, a central RNA-binding domain, and carboxy-terminal binding sites for the other major components of the RNA degradosome.
Keywords: RNA degradation, ribonuclease E, protein–protein interactions, RNA degradosome, DEAD-box RNA helicase, RhlB activation
mRNA decay in Escherichia coli is mediated by the combined action of endo- and exoribonucleases (for review, see Belasco and Higgins 1988; Ehretsmann et al. 1992; Nierlich and Murakawa 1996). Two of the principal nucleases, ribonuclease E (RNase E) and polynucleotide phosphorylase (PNPase), have been shown to be components of a multienzyme complex, the RNA degradosome (Carpousis et al. 1994; Py et al. 1994, 1996). Although RNase E and PNPase are functional by themselves, their association in a complex could coordinate their activities in the degradation of RNA (Cohen 1995; Xu and Cohen 1995; Braun et al. 1996). The two other major components of the degradosome were identified as enolase and the DEAD-box helicase RhlB (Miczak et al. 1996; Py et al. 1996). In vitro experiments have shown that RhlB in the degradosome can facilitate PNPase-mediated degradation presumably by unwinding RNA stem–loop structures (Py et al. 1996; E. Blum, A.J. Carpousis, and C.F. Higgins, in prep.). Enolase is a glycolytic enzyme whose role in mRNA decay still remains to be elucidated. Plausible functions include the regulation of degradosome activity in response to growth conditions.
Besides the major components, preparations of the RNA degradosome contain several other proteins present in nonstoichiometric amounts. These include the chaperones DnaK and GroEL, known to aid protein folding, that could have a role in the assembly of the RNA degradosome (Miczak et al. 1996; Blum et al. 1997). Polyphosphate kinase, which can reversibly catalyze the formation of polyphosphate from ATP, is another enzyme associated with the degradosome. It has been suggested that the regulation of polyphosphate levels may be involved in controlling the activity of the RNA degradosome (Blum et al. 1997).
Recently, several other degradosome-like complexes have been identified in eukaryotic cells. A PNPase homolog is part of a multiprotein complex implicated in the regulation of chloroplast message stability (Hayes et al. 1996). The mtEXO complex and the exosome, involved in RNA processing and degradation, are two other complexes described in yeast and its mitochondria (Margossian et al. 1996; Mitchell et al. 1997). One of the ribonucleases found in the exosome has a human homolog, suggesting that a similar complex could also exist in animal cells. In addition to the 3′ → 5′ exoribonuclease activity, the mtEXO complex contains a putative DExH-box helicase. Similarly, two ATP-dependent RNA helicases may be associated with the exosome (Anderson and Parker 1998; de la Cruz et al. 1998). The identification of multiprotein complexes in bacteria, plants, and yeast suggests that assemblies of ribonucleases with other enzymes such as RNA helicases could be a common feature in RNA processing and decay.
E. coli RNase E is a large protein containing 1061 amino acids (Cormack et al. 1993; Casarégola et al. 1992). The amino-terminal half of the protein contains the catalytic site for the single-strand-specific endonuclease (ENDO) activity (Taraseviciene et al. 1995; McDowall and Cohen 1996). It was discovered recently that this region also contains a 3′ polyadenylate or polyuridylate nuclease (PAUN) activity (Huang et al. 1998). The carboxy-terminal half (CTH) of RNase E is characterized by regions of different amino acid composition including proline-rich, highly charged, and acidic segments (Claverie-Martin et al. 1991; Casarégola et al. 1992). The highly charged segment contains the previously described arginine-rich RNA binding domain (AR–RBD) (Cormack et al. 1993; Taraseviciene et al. 1995; McDowall and Cohen 1996). The AR–RBD is not necessary for ENDO or PAUN activity (McDowall and Cohen 1996; Huang et al. 1998). In this study we have characterized protein–protein interactions within the degradosome. We show that the CTH of RNase E contains binding sites for each of the three other major degradosomal components, PNPase, RhlB, and enolase. In addition, a polypeptide from the highly charged region of RNase E, containing the RhlB binding site and the AR–RBD, strongly stimulates RhlB activity. A model for the regulation of the RNA helicase activity within the degradosome is presented.
Results
The RNA helicase RhlB binds to the highly charged region of RNase E
We used Far Western blotting in our initial analysis of the protein interactions within the degradosome. Each of the degradosomal proteins were expressed by the pET system, separated by SDS–PAGE, transferred to a membrane, and renatured in situ. In an initial experiment using RNase E as a probe, we detected an interaction with the RNA helicase RhlB, but not enolase or PNPase (data not shown). To localize the RhlB binding site of RNase E, different polypeptides encompassing the entire RNase E protein were tested. Figure 1A shows a Coomassie-stained SDS–polyacrylamide gel of E. coli extracts in which these polypeptides were lightly overexpressed by a limited induction of <30 min (lanes 4–9). In Figure 1C, the blotted proteins were incubated with RhlB in solution, and the bound protein was detected by use of a rabbit anti-RhlB serum. As expected, the overexpressed RhlB transferred to the membrane is detected in lane 2. Lane 3 demonstrates that the RhlB probe interacts with full-length RNase E on the membrane, confirming our initial reciprocal binding assay. Many proteins in lane 3 do not bind RhlB (cf. Fig.1, A and C) showing the specificity of the interaction under the conditions used. The interaction of RhlB with RneB, C, and D (lanes 5–7), but not RneA (lane 4), maps the binding site to the central region of RNase E (Fig. 1D). Two smaller polypeptides, RneHC1 and RneHC2, also interact with RhlB (lanes 8,9), further limiting the binding site to the highly charged region of RNase E from residues 628–843. The highly charged composition of these polypeptides is conferred by a large region containing both positively and negatively charged residues (48% mixed charged), a proline-rich region (17% mixed charged) and a short arginine-rich stretch (43% positively charged).
Figure 1.


RhlB interacts with the highly charged region of RNase E. (A) Crude extracts of BL21(DE3) containing the control vector pET11a (lane 1) or plasmids overexpressing RhlB, RNase E, RneA, RneB, RneC, RneD, RneHC1, and RneHC2 (lanes 2–9) were analyzed on a 9% SDS-polyacrylamide gel and stained with Coomassie blue. (C) For the Far Western analysis, the proteins were blotted to a membrane, incubated with RhlB in solution, and probed with an anti-RhlB antibody. The RhlB in solution was partially purified from E. coli with a pET derivative expressing RhlB. (B) A control in which the membrane was incubated with a mock preparation from E. coli containing the pET11a vector. In A, protein molecular masses (kD) are marked at left. The position of the recombinant proteins was determined with Ponceau red and is indicated by arrowheads. (D) A schematic representation of the RNase E polypeptides overexpressed by the pET system. The carboxy-terminal half can be roughly divided, by amino acid content, into a proline-rich segment (residues 524–568, 33% proline), a highly charged (HC) domain (residues 600–820, 45% charged), and an acidic terminal part rich in proline residues (845–1061, 20% acidic, 11% proline). The highly charged domain consists of three successive regions: a mixed charged region alternating arginine and acidic residues (600–738), a second proline-rich segment (739–778), and a short region mainly charged in arginine (789–820).
In Figure 1C, the RhlB protein used as a probe was partially purified (see Materials and Methods). This preparation was >than 70% RhlB by Coomassie-stained SDS–PAGE (not shown). The control experiment in Figure 1B excludes any possibility that the anti-RhlB serum cross-reacts with RNase E. The signal below RhlB (Fig. 1B,C) is the result of a reaction of the anti-RhlB serum with another protein present in all the extracts. It is unlikely that the RNase E-RhlB interaction detected here is caused by an RNA bridge. First, the bulk nucleic acid was removed by precipitation with polyethyleneimine (PEI) during the preparation of the RhlB probe. Second, the blots were incubated in the presence of pancreatic ribonuclease. Because the RhlB probe used in these experiments is not completely pure, we cannot exclude the possibility that the interaction with RNase E is mediated by another protein. However, the yeast two-hybrid analysis presented next argues for a direct interaction between these two proteins.
Protein interactions detected by the yeast two-hybrid system
We also examined the interactions between the major proteins of the RNA degradosome using the yeast two-hybrid system that is based on a transcription activation assay (Golemis et al. 1994). RNase E was divided into three regions: amino terminal (1–528), central (500–752), and carboxy terminal (734–1061). The entire protein was used for PNPase, RhlB, and enolase. These proteins were fused to either the DNA-binding or the transcriptional activation domains. As shown in Table 1, all of the possible pair-wise combinations were tested. Six of the pairs activated transcription: (1) RNase E amino-terminal region with itself; (2) RNase E central region with itself; (3) RNase E central region with RhlB; (4) RNase E carboxy-terminal region with RhlB; (5) RNase E carboxy-terminal region with enolase; and (6) enolase with itself. For the three non-self interactions, each of the two possible pairwise combinations activated transcription. The interaction of the central (500–752) and carboxy-terminal (734–1061) regions of RNase E with RhlB confirms the results of the Far Western analysis and provides unambiguous support for a direct interaction between RNase E and RhlB. The interaction with both regions suggests that important determinants for RhlB binding are located in the small overlap between the two constructs (734–752). This interpretation is supported by the in vivo analysis described below. The association of enolase with the carboxy-terminal region (734–1061) of RNase E, but not with PNPase, was unexpected because enolase was shown previously to copurify with PNPase (see Discussion). However, no interaction was detected between enolase and PNPase by use of the two-hybrid system or by Far Western blotting (data not shown), and the in vivo analysis described below also argues against a direct interaction between PNPase and enolase. The enolase self reaction is consistent with a previous biochemical study showing that native enolase is a dimer (Spring and Wold 1971).
Table 1.
Yeast two-hybrid analysis of interactions between the components of the RNA degradosome
| Activation domain
|
DNA-binding domain
|
|||||
|---|---|---|---|---|---|---|
| RNase E
|
RhlB
|
enolase
|
PNPase
|
|||
| amino-terminal
|
central
|
carboxy-terminal
|
||||
| RNase E amino-terminal | + | − | − | − | − | − |
| central | − | + | − | + | − | − |
| carboxy-terminal | − | − | − | + | + | − |
| RhlB | − | + | + | − | − | − |
| enolase | − | − | + | − | xa | − |
| PNPase | − | − | − | − | − | − |
The regions of RNase E correspond to 1–528, amino-terminal; 500–752, central; 734–1061, carboxy-terminal. The entire protein was used for RhlB, enolase, and PNPase. All of the positive interactions (+) were Lac+ and Leu+ on the appropriate indicator plates. All the negative interactions (−) were Lac− and Leu−.
For the enolase–enolase result (x), Leu+ was detected but Lac+ was only observed when the plasmid with the DNA binding domain was introduced after the plasmid with the activation domain.
Construction of an E. coli strain for the conditional expression of RNase E mutants
We introduced an amber stop mutation at the first tyrosine codon of the rne gene in an E. coli strain carrying a temperature-sensitive amber suppressor (Fig. 2A). The resulting strain, pBRN1, grew at 30°C but not at 42°C, the nonpermissive temperature. This conditional growth was expected because RNase E is an essential protein. Growth at 42°C was restored by complementation with a low-copy-number plasmid, pAM–rne, encoding the intact rne gene. A set of deletions in the carboxy-terminal region of RNase E was constructed by inverse PCR of the pAM–rne plasmid (Fig. 2B). These deletions were designed to remove the proline-rich, the highly charged, and the acidic regions of RNase E. All the deletions except the largest, RneΔ15, were able to complement the rne amber strain at 42°C. This result confirms the report that E. coli strains with deletions of the CTH of RNase E are viable (Kido et al. 1996). Figure 2C shows that the plasmid encoded RNase E (lane 1) or the mutant proteins (lanes 2–6) are detected by Western blotting at the nonpermissive temperature (RneΔ16 and RneΔ18 not shown). As expected, the endogenous RNase E protein was not detected under these conditions.
Figure 2.


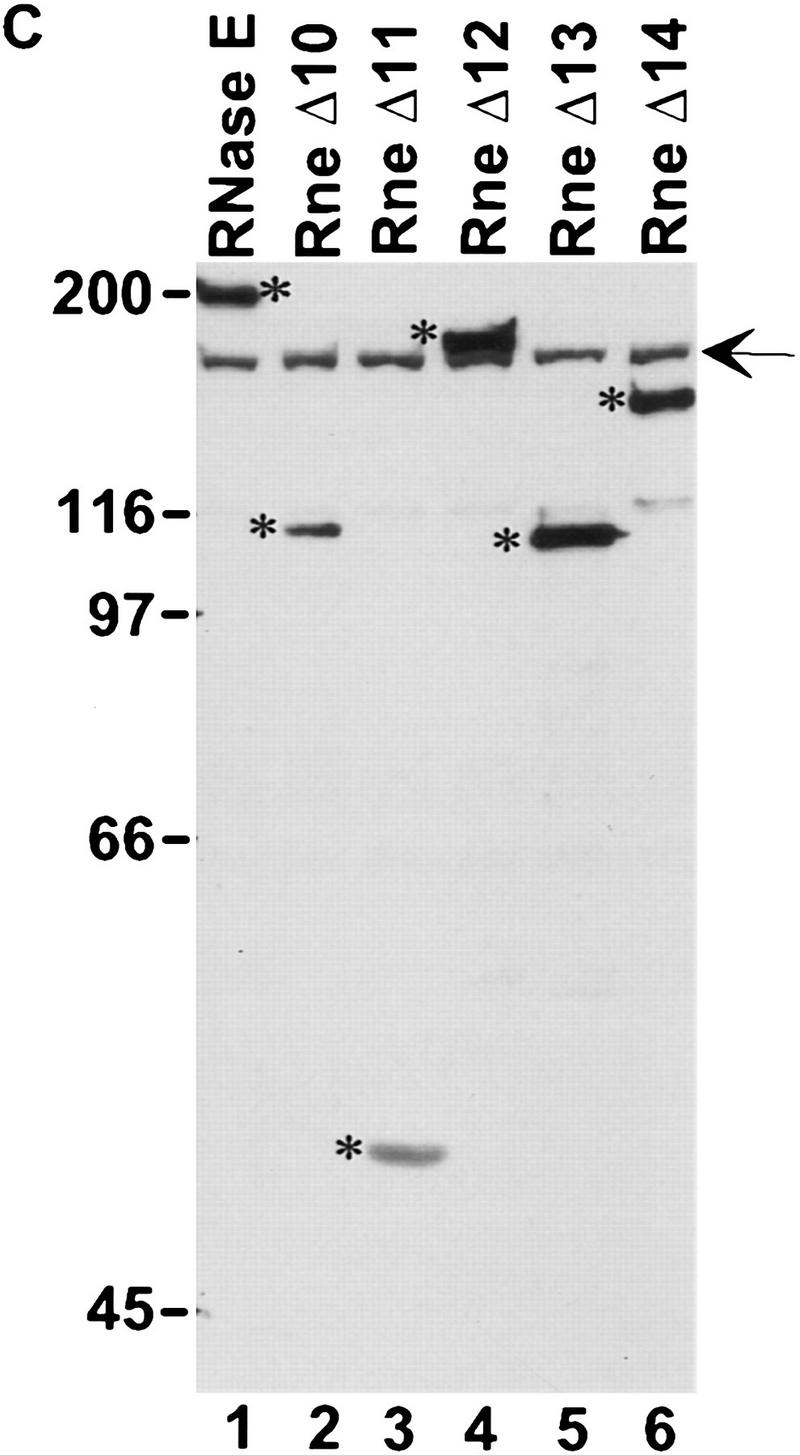
Construction of E. coli strains expressing various deletions in RNase E. (A) A schematic representation of the experimental system. The PBRN1 strain carries the rne gene with an amber mutation in its first tyrosine codon and the temperature-sensitive tRNA suppressor supFts that inserts tyrosine. At 30°C, wild-type RNase E protein is produced because of the suppression by supFts. At 42°C, a polypeptide containing only the first 25 amino acids of RNase E is produced and growth is supported by a plasmid encoding the wild-type RNase E (pAM–rne). (B) The structure of the mutant proteins with the boundaries of the deletions noted in parentheses. The messages encoding these proteins, expressed from the rne promoter (Prne), contain the natural 5′ and 3′ UTR encoded by the rne gene. PBRN1 containing pAM–rne and its derivatives was grown at 42°C to the stationary phase. Total cell extracts were analyzed by Western blotting with an anti-RNase E antibody. (C) Expression of RNase E and mutant peptides at 42°C. The RNase E polypeptides are marked by asterisks. The signal indicated by the arrow is the result of a cross-reaction of the antiserum with another protein present in all the extracts.
Analysis of the composition of the degradosome by coimmunopurification
In an initial experiment with the pAM–rne plasmid expressing wild-type RNase E in the PBRN1 strain, it was difficult to detect the coimmunopurification of RhlB with RNase E. RhlB is probably poorly expressed because of the lack of a recognizable Shine–Dalgarno translation initiation sequence. The protein was not detected by two-dimensional gel electrophoresis of total proteins even when multiple copies of the gene are present (Kalman et al. 1991). We introduced into the PBRN1 strain a medium-copy-number plasmid with rhlB under the control of the lac transcription and translation signals. The basal expression without IPTG induction was sufficient to permit easy detection of RhlB by Western blotting. All the results reported below were obtained in the presence of this plasmid.
In Figure 3, coimmunopurifications were performed by use of rabbit antibodies, against RNase E (A,C) or PNPase (B), which were cross-linked to protein A–Sepharose beads. The immunopurified complexes were released with SDS, separated by SDS-PAGE, and visualized by silver staining (A,B). The identity of PNPase, RhlB, and enolase was verified by Western blotting (A–C). Figure 3A, lane 3, shows that enolase, PNPase, and RhlB efficiently copurify with the full-length RNase E. For comparison, lane 1 shows a preparation of the RNA degradosome purified by ion exchange chromatography and glycerol gradient sedimentation (Py et al. 1996). Lane 2 is a control showing the background using beads with preimmune serum. All of the deletions in the CTH of RNase E affected the composition of the RNA degradosome except RneΔ12 (lane 6) in which only the first proline-rich segment of RNase E was deleted. The changes in composition range from subcomplexes missing PNPase (RneΔ10) or RhlB and enolase (RneΔ13 and RneΔ14) to no degradosomal proteins associated with RNase E (RneΔ11).
Figure 3.


Analysis of the composition of the RNA degradosome. The PBRN1 strain, containing pAM–rne or its derivatives and pAPT–rhlB was grown at 42°C to stationary phase.Plasmid pAPT–rhlB is a p15A-derived plasmid expressing RhlB under the control of the Plac expression signals. Protein extracts were prepared as described in the Materials and Methods. Anti-RNase E antibody (A,C) or anti-PNPase antibody (B), cross-linked to protein A–Sepharose beads, was used to immunopurify the protein complexes. The composition of the immunopurified protein complexes was analyzed by SDS-PAGE and silver staining (top, A, B) or by Western blotting (bottom, A–C). (Lane 1) The RNA degradosome preparation from Py et al. (1996). (Lane 2) A control immunopurification using the preimmune serum for each antibody. The RNase E protein and its mutants are marked with asterisks. In B, X indicates an unrelated protein present in all the cell extracts that react with the anti-PNPase antibody. The molecular masses of markers (lane M) are indicated at left (in kD).
The association of RhlB and enolase with RNase E
With RneΔ10 and RneΔ12 (Fig. 3A, lanes 4,6), RhlB and enolase still copurify with RNase E, showing that neither the acidic carboxyl terminus, nor the first proline-rich region, are essential for the presence of these proteins in the RNA degradosome. In contrast, they do not copurify with RneΔ11, RneΔ13, and RneΔ14. These have in common a deletion (636–845) covering a large part of the highly charged region of RNase E (see Fig. 2B). These in vivo results agree with experiments presented above showing that polypeptides from this region are sufficient to bind to RhlB. Although the copurification of enolase with the RNase E mutants is the same as for RhlB, it is unlikely that the two proteins interact directly. First, using an antibody against enolase, RhlB does not copurify in the RneΔ11 strain (data not shown). Furthermore, the two-hybrid analysis (see above) revealed a direct interaction between enolase and the carboxy-terminal part of RNase E (734–1061). Taken together, these data argue for an enolase binding site between residues 734–845 of RNase E. Most of this region was deleted in RneΔ16 (739–845) to test the interaction with enolase. As shown by the Western analysis in Fig. 3C, enolase does not copurify with RneΔ16. However, RhlB still copurifies. This result shows clearly that the second half of the highly charged region of RNase E (residues 739–845) is important for the interaction with enolase.
In the yeast two-hybrid analysis, RhlB interacted with the central (500–752) and carboxy-terminal (734–1061) regions of RNase E (Table 1) suggesting that the RhlB binding site spans the overlap (734–752). When most of this region is deleted in RneΔ16 (739–845), RhlB still efficiently copurifies with RNase E (Fig. 3C, lane 4) indicating that residues 739–752 are not critical for RhlB binding. However, with the slightly larger deletion, RneΔ18 (728–845), the interaction with RhlB was disrupted (lane 5), showing that residues necessary for the binding are located in the small segment, from 734 to 738 (SVAEE), in the highly charged region of RNase E.
The association of PNPase with RNase E
PNPase copurifies with all of the RNase E deletions except RneΔ10 and RneΔ11 (Fig. 3A, lanes 4,5). The loss of interaction with PNPase in the RneΔ11 strain confirms the report that the CTH of RNase E is required for the interaction with PNPase (Kido et al. 1996). The RneΔ10 strain permits a more precise mapping of the interaction site to the acidic segment in the last 217 residues of RNase E. This result was confirmed by immunopurification of PNPase (Fig. 3B). Full-length RNase E, enolase, and RhlB copurified with PNPase as expected (lane 3). The RneΔ10 and RneΔ11 polypeptides did not copurify with PNPase (lanes 4,5). Enolase and RhlB were also not detected. Moreover, RneΔ13 (lane 6), which still contains the last 217 amino acids of RNase E, copurified with PNPase, but not enolase and RhlB. Thus, a direct interaction between PNPase and enolase or RhlB cannot be detected, indicating that their association in the degradosome is mediated through their independent interaction with the CTH of RNase E.
The association of DnaK with RNase E
Additional minor polypeptides were shown previously to copurify with the RNA degradosome (Carpousis et al. 1994; Py et al. 1996). One of these components was identified as DnaK in the degradosome purified by use of a FLAG-tagged RNase E and in our degradosome preparations (Miczack et al. 1996; Blum et al. 1997). As shown in Figure 3 (A,B), DnaK was detected by silver staining and Western analysis in our purified degradosome preparation (lane 1) and also by immunopurification of full-length RNase E (lane 3). DnaK efficiently copurified with RneΔ10, a deletion of the acidic carboxyl terminus of RNase E which disrupts the association with PNPase (Fig. 3A, lane 4), yet did not coimmunopurify with PNPase in the RneΔ10 background (Fig. 3B, lane 4). These results demonstrate that DnaK does not interact directly with PNPase. DnaK did not copurify with RneΔ11 (Fig. 3A, lane 5), implicating the central part of RNase E in the association of DnaK with the degradosome. Whether DnaK interacts directly with RNase E, or through RhlB and enolase, remains to be clarified.
The interaction with the highly charged region of RNase E stimulates RhlB activity
Our results show that RhlB and RNase E physically interact. Because we found previously that RhlB by itself had no detectable ATPase activity (Py et al. 1996), we tested the effect of RNase E binding on RhlB activity. Preliminary experiments with the full-length RNase E and the RneD polypeptide suggested that these proteins can stimulate RhlB activity. Figure 4 shows an assay of RhlB ATPase activity in the presence of increasing amounts of RneHC2 [(HC) highly charged], the smallest RNase E polypeptide shown to interact with RhlB by Far Western analysis. As a control, we used a mutant, RhlB*, in which the DEAD motif was changed to DKAD. A mutation in the same position in rabbit eIF4A, another DEAD-box RNA helicase, is known to abolish ATPase activity (Pause and Sonenberg 1992). As shown in Figure 4, no significant activity was detected for the RhlB mutant in the presence of saturating amounts of RneHC2, even though RhlB* binds to RneHC2 as revealed by Far Western blotting (data not shown). These results indicate that RneHC2 is an autonomous protein domain sufficient to mediate both the interaction with the RNA helicase and the stimulation of its ATPase activity.
Figure 4.

The RneHC2 polypeptide stimulates RhlB ATPase activity. The specific ATPase activity of wild-type (□) and mutant (♦) RhlB protein was determined in the presence of increasing concentrations of the RneHC2 polypeptide. The values are the average of duplicate assays. The reactions, containing 1 μg of RhlB and 0–2 μg of the RneHC2 polypeptide, were as described (Py et al. 1996), except that the yeast RNA was increased to 200 μg/ml. Less than 15% of the ATP was converted to ADP and Pi under the conditions of maximal activity. RhlB* carries the E166K point mutation in the DEAD motif (DEAD→DKAD). RhlB, RhlB*, and RneHC2 were partially purified from cells overexpressing these proteins. Each protein was judged to be at least 70% pure by SDS-PAGE. Protein concentrations were determined by Lowry assay using a BSA standard.
In Figure 4, the specific activity for wild-type RhlB is 300 U/mg with saturating RneHC2. This value is in reasonable agreement with the previous measurement of 190 U/mg in the RNA degradosome (Py et al. 1996). Without RneHC2 activation, the specific activity is <20 U/mg showing at least a 15-fold stimulation. From Figure 4, we estimate that the activity saturates at ∼0.6 μg of RneHC2 in a reaction containing 1 μg of RhlB. Correcting for their molecular weights, this corresponds to one molecule of RneHC2 per molecule of RhlB.
Discussion
The carboxy-terminal half of RNase E organizes the degradosomal protein interactions
The protein interactions between the components of the RNA degradosome were investigated by three complementary techniques: Far Western blotting, the yeast two-hybrid assay, and coimmunopurification. The Far Western and two-hybrid analyses were particularly useful in identifying specific regions of interaction within RNase E, whereas the coimmunopurification experiments provided direct information about the organization of the degradosome in the bacterial cell. The main conclusion of the combined approaches is that the CTH of RNase E constitutes the structural core of the degradosome around which RhlB, enolase, and PNPase assemble (Fig. 5A). Our results clearly demonstrate that the CTH of RNase E contains distinct binding sites for each of these proteins. In addition, we did not detect any direct interactions between RhlB, enolase, and PNPase. By deleting their binding sites in the CTH of RNase E, it was possible to generate subcomplexes of the degradosome containing three components (RNase E-RhlB–enolase and RNase E-RhlB–PNPase) or two components (RNase E-PNPase and RNase E-RhlB). This shows that the individual deletions do not significantly affect the other binding sites, revealing a multidomain structural organization of the CTH of RNase E.
Figure 5.
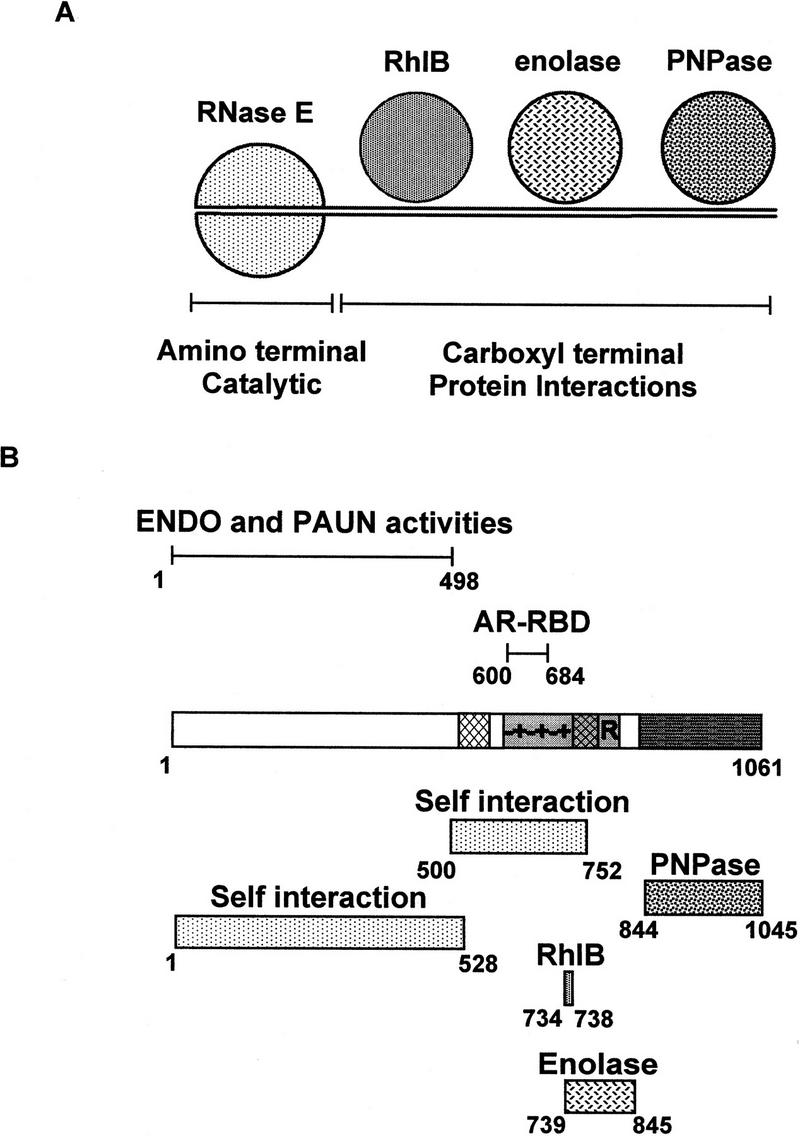
Organization of the protein interactions within the RNA degradosome. (A) Cartoon of the E. coli RNA degradosome illustrating the central role of the CTH of RNase E in the protein interactions. The actual stoichiometry and structure of the components within the degradosome are not certain. It seems reasonable to suppose that RNase E is dimeric although a trimeric or higher order structure is possible (see Discussion). (B) Summary diagram illustrating the known activities and binding regions in RNase E.
Only the amino-terminal half of RNase E, known to contain the ENDO activity, is essential for cell viability (Jain and Belasco 1995; Taraseviciene et al. 1995). Nevertheless, we believe that the integrity of the degradosome is important for normal mRNA turnover. Several E. coli mutants expressing an RNase E that is missing the CTH of the protein were obtained in a selection for extragenic suppressors of a temperature-sensitive mukB allele (Kido et al. 1996). The suppressor strains overexpress the defective MukB protein twofold, apparently caused by the slower turnover of the mukB message. In our strains, the deletion of the CTH of RNase E (RneΔ11) leads to a threefold slower degradation of RNA1 (N.F. Vanzo and A.J. Carpousis, unpubl.), an antisense RNA whose degradation depends on both RNase E and PNPase (Cohen 1995; Xu and Cohen 1995). Whether these effects are caused by the disruption of the RNA degradosome or defective RNase E activity remains to be clarified.
Protein interactions in the RNA degradosome
Figure 5B summarizes our current understanding of the structural and functional features of RNase E. The regions of RNase E identified here, involved in the interaction with itself or the other components of the degradosome, are indicated in the lower part of Figure 5B. The best localized site is that of RhlB. An interaction with the central region of RNase E was detected by Far Western analysis and a core site from 734 to 738 was determined on the basis of two-hybrid and coimmunopurification results. This stretch of five residues (SVAEE) lies at the carboxy-terminal end of the mixed charge region in the highly charged domain of RNase E. Our in vivo results indicate that the core site is essential for RhlB association with the degradosome, but flanking residues could also be important for the structure of this site. The enolase binding site (739–845), defined by a combination of two-hybrid and coimmunopurification experiments, includes the proline- and arginine-rich segments of the highly charged region. The in vivo analysis of the degradosome has permitted us to restrict the PNPase binding site to residues 844–1045 in the acidic carboxy-terminal part of RNase E. For enolase and PNPase, the actual binding sites could be smaller than defined here and further experiments will be required to identify the critical residues for these interactions. We were not able to detect the RNase E-PNPase interaction by Far Western blotting or two-hybrid analysis. Highly purified, enzymatically active PNPase is a trimer of the 78-kD chains, forming a large 234-kD native protein (Littauer and Soreq 1982). This native trimeric state could be critical for the interaction with RNase E. In contrast to the experiments in the bacterial cell where PNPase can properly fold and assemble, its misfolding on a membrane or in yeast could account for the failure to detect an interaction. Our results highlight the importance of using a variety of techniques to identify the proteins interactions within a complex such as the RNA degradosome.
None of these methods have detected a direct interaction between PNPase and enolase. Because of the identification of the β-subunit of PNPase as enolase (Py et al. 1996), this result is surprising. The β-subunit copurifies with PNPase under certain conditions, but it is not essential for phosphorylase activity (Portier 1975; Soreq and Littauer 1977). Our in vivo experiments clearly show that enolase fails to copurify with PNPase in the absence of RNase E, demonstrating that RNase E serves as a bridge between these two proteins. It is known that RNase E is very sensitive to proteolysis during purification (Carpousis et al. 1994). We believe that the copurification of enolase with PNPase reported previously could be mediated by degradation products from the CTH of RNase E. Because the enolase and PNPase binding sites are next to each other in the CTH of RNase E, a relatively small RNase E polypeptide of 100–300 amino acids could bind both proteins.
The self interactions of the amino and central regions of RNase E detected in our two-hybrid analysis is the first direct evidence for its capacity to oligomerize. A previous analysis of the degradation of the S20 ribosomal protein message suggested that native RNase E is composed of at least two active subunits (Mackie et al. 1997). Many enzymes are only active in their multimeric native state and the oligomerization of the amino-terminal half of RNase E could be necessary for the ENDO or PAUN activities. The self interaction of the central region suggests that the CTH of RNase E may also be oligomeric. Whether this oligomerization is important for the protein interactions remains to be determined. The quaternary structure of RNase E in the complex is unsure as a result of the significant heterogeneity in the degradosome preparations. Carpousis et al. (1994) suggested that RNase E was dimeric on the basis of the stoichiometry of the degradosomal components in an immunoprecipitate, and an estimation of the molecular weight of the complex by sedimentation.
Activation of the RNA helicase RhlB by the highly charged region of RNase E
On the basis of a sequence analysis of its gene, RhlB was initially identified as a member of the DEAD-box family of ATP-dependent RNA helicases, known to unwind double-stranded regions (Kalman et al. 1991). Consistent with this identification, in vitro experiments showed that RhlB in the degradosome facilitates the PNPase-mediated degradation of structured RNA in an ATP-dependent reaction (Py et al. 1996). In addition, the purified RNA degradosome has an RNA-dependent ATPase, a characteristic activity of the DEAD-box proteins. Nevertheless, overexpressed and partially purified RhlB does not by itself have a detectable ATPase activity. The work described here shows that RhlB interacts with the highly charged region of RNase E and that this interaction stimulates its activity at least 15-fold. The RneHC2 polypeptide, containing the core RhlB binding site, can fully activate RhlB to a specific activity comparable with that previously measured in the RNA degradosome. This stimulation saturates at a stoichiometry of one molecule of RneHC2 per molecule of RhlB.
RhlB is the second example of a DEAD-box helicase stimulated by a protein partner. The eukaryotic translation factor eIF4B stimulates the RNA unwinding activity of eIF4A, the prototype of the DEAD-box family (Rozen et al. 1990; Pause et al. 1992). Members of this family share a core region of ∼300 amino acids containing eight highly conserved motifs (Schmid and Linder 1992). It has been proposed that this core domain is insufficient for RNA helicase activity and that an additional RNA-binding site is required (Gibson and Thompson 1994). This could be provided by a domain adjacent to the core or by a protein partner. eIF4B has been suggested to supply the additional RNA-binding domain for the activation of eIF4A (Milburn et al. 1990; Pause et al. 1992). RhlB contains an arginine-rich stretch at the carboxyl terminus that is predicted by primary sequence analysis (Gascuel and Golmard 1988) to be in a random coil conformation. The interaction with RneHC2 could fold this region into a functional RNA-binding site, activating the helicase. The other possibility is that RneHC2, which contains most of the AR–RBD, supplies the additional RNA-binding site. This would directly implicate the AR–RBD, which is not required for ENDO or PAUN activity, in the regulation and specificity of RhlB. Recent experiments with purified RhlB and RneHC2 demonstrate the unwinding of short RNA duplexes in an ATP-dependent reaction (N.F. Vanzo and A.J. Carpousis, unpubl.). Thus, it should be possible to directly examine the mechanism of stimulation of the RhlB helicase activity by the highly charged region of RNase E.
There are now many examples of enzymes, acting in the same biological process, that are associated in a complex (see Alberts 1998 and references therein). Recently, degradosome-like complexes have also been described in eukaryotes (see introductory section). It is evident that these sophisticated protein machines offer the possibility to regulate and coordinate the activities of the individual components. The activation of the RNA helicase RhlB by RNase E, demonstrated here, is one example of this type of control.
Materials and methods
Bacterial strains and plasmids
The XA103 strain [nalA rif argE(am) supF metB thi ara Δ(lac-pro)] and the supFts strain PB4144 (KH5402; Kimura et al. 1979) were kindly provided by J. Miller (UCLA) and D. Lane (CNRS), respectively. AC23 (ams zce-726::Tn10), a derivative of MC1061, is isogenic with the previously described CH1828 strain (Mudd et al. 1990). The wild-type rne gene of XA103 was substituted by the ams allele by P1 transduction using AC23 strain as donor. A streptomycin-resistant derivative of XA103 ams was isolated by plating on 200 μg/ml of the antibiotic. The resulting strain was named XA103a.
The E. coli strain used for most of DNA manipulations in this study was DH5αF′ (F′/endA1 hsdR17 (rk−mk+) supE44 thi-1 recA1 gyrA (Nalr) relA1 Δ(lacZYA–argF)U169 deoR (φ80dlacΔ(lacZ)M15). The BL21(DE3) strain (Novagen) was used for protein overexpression. To overexpress PNPase, the E. coli pnp coding region was amplified by PCR with pfu DNA polymerase (Stratagene) by use of primers pnp1 and pnp2 (Table 3, below). This PCR product was cloned into pUC18 and then into pET11a, as described for the pcnB gene by Raynal et al. (1996). The pET11a derivatives for the overexpression of enolase and RhlB were constructed previously (Py et al. 1996) by the same procedure.
Table 3.
Primers used in this study
| Name
|
Sequence (from 5′ to 3′)a
|
|---|---|
| rne215 | ggatccTACGGAATAACCCATTTTGCCCGAC |
| rne541 | atATGAAAAGAATGTTAATCAACGCAACTCAGC |
| rne627 | GATATCCAGGTCcTACAGACGCTGCCC |
| rne1192 | ATGCGCGAACGATTACGTTGCTCT |
| rne2041 | ATGCTGCCGAAGCTGCATGAAGAA |
| rne2064 | TTCTTCATGCAGCTTCGGCAGCATG |
| rne2422 | GAACGTACTGAAGGCAGCGATAATC |
| rne2445 | ATTATCGCTGCCTTCAGTACGTTC |
| rne2721 | CTGATTGAGCTGACGCTGTTTACGACGCGGC |
| rne2754 | TTCTTCGGCTACGCTTTGCTCGTAACGC |
| rne3069 | CCAGACTTTGCCAGAGGCCAGTTC |
| rne3076 | TATCCAATTGTACGTCCGCAAGATGTAC |
| rne3676 | GCAACACGTCATGCCTCTGCCG |
| rneYSL1 | GAGTAAgaattcATGAAAAGAATGTTAATC |
| rneYSL2 | GCGCGctcgagTTACGCAGGTTGTTCCGGACGCTTAC |
| rneYSL3 | CCAACCgaattcTACATGCTGCCGAAGCTG |
| rneYSL4 | TTCCTGctcgagTTATTCGGCAGCGACAGTTTC |
| rneYSL5 | CGTTACgaattcAGCGTAGCCGAAGAAGCG |
| rneYSL6 | ACTTTGctcgagTTATTAtTCcACAGGTTG |
| pnp1 | atATGCTTAATCCGATCGTTCGTAAATTCC |
| pnp2 | TTACTCGCCCTGTTCAGCAGCC |
| pnpYSL1 | GGGCGgaattcATGCTTAATCCGATCGTT |
| pnpYSL2 | GGGGGctcgagTTACTCGCCCTGTTCAGC |
| rhlB607K | ATTCAGGTGGTGGTACTGGACaAAGCCGATCGCA |
| rhlB1282R | ATTTGCTTACCGGAATTGAGT |
| rhlBYSL1 | GGGCGgaattcATGAGCAAAACACATTTAACAG |
| rhlBYSL2 | GGGGGctcgagTTAACCTGAACGACGACGATTACGCGG |
| enoYSL1 | CTTGAGGggatccTAATGTCCAAAATCGTAAAAATC |
| enoYSL2 | CTAGTTctcgagTCAGATAAAGTCAGTCTTATG |
| enoYSL3 | CTTGAGGAActcgagATGTCCAAAATCGTAAAAATC |
| enoYSL4 | GCTAGTTctcgagTCAGATAAAGTCAGTCTTATG |
The lowercase letters, showing the substitutions introduced to manufacture restriction sites, prevent secondary structure formation within the primer or create mutations.
Vectors encoding polypeptides derived from the rne gene
The plasmid pET-rne overexpressing full-length RNase E protein was constructed as follows. First, a 3.5-kb ApoI–ApoI chromosomal fragment containing the entire rne gene was cloned in the EcoRI site of the pET11a vector. A clone having the rne gene in the same orientation as the T7 RNA polymerase transcription gave the pET–rne1 plasmid. Second, a PCR-generated fragment encoding residues 1–217 of the RNase E protein was cloned into the SmaI site of pUC18 vector to give the pUC–rne1 plasmid. The primers for PCR were rne541 and rne1192 (Table 3, below). After sequencing, the NdeI–HindIII rne fragment was excised and ligated to the pET–rne1 plasmid digested by the same enzymes to make the pET–rne plasmid. Other constructions are as shown in Table 2 and the primers for PCR are listed in Table 3. Polypeptides RneB, RneD, RneHC1, and RneHC2 are translational fusions with the first 13 residues of bacteriophage T7 gene 10 protein. To construct the pET–rneD plasmid, a HindIII–ApoI rne fragment was excised from pET–rne plasmid and subcloned into pGEM–7Zf vector (Promega) digested by HindIII–EcoRI to give pGEM–rneD plasmid. This plasmid was digested by BamHI and ApoI, liberating BamHI–BamHI and BamHI–ApoI rne fragments. Both were ligated with the pET11b vector digested by BamHI and EcoRI. A clone with the correct orientation of the BamHI fragment was chosen on the basis of restriction analysis.
Table 2.
Plasmids expressing RNase E polypeptides
| Vector (restriction sites)
|
rne insert
|
Plasmid (residues encoded)
|
|---|---|---|
| pET11a (NdeI–EcoRI) | NdeI–ApoI fragment | pET–rne (1–1061) |
| pET11a (NdeI–BamHI) | NdeI–BstYI from pET–rne | pET–rneA (1–263) |
| pET11c (BamHI) | BstYI–BamHI from pET–rne | pET–rneB (263–844) |
| pET11a (NdeI–BamHI) | NdeI–BamHI from pET–rne | pET–rneC (1–844) |
| pET11b (BamHI–EcoRI) | BamHI–ApoI from pGEM–rneD | pET–rneD (185–1061) |
| pET11c (BamHI filled) | PCR-generated fragmenta | pET–rneHC1 (501–843) |
| pET11c (BamHI filled) | PCR-generated fragmentb | pET–rneHC2 (628–843) |
Primers rne2041 and rne3069.
Primers rne2422 and rne3069.
Protein overexpression and preparation of extracts
Crude protein extracts for the blots in the Far Western analysis were prepared as follows. A fresh overnight colony of BL21(DE3) containing the appropriate plasmid was grown at 37°C in LB–ampicillin to OD600nm = 0.4. The culture was then shifted to 30°C to optimize solubilization of overexpressed proteins and induced from 20–30 min with 1 mm IPTG. Lysates were prepared as in Carpousis et al. (1994) with some modifications. Briefly, the cultures were concentrated 25-fold and suspended in the lysosyme–EDTA buffer, and the cells were broken with a freeze–thaw step. The DNase–Triton buffer was added, and the suspension was incubated for 30 min with agitation and then adjusted to 1m NH4Cl.
To obtain partially purified proteins as probes for the Far Western analysis, and for the RhlB ATPase assay, crude extracts were prepared as described above except that the induction time was for 2 hr. The extracts were clarified by centrifugation (15 min, 10,000g), the supernatants were collected and PEI was added slowly with gentle agitation to a final concentration of 0.2% (Gegenheimer 1990). The precipitated nucleic acid was removed by centrifugation (15 min, 10,000g), the PEI was removed by precipitating proteins with 60% ammonium sulfate, and the pellets were suspended in buffer A containing 200 mm NaCl (Carpousis et al. 1994).
Western and Far Western analysis
Protein gels and Western blotting were performed as described in Raynal et al. (1996) except that gels were electroblotted with a carbonate transfer buffer (Dunn 1986) for 12 hr at 60 mA. Polyclonal antibodies against the purified components of the RNA degradosome were raised in rabbits (Eurogentec). Polyclonal rabbit antiserum against E. coli DnaK was a gift from Bernd Bukau (University of Freiburg, Germany). For Far Western analysis, the blots were placed in TEN 50 buffer (Cormack et al. 1993) and stored at 4°C for a minimum of 5 days to permit in situ renaturation of proteins. Filters were blocked for 1 hr in HHB (HEPES-hybridization buffer; 20 mm HEPES at pH 7.7, 75 mm KCl, 0.1 mm EDTA, 2.5 mm MgCl2,1 mm DTT, 0.05 % Triton) containing 5% nonfat milk. Membranes were then incubated overnight at 4°C in HHB supplemented with 1% nonfat milk, RNase A (0.2 μg/ml final), and the appropriate partially purified protein. Filters were washed three times for 10 min with HBB supplemented with 1% nonfat milk and submitted to Western analysis.
Yeast plasmids
The plasmid vectors used for yeast two-hybrid analysis, pEG202, pJG4-5, pJK101, pRFHM-1, pSH17-4, and pSH18-34, were provided by Roger Brent and are described in Golemis et al. (1994). pBluescript K+ was from Stratagene. Genes encoding components of the degradosome were amplified with Vent polymerase (New England Biolabs) and inserted into either pEG202 (to generate bait plasmids; pB series) or into pJG4-5 (to generate activation plasmids; pA series). Restriction sites were added to aid cloning. The pB series of plasmids result in a fusion between the protein of interest and the DNA-binding protein LexA. The pA series of plasmids result in a fusion between the protein of interest and an acidic domain (acid blob), which functions as a portable transcriptional activation motif.
Plasmid pBP1, containing the E. coli pnp gene, was constructed by PCR with primers pnpYSL1 and pnpYSL2, cloning the EcoRI- and XhoI-digested PCR product into pEG202 digested with the same enzymes. Plasmid pBB, which expresses RhlB, was constructed similarly with primers rhlBYSL1 and rhlBYSL2. For pBEN, which expresses residues 1–528 of the amino-terminal domain of RNase E, primers rneYSL1 and rneYSL2 were used. For pBECN, which expresses the central domain of RNase E (residues 500–752), primers rneYSL3 and rneYSL4 were used. For pBEC, which expresses the carboxy-terminal domain of RNase E (residues 734–1061), primers rneYSL5 and rneYSL6 were used. These fragments were all cloned into pEG202 digested with EcoRI and XhoI. Plasmid pBENO, which expresses the enolase gene, was amplified with primers enoYSL1 and enoYSL2 and cloned into pEG202 cut with BamHI and XhoI.
Plasmids pAP expressing PNPase, pAB expressing RhlB, pAEN expressing RNase E amino-terminal domain, pAECN expressing RNase E central domain, and pAEC expressing RNase E carboxy-terminal domain used the same amplified genes as the pB vectors, but cloned into pJG4-5 cut with EcoRI and XhoI. Plasmid pAENO, expressing enolase, was made with primers enoYSL3 and enoYSL4, then cloned into pJG4-5 cut with XhoI. Some of the inserts were blunt-end ligated into pBluescript K+ before being subcloned into their appropriate yeast two-hybrid vectors.
Yeast two-hybrid analysis
The two-hybrid method used here is based on the LexA–B42 acid blob system (Gyuris et al. 1993). The two reporter genes are LexAop–LEU2 and LexAop–LacZ. EGY48 (ura3, trp1, his3, lexA operator–LEU2) was the yeast strain used throughout. Cells were grown in YPD or complete minimal (CM) dropout media as described by Golemis et al. (1994) except for X-gal plates that contained 0.07 m of potassium phosphate (pH 7.0), and 0.04 μg/ml X-gal (Sambrook et al. 1989). Yeast transformations were performed according to Gietz et al. (1992), and nutritional selection was maintained throughout.
EGY48 was cotransformed with pSH18-34 and the pB series. Plasmids from the pA series were then introduced into these transformed yeast and selected at 30°C on solid CM–glucose (−Ura, −His, and −Trp). Six colonies from each transformation were streaked on CM–glucose (−Ura, −His, and −Trp), restreaked on the test plates, and observed for 3 days. The interaction trap testing and controls (e.g., checking that the pB series did not autoactivate the reporter genes and were able to repress the LexA operators) were as described by Golemis et al. (1994), except that the tests were carried out on plates instead of using the liquid β-galactosidase filter assay. A positive interaction was interpreted as those transformants that turned blue on CM–galactose-X-gal (−Ura, −His, and −Trp) and were able to grow on CM–galactose (−Ura, −His, and −Trp) without leucine.
Construction of the PBRN1 strain
Introduction of the amber mutation was carried out in two steps. First, we cloned into the thermosensible pSC101-based vector pFC13 (Cornet et al. 1994) the rne gene in which the first tyrosine codon (TAT), encoding amino acid 25 of RNase E, was changed to an amber stop codon (TAG). Then, we used this plasmid to introduce the tyr25am mutation on the chromosome by homologous recombination.
Introduction of the tyr25am mutation in the rne gene and cloning into the pFC13 vector was carried out in several steps. First, a BamHI–EcoRV rne fragment containing the amber mutation was constructed by PCR-directed mutagenesis. The downstream mutagenic primer was rne627 (lowercase c creates the amber mutation TAG, Table 3). The upstream primer was rne215. The former hybridizes near the RNase E processing site in the 5′ UTR rne gene and creates a BamHI site (lowercase letters, Table 3) at the 5′ end of the PCR product. Amplification was carried out with Pfu DNA polymerase as recommended by Stratagene by use of pET–rne1 as the template. This PCR product was cloned into the SmaI site of pUC18 and sequenced. The plasmid pUC–rne1, constructed as described above (with the NdeI–HindIII rne fragment in the opposite sense of Plac), was digested by BamHI and EcoRV, and the excised region was replaced by the BamHI–EcoRV fragment carrying the tyr25am mutation. A BamHI–HindIII rne fragment was purified and ligated together with the HindIII–XhoI carboxy-terminal rne fragment (from the pGEM–rneD plasmid) into the pFC13 vector digested by BamHI–XhoI. The resulting plasmid named pFC–rne was used to introduce the amber mutation on the chromosome of XA103a strain by homologous recombination. This strain has a thermosensible phenotype provided by the ams allele and carries a constitutive supF suppressor. After transformation, the selection steps were as described previously (Cornet et al. 1994). Only recombinants carrying the amber allele were able to grow at 42°C. The tyr25am mutation was transferred into PB4144 strain by P1 transduction by use of the Tn10 marker that is linked to the rne loci. A tetracycline-resistant transductant with a thermosensible phenotype at 42°C was selected and the presence of the amber mutation was confirmed by PCR amplification and sequencing. A recA− derivative of this strain was constructed by P1 transduction by use of GY8382 strain as the donor (Sommer et al. 1998). The recA− phenotype was checked by sensitivity to UV irradiation. This strain was named PBRN1.
Construction of deletions within the rne gene
The pAM–rne plasmid was constructed as follows. A PstI–PstI chromosomal fragment encoding the entire RNase E protein was cloned into the PstI site of pGB2-derived plasmid pAM238 (Churchward et al. 1984) containing a polylinker upstream of the lac promoter (J.-P. Bouché, unpubl.) and the ampicillin resistance gene. All rne internal deletions were generated by use of inverse PCR (Hemsley et al. 1989). In each PCR reaction, the pAM–rne plasmid was amplified with primer sets located back to back on the rne sequence. The upstream oligonucleotides used to construct RneΔ10 and RneΔ11 were rne3069 and rne2064, respectively, and the downstream oligonucleotide was rne3676 (Table 3). For RneΔ12, the oligonucleotides were rne2064 and rne2422. For RneΔ15, the oligonucleotides were rne1192 and rne3676. The upstream primers for RneΔ13, RneΔ14, RneΔ16, and RneΔ18 were rne2064, rne2445, rne2754, and rne2721, respectively. The same downstream primer, rne3076, was used for these deletions. PCR to amplify long templates was performed as in Cheng et al. (1994) except that Taq (Appligene) and Pfu DNA polymerase (Stratagene) were used in a ratio of 2.5–0.05 units. PCR products were gel purified, 3′-end blunted and 5′-end phosphorylated with T4 DNA polymerase and T4 polynucleotide kinase (Biolabs), self-ligated, and used to transform DH5α. Two independent clones, containing the appropriate deletion, complementing the PBRN1 strain at 42°C and expressing a mutant protein of the correct size, were further analyzed in the coimmunopurification experiments. In each case, both clones gave the same result and only one is presented in Figure 3.
Immunopurification of the RNA degradosome from the PBRN1 strain
Cultures (100 ml) were grown at 42°C to stationary phase. Partially purified protein extracts were prepared as described above for the overexpressed degradosomal proteins. The ammonium sulfate pellets were suspended in 200 μl of Buffer A (with 300 mm NaCl throughout this protocol) (Carpousis et al. 1994). The protein concentration was estimated by the optical density at 280 nm.
Anti-RNase E or anti-PNPase antibodies were cross-linked to protein A–Sepharose (Pharmacia) as follows. All incubations were at 4°C with gentle agitation. The beads were incubated for 1 hr with the polyclonal rabbit antiserum, washed three times with 10 vol of CLB (cross-linking buffer–20 mm Na2HPO4, 5 mm NaH2PO4, 0.2 mm NaCl, 0.5 mm EDTA), suspended in 5 vol of CLB containing 1% glutaraldehyde and incubated for another hour, and washed three times with 10 vol of CLB and twice with 10 vol of buffer A. These beads were then incubated with 2 mg of protein for 2 hr, washed three times with buffer A, suspended in 200 μl of SDS-PAGE loading buffer without β-mercaptoethanol, and heated for 15 min at 55°C. These conditions prevented the release of the antibodies. The beads were removed by centrifugation, and the supernatants were boiled for 3 min with β-mercaptoethanol to denature the proteins completely. Silver staining was performed as in Monod et al. (1997).
Mutagenesis of the RhlB DEAD motif to DKAD
The mutation was created by oligonucleotide-directed mutagenesis. A PCR product encoding amino acids 159–383 of the RhlB protein was generated with primers rhlB1282R and rhlB607K (see Table 3; the lowercase a shows the substitution that creates the E166K mutation). After cloning, this product was sequenced then substituted for the corresponding BstXI–BglII fragment in pET–rhlB.
Acknowledgments
For helpful technical advice, we thank Paul Watt (yeast two-hybrid system) and Jean-Yves Bouet (E. coli strain construction). We also thank Marc Dreyfus, Pascal Lopez, Ben Luisi, Kenneth McDowall, and Martyn Symmons for discussions about various aspects of this research; Tamas Kiss for critically reading the manuscript; and Pat Dennis, who during the early stages of this work, sequenced the insert in pET11–rhlB and found a PCR induced mutation that was subsequently corrected. C.F.H. is a Howard Hughes International Research Scholar. The research in Toulouse was supported by the CNRS. Additional funding was from the Association pour la Recherche sur le Cancer, the Conseil Régional Midi Pyrénées, the Biotechnology and Biological Sciences Research Council, the Imperial Cancer Research Fund, and the European Union.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
It has been demonstrated, using a Flag-tagged protein, that the carboxy-terminal half of RNase E (residues 530–1061) is necessary and sufficient for the interactions with RhlB, enolase, and PNPase (V.R. Kaberdin, A. Miczak, J.S. Jakobsen, S. Lin-Chao, K.J. McDowall, and A. von Gabain. 1998. Proc. Natl. Acad. Sci., in press).
Footnotes
E-MAIL Carpousi@ibcg.biotoul.fr; FAX (033) 05.61.33.58.86.
References
- Alberts B. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell. 1998;92:291–294. doi: 10.1016/s0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- Anderson JSJ, Parker R. The 3′ to 5′ degradation of yeast mRNA is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J. 1998;17:1497–1506. doi: 10.1093/emboj/17.5.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belasco JG, Higgins CF. Mechanisms of mRNA decay in bacteria: A perspective. Gene. 1988;72:15–23. doi: 10.1016/0378-1119(88)90123-0. [DOI] [PubMed] [Google Scholar]
- Blum E, Py B, Carpousis AJ, Higgins CF. Polyphosphate kinase is a component of the Escherichia coli RNA degradosome. Mol Microbiol. 1997;26:387–398. doi: 10.1046/j.1365-2958.1997.5901947.x. [DOI] [PubMed] [Google Scholar]
- Braun F, Hajnsdorf E, Régnier P. Polynucleotide phosphorylase is required for the rapid degradation of the RNase E-processed rpsO mRNA of Escherichia coli devoid of its 3′ hairpin. Mol Microbiol. 1996;19:997–1005. doi: 10.1046/j.1365-2958.1996.440971.x. [DOI] [PubMed] [Google Scholar]
- Carpousis AJ, Van Houwe G, Ehretsmann C, Krisch HM. Copurification of E. coli RNase E and PNPase: Evidence for a specific association between two enzymes important in RNA processing and degradation. Cell. 1994;76:889–900. doi: 10.1016/0092-8674(94)90363-8. [DOI] [PubMed] [Google Scholar]
- Casarégola S, Jacq A, Laoudj D, McGurk G, Margarson S, Tempête M, Norris V, Holland IB. Cloning and analysis of the entire E. coli ams gene. Ams is identical to hmp1 and encodes a 114 kDa protein that migrates as a 180 kDa protein. J Mol Biol. 1992;228:30–40. doi: 10.1016/0022-2836(92)90489-7. [Corrigendum 238: 867.] [DOI] [PubMed] [Google Scholar]
- Cheng S, Fockler C, Barnes WM, Higuchi R. Effective amplification of long targets from cloned inserts and human genomic DNA. Proc Natl Acad Sci. 1994;91:5695–5699. doi: 10.1073/pnas.91.12.5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchward G, Belin D, Nagamine Y. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene. 1984;31:165–171. doi: 10.1016/0378-1119(84)90207-5. [DOI] [PubMed] [Google Scholar]
- Claverie-Martin F, Diaz-Torres MR, Yancey SD, Kushner SE. Analysis of the altered mRNA stability (ams) gene from E. coli. J Biol Chem. 1991;266:2843–2851. [PubMed] [Google Scholar]
- Cohen SN. Surprises at the 3′ end of prokaryotic RNA. Cell. 1995;80:829–832. doi: 10.1016/0092-8674(95)90284-8. [DOI] [PubMed] [Google Scholar]
- Cormack RS, Genereaux JL, Mackie GA. RNase E activity is conferred by a single polypeptide: Overexpression, purification, and properties of the ams/rne hmp 1 gene product. Proc Natl Acad Sci. 1993;90:9006–9010. doi: 10.1073/pnas.90.19.9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornet F, Mortier I, Patte J, Louarn JM. Plasmid pSC101 harbors a recombination site, psi, which is able to resolve plasmid multimers and to substitute for the analogous chromosomal Escherichia coli site dif. J Bact. 1994;176:3188–3195. doi: 10.1128/jb.176.11.3188-3195.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz J, Kressler D, Tollervey D, Linder P. Dob1p (Mtr4p) is a putative ATP-dependent RNA helicase required for the 3′ end formation of 5.8s rRNA in Saccharomyces cerevisiae. EMBO J. 1998;17:1128–1140. doi: 10.1093/emboj/17.4.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn SD. Effects of the modification of transfer buffer composition and the renaturation of proteins in gels on the recognition of proteins on Western blots by monoclonal antibodies. Anal Biochem. 1986;157:144–153. doi: 10.1016/0003-2697(86)90207-1. [DOI] [PubMed] [Google Scholar]
- Ehretsmann C, Carpousis AJ, Krisch HM. Messenger RNA degradation in procaryotes. FASEB J. 1992;6:3186–3192. doi: 10.1096/fasebj.6.13.1397840. [DOI] [PubMed] [Google Scholar]
- Gascuel O, Golmard JL. A simple method for predicting the secondary structure of globular proteins: Implications and accuracy. Comput Appl Biosci. 1988;4:357–365. doi: 10.1093/bioinformatics/4.3.357. [DOI] [PubMed] [Google Scholar]
- Gegenheimer P. Preparation of extracts from plants. Methods Enzymol. 1990;182:174–193. doi: 10.1016/0076-6879(90)82016-u. [DOI] [PubMed] [Google Scholar]
- Gibson TJ, Thompson JD. Detection of dsRNA-binding domains in RNA helicase A and Drosophila maleless: Implications for monomeric RNA helicases. Nucleic Acids Res. 1994;22:2552–2556. doi: 10.1093/nar/22.13.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz D, St. Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golemis EA, Gyuris J, Brent R. Interaction trap/two hybrid system to identify interacting proteins. In: Ausubel FM, editor. Current protocols in molecular biology. 2, 13.14. New York, NY: Wiley; 1994. [DOI] [PubMed] [Google Scholar]
- Gyuris J, Golemis EA, Chertkov H, Brent R. Cdi1, a human G1 and S phase protein phophatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- Hayes R, Kudla J, Schuster G, Gabay L, Maliga P, Gruissem W. Chloroplast mRNA 3′-end processing by a high molecular encoded RNA binding proteins. EMBO J. 1996;15:1132–1141. [PMC free article] [PubMed] [Google Scholar]
- Hemsley A, Arnheim N, Toney MD, Cortopassi G, Galas DJ. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 1989;17:6545–6551. doi: 10.1093/nar/17.16.6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Liao J, Cohen SN. Poly(A)- and poly(U)-specific RNA 3′ tail shortening by E. coli ribonuclease E. Nature. 1998;391:99–102. doi: 10.1038/34219. [DOI] [PubMed] [Google Scholar]
- Jain C, Belasco JG. RNase E auto-regulates its synthesis by controlling the degradation rate of its own mRNA in Escherichia coli: Unusual sensitivity of the rne transcript to RNase E activity. Genes & Dev. 1995;9:84–96. doi: 10.1101/gad.9.1.84. [DOI] [PubMed] [Google Scholar]
- Kalman M, Murphy H, Cashel M. rhlB, a new Escherichia coli K-12 gene with an RNA helicase-like protein sequence motif, one of at least five such possible genes in a prokaryote. New Biologist. 1991;3:886–895. [PubMed] [Google Scholar]
- Kido M, Yamanaka K, Mitani T, Niki H, Ogura T, Hiraga S. RNase E polypeptides lacking a carboxyl-terminal half suppress a mukB mutation in Escherichia coli. J Bacteriol. 1996;178:3917–3925. doi: 10.1128/jb.178.13.3917-3925.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Miki T, Hiraga S, Nagata T, Yura T. Conditionally lethal amber mutations in the dnaA region of the Escherichia coli chromosome that affect chromosome replication. J Bacteriol. 1979;140:825–834. doi: 10.1128/jb.140.3.825-834.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littauer UZ, Soreq H. Polynucleotide phosphorylase. Enzymes. 1982;15:517–533. [Google Scholar]
- Mackie GA, Genereaux JL, Masterman SK. Modulation of the activity of RNase E in vitro by RNA sequences and secondary structures 5′ to cleavage sites. J Biol Chem. 1997;272:609–616. [PubMed] [Google Scholar]
- Margossian SP, Li H, Zassenhaus HP, Butow RA. The DExH box protein Suv3p is a component of a yeast mitochondrial 3′ to 5′ exoribonuclease that suppresses group I intron toxicity. Cell. 1996;84:199–209. doi: 10.1016/s0092-8674(00)80975-7. [DOI] [PubMed] [Google Scholar]
- McDowall KJ, Cohen SN. The N-terminal domain of the rne gene product has RNase E activity and is non-overlapping with the arginine-rich RNA-binding site. J Mol Biol. 1996;255:349–355. doi: 10.1006/jmbi.1996.0027. [DOI] [PubMed] [Google Scholar]
- Miczak A, Kaberdin VR, Wei CL, Lin-Chao S. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc Natl Acad Sci. 1996;93:3865–3869. doi: 10.1073/pnas.93.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milburn SC, Hershey JWB, Davies MV, Kelleher K, Kaufman RJ. Cloning and expression of eukaryotic initiation factor 4B cDNA: Sequence determination identifies a common RNA recognition motif. EMBO J. 1990;9:2783–2790. doi: 10.1002/j.1460-2075.1990.tb07466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P, Petfalski E, Shevchenko A, Mann M, Tollervey D. The exosome: A conserved eukaryotic RNA processing complex containing multiple 3′-5′ exoribonucleases. Cell. 1997;91:457–466. doi: 10.1016/s0092-8674(00)80432-8. [DOI] [PubMed] [Google Scholar]
- Monod C, Repoila F, Kutateladze M, Tétart F, Krisch HM. The genome of the pseudo T-even bacteriophages, a diverse group that resembles T4. J Mol Biol. 1997;267:237–249. doi: 10.1006/jmbi.1996.0867. [DOI] [PubMed] [Google Scholar]
- Mudd EA, Krisch HM, Higgins CF. RNase E, and endoribonuclease, has a general role in the chemical decay of Escherichia coli mRNA: Evidence that rne and ams are the same genetic locus. Mol Microbiol. 1990;4:2127–2135. doi: 10.1111/j.1365-2958.1990.tb00574.x. [DOI] [PubMed] [Google Scholar]
- Nierlich DP, Murakawa GJ. The decay of bacterial messenger RNA. Prog Nucleic Acids Res Mol Biol. 1996;52:153–216. doi: 10.1016/s0079-6603(08)60967-8. [DOI] [PubMed] [Google Scholar]
- Pause A, Sonenberg N. Mutational analysis of a DEAD box RNA helicase: The mammalian translation initiation factor eIF4A. EMBO J. 1992;11:2643–2654. doi: 10.1002/j.1460-2075.1992.tb05330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portier C. Quaternary structure of polynucleotide phosphorylase from E. coli: Evidence for a complex between two types of polypeptide chains. Eur J Biochem. 1975;55:573–585. doi: 10.1111/j.1432-1033.1975.tb02194.x. [DOI] [PubMed] [Google Scholar]
- Py B, Causton H, Mudd EA, Higgins CF. A protein complex mediating mRNA degradation in Escherichia coli. Mol Microbiol. 1994;14:717–729. doi: 10.1111/j.1365-2958.1994.tb01309.x. [DOI] [PubMed] [Google Scholar]
- Py B, Higgins CF, Krisch HM, Carpousis AJ. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature. 1996;381:169–172. doi: 10.1038/381169a0. [DOI] [PubMed] [Google Scholar]
- Raynal LC, Krisch HM, Carpousis AJ. Bacterial poly(A) polymerase: An enzyme that modulates RNA stability. Biochimie. 1996;78:390–398. doi: 10.1016/0300-9084(96)84745-6. [DOI] [PubMed] [Google Scholar]
- Rozen F, Edery I, Meerovitch K, Dever TE, Merrick WC, Sonenberg N. Bidirectional RNA helicase activity of eucaryotic translation initiation factors 4A and 4B. Mol Cell Biol. 1990;10:1134–1144. doi: 10.1128/mcb.10.3.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schmid SR, Linder P. D-E-A-D protein family of putative RNA helicase. Mol Microbiol. 1992;6:283–292. doi: 10.1111/j.1365-2958.1992.tb01470.x. [DOI] [PubMed] [Google Scholar]
- Sommer S, Boudsocq F, Devoret R, Bailone A. Specific RecA amino acid changes affect RecA-UmuD′C interaction. Mol Microbiol. 1998;28:281–292. doi: 10.1046/j.1365-2958.1998.00803.x. [DOI] [PubMed] [Google Scholar]
- Soreq H, Littauer UZ. Purification and characterization of polynucleotide phosphorylase from Escherichia coli. J Biol Chem. 1977;252:6885–6888. [PubMed] [Google Scholar]
- Spring TG, Wold F. The purification and characterization of Escherichia coli enolase. J Biol Chem. 1971;246:6797–6802. [PubMed] [Google Scholar]
- Taraseviciene L, Björk GR, Uhlin BE. Evidence for an RNA binding region in the Escherichia coli processing endoribonuclease RNase E. J Biol Chem. 1995;270:26391–26398. doi: 10.1074/jbc.270.44.26391. [DOI] [PubMed] [Google Scholar]
- Xu F, Cohen SN. RNA degradation in Escherichia coli regulated by 3′ adenylation and 5′ phosphorylation. Nature. 1995;374:180–183. doi: 10.1038/374180a0. [DOI] [PubMed] [Google Scholar]