

Abstract
The inference of genetic regulatory networks from global measurements of gene expressions is an important problem in computational biology. Recent studies suggest that such dynamical molecular systems are poised at a critical phase transition between an ordered and a disordered phase, affording the ability to balance stability and adaptability while coordinating complex macroscopic behavior. We investigate whether incorporating this dynamical system-wide property as an assumption in the inference process is beneficial in terms of reducing the inference error of the designed network. Using Boolean networks, for which there are well-defined notions of ordered, critical, and chaotic dynamical regimes as well as well-studied inference procedures, we analyze the expected inference error relative to deviations in the networks' dynamical regimes from the assumption of criticality. We demonstrate that taking criticality into account via a penalty term in the inference procedure improves the accuracy of prediction both in terms of state transitions and network wiring, particularly for small sample sizes.
1. Introduction
The execution of various developmental and physiological processes in cells is carried out by complex biomolecular systems. Such systems are dynamic in that they are able to change states in response to environmental cues and exhibit multiple steady states, which define different cellular functional states or cell types.
The massively parallel dynamics of complex molecular networks furnish the cell with the ability to process information from its environment and mount appropriate responses. To be able to stably execute cellular functions in a variable environment while being responsive to specific changes in the environment, such as the activation of immune cells upon exposure to pathogens or their components, the cell needs to strike a balance between robustness and adaptability.
Theoretical considerations and computational studies suggest that many types of complex dynamical systems can indeed strike such an optimal balance, under a variety of criteria, when they are operating close to a critical phase transition between an ordered and a disordered dynamical regime [1–3]. There is also accumulating evidence that living systems, as manifestations of their underlying networks of molecular interactions, are poised at the critical boundary between an organized and a disorganized state, indicating that cellular information processing is optimal in the critical regime, affording the cell with the ability to exhibit complex coordinated macroscopic behavior [4–8]. Studies of human brain oscillations [9], computer network traffic and the Internet [10,11], financial markets [12], forest fires [13], neuronal networks supporting our senses [14], and biological macroevolution have also revealed critical dynamics [15].
A key goal in systems biology research is to characterize the molecular mechanisms governing specific cellular behaviors and processes. This typically entails selecting a model class for representing the system structure and state dynamics, followed by the application of computational or statistical inference procedures for revealing the model structure from measurement data [16]. Multiple types of data can be potentially used for elucidating the structure of molecular networks, such as transcriptional regulatory networks, including genome wide transcriptional profiling with DNA microarrays or other high-throughput technologies, chromatin immunoprecipitation-on-chip (ChIP-on-chip) for identifying DNA sequences occupied by specific DNA binding proteins, computational predictions of transcription factor binding sites based on promoter sequence analysis, and other sources of evidence for molecular interactions [17,18]. The inference of genetic networks is particularly challenging in the face of small sample sizes, particularly because the number of variables in the system (e.g., genes) typically greatly outnumbers the number of observations. Thus, estimates of the errors of a given model, which themselves are determined from the measurement data, can be highly variable and untrustworthy.
Any prior knowledge about the network structure, architecture, or dynamical rules is likely to improve the accuracy of the inference, especially in a small sample size scenario. If biological networks are indeed critical, a key question is whether this knowledge can be used to improve the inference of network structure and dynamics from measurements. We investigated this question using the class of Boolean networks as models of genetic regulatory networks.
Boolean networks and the more general class of probabilistic Boolean networks are popular approaches for modeling genetic networks, as these model classes capture multivariate nonlinear relationships between the elements of the system and are capable of exhibiting complex dynamics [5,16,19–21]. Boolean network models have been constructed for a number of biomolecular systems, including the yeast cell cycle [22,23], mammalian cell cycle [24], Drosophila segment polarity network [25], regulatory networks of E. coli metabolism [26], and Arabidopsis flower morphogenesis [27–29].
At the same time, these model classes have been studied extensively regarding the relationships between their structure and dynamics. Particularly in the case of Boolean networks, dynamical phase transitions from the ordered to the disordered regime and the critical phase transition boundary have been characterized analytically for random ensembles of networks [30–34]. This makes these models attractive for investigating the relationships between structure and dynamics [35,36].
In particular, the so-called average sensitivity was shown to be an order parameter for Boolean networks [31]. The average sensitivity, which can be computed directly from the Boolean functions specifying the update rules (i.e., state transitions) of the network, measures the average response of the system to a minimal transient perturbation and is equivalent to the Lyapunov exponent [33]. There have been a number of approaches for inferring Boolean and probabilistic Boolean networks from gene expression measurement data [20,21,37–44].
We address the relationship between the dynamical regime of a network, as measured by the average sensitivity, and the inference of the network from data. We study whether the assumption of criticality, embedded in the inference objective function as a penalty term, improves the inference of Boolean network models. We find that for small sample sizes the assumption is beneficial, while for large sample sizes, the performance gain decreases gradually with increasing sample size. This is the kind of behavior that one hopes for when using penalty terms.
This paper is organized as follows. In Section 2, we give a brief definition of Boolean Networks and the concept of sensitivity. Then in Section 3, three measures used in this paper to evaluate the performance of the predicted networks are introduced, and a theoretical analysis of the relationship between the expected error and the sensitivity deviation is presented. Based on this analysis, an objective function is proposed to be used for the inference process in Section 4, while the simulation results are presented in Section 5.
2. Background and Definitions
2.1. Boolean Networks
A Boolean network 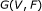 is defined by a set of nodes
is defined by a set of nodes 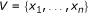 ,
, 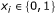 and a set of Boolean functions
and a set of Boolean functions 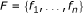 ,
, 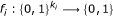 . Each node
. Each node 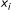 represents the expression state of the gene
represents the expression state of the gene 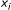 , where
, where 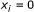 means that the gene is OFF, and
means that the gene is OFF, and 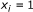 means it is ON. Each Boolean function
means it is ON. Each Boolean function 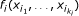 with
with 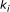 specific input nodes is assigned to node
specific input nodes is assigned to node 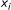 and is used to update its value. Under the synchronous updating scheme, all genes are updated simultaneously according to their corresponding update functions. The network's state at time
and is used to update its value. Under the synchronous updating scheme, all genes are updated simultaneously according to their corresponding update functions. The network's state at time  is represented by a vector
is represented by a vector 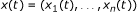 and, in the absence of noise, the system transitions from state to state in a deterministic manner.
and, in the absence of noise, the system transitions from state to state in a deterministic manner.
2.2. Sensitivity
The activity of gene 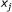 in function
in function 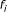 is defined as
is defined as
| (1) |
where 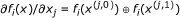 is the partial derivative of
is the partial derivative of 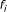 with respect to
with respect to 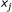 ,
,  is addition modulo 2, and
is addition modulo 2, and 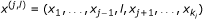 ,
, 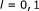 [31]. Note that the activity is equivalent to the expectation of the partial derivative with respect to the uniform distribution. Since the partial derivative is itself a Boolean function, its expectation is equal to the probability that a change in the
[31]. Note that the activity is equivalent to the expectation of the partial derivative with respect to the uniform distribution. Since the partial derivative is itself a Boolean function, its expectation is equal to the probability that a change in the 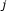 th input causes a change in the output of the function, and hence the activity is a number between zero and one. The average sensitivity of a function
th input causes a change in the output of the function, and hence the activity is a number between zero and one. The average sensitivity of a function 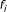 equals the sum of the activities of its input variables:
equals the sum of the activities of its input variables:
| (2) |
In the context of random Boolean networks (RBNs), which are frequently used to study dynamics of regulatory network models, another important parameter is the bias  of a function
of a function  , which is defined to be the probability that the function takes on the value 1. A random Boolean function with bias
, which is defined to be the probability that the function takes on the value 1. A random Boolean function with bias  can be generated by flipping a
can be generated by flipping a  -biased coin
-biased coin 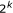 times and filling in the truth table. In other words, the truth table is a realization of
times and filling in the truth table. In other words, the truth table is a realization of 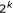 independent Bernoulli (
independent Bernoulli ( ) random variables. For a function
) random variables. For a function 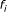 with bias
with bias 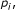 the expectation of its average sensitivity is
the expectation of its average sensitivity is
| (3) |
The sensitivity of a Boolean network is then defined as
| (4) |
Sensitivity is in fact a global dynamical parameter that captures how a one-bit perturbation spreads throughout the network and its expectation under the random Boolean network model is equivalent to the well-known phase transition curve [31].
3. Error Analysis
3.1. Performance Measures
There are several ways to measure the performance of an inference method by comparing the state transitions, wiring, or sensitivities with the original network. In this paper, we will use three measures that are described below.
3.1.1. The State Transition Error
This quantity generally shows the fraction of outputs that are incorrectly predicted, and it can be defined as
| (5) |
where 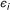 denotes the normalized error of the predicted function
denotes the normalized error of the predicted function 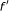 . Additionally,
. Additionally, 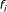 and
and 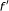 are extended such that they are functions of all the variables (instead of
are extended such that they are functions of all the variables (instead of 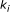 variables) by adding fictitious (i.e., dummy) variables.
variables) by adding fictitious (i.e., dummy) variables.
3.1.2. The Receiver Operating Characteristic (ROC)
This measurement has been widely used in classification problems. An ROC space is defined by the false positive ratio (FPR) and the true positive ratio (TPR) plotted on the  - and
- and  -axes, respectively, which depicts the relative tradeoffs between true positives and false positives. The FPR and TPR are defined as
-axes, respectively, which depicts the relative tradeoffs between true positives and false positives. The FPR and TPR are defined as
| (6) |
where TP and FP represent true positive and false positive instances, respectively, while  and
and  represent the total positive and negative instances, respectively. We will use the ROC distributions to evaluate the accuracy of "wiring" (i.e., the specific input nodes assigned to each node) for the inferred network.
represent the total positive and negative instances, respectively. We will use the ROC distributions to evaluate the accuracy of "wiring" (i.e., the specific input nodes assigned to each node) for the inferred network.
3.1.3. The Sensitivity Error
The sensitivity error measures the deviation in the sensitivity of a predicted network and is defined as
| (7) |
where 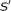 is the sensitivity of the predicted network.
is the sensitivity of the predicted network.
3.2. Analysis of Expected Error
All Boolean networks with a fixed number of genes  can be grouped into different families according to the network's sensitivity. Assuming that
can be grouped into different families according to the network's sensitivity. Assuming that 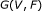 and
and 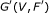 are the original network and another random network,
are the original network and another random network,  and
and 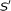 are their sensitivities, respectively, and
are their sensitivities, respectively, and 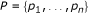 and
and 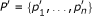 are the biases with which functions
are the biases with which functions 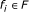 and
and 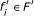 are generated. Let
are generated. Let 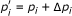 . The expectation of the state transition error between them can be written as
. The expectation of the state transition error between them can be written as
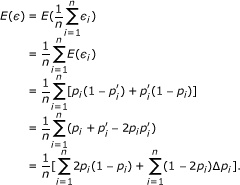 |
(8) |
Using the relationship between sensitivity and bias in Section 2.2, we have
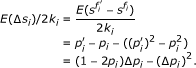 |
(9) |
Then,
| (10) |
If we further assume that both networks' connectivity is constant, 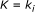 (
(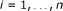 ), then
), then
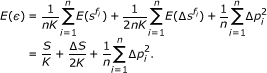 |
(11) |
This means that the expectation of the state transition error 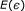 generally depends on the original network's connectivity
generally depends on the original network's connectivity  , its sensitivity
, its sensitivity  , the sensitivity deviation
, the sensitivity deviation 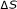 , and the mean quadratic terms of the bias deviation
, and the mean quadratic terms of the bias deviation 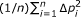 .
.
(1) If 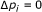 , then
, then 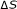 will be 0. In this case, each function
will be 0. In this case, each function 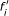 keeps the same bias with that of the original network, and then
keeps the same bias with that of the original network, and then
| (12) |
(2) If 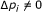 and
and 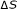 is 0, the predicted network still stays in the same sensitivity class, and
is 0, the predicted network still stays in the same sensitivity class, and
| (13) |
(3) If 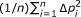 is relatively small compared with
is relatively small compared with 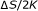 , we can treat it as a constant
, we can treat it as a constant  . Then,
. Then,
| (14) |
In this case, 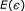 will have a linear relationship with
will have a linear relationship with 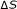 . This indicates that
. This indicates that 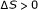 will surely introduce additional error
will surely introduce additional error 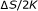 . Our simulations indicate that the inference method we use (best-fit, see below) yields a network with
. Our simulations indicate that the inference method we use (best-fit, see below) yields a network with 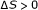 in most cases.
in most cases.
4. Inference Method
To infer a Boolean network, for each target node we need to apply some optimization criterion to each set of input variables and Boolean function on those input variables and then choose the variable set and corresponding Boolean function that minimizes the objective function. The first step in our proposed procedure is to find variable sets and Boolean functions that provide good target prediction. Based upon time-series observations, given a target node 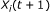 and an input-node vector
and an input-node vector 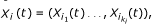 the best predictor,
the best predictor, 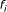 , minimizes the error,
, minimizes the error, 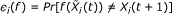 , among all possible predictors
, among all possible predictors  . Finding the best predictor for a given node means finding the minimal error among all Boolean functions over all input-variable combinations. We consider three variable combinations. Since we will optimize via an objective function containing a sensitivity penalty term, we will select a collection of input-variable sets and select the minimal-error Boolean function over each input-variable set. This is accomplished by using the plugin (resubstitution) estimate of the error
. Finding the best predictor for a given node means finding the minimal error among all Boolean functions over all input-variable combinations. We consider three variable combinations. Since we will optimize via an objective function containing a sensitivity penalty term, we will select a collection of input-variable sets and select the minimal-error Boolean function over each input-variable set. This is accomplished by using the plugin (resubstitution) estimate of the error 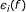 , which is given by the number of times
, which is given by the number of times 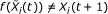 in the data divided by the number of times, the pair
in the data divided by the number of times, the pair 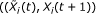 is observed in the data. This procedure is equivalent to the best-fit extension method [45]. We make use of an efficient algorithm for solving the best-fit extension problem and finding all functions having error smaller than a given threshold [37]. We then select the four best variable sets and corresponding Boolean functions as candidates. The limitation of four candidates is based on computational considerations; in principle, there is no such limitation.
is observed in the data. This procedure is equivalent to the best-fit extension method [45]. We make use of an efficient algorithm for solving the best-fit extension problem and finding all functions having error smaller than a given threshold [37]. We then select the four best variable sets and corresponding Boolean functions as candidates. The limitation of four candidates is based on computational considerations; in principle, there is no such limitation.
Because we have a small data sample, if we were to use the resubstitution error estimates employed for variable selection as error estimates for the best Boolean functions, we would expect optimistic estimates. Hence, for each selected variable set, we estimate the error of the corresponding Boolean function via the .632 bootstrap [46,47]. A bootstrap sample consists of  equally likely draws with replacement from the original sample consisting of
equally likely draws with replacement from the original sample consisting of  data pairs
data pairs 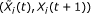 . For the zero-bootstrap estimator,
. For the zero-bootstrap estimator, 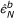 , the function is designed on the bootstrap sample and tested on the points left out, this is done repeatedly, and the bootstrap estimate is the average error made on the left-out points.
, the function is designed on the bootstrap sample and tested on the points left out, this is done repeatedly, and the bootstrap estimate is the average error made on the left-out points. 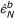 tends to be a high-biased estimator of the true error, since the number of points available for design is on average only 0.632
tends to be a high-biased estimator of the true error, since the number of points available for design is on average only 0.632  . The .632 bootstrap estimator attempts to correct this bias via a weighted average,
. The .632 bootstrap estimator attempts to correct this bias via a weighted average,
| (15) |
where 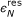 is the original resubstitution estimate.
is the original resubstitution estimate.
We summarize the procedure as follows.
(1) For each three-variable set  , do the following.
, do the following.
(i) Compute the resubstitution errors of all Boolean functions using the full sample data set.
(ii) Choose the Boolean function, 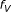 , possessing the lowest resubstitution error as the corresponding function for
, possessing the lowest resubstitution error as the corresponding function for  .
.
(iii) Bootstrap the sample and compute the zero-bootstrap error estimate for 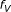 .
.
(iv) Compute the .632 bootstrap error estimate for 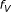 using the computed resubstitution and zero-bootstrap estimates.
using the computed resubstitution and zero-bootstrap estimates.
(2) Select the four input-variable sets whose corresponding functions possess the lowest .632 bootstrap estimates.
This procedure is the same as the one used in [48] to evaluate the impact of different error estimators on feature-set ranking. It was demonstrated there that the bootstrap tends to outperform cross-validation methods in choosing good feature sets. While it was also observed that bolstering tends to be slightly better than bootstrap, bolstering cannot be applied in discrete settings, so it is not a viable option in our context.
Motivated by the analysis in Section 3, we refine the inference process by incorporating the sensitivity. We construct an objective function
| (16) |
where 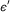 represents the bootstrap-estimated error of the previously selected Boolean function and
represents the bootstrap-estimated error of the previously selected Boolean function and 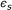 is the sensitivity error. The first item represents the prediction error, while the second represents the "structural error" associated with general network dynamics. Our hypothesis is that a better inference should have a small error in both state transition and sensitivity, and consequently, the value of its objective function
is the sensitivity error. The first item represents the prediction error, while the second represents the "structural error" associated with general network dynamics. Our hypothesis is that a better inference should have a small error in both state transition and sensitivity, and consequently, the value of its objective function 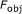 should be minimal. Of the four input-variable sets selected via prediction error for a target node, we use the one with minimal objective function
should be minimal. Of the four input-variable sets selected via prediction error for a target node, we use the one with minimal objective function 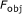 for network construction.
for network construction.
5. Simulation Results
All simulations are performed for random Boolean networks with 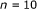 and
and 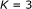 . For a given BN, we randomly generate
. For a given BN, we randomly generate 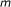 pairs of input and output states. We also consider the effect of noise, with 5% noise added to the output states of each gene by flipping its value with probability 0.05.
pairs of input and output states. We also consider the effect of noise, with 5% noise added to the output states of each gene by flipping its value with probability 0.05.
From the perspective of network inference, performance is best characterized via a distance function between the ground-truth network and the inferred network, more specifically, by the expected distance between the ground-truth and inferred network as estimated by applying the inference to a random sample of ground-truth networks [49]. In our case, we have chosen the normalized state-transition error as the distance between the networks.
First, we investigate the performance of the new method on networks with different sensitivities, 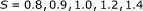 , on sample sizes ranging from 10 to 40. There are total of 200 networks for each value of the sensitivity. The left columns of Figures 1–5 are the histograms of the distribution of the true state-transition error (Section 3.1.1) for both the traditional best-fit method (combined with .632 bootstrap) and the new proposed method. They show that the proposed method reduces this error dramatically in small sample situations. As sample size increases, the performance of both methods becomes closer. The middle columns of Figures 1–5 are the histograms of the distribution of the sensitivity error (Section 3.1.3). As can be seen, the best-fit method usually ends up with a network with larger sensitivity in small sample cases, while the proposed method can find a network operating in the same or nearby dynamic regime. The right columns of Figures 1–5 are the ROC distributions of both methods (Section 3.1.2). The proposed method has approximately the same TPR as the best-fit method but with a lower FPR. This means that the recovered connections will have higher reliability. Figure 6 shows the mean error in state transition and sensitivity under different samples sizes, for zero noise and 5% noise.
, on sample sizes ranging from 10 to 40. There are total of 200 networks for each value of the sensitivity. The left columns of Figures 1–5 are the histograms of the distribution of the true state-transition error (Section 3.1.1) for both the traditional best-fit method (combined with .632 bootstrap) and the new proposed method. They show that the proposed method reduces this error dramatically in small sample situations. As sample size increases, the performance of both methods becomes closer. The middle columns of Figures 1–5 are the histograms of the distribution of the sensitivity error (Section 3.1.3). As can be seen, the best-fit method usually ends up with a network with larger sensitivity in small sample cases, while the proposed method can find a network operating in the same or nearby dynamic regime. The right columns of Figures 1–5 are the ROC distributions of both methods (Section 3.1.2). The proposed method has approximately the same TPR as the best-fit method but with a lower FPR. This means that the recovered connections will have higher reliability. Figure 6 shows the mean error in state transition and sensitivity under different samples sizes, for zero noise and 5% noise.
Figure 1.

Histograms of the true error in state transition and in sensitivity and the ROC distribution for the 1000 random BNs under sample size 10.
Figure 5.

Histograms of the true error in state transition and in sensitivity and the ROC distribution for the 1000 random BNs under sample size 40.
Figure 6.

Mean state transition and sensitivity error for sample sizes ranging from 10 to 40, computed with zero noise and 5% noise.
Figure 2.

Histograms of the true error in state transition and in sensitivity and the ROC distribution for the 1000 random BNs under sample size 15.
Figure 3.

Histograms of the true error in state transition and in sensitivity and the ROC distribution for the 1000 random BNs under sample size 20.
Figure 4.

Histograms of the true error in state transition and in sensitivity and the ROC distribution for the 1000 random BNs under sample size 30.
In practice, we do not know the network sensitivity, so that the assumed value in the inference procedure may not agree with the actual value. Hence, the inference procedure must be robust to this difference. Under the assumption of criticality for living systems, it is natural to set 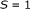 in the inference procedure. Moreover, assuming that a living system remains near the border between order and disorder, the true sensitivity of gene regulatory networks will remain close to 1 under the Boolean formalism. Thus, to investigate robustness, we generated 1000 networks with sensitivities
in the inference procedure. Moreover, assuming that a living system remains near the border between order and disorder, the true sensitivity of gene regulatory networks will remain close to 1 under the Boolean formalism. Thus, to investigate robustness, we generated 1000 networks with sensitivities 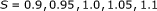 , and then inferred them using
, and then inferred them using 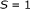 with the proposed method. The mean state transition errors of both methods are shown in Figure 7.
with the proposed method. The mean state transition errors of both methods are shown in Figure 7.
Figure 7.

Mean state transition error of different sensitivity deviation for sample sizes ranging from 10 to 70, computed with zero noise and 5% noise.
When the actual sensitivity is 1, the method helps for small samples and the performances become close for large samples, analogous to Figure 6. When the true network deviates from the modelling assumption, 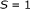 , the proposed method helps for small samples and results in some loss of performance for large samples. This kind of behavior is what one would expect with an objective function that augments the error. In effect, the sensitivity is a penalty term in the objective function that is there to impose constraint on the optimization. In our case, when the true sensitivity is not equal to 1, the sensitivity constraint
, the proposed method helps for small samples and results in some loss of performance for large samples. This kind of behavior is what one would expect with an objective function that augments the error. In effect, the sensitivity is a penalty term in the objective function that is there to impose constraint on the optimization. In our case, when the true sensitivity is not equal to 1, the sensitivity constraint 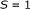 yields smaller sensitivity error than the best-fit method in small sample situations, while the sensitivity error of the best-fit method is smaller for large samples. In sum, the constraint is beneficial for small samples.
yields smaller sensitivity error than the best-fit method in small sample situations, while the sensitivity error of the best-fit method is smaller for large samples. In sum, the constraint is beneficial for small samples.
Finally, the performance of the new method with regard to wiring for small sensitivity deviation is presented in Figure 8. It shows that the new method can achieve the same TPR with a lower FPR under a small sensitivity deviation in small sample situations.
Figure 8.

The distribution of ROC with sensitivity deviation from 0.9 to 1.1 for sample sizes 10, 15, and 20, computed with zero noise and 5% noise.
6. Conclusions
Sensitivity is a global structural parameter of a network which captures the network's operating dynamic behavior: ordered, critical, or chaotic. Recent evidence suggests that living systems operate at the critical phase transition between ordered and chaotic regimes. In this paper, we have proposed a method to use this dynamic information to improve the inference of Boolean networks from observations of input-output relationships. First, we have analyzed the relationship between the expectation of the error and the deviation of sensitivity, showing that these quantities are strongly correlated with each other. Based on this observation, an objective function is proposed to refine the inference approach based on the best-fit method. The simulation results demonstrate that the proposed method can improve the predicted results both in terms of state transitions, sensitivity, and network wiring. The improvement is particularly evident in small sample size settings. As the sample size increases, the performance of both methods becomes similar. In practice, where one does not know the sensitivity of the true network, we have assumed it to be 1, the critical value, and investigated inference performance relative to its robustness to the true sensitivity deviating from 1. For small samples, the kind we are interested in when using such a penalty approach, the proposed method continues to outperform the best-fit method.
For practical applications, one can apply an optimization strategy, such as genetic algorithms, to attain suboptimal solutions instead of the brute force searching strategy used in this paper. As the final chosen function for each gene generally lies within the top three candidates in our simulations, one can just select from a few top candidate functions for each gene instead of using all of the possible 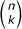 candidates. Finally, it should be noted that the ideas presented here could also be incorporated into other inference methods, such as the ones in [40,41].
candidates. Finally, it should be noted that the ideas presented here could also be incorporated into other inference methods, such as the ones in [40,41].
Contributor Information
Wenbin Liu, Email: wliu@systemsbiology.org.
Harri Lähdesmäki, Email: harri.lahdesmaki@tut.fi.
Edward R Dougherty, Email: edward@ece.tamu.edu.
Ilya Shmulevich, Email: is@ieee.org.
Acknowledgments
Support from NIGMS GM072855 (I.S.), P50-GM076547 (I.S.), NSF CCF-0514644 (E.D.), NCI R01 CA-104620 (E.D.), NSFC under no. 60403002 (W.L.), NSF of Zhejiang province under nos. Y106654 and Y405553 (W.L.) is gratefully acknowledged.
References
- Langton CG. Computation at the edge of chaos: phase transitions and emergent computation. Physica D. 1990;42(1–3):12–37. [Google Scholar]
- Krawitz P, Shmulevich I. Basin entropy in Boolean network ensembles. Physical Review Letters. 2007;98(15) doi: 10.1103/PhysRevLett.98.158701. [DOI] [PubMed] [Google Scholar]
- Packard NH. In: Dynamic Patterns in Complex Systems. Kelso JAS, Mandell AJ, Shlesinger MF, editor. World Scientific, Singapore; 1988. Adaptation towards the edge of chaos; pp. 293–301. [Google Scholar]
- Rämö P, Kesseli J, Yli-Harja O. Perturbation avalanches and criticality in gene regulatory networks. Journal of Theoretical Biology. 2006;242(1):164–170. doi: 10.1016/j.jtbi.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Kauffman SA. The Origins of Order: Self-Organization and Selection in Evolution. Oxford University Press, New York, NY, USA; 1993. [Google Scholar]
- Shmulevich I, Kauffman SA, Aldana M. Eukaryotic cells are dynamically ordered or critical but not chaotic. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(38):13439–13444. doi: 10.1073/pnas.0506771102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra R, Villani M, Semeria A. Genetic network models and statistical properties of gene expression data in knock-out experiments. Journal of Theoretical Biology. 2004;227(1):149–157. doi: 10.1016/j.jtbi.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Nykter M, Price ND, Aldana M. et al. Gene expression dynamics in the macrophage exhibit criticality. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(6):1897–1900. doi: 10.1073/pnas.0711525105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkenkaer-Hansen K, Nikouline VV, Palva JM, Ilmoniemi RJ. Long-range temporal correlations and scaling behavior in human brain oscillations. Journal of Neuroscience. 2001;21(4):1370–1377. doi: 10.1523/JNEUROSCI.21-04-01370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde S, Solé RV. Self-organized critical traffic in parallel computer networks. Physica A. 2002;312(3-4):636–648. doi: 10.1016/S0378-4371(02)00872-5. [DOI] [Google Scholar]
- Fukuda K, Takayasu H, Takayasu M. Origin of critical behavior in Ethernet traffic. Physica A. 2000;287(1-2):289–301. doi: 10.1016/S0378-4371(00)00452-0. [DOI] [Google Scholar]
- Lux T, Marchesi M. Scaling and criticality in a stochastic multi-agent model of a financial market. Nature. 1999;397(6719):498–500. doi: 10.1038/17290. [DOI] [Google Scholar]
- Malamud BD, Morein G, Turcotte DL. Forest fires: an example of self-organized critical behavior. Science. 1998;281(5384):1840–1842. doi: 10.1126/science.281.5384.1840. [DOI] [PubMed] [Google Scholar]
- Kinouchi O, Copelli M. Optimal dynamical range of excitable networks at criticality. Nature Physics. 2006;2(5):348–352. doi: 10.1038/nphys289. [DOI] [Google Scholar]
- Sneppen K, Bak P, Flyvbjerg H, Jensen MH. Evolution as a self-organized critical phenomenon. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(11):5209–5213. doi: 10.1073/pnas.92.11.5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmulevich I, Dougherty ER. Genomic Signal Processing. Princeton University Press, Princeton, NJ, USA; 2007. (Princeton Series in Applied Mathematics). [Google Scholar]
- Ramsey SA, Klemm SL, Zak DE. et al. Uncovering a macrophage transcriptional program by integrating evidence from motif scanning and expression dynamics. PLoS Computational Biology. 2008;4(3, e1000021):1–25. doi: 10.1371/journal.pcbi.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lähdesmäki H, Rust AG, Shmulevich I. Probabilistic inference of transcription factor binding from multiple data sources. PLoS ONE. 2008;3(3):e1820. doi: 10.1371/journal.pone.0001820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman S. Homeostasis and differentiation in random genetic control networks. Nature. 1969;224(5215):177–178. doi: 10.1038/224177a0. [DOI] [PubMed] [Google Scholar]
- Shmulevich I, Dougherty ER, Kim S, Zhang W. Probabilistic Boolean networks: a rule-based uncertainty model for gene regulatory networks. Bioinformatics. 2002;18(2):261–274. doi: 10.1093/bioinformatics/18.2.261. [DOI] [PubMed] [Google Scholar]
- Shmulevich I, Dougherty ER, Zhang W. From Boolean to probabilistic Boolean networks as models of genetic regulatory networks. Proceedings of the IEEE. 2002;90(11):1778–1792. doi: 10.1109/JPROC.2002.804686. [DOI] [Google Scholar]
- Li F, Long T, Lu Y, Ouyang Q, Tang C. The yeast cell-cycle network is robustly designed. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(14):4781–4786. doi: 10.1073/pnas.0305937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidich MI, Bornholdt S. Boolean network model predicts cell cycle sequence of fission yeast. PLoS ONE. 2008;3(2):e1672. doi: 10.1371/journal.pone.0001672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauré A, Naldi A, Chaouiya C, Thieffry D. Dynamical analysis of a generic Boolean model for the control of the mammalian cell cycle. Bioinformatics. 2006;22(14):e124–e131. doi: 10.1093/bioinformatics/btl210. [DOI] [PubMed] [Google Scholar]
- Albert R, Othmer HG. The topology of the regulatory interactions predicts the expression pattern of the segment polarity genes in Drosophila melanogaster. Journal of Theoretical Biology. 2003;223(1):1–18. doi: 10.1016/S0022-5193(03)00035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samal A, Jain S. The regulatory network of E. coli metabolism as a Boolean dynamical system exhibits both homeostasis and flexibility of response. BMC Systems Biology. 2008;2, article 21 doi: 10.1186/1752-0509-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Soto C, Padilla-Longoria P, Alvarez-Buylla ER. A gene regulatory network model for cell-fate determination during Arabidopsis thaliana flower development that is robust and recovers experimental gene expression profiles. The Plant Cell. 2004;16(11):2923–2939. doi: 10.1105/tpc.104.021725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza L, Alvarez-Buylla ER. Dynamics of the genetic regulatory network for Arabidopsis thaliana flower morphogenesis. Journal of Theoretical Biology. 1998;193(2):307–319. doi: 10.1006/jtbi.1998.0701. [DOI] [PubMed] [Google Scholar]
- Mendoza L, Thieffry D, Alvarez-Buylla ER. Genetic control of flower morphogenesis in Arabidopsis thaliana: a logical analysis. Bioinformatics. 1999;15(7-8):593–606. doi: 10.1093/bioinformatics/15.7.593. [DOI] [PubMed] [Google Scholar]
- Derrida B, Pomeau Y. Random networks of automata: a simple annealed approximation. Europhysics Letters. 1986;1(2):45–49. doi: 10.1209/0295-5075/1/2/001. [DOI] [Google Scholar]
- Shmulevich I, Kauffman SA. Activities and sensitivities in Boolean network models. Physical Review Letters. 2004;93(4) doi: 10.1103/PhysRevLett.93.048701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldana M, Coppersmith S, Kadanoff LP. In: Perspectives and Problems in Nonlinear Science. Kaplan E, Marsden JE, Sreenivasan KR, editor. Springer, New York, NY, USA; 2002. Boolean dynamics with random couplings; pp. 23–89. [Google Scholar]
- Luque B, Solé RV. Lyapunov exponents in random Boolean networks. Physica A. 2000;284(1–4):33–45. [Google Scholar]
- Luque B, Solé RV. Phase transitions in random networks: simple analytic determination of critical points. Physical Review E. 1997;55(1):257–260. doi: 10.1103/PhysRevE.55.257. [DOI] [Google Scholar]
- Nochomovitz YD, Li H. Highly designable phenotypes and mutational buffers emerge from a systematic mapping between network topology and dynamic output. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(11):4180–4185. doi: 10.1073/pnas.0507032103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykter M, Price ND, Larjo A. et al. Critical networks exhibit maximal information diversity in structure-dynamics relationships. Physical Review Letters. 2008;100(5) doi: 10.1103/PhysRevLett.100.058702. [DOI] [PubMed] [Google Scholar]
- Lähdesmäki H, Shmulevich I, Yli-Harja O. On learning gene regulatory networks under the Boolean network model. Machine Learning. 2003;52(1-2):147–167. [Google Scholar]
- Zhou X, Wang X, Dougherty ER. Gene prediction using multinomial probit regression with Bayesian gene selection. EURASIP Journal on Applied Signal Processing. 2004;2004(1):115–124. doi: 10.1155/S1110865704309157. [DOI] [Google Scholar]
- Zhao W, Serpedin E, Dougherty ER. Inferring gene regulatory networks from time series data using the minimum description length principle. Bioinformatics. 2006;22(17):2129–2135. doi: 10.1093/bioinformatics/btl364. [DOI] [PubMed] [Google Scholar]
- Marshall S, Yu L, Xiao Y, Dougherty ER. Inference of a probabilistic Boolean network from a single observed temporal sequence. EURASIP Journal on Bioinformatics and Systems Biology. 2007;2007 doi: 10.1155/2007/32454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal R, Ivanov I, Datta A, Bittner ML, Dougherty ER. Generating Boolean networks with a prescribed attractor structure. Bioinformatics. 2005;21(21):4021–4025. doi: 10.1093/bioinformatics/bti664. [DOI] [PubMed] [Google Scholar]
- Tabus I, Astola J. On the use of MDL principle in gene expression prediction. EURASIP Journal on Applied Signal Processing. 2001;2001(4):297–303. doi: 10.1155/S1110865701000270. [DOI] [Google Scholar]
- Akutsu T, Miyano S, Kuhara S. Inferring qualitative relations in genetic networks and metabolic pathways. Bioinformatics. 2000;16(8):727–734. doi: 10.1093/bioinformatics/16.8.727. [DOI] [PubMed] [Google Scholar]
- Akutsu T, Miyano S, Kuhara S. Algorithms for identifying Boolean networks and related biological networks based on matrix multiplication and fingerprint function. Journal of Computational Biology. 2000;7(3-4):331–343. doi: 10.1089/106652700750050817. [DOI] [PubMed] [Google Scholar]
- Boros E, Ibaraki T, Makino K. Error-free and best-fit extensions of partially defined Boolean functions. Information and Computation. 1998;140(2):254–283. doi: 10.1006/inco.1997.2687. [DOI] [Google Scholar]
- Efron B. Estimating the error rate of a prediction rule: improvement on cross-validation. Journal of the American Statistical Association. 1983;78(382):316–331. doi: 10.2307/2288636. [DOI] [Google Scholar]
- Efron B, Tibshirani RJ. An Introduction to the Bootstrap. Vol. 57. Chapman & Hall, New York, NY, USA; 1993. (Monographs on Statistics and Applied Probability). [Google Scholar]
- Sima C, Attoor S, Brag-Neto U, Lowey J, Suh E, Dougherty ER. Impact of error estimation on feature selection. Pattern Recognition. 2005;38(12):2472–2482. doi: 10.1016/j.patcog.2005.03.026. [DOI] [Google Scholar]
- Dougherty ER. Validation of inference procedures for gene regulatory networks. Current Genomics. 2007;8(6):351–359. doi: 10.2174/138920207783406505. [DOI] [PMC free article] [PubMed] [Google Scholar]