

Abstract
The Hedgehog (Hh) family of secreted proteins controls many aspects of animal development. In Drosophila, Hh transduces its signal via Cubitus interruptus (Ci), a transcription factor present in two forms: a full-length activator and a carboxy-terminally truncated repressor that is derived from the full-length form by proteolytic processing. The proteolytic processing of Ci is promoted by the activities of protein kinase A (PKA) and Slimb, whereas it is inhibited by Hh. Here we show that PKA inhibits the activity of the full-length Ci in addition to its role in regulating Ci proteolysis. Whereas Ci processing is blocked in both PKA and slimb mutant cells, the accumulated full-length Ci becomes activated only in PKA but not in slimb mutant cells. Moreover, PKA inhibits an uncleavable activator form of Ci. These observations suggest that PKA regulates the activity of the full-length Ci independent of its proteolytic processing. We also provide evidence that PKA regulates both the proteolytic processing and transcriptional activity of Ci by directly phosphorylating Ci. We propose that phosphorylation of Ci by PKA has two separable roles: (1) It blocks the transcription activity of the full-length activator form of Ci, and (2) it targets Ci for Slimb-mediated proteolytic processing to generate the truncated form that functions as a repressor.
Keywords: PKA, Ci, Hedgehog, Slimb, proteolysis, limb development
Secreted proteins of the Hedgehog (Hh) family govern cell growth and patterning in a wide variety of developmental processes in both vertebrates and invertebrates (Ingham 1994; Hammerschmidt et al. 1997). Moreover, mutations in components of the Hh signaling pathway have been implicated in many human disorders including cancers (Dean 1996; Goodrich and Scott 1998). In Drosophila, Hh is required at multiple developmental stages and is responsible for patterning embryonic segments and adult structures such as wings, legs, and eyes (Ingham 1994; Hammerschmidt et al. 1997). In the developing wings, posterior (P) compartment cells express and secrete Hh proteins that diffuse into the anterior (A) compartment and induce neighboring A compartment cells to express decapentaplegic (dpp), which encodes a member of the transforming growth factor-β/bone morphogenetic protein (TGFβ/BMP) family of secreted proteins (Basler and Struhl 1994; Capdevila and Guerrero 1994; Tabata and Kornberg 1994). Dpp then diffuses into both A and P compartments and functions as a long-range morphogen to control the growth and patterning of cells in the whole wing (Lecuit et al. 1996; Nellen et al. 1996). Although the long-range organizing influence of Hh is mediated primarily by Dpp, Hh functions as a local morphogen to specify patterns near the A/P compartment boundary by activating other genes including patched (ptc) and engrailed (en) (Strigini and Cohen 1997).
Hh transduces its signal through a receptor complex containing the transmembrane protein Ptc and the serpentine protein Smoothened (Smo) (Alcedo and Noll 1997; Ingham 1998). In the absence of Hh, Ptc inhibits the activity of Smo. Hh directly binds to Ptc and alleviates this repression. Smo then signals to activate Cubitus interruptus (Ci), a member of the Gli family of zinc finger transcription factors (Ruiz i Altaba 1997). In imaginal disc development, Ci has dual roles that are carried out by two distinct forms of Ci. In A compartment cells distant from the A/P compartment boundary, Ci is proteolytically cleaved to form a carboxy-terminally truncated form (Ci75) (Aza-Blanc et al. 1997). This form of Ci functions as a repressor and blocks the expression of Hh responsive genes such as dpp (Dominguez et al. 1996; Methot and Basler 1999) . In A compartment cells near the A/P compartment boundary, Hh signaling prevents the cleavage of Ci and stimulates the activity of accumulated full-length Ci (Aza-Blanc et al. 1997; Methot and Basler 1999). The full-length form of Ci functions as a transcriptional activator and controls the expression of Hh target genes such as ptc and en (Alexandre et al. 1996; Dominguez et al. 1996; Methot and Basler 1999).
Proteolytic processing of Ci requires the activities of protein kinase A (PKA) and Slimb, an F-box/WD40 repeat-containing protein (Johnson et al. 1995; Jiang and Struhl 1998; Ohlmeyer and Kalderon 1998). Eliminating PKA or Slimb function abrogates Ci proteolysis, so that no truncated Ci repressor forms and full-length Ci accumulates, and as a consequence, Hh signaling is activated (Jiang and Struhl 1995, 1998; Lepage et al. 1995; Li et al. 1995; Pan and Rubin 1995; Strutt et al. 1995; Theodosiou et al. 1998). Ci contains multiple PKA phosphorylation consensus sites in its carboxy-terminal region, implying that Ci is a PKA substrate (Orenic et al. 1990). In support of this, mutating the PKA phosphorylation sites blocks Ci processing in tissue culture cells, suggesting that phosphorylation of Ci by PKA targets Ci for proteolytic processing (Chen et al. 1998). Recent studies suggest that the vertebrate homolog of Slimb, βTRCP, specifies the substrates of the so-called SCF (Skp1, Cdc53, and F-box) ubiquitin ligase complex. This complex targets phosphorylated substrates (such as IκB and β-catenin) for ubiquitination followed by proteasome-mediated proteolysis (Maniatis 1999). By analogy, it has been proposed that Slimb acts downstream of PKA to target phosphorylated Ci for ubiquitin/proteasome-mediated proteolysis (Jiang and Struhl 1998; Maniatis 1999).
Here we provide in vivo evidence that phosphorylation of Ci by PKA not only targets Ci for Slimb-mediated proteolysis but also inhibits the activity of the full-length activator form of Ci independent of its processing. We show that although Ci processing is blocked both in PKA and slimb mutant cells, the accumulated full-length form of Ci is only activated in PKA but not in slimb mutant cells. Moreover, PKA can inhibit the activity of an uncleavable activator form of Ci. We demonstrate that both the proteolytic processing and the transcriptional activity of the full-length Ci are regulated by PKA phosphorylation of Ci. Finally, we show that PKA and Su(fu) act through distinct mechanisms to inhibit the activity of the full-length Ci.
Results
High levels of constitutive PKA activity do not promote Ci processing in slimb mutant cells
PKA and Slimb are both required for proteolytic processing of Ci (Jiang and Struhl 1998). To determine the epistatic relationship between PKA and Slimb in regulating Ci processing, we generated slimb mutant clones that express high levels of a constitutively active form of PKA (mC*, Li et al. 1995) and assayed them for Ci processing by immunostaining with a Ci antibody (2A1), which recognizes the full-length but not the truncated form of Ci (Motzny and Holmgren 1995; Aza-Blanc et al. 1997). mC* was expressed from a UAS-mC* transgene under the control of a Gal4 driver line, MS1096, that expresses high levels of Gal4 almost uniformly in the wing pouch region (Fig. 1C, Capdevila and Guerrero 1994). It has been shown that overexpression of mC* at high levels in wing discs can override Hh signaling and block ptc expression (Li et al. 1995). Consistent with this observation, overexpression of mC* using the MS1096 Gal4 driver line blocks the accumulation of full-length Ci as well as the expression of ptc–lacZ induced by Hh (Fig. 1B, see below), suggesting that high levels of PKA activity act dominantly to promote Ci processing. However, overexpression of mC* does not promote Ci processing in the absence of slimb function, as anteriorly situated slimb mutant cells that express MS1096/UAS-mC* accumulate high levels of full-length Ci (Fig. 1D–E). These observations are consistent with Slimb acting either downstream of or parallel to PKA in regulating the proteolytic processing of Ci.
Figure 1.
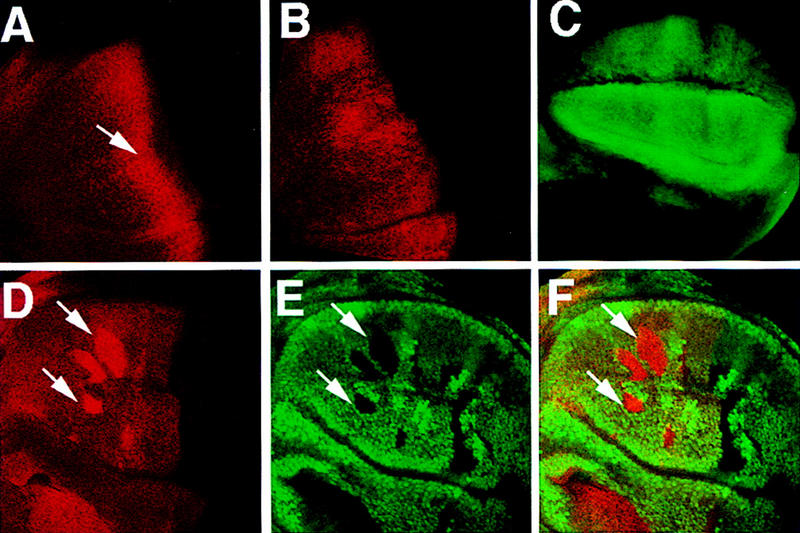
High levels of constitutive PKA activity do not promote Ci processing in slimb mutant cells. (A) A wild-type late third-instar wing disc stained with an anti-Ci antibody (2A1) that recognizes the full-length form of Ci. In this and subsequent figures, all of the wing discs are shown with anterior to the left and ventral up. The arrow indicates the accumulation of full-length Ci in A compartment cells near the A/P compartment boundary. (B) A late third-instar wing disc expressing UAS–mC* with MS1096 and stained with 2A1. High levels of constitutive PKA activity block the accumulation of full-length Ci in A compartment cells near the A/P compartment boundary. (C) A late third-instar wing disc expressing UAS–GFP with MS1096. GFP is expressed almost uniformly in the wing pouch region with slightly higher levels in dorsal compartment cells. The GFP expression reflects the pattern of Gal4 expression driven by the MS1096 Gal4 line. (D–F) A late third-instar wing disc containing slimb clones and expressing UAS–mC* with MS1096 and doubly stained with 2A1 (red in D and F) and a Myc antibody (green in E and F). slimb mutant cells (marked by the lack of Myc–GFP expression and indicated by the arrows) accumulate high levels of full-length Ci even though they express high levels of mC*.
Ci is activated in PKA but not in slimb mutant cells
Previous genetic studies showed that anteriorly situated PKA and slimb mutant clones induced limb duplication as well as ectopic dpp expression (Jiang and Struhl 1995, 1998; Lepage et al. 1995; Li et al. 1995; Pan and Rubin 1995; Theodosiou et al. 1998). However, close examination of phenotypes associated with PKA and slimb mutant clones revealed interesting differences. The supernumerary wings induced by slimb mutant clones are on the average smaller than those induced by PKA mutant clones (Fig. 2, cf. B with C and D). Moreover, the supernumerary wings induced by slimb clones often contain incomplete vein structures (Fig. 2D). In the supernumerary wings, slimb mutant cells contribute to ectopic vein 3 whereas PKA mutant cells contribute to intervein tissues that normally form posteriorly to vein 3 (Fig. 2A–D). Thus the supernumerary wings induced by slimb mutant clones resemble more closely those induced by low levels of dpp expression than those induced by ectopic hh expression (Basler and Struhl 1994; Zecca et al. 1995). These observations imply that not all of the outputs of Hh signaling are induced in slimb mutant cells.
Figure 2.
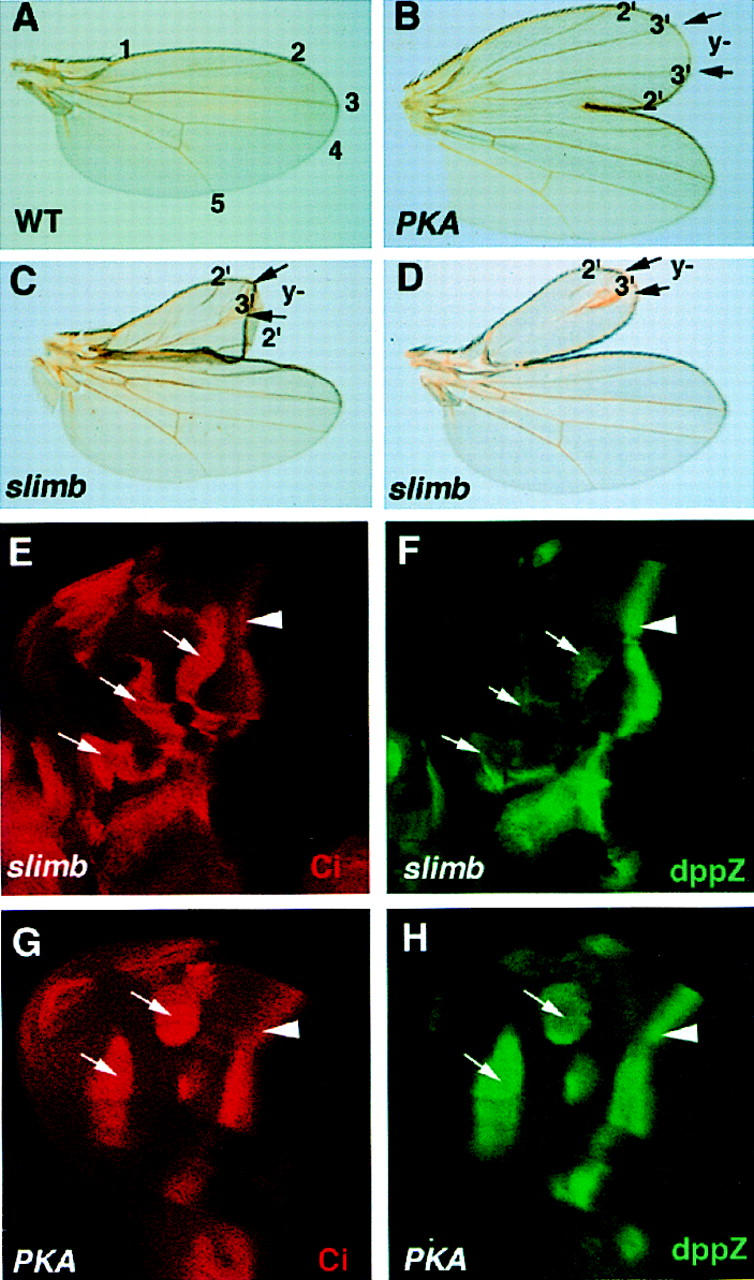
Different phenotypes induced by PKA and slimb mutant clones. (A) A wild-type adult wing with its longitudinal veins indicated by numbers. (B–D) Supernumerary wings organized by a clone of PKA mutant cells (B) or a clone of slimb mutant cells (C,D). Both PKA and slimb mutant cells are marked by y− and are situated between two arrows along the wing margin. In the supernumerary wings, PKA mutant cells form intervein tissues flanked by ectopic vein3; slimb mutant cells form ectopic vein3. The supernumerary wings organized by slimb clones are smaller than that induced by the PKA clone and contain incomplete veins (C,D). (E,F) A late third-instar wing disc carrying slimb clones and showing the accumulation of full-length Ci (E) and dpp–lacZ expression (F). Anteriorly situated slimb mutant clones (indicated by the arrows) accumulate high levels of full-length Ci and express dpp–lacZ at levels lower than those of the endogenous dpp–lacZ at the A/P compartment boundary (arrowhead). (G,H) A late third-instar wing disc carrying PKA mutant clones and showing Ci accumulation (G) and dpp–lacZ expression (H). Anteriorly situated PKA mutant clones (arrows) accumulate high levels of full-length Ci and express dpp–lacZ at levels comparable to those of the endogenous dpp–lacZ (arrowhead).
To compare slimb and PKA mutant phenotypes molecularly, we examined the expression of dpp–lacZ reporter genes in PKA and slimb mutant clones. Although both slimb and PKA mutant clones accumulate full-length Ci at comparable levels (Fig. 2E,G), slimb mutant clones ectopically express dpp–lacZ at levels lower than those of endogenous dpp–lacZ at the A/P compartment boundary (Fig. 2F), whereas PKA mutant cells fully activate dpp–lacZ (Fig. 2H). The low levels of dpp–lacZ expression observed in slimb mutant clones may explain the smaller size of the supernumerary wings induced by slimb mutant clones, as Dpp functions in a concentration-dependent manner to control the growth and patterning of the wings (Zecca et al. 1995; Lecuit et al. 1996; Nellen et al. 1996).
We next examined whether genes regulated by the full-length activator form of Ci, such as ptc and en, are expressed in slimb mutant cells. Anteriorly situated slimb mutant clones in the wing pouch region fail to induce ectopic expression of ptc–lacZ (Fig. 3B), nor do they ectopically express en (data not shown). In contrast, PKA mutant clones situated in similar regions activate ptc–lacZ (Fig. 3A) and express low levels of en (Ohlmeyer and Kalderon 1998). Because ptc and en are regulated by the full-length activator form of Ci, these results suggest that the full-length Ci that accumulates in slimb mutant cells is transcriptionally silent. The low levels of ectopic dpp–lacZ expression observed in slimb mutant cells are likely due to derepression of dpp, as Ci processing, which generates the repressor form of Ci, is blocked in slimb mutant cells. Although the result shown in Figure 3B was obtained using a hypomorphic slimb allele, slimb1, similar result was obtained with a slimb null allele, slimbP (Jiang and Struhl 1998; data not shown). This argues against the possibility that the full-length Ci accumulated in slimb mutant cells is inactive because it is trapped in an SCF complex containing Slimb.
Figure 3.
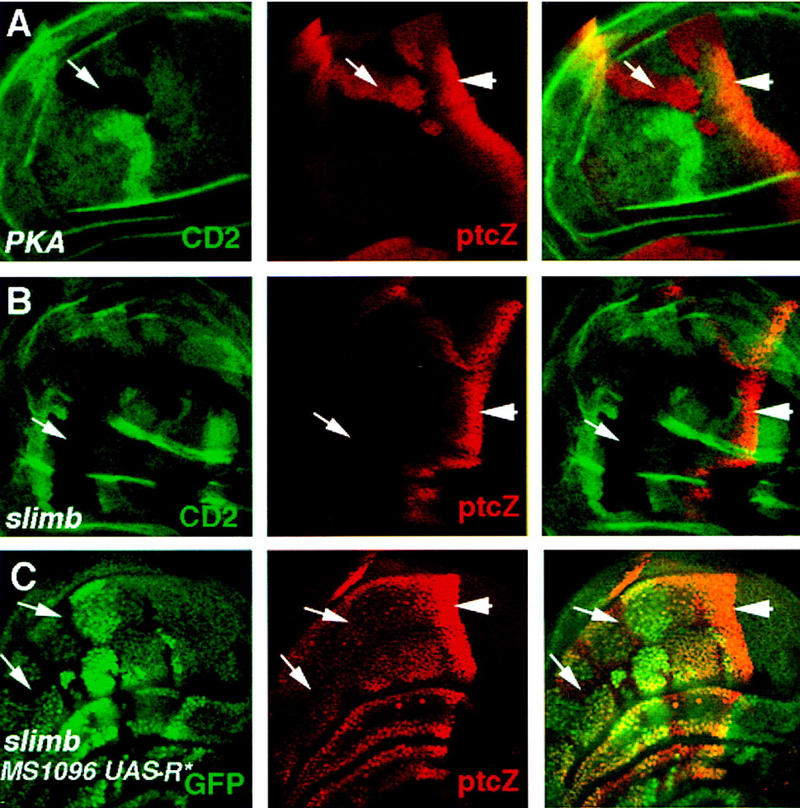
PKA but not slimb mutant cells ectopically express ptc–lacZ. (A,B) Late third-instar wing discs carrying PKA (A) or slimb (B) mutant clones and doubly stained for the marker gene (CD2) expression (green in left and right panels) and ptc–lacZ expression (red in middle and right panels). Both PKA and slimb mutant clones are marked by the lack of CD2 expression. Anteriorly situated PKA mutant cells ectopically express ptc–lacZ (arrow in A); slimb mutant cells do not (arrow in B). (C) A late third-instar wing disc carrying slimb mutant clones and expressing MS1096/UAS–R*. The disc was doubly stained for GFP expression (green in left and right panels) and ptc–lacZ expression (red in middle and right panels). slimb mutant cells are marked by the lack of GFP expression (arrows). Anteriorly situated slimb mutant cells with reduced PKA activity express ptc–lacZ. The levels of ectopic ptc–lacZ expression are lower than those at the compartment boundary because expressing UAS–R* only partially eliminates PKA activity.
Two possible models might explain the differences between PKA and slimb mutant phenotypes. In addition to its role in regulating Ci proteolysis, Slimb might play a positive role in transducing Hh signal. Alternatively, PKA might repress the activity of the full-length activator form of Ci even when Ci processing is blocked. To distinguish between these two possibilities, we examined the activity of Ci in cells deficient for both Slimb and PKA activities. To generate PKA and slimb double mutant cells, we induced slimb mutant clones in wing discs expressing a mutant PKA regulatory subunit, R*, from a UAS-R* transgene under the control of the MS1096 Gal4 line. Expression of R* reduces PKA catalytic activity by sequestering the catalytic subunit in the inactive holoenzyme (Li et al. 1995; Ohlmeyer and Kalderon 1997). As shown in Figure 3C, slimb mutant cells expressing R* activate ptc–lacZ expression, suggesting that slimb mutant cells are competent to activate ptc–lacZ but are normally blocked by PKA. This observation argues against a positive role for slimb in activating Ci and suggests an inhibitory role for PKA in keeping the full-length form of Ci inactive prior to its proteolytic processing.
PKA inhibits an uncleavable activator form of Ci
Overexpression of a constitutively active form of PKA (mC*) at high levels can override Hh signaling to block Hh-mediated activation of ptc (Li et al. 1995). In agreement with this previous observation, wing discs expressing UAS-mC* under the control of the MS1096 Gal4 driver line showed disruption in ptc–lacZ expression domain at the A/P compartment boundary (Fig. 4B). To obtain further evidence that PKA inhibits the activator form of Ci independent of its ability to promote Ci processing, we asked whether overexpression of mC* could suppress the activity of an uncleavable activator form of Ci, CiU, which contains a small deletion (from amino acid 612 to amino acid 760) that removes the cleavage site (Methot and Basler 1999). It has been shown that CiU cannot be processed to generate the repressor form of Ci but can still mediate Hh-dependent activation of target genes (Methot and Basler 1999). We coexpressed mC* and a HA-tagged form of CiU (HACiU) using the MS1096 Gal4 line and determined whether mC* could suppress the activity of HACiU. Wing discs expressing MS1096/HACiU ectopically expressed ptc–lacZ at uniformly high levels in the P compartment (Fig. 4C), consistent with the previous observation that the activity of CiU depends on Hh signaling (Methot and Basler 1999). In contrast, wing discs coexpressing HACiU and mC* showed reduction in the levels of ptc–lacZ expression, which is more evident in A compartment cells near the compartment boundary (Fig. 4D). This observation suggests that overexpression of mC* can override Hh signaling to suppress the activity of CiU even though this form of Ci cannot be processed. Taken together, both loss-of-function and gain-of-function studies provide strong evidence that PKA negatively regulates the transcriptional activity of the full-length Ci independent of Ci cleavage.
Figure 4.
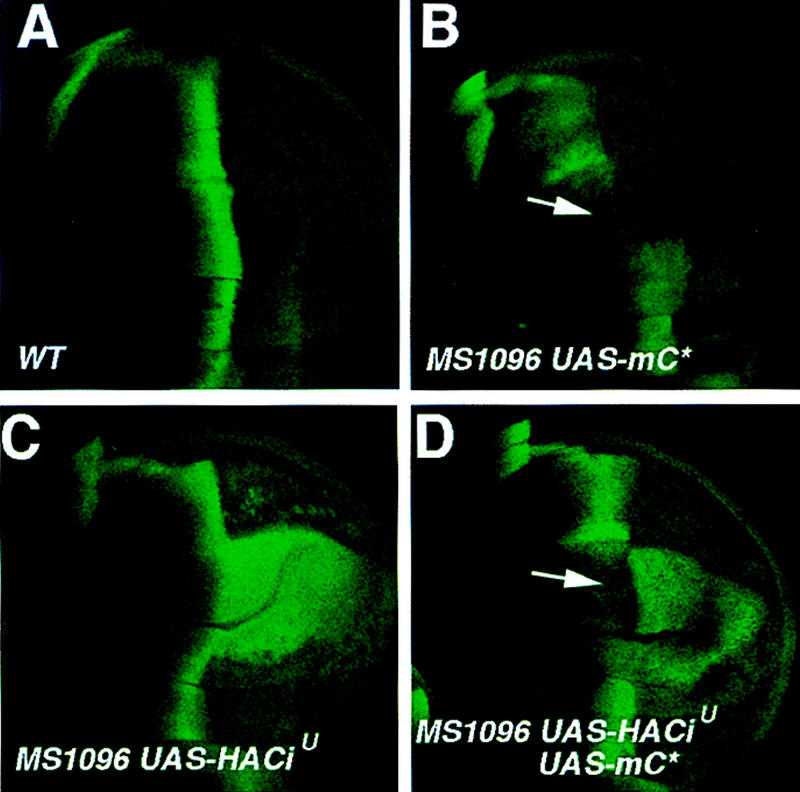
PKA inhibits the activity of an uncleavable activator form of Ci. Wing discs in all panels were stained for ptc–lacZ expression. (A) A late third-instar wing disc showing the wild-type ptc–lacZ expression. (B) A wing disc expressing a constitutively active form of PKA catalytic subunit (mC*) under the control of the MS1096 Gal4 line. ptc–lacZ expression is suppressed in the wing pouch region (arrow). (C) A wing disc expressing an uncleavable Ci (HACiU) with MS1096. ptc–lacZ is ectopically expressed in P but not in A compartment cells away from the compartment boundary. (D) A wing disc expressing UAS–HACiU and UAS–mC* under the control of the MS1096 Gal4 line. Ectopic ptc–lacZ expression induced by HACiU is partially suppressed by coexpression of mC*. The arrow indicates a gap in the ptc–lacZ expression domain. The suppression of ptc–lacZ expression is more evident in A compartment cells near the A/P compartment boundary (indicated by the arrow).
Effects of mutating PKA phosphorylation sites on Ci processing and activity in vivo
To determine whether the PKA phosphorylation sites are required for Ci proteolytic processing in vivo, we generated transformants expressing wild-type HACi or mutant HACi−3P under the control of UAS promoter. HACi−3P has three PKA phosphorylation sites mutated to Ala (S838A, S856A, and S892A). HACi or HACi−3P was expressed in developing wings using the MS1096 Gal4 driver line. Cell extracts were prepared from corresponding wing discs and analyzed by Western blot using an HA-specific antibody. As shown in Figure 5A, HACi was processed to generate the truncated form of Ci (Ci75) whereas HACi−3P was no longer processed and accumulated only as the full-length form, suggesting that PKA phosphorylation at the carboxy-terminal region of Ci mediates its processing in vivo.
Figure 5.
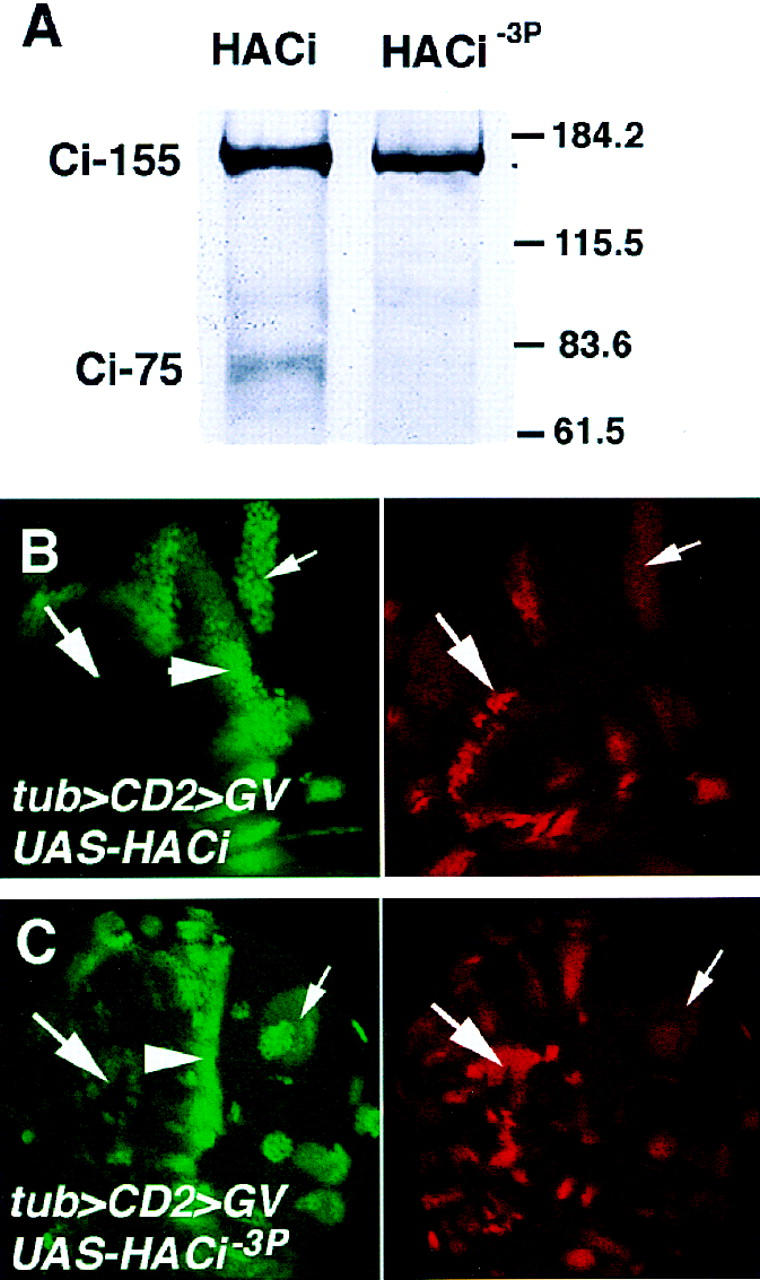
Effects of mutating the PKA phosphorylation sites in Ci on its processing and activity. (A) Cell extracts from wing discs expressing either HACi or HACi−3P with MS1096 were probed with an anti-HA antibody. HACi is processed to form the truncated form (Ci-75) whereas HACi−3P is not. (B,C) Wing discs expressing HACi (B) or HACi−3P (C) under the control of tub>CD2>GV. The discs were doubly labeled to show ptc–lacZ expression (left) and HA staining (right). Only P (small arrow) but not A (big arrow) compartment cells that express HACi induce ectopic expression of ptc–lacZ (B). In contrast, both A and P compartment cells that express HACi−3P ectopically activate ptc–lacZ, although the levels of ptc–lacZ expression in A compartment cells (big arrow in C) are lower than those in P compartment cells (small arrow in C) or at the compartment boundary (arrowhead).
We next investigated whether mutating the PKA phosphorylation sites in Ci altered its activity. To do this, we expressed HACi or HACi−3P in wing discs and compared their transcription activity by assaying for ptc–lacZ expression. The upstream activating sequence (UAS) transgenes were expressed using a weak Gal4 driver line, tubulin>CD2>GV, which contains a flp-out cassette between a tubulin promoter and a Gal4-VP16 fusion gene (Struhl and Adachi 1998). The Gal4-VP16 protein contains a point mutation in the VP16 activating domain to reduce its activity. After removal of the flp-out cassette by Flp-mediated recombination, Gal4-VP16 is expressed under the control of the ubiquitous tubulin promoter. As shown in Figure 5B, clones of cells expressing tub>GV/UAS-HACi activated ptc–lacZ when they were situated in the P compartment but failed to do so when they were situated in the A compartment away from the A/P compartment boundary, suggesting that the transcriptional activity of the exogenously expressed wild-type Ci depends on Hh. In contrast, A compartment cells expressing tub>GV/HACi−3P ectopically activated ptc–lacZ, albeit at levels lower than those of endogenous ptc–lacZ or those induced by P compartment cells expressing tub>GV/UAS-HACi−3P (Fig. 5C). We examined four independent transformant lines for HACi and five independent lines for HACi−3P. None of the HACi lines activated ptc–lacZ in A compartment cells away from the A/P compartment boundary whereas all of the HACi−3P lines induced ectopic ptc–lacZ in A compartment cells although the levels of ectopic ptc–lacZ varied from line to line. In addition, we compared the levels of exogenously expressed HACi and HACi−3P from multiple transformed lines by staining wing discs with an anti-HA antibody. We found that HACi and HACi−3P were expressed at comparable levels (Fig. 5B,C), suggesting that the difference in the activity between the wild-type and mutant form of Ci is due to mutations in the PKA phosphorylation sites rather than difference in the expression levels. Thus, mutating the PKA phosphorylation sites in Ci blocks Ci processing and renders Ci constitutively active. This suggests that PKA antagonizes Hh signaling by directly phosphorylating Ci. We note that the activity of HACi−3P is still regulated by Hh, as P compartment cells expressing HACi−3P activated ptc–lacZ at levels higher than those induced by A compartment cells expressing the same form of Ci (Fig. 5C). A likely explanation is that other negative regulators of the Hh pathway, such as Su(fu) and Costal2 (Cos2), may inhibit the activity of HACi−3P in A compartment cells (Robbins et al. 1997; Sisson et al. 1997; Ohlmeyer and Kalderon 1998).
PKA phosphorylation of Ci mediates repression of the activator form of Ci
Although the above experiments strongly suggest that PKA blocks Hh signaling by directly phosphorylating Ci and targeting it for proteolysis, they do not address whether such phosphorylation also regulates the activity of the full-length activator form of Ci. It is possible that phosphorylation of Ci might only serve to target Ci for processing whereas the observed processing-independent inhibition of Ci activity by PKA might be indirect. Alternatively, phosphorylation of Ci by PKA may block the activity of the full-length activator form of Ci independent of Ci processing. To distinguish between these two possibilities, we investigated whether mutating PKA phosphorylation sites in the uncleavable activator form of Ci (CiU) could alter its activity. We mutated the same three PKA phosphorylation sites (S838A, S856A, and S892A) in HACiU to generate HACiU−3P, and expressed UAS-HACiU or UAS-HACiU−3P in wings using the MS1096 Gal4 line and assayed for ptc–lacZ expression. Consistent with previous observations, ubiquitously expressing CiU in the wing pouch region induced ectopic ptc–lacZ expression only in the P compartment but not in the A compartment (Fig. 6A, Methot and Basler 1999). This suggests that the activity of CiU is still inhibited in A compartment cells even though it is not processed and that this inhibition is alleviated by Hh signaling in P compartment cells. In contrast, expressing HACiU−3P at comparable levels in the wing pouch region induced ectopic ptc–lacZ expression in both A and P compartments (Fig. 6B). We examined four independent transformant lines for CiU and six independent transformant lines for CiU−3P. None of the CiU lines gave rise to appreciable ectopic ptc–lacZ expression in the A compartment whereas all the CiU−3P lines ectopically activated ptc–lacZ in both A and P compartments. Thus, mutating PKA phosphorylation sites in HACiU alleviates the inhibition of HACiU activity in A compartment cells, suggesting that PKA blocks the transcriptional activity of this uncleavable form of Ci by directly phosphorylating its carboxy-terminal region. We note that the levels of ectopic ptc–lacZ expression induced by CiU−3P are lower in A compartment cells than in P compartment cells, suggesting that CiU−3P may still be regulated by other inhibitory components in the Hh pathway.
Figure 6.
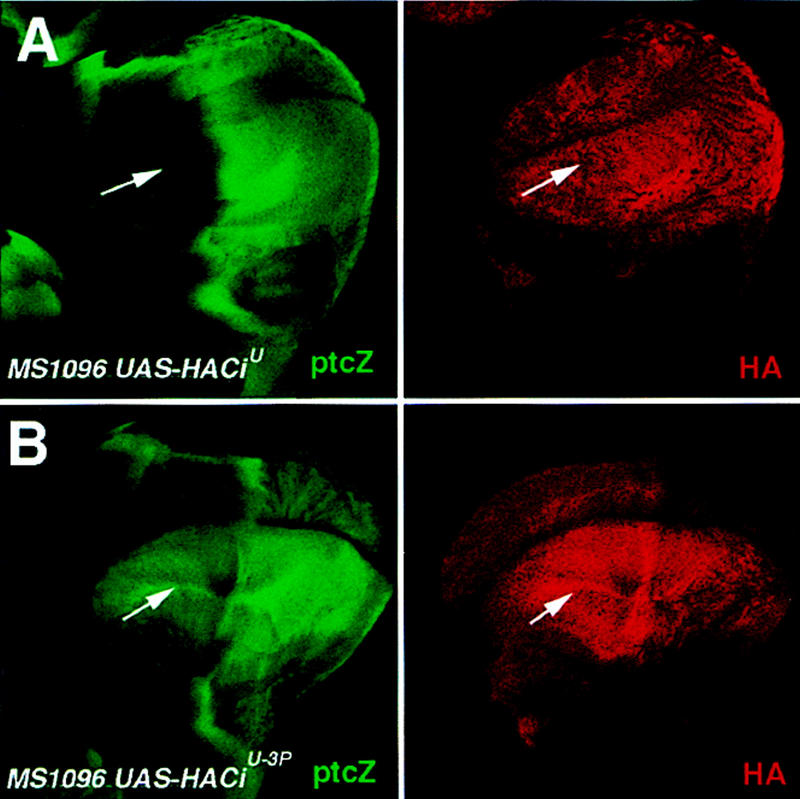
Mutating PKA phosphorylation sites in CiU alters its activity. Wing discs expressing UAS–HACiU (A) or UAS–HACiU−3P (B) under the control of the MS1096 Gal4 line were doubly stained for ptc–lacZ expression (green) and HA expression (red). HACiU induces ectopic ptc–lacZ expression only in P but not in A compartment cells (A). In contrast, HACiU−3P activates ptc–lacZ in both A and P compartment cells (B). Note that the levels of ptc–lacZ in A compartment cells are lower than those in P compartment cells (B).
Inhibition of Ci transcriptional activity by PKA in slimb Su(fu) double mutant cells
Su(fu) has been implicated as an inhibitor of Ci transcriptional activity (Alves et al. 1998; Monnier et al. 1998; Ohlmeyer and Kalderon 1998). To explore the relationship between PKA and Su(fu) in regulating the full-length Ci, we examined the expression of ptc–lacZ and en in slimb Su(fu) double mutant and slimb Su(fu) PKA triple mutant cells. We generated slimb Su(fu) PKA triple mutant cells by inducing slimb mutant clones in wing discs homozygous for Su(fu) and expressing UAS-R* under the control of the MS1096 Gal4 line. Expressing UAS-R* dramatically reduces but does not completely eliminate PKA activity. Although anteriorly situated slimb singly mutant clones do not induce ectopic ptc–lacZ expression (Fig. 3B), slimb Su(fu) double mutant clones located in similar regions ectopically expressed ptc–lacZ, albeit at levels lower than those of the endogenous ptc–lacZ at the A/P compartment boundary (Fig. 7A). This observation suggests that Su(fu) also contributes to the suppression of Ci activity in slimb mutant cells. However, the relatively low levels of ectopic ptc–lacZ in slimb Su(fu) clones imply that Ci is not fully activated. Consistent with this, slimb Su(fu) double mutant cells failed to activate en (Fig. 7C), which is normally activated only by the highest levels of Hh signaling (Strigini and Cohen 1997). In contrast, anteriorly situated slimb Su(fu) double mutant cells with reduced PKA activity expressed ptc–lacZ at levels comparable to those at the A/P compartment boundary (Fig. 7B). Moreover, they ectopically expressed en (Fig. 7D). These results suggest that PKA inhibits the activity of the full-length Ci independent of Su(fu) and that PKA and Su(fu) act in parallel to block the activity of the full-length Ci.
Figure 7.
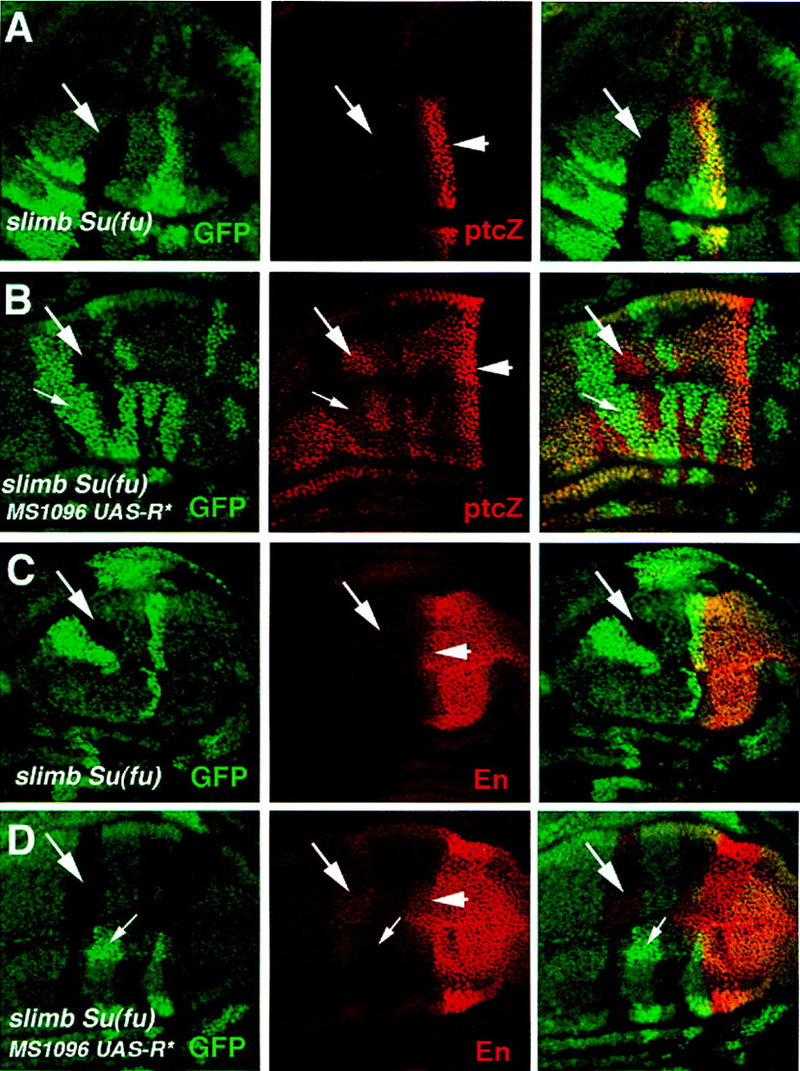
PKA inhibits Ci activity in slimb Su(fu) double mutant cells. Late third-instar wing discs carrying slimb Su(fu) double mutant clones with (B,D) or without (A,C) the expression of a mutant form of PKA regulatory subunit (R*). The wing discs were doubly stained for GFP expression (green) and ptc–lacZ expression (red in A and B) or En expression (red in C and D). slimb Su(fu) double mutant clones are marked by the lack of GFP expression (big arrows); the twin clones, which are homozygous for the wild-type slimb and Su(fu) genes, are marked by high levels of GFP expression (small arrows in B and D). slimb Su(fu) double mutant cells situated in the A compartment ectopically express low levels of ptc–lacZ (arrow in A) and do not express En (arrow in C). slimb Su(fu) mutant cells expressing MS1096/UAS–R* ectopically activate ptc–lacZ at high levels (big arrow in B) as well as En (big arrow in D). Cells expressing MS1096/UAS–R* but homozygous for the wild-type slimb and Su(fu) genes activate ptc–lacZ at low levels (small arrow in B) and do not express En (small arrow in D). Arrowheads in the middle panels indicate the A/P compartment boundary.
Discussion
Previous studies in Drosophila and vertebrate model organisms identified PKA as a conserved negative regulator in the Hh signal transduction pathway (for review, see Ingham 1998). Here we investigated the mechanism by which PKA antagonizes Hh signaling activity in vivo. We demonstrate that the transcription factor Ci, which mediates Hh signaling, is a target for PKA. We show that in addition to targeting Ci for proteolytic processing, which generates the repressor form of Ci, PKA also inhibits the activity of the full-length activator form of Ci. Moreover, we provide evidence that both regulatory events are controlled via phosphorylation of Ci by PKA.
A recent study by Methot and Basler (1999) demonstrated the existence of distinct activator and repressor forms of Ci. These two forms play separable roles in Drosophila limb development by regulating different sets of Hh target genes. In the developing wing, the expression of ptc and en appears to be exclusively controlled by the activator form of Ci whereas dpp expression is governed by both the activator and repressor forms. Interestingly, preventing Ci proteolysis with an uncleavable form of Ci is not sufficient to convert Ci into a constitutive activator, suggesting that the full-length activator form of Ci encounters additional regulatory block(s) that need to be alleviated by Hh signaling (Methot and Basler 1999). Here we provide evidence that PKA activity exerts such a block. Our initial evidence that PKA regulates the activator form of Ci came from a close examination of PKA and slimb mutant phenotypes. In slimb mutant cells, Ci processing is nearly abolished, and, as a consequence, full-length Ci accumulates. However, the expression of ptc–lacZ and en is not induced, suggesting that the full-length form of Ci that accumulates in slimb mutant cells is transcriptionally silent. In contrast, PKA mutant cells express ptc–lacZ and en even though they accumulate full-length Ci at comparable levels as slimb mutant cells. This suggests that the full-length Ci that accumulates in PKA mutant cells is transcriptionally active. Furthermore, slimb mutant cells with reduced PKA activity ectopically express ptc–lacZ, arguing that the lack of Ci activity in slimb mutant cells is due to an inhibitory role of PKA rather than a positive requirement for Slimb in the Hh signaling pathway. Further evidence that PKA regulates the activator form of Ci independent of its processing came from the gain-of-function studies. We showed that the ectopic expression of ptc–lacZ induced by the uncleavable activator form of Ci (CiU) was suppressed by overexpression of a constitutively active form of PKA (mC*).
In support of the view that Ci is a direct target for PKA in regulating Hh signaling, we found that a modified form of Ci with three PKA phosphorylation consensus sites mutated was not processed but exhibited constitutive activity when expressed in the developing wings. Although these observations suggest that PKA antagonizes Hh signaling by directly phosphorylating Ci and targeting it for proteolysis, they do not to address whether phosphorylation of Ci by PKA also regulates the activity of the full-length activator form of Ci. The low levels of constitutive activity exhibited by the PKA phosphorylation-deficient form of Ci could be secondary to the lack of Ci processing, which results in a dramatic increase in the levels of the full-length activator form of Ci, as it has been shown that overexpression of a full-length wild type form of Ci can activate ptc expression in wing discs (Alexandre et al. 1996). To define the role of PKA phosphorylation in regulating the activity of the full-length Ci, we took advantage of the uncleavable activator form of Ci (CiU) (Methot and Basler 1999). We found that mutating multiple PKA phosphorylation sites in CiU dramatically altered its transcriptional activity and rendered it constitutively active (Fig. 6). This observation suggests that the activity of CiU is normally blocked by PKA phosphorylation even though its processing is impaired. This result provides compelling evidence that PKA phosphorylation of Ci inhibits the activator form of Ci independent of its role in promoting Ci processing. Taken together, our results suggest the following working model for the inhibitory function of PKA in the Hh pathway (Fig. 8). We propose that PKA phosphorylation of Ci in its carboxy-terminal region has two separable roles: (1) It blocks the activity of the full-length activator form of Ci, and (2) it targets the full-length Ci for Slimb-mediated proteolysis to generate the truncated repressor form of Ci. Such a dual regulation ensures that only the repressor form of Ci is active in the absence of any Hh signaling. This model accounts for the difference between PKA and slimb mutant phenotypes. In slimb mutant cells, Ci is not processed to the repressor form but accumulates in an inactive phosphorylated form, and, as a consequence, dpp is derepressed at low levels but ptc and en are not activated. In PKA mutant cells, however, Ci accumulates in an unphosphorylated or hypophosphorylated active form, and, as a consequence, ptc and en are activated.
Figure 8.

Model for dual regulation of Ci by PKA. Phosphorylation of Ci in its carboxy-terminal region by PKA keeps the full-length Ci (Ci155) in an inactive form and targets it for Slimb-mediated proteolytic processing to generate the truncated repressor form (Ci75). Su(fu) and Cos2 also negatively regulate Ci by forming a complex with Ci (see text for details).
How phosphorylation of Ci regulates its activity and proteolytic processing remained to be explored. It has been proposed that Su(fu) attenuates Hh signaling activity by blocking a maturation step that converts Ci into a short-lived nuclear transcriptional activator (Ohlmeyer and Kalderon 1998). Our analyses of slimb Su(fu) double mutant and slimb Su(fu) PKA triple mutant phenotypes suggest that inhibition of Ci activity by PKA is independent of Su(fu). When Ci processing is blocked, removing Su(fu) only partially stimulates the activity of the full-length Ci whereas simultaneously removing Su(fu) function and reducing PKA activity leads to virtually full activation of Ci (Fig. 7). These observations suggest that PKA and Su(fu) act in parallel through independent mechanisms to regulate the activity of the full-length Ci. In slimb Su(fu) double mutant cells, the majority of unprocessed full-length Ci appears to be transformed into a labile nuclear form (Ohlmeyer and Kalderon 1998), and yet the activity of this nuclear form of Ci seems to be inhibited by PKA (Fig. 7). This implies that PKA might inhibit Ci at a step after it enters the nucleus. For example, phosphorylation of Ci by PKA might prevent the formation of an active Ci transcription complex or might attenuate its ability to bind DNA. Another possible mechanism by which PKA exerts its influence on Ci is to regulate its nuclear trafficking. It has been shown recently that Hh signaling increases the nuclear import of full-length Ci (Chen et al. 1999). As PKA and Hh act antagonistically, it is possible that PKA phosphorylation of Ci might tether the full-length Ci in the cytoplasm in the absence of Hh signaling.
Su(fu), Cos2, and the Ser/Thr kinase Fu form a multiprotein complex with Ci and the complex associates with microtubules in a manner regulated by Hh (Robbins et al. 1997; Sisson et al. 1997; Monnier et al. 1998). It has been proposed that the assembly of the microtubule-associated Ci complex is critical for inhibiting Ci activity, possibly by tethering Ci in the cytoplasm (Chen et al. 1999). The relationship between PKA phosphorylation and the formation of Ci complex is not known. It is not clear whether they are two independent processes or whether one step might regulate the other. The nearly identical phenotypes caused by loss of PKA or Cos2 function in limb development suggest that these two regulatory events might be intimately related. For example, Cos2 might target Ci for efficient phosphorylation by PKA, allowing PKA to exert its negative regulation on Ci. Alternatively, phosphorylation of Ci by PKA might regulate the complex formation, allowing Cos2 to exert its influence on Ci. Further genetic and biochemical studies are required to resolve this important issue.
It has been proposed that phosphorylation of Ci by PKA allows Slimb to bind Ci and target it for ubiquitin/proteasome-mediated proteolysis (Jiang and Struhl 1998). The epistatic relationship between PKA and Slimb defined by this study is consistent with this hypothesis (Fig. 1). Moreover, it has been shown recently that proteasome is involved in Ci proteolytic processing (Chen et al. 1999). However, no evidence has been obtained to indicate that Ci is ubiquitinated (Chen et al. 1999). It is possible that the polyubiquitin chains added to Ci might be unstable and thus might escape detection. Alternatively, the proteolytic processing of Ci might not be directly mediated by ubiquitination, and Slimb might regulate Ci processing indirectly. For example, the SCFSlimb might promote the ubiquitination and degradation of an inhibitor of a protease that cleaves Ci.
Another important question that remains largely unanswered is how Hh antagonizes PKA. The structural similarity between Smo and G protein-coupled seven-transmembrane receptors suggests that Hh signaling might antagonize PKA by down-regulating its cAMP dependent kinase activity (Alcedo et al. 1996). However, the observations that a constitutively active cAMP independent form of PKA (mC*) can rescue PKA mutant phenotypes without perturbing normal Hh signaling both in embryos and in imaginal discs strongly argue against this possibility (Jiang and Struhl 1995; Li et al. 1995; Ohlmeyer and Kalderon 1997). The finding that high but not low levels of mC* are able to override Hh signaling is more consistent with a model in which Hh and PKA act competitively and antagonistically on Ci (Jiang and Struhl 1995; Li et al. 1995). For example, Hh may activate a phosphatase that removes the phosphates added to Ci by PKA. In support of this view, pharmacological evidence suggests that Hh stimulates target gene expression via a PP2A-like phosphatase in tissue culture cells (Krishnan et al. 1997; Chen et al. 1999). However, there is no genetic evidence for the involvement of a phosphatase in the Hh pathway.
In vertebrates, Hh signaling is mediated by three members of the Gli family of transcription factors: Gli1, Gli2, and Gli3 (Ruiz i Altaba 1997). Like Ci, all three Gli proteins contain multiple PKA phosphorylation consensus sites at conserved positions, so they are likely to be direct targets for PKA regulation in the vertebrate Hh signaling pathway. Among the three Gli proteins, Gli3 is both structurally and functionally related to Ci. Gli3 has been implicated to have both activator and repressor function depending on the developmental contexts (Buscher et al. 1997; Motoyama et al. 1998; Ruiz i Altaba 1998, 1999; Dai et al. 1999). Moreover, PKA appears to promote Gli3 processing to generate a putative repressor form (Dai et al. 1999; Ruiz i Altaba 1999). Thus, the mechanism by which PKA targets Ci for Slimb-mediated processing may well be conserved from invertebrates to vertebrates. Gli1 and Gli2 appear to function mainly as positive regulators in the vertebrate Hh signaling pathway (Lee et al. 1997; Ding et al. 1998; Matise et al. 1998). Unlike Gli3 and Ci, Gli1 and Gli2 do not undergo PKA-dependent processing (Dai et al. 1999; Ruiz i Altaba 1999), however, their activities are likely to be regulated by PKA. For example, it has been shown that overexpression of a constitutive active form of PKA represses the transcriptional activity of Gli1 in mammalian culture cells (Ruiz i Altaba 1999). Thus, whereas the mechanism by which PKA regulates Ci processing may only apply to Gli3, the processing-independent inhibitory mechanism defined by this study may well apply to all three Gli proteins and is likely to be a more general mechanism by which PKA negatively regulates Hh signaling.
Material and methods
Transgenes and mutations
To construct UAS–HACi, a double HA-epitope tag was added to the amino terminus of Ci at the MluI site, and the HA-tagged Ci was inserted between the KpnI and XbaI sites in the pUAST vector (Brand and Perrimon 1993). UAS–HACi−3P was derived from UAS–HACi with three PKA phosphorylation consensus sites mutated (S838A, S856A, and S892A) by PCR-based site-directed mutagenesis. UAS–HACi−3P was derived from UAS–HACiU (Methot and Basler 1999), with the same three PKA sites mutated as for HACi−3P. UAS-R* and UAS-mC* were described in Li et al. (1995). Reporter genes used for this study were dpp–lacZ (Blackman et al. 1991) and ptc–lacZ (Chen and Struhl 1997); Gal4 lines were MS1096 (Capidevila and Guerrero 1994); tub>CD2>GV (generated by Konrad Basler, as cited in Struhl and Adachi 1998); marker genes were hsp–CD2 (Jiang and Struhl 1995) and hsp–myc–GFP (Jiang and Struhl 1998). slimb1 is a hypomorphic allele that is specific for the Hh pathway (Jiang and Struhl 1998), DC0E95 is an apparently null allele of the DC0 gene that encodes the catalytic subunit of PKA (Jiang and Struhl 1998), and Su(fu)LP is a null allele (Preat 1992).
Generating clones of marked cells
Clones of mutant cells were generated by FLP/FRT-mediated mitotic recombination (Xu and Rubin 1993). Genotypes for generating clones are as follows: (1) slimb mutant clones: y hsp–flp.1/y or Y; FRT82B slimb1/FRT82B hsp–CD2, y+ (or hsp–myc–GFP, w+) with or without dpp–lacZ or ptc–lacZ on the second chromosome. (2) PKA mutant clones: y hsp–flp.1/y or Y; DC0E95 stc FRT39, w+/ hsp–CD2, y+ FRT39, w+ with or without dpp–lacZ or ptc–lacZ on the third chromosome. (3) slimb Su(fu) mutant clones: y hsp–flp.1/y or Y; FRT82B Su(fu)LP slimb1/FRT82B, hsp–myc–GFP, w+. (4) slimb mutant clones expressing mC* yw MS1096 hsp–flp/y or Y; UAS-mC*/ptc–lacZ; FRT82B slimb1/FRT82B, hsp–myc–GFP, w+. (5) slimb mutant clones expressing R*: yw MS1096 hsp–flp/y or Y; UAS–R*/ptc–lacZ; FRT82B slimb1/FRT82B hsp–myc–GFP, w+. (6) slimb Su(fu) mutant clones expressing R*: yw MS1096 hsp–flp/y or Y; UAS–R*/ptc-lacZ; FRT82B Su(fu)LP slimb1/FRT82B, hsp–myc–GFP, w+.
Imaginal disc staining and Western blot analysis
Standard protocols for immunofluorescence staining and Western blot analysis of imaginal discs were used (Jiang and Struhl 1998). Primary antibodies used for this study are: monoclonal rat anti-Ci antibody, 2A1(Motzny and Holmgren 1995); mouse anti-HA antibody (Santa Cruz); rabbit anti-βgal (Cappel); mouse anti-Myc (Santa Cruz); monoclonal mouse anti-CD2, OX34 (Seretec); monoclonal mouse anti-En (DSHB, University of Iowa).
Acknowledgments
We thank Drs. Dan Kalderon, Konrad Basler, and Gary Struhl for providing plasmids and fly stocks. We are grateful to Drs. James Chen, Jon Graff, Mark Henkemeyer, and D.J. Pan for critically reading the manuscript. This work was initiated in Gary Struhl's laboratory. J.J. thanks Dr. Gary Struhl for his generous support and Atsuko Adachi for injecting some of the constructs used in this study. J.J. is a Searle Scholar supported by the Chicago Community Trust, a Eugene McDermott Endowed Scholar in Biomedical Research, and a Leukemia Society Special Fellow.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jiang@utsw.swmed.edu; FAX (214) 648-1960.
References
- Alcedo J, Noll M. Hedgehog and its Patched-Smoothened receptor complex: a Novel signalling mechanism at the cell surface. Biol Cenm. 1997;378:583–590. doi: 10.1515/bchm.1997.378.7.583. [DOI] [PubMed] [Google Scholar]
- Alcedo J, Ayzenzon M, Von Ohlen T, Noll M, Hooper JE. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the Hedgehog signal. Cell. 1996;86:231–232. doi: 10.1016/s0092-8674(00)80094-x. [DOI] [PubMed] [Google Scholar]
- Alexandre C, Jacinto A, Ingham PW. Transcriptional activation of Hedgehog target genes in Drosophila is mediated directly by the Cubitus interruptus protein, a member of the GLI family of the zinc finger DNA-binding proteins. Genes & Dev. 1996;10:2003–2013. doi: 10.1101/gad.10.16.2003. [DOI] [PubMed] [Google Scholar]
- Alves G, Limbourg-Bouchon B, Tricoire H, Brissard-Zahraoui J, Lamour-Isnard C, Busson D. Modulation of Hedgehog target gene expression by the Fused serine-threonine kinase in wing imaginal discs. Mech Dev. 1998;78:17–31. doi: 10.1016/s0925-4773(98)00130-0. [DOI] [PubMed] [Google Scholar]
- Aza-Blanc P, Ramirez-Weber F, Laget M, Schwartz C, Kornberg T. Proteolysis that is inhibited by Hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell. 1997;89:1043–1053. doi: 10.1016/s0092-8674(00)80292-5. [DOI] [PubMed] [Google Scholar]
- Basler K, Struhl G. Compartment boundaries and the control of Drosophila limb pattern by hedgehog protein. Nature. 1994;368:208–214. doi: 10.1038/368208a0. [DOI] [PubMed] [Google Scholar]
- Blackman RK, Sanicola M, Raftery LA, Gillevet T, Gelbart WM. An extensive 3′ cis-regulatory region directs the imaginal disk expression of decapentaplegic, a member of the TGF-beta family in Drosophila. Development. 1991;111:657–666. doi: 10.1242/dev.111.3.657. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Buscher D, Bosse B, Heymer J, Ruther U. Evidence for genetic control of Sonic hedgehog by Gli3 in mouse limb development. Mech Dev. 1997;62:175–182. doi: 10.1016/s0925-4773(97)00656-4. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Guerrero I. Targeted expression of the signalling molecule decapentaplegic induces pattern duplications and growth alterations in Drosophila wings. EMBO J. 1994;13:4459–4468. doi: 10.1002/j.1460-2075.1994.tb06768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Struhl S. Dual roles for patched in sequestering and transducing hedgehog. Cell. 1996;87:553–563. doi: 10.1016/s0092-8674(00)81374-4. [DOI] [PubMed] [Google Scholar]
- Chen CH, von Kessler DP, Park W, Wang B, Ma Y, Beachy PA. Nuclear trafficking of Cubitus interruptus in the transcriptional regulation of Hedgehog target gene expression. Cell. 1999;98:305–316. doi: 10.1016/s0092-8674(00)81960-1. [DOI] [PubMed] [Google Scholar]
- Chen Y, Gallaher N, Goodman RH, Smolik SM. Protein kinase A directly regulates the activity and proteolysis of cubitus interruptus. Proc Natl Acad Sci. 1998;95:2349–2354. doi: 10.1073/pnas.95.5.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Akimaru H, Tanaka T, Maekawa T, Nakafuku M, Ishii S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J Biol Chem. 1999;274:8143–8152. doi: 10.1074/jbc.274.12.8143. [DOI] [PubMed] [Google Scholar]
- Dean M. Polarity, proliferation and the hedgehog pathway. Nat Genet. 1996;14:245–247. doi: 10.1038/ng1196-245. [DOI] [PubMed] [Google Scholar]
- Ding Q, Motoyama J, Gasca S, Mo R, Sasaki H, Rossant J, Hui CC. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development. 1998;125:2533–2543. doi: 10.1242/dev.125.14.2533. [DOI] [PubMed] [Google Scholar]
- Dominguez M, Brunner M, Hafen E, Basler K. Sending and receiving the Hedgehog signal: Control by the Drosophila Gli protein Cubitus interruptus. Science. 1996;272:1621–1625. doi: 10.1126/science.272.5268.1621. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Scott MP. Hedgehog and patched in neural development and disease. Neuron. 1998;21:1243–1257. doi: 10.1016/s0896-6273(00)80645-5. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M, Brook A, McMahon A. The world according to Hedgehog. Trends Genet. 1997;13:14–21. doi: 10.1016/s0168-9525(96)10051-2. [DOI] [PubMed] [Google Scholar]
- Ingham PW. Hedgehog points the way. Curr Biol. 1994;4:347–350. doi: 10.1016/s0960-9822(00)00076-2. [DOI] [PubMed] [Google Scholar]
- ————— Transducing Hedgehog: The story so far. EMBO J. 1998;17:3505–3511. doi: 10.1093/emboj/17.13.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Struhl G. Protein kinase A and Hedgehog signalling in Drosophila limb development. Cell. 1995;80:563–572. doi: 10.1016/0092-8674(95)90510-3. [DOI] [PubMed] [Google Scholar]
- ————— Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature. 1998;391:493–496. doi: 10.1038/35154. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Grenier JK, Scott MP. patched overexpression alters wing disc size and pattern: Transcriptional and post-transcriptional effects on hedgehog targets. Development. 1995;121:4161–4170. doi: 10.1242/dev.121.12.4161. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Pereira FA, Qiu Y, Chen CH, Beachy PA, Tsai SY, Tsai MJ. Mediation of Sonic hedgehog-induced expression of COUP-TFII by a protein phosphatase. Science. 1997;278:1947–1950. doi: 10.1126/science.278.5345.1947. [DOI] [PubMed] [Google Scholar]
- Lecuit T, Brook WJ, Ng M, Callega M, Sun H, Cohen SM. Two distinct mechanisms for long-range patterning by Decapentaplegic in the Drosophila wing. Nature. 1996;381:387–393. doi: 10.1038/381387a0. [DOI] [PubMed] [Google Scholar]
- Lee J, Platt KA, Censullo P, Ruiz i Altaba A. Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development. 1997;124:2537–2552. doi: 10.1242/dev.124.13.2537. [DOI] [PubMed] [Google Scholar]
- Lepage T, Cohen SM, Diaz-Benjumea FJ, Parkhurst SM. Signal transduction by cAMP-dependent protein kinase A in Drosophila limb patterning. Nature. 1995;373:711–715. doi: 10.1038/373711a0. [DOI] [PubMed] [Google Scholar]
- Li W, Ohlmeyer JT, Lane ME, Kalderon D. Function of protein kinase A in hedghehog signal transduction and Drosophila imaginal disc development. Cell. 1995;80:553–562. doi: 10.1016/0092-8674(95)90509-x. [DOI] [PubMed] [Google Scholar]
- Maniatis T. A ubiquitin ligase complex essential for the NF-κB, Wnt/Wingless, and Hedgehog signaling pathways. Genes & Dev. 1999;13:505–510. doi: 10.1101/gad.13.5.505. [DOI] [PubMed] [Google Scholar]
- Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development. 1998;125:2759–2770. doi: 10.1242/dev.125.15.2759. [DOI] [PubMed] [Google Scholar]
- Methot N, Basler K. Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell. 1999;96:819–831. doi: 10.1016/s0092-8674(00)80592-9. [DOI] [PubMed] [Google Scholar]
- Monnier V, Dussillol F, Alves G, Lamour-Isnard C, Plessis A. Suppressor of fused links fused and Cubitus interruptus on the hedgehog signalling pathway. Curr Biol. 1998;8:583–586. doi: 10.1016/s0960-9822(98)70227-1. [DOI] [PubMed] [Google Scholar]
- Motoyama J, Liu J, Mo R, Ding Q, Post M, Hui CC. Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat Genet. 1998;20:54–57. doi: 10.1038/1711. [DOI] [PubMed] [Google Scholar]
- Motzny CK, Holmgren R. The Drosophila cubitus interruptus protein and its role in the wingless and hedgehog signal transduction pathways. Mech Dev. 1995;52:137–150. doi: 10.1016/0925-4773(95)00397-j. [DOI] [PubMed] [Google Scholar]
- Nellen D, Burke R, Struhl G, Basler K. Direct and long-range action of a DPP morphogen gradient. Cell. 1996;85:357–368. doi: 10.1016/s0092-8674(00)81114-9. [DOI] [PubMed] [Google Scholar]
- Ohlmeyer JT, Kalderon D. Dual pathways for induction of wingless expression by protein kinase A and Hedgehog in Drosophila embryos. Genes & Dev. 1997;11:2250–2258. doi: 10.1101/gad.11.17.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Hedgehog stimulates maturation of Cubitus interruptus into a labile transcriptional activator. Nature. 1998;396:749–753. doi: 10.1038/25533. [DOI] [PubMed] [Google Scholar]
- Orenic TV, Slusarski DC, Kroll KL, Holmgren RA. Cloning and characterization of the segment polarity gene cubitus interruptus Dominant of Drosophila. Genes & Dev. 1990;4:1053–1067. doi: 10.1101/gad.4.6.1053. [DOI] [PubMed] [Google Scholar]
- Pan D, Rubin GM. cAMP-dependent protein kinase and hedgehog act antagonistically in regulating decapentaplegic transcription in Drosophila imaginal discs. Cell. 1995;80:543–552. doi: 10.1016/0092-8674(95)90508-1. [DOI] [PubMed] [Google Scholar]
- Preat T. Characterization of Suppressor of fused, a complete suppressor of the fused segment polarity gene of Drosophila melanogaster. Genetics. 1992;132:725–736. doi: 10.1093/genetics/132.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins DJ, Nybakken KE, Kobayashi R, Sisson JC, Bishop JM, Therond PP. Hedgehog elicits signal transduction by means of a large complex containing the kinesin-related protein costal2. Cell. 1997;90:225–234. doi: 10.1016/s0092-8674(00)80331-1. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. Catching a Gli-mpse of Hedgehog. Cell. 1997;90:193–196. doi: 10.1016/s0092-8674(00)80325-6. [DOI] [PubMed] [Google Scholar]
- ————— Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development. 1998;125:2203–2212. doi: 10.1242/dev.125.12.2203. [DOI] [PubMed] [Google Scholar]
- ————— Gli proteins encode context-dependent positive and negative functions: Implications for development and disease. Development. 1999;126:3205–3216. doi: 10.1242/dev.126.14.3205. [DOI] [PubMed] [Google Scholar]
- Sisson JC, Ho KS, Suyama K, Scott MP. Costal2, a novel kinesin-related protein in the Hedgehog signaling pathway. Cell. 1997;90:235–245. doi: 10.1016/s0092-8674(00)80332-3. [DOI] [PubMed] [Google Scholar]
- Strigini M, Cohen SM. A Hedgehog activity gradient contributes to AP axial patterning of the Drosophila wing. Development. 1997;124:4697–4705. doi: 10.1242/dev.124.22.4697. [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A. Nuclear access and action of notch in vivo. Cell. 1998;93:649–660. doi: 10.1016/s0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- Struhl G, Basler K. Organizing activity of wingless protein in Drosophila. Cell. 1993;72:527–540. doi: 10.1016/0092-8674(93)90072-x. [DOI] [PubMed] [Google Scholar]
- Strutt DI, Wiersdorff V, Mlodzik M. Regulation of furrow progression in the Drosophila eye by cAMP-dependent protein kinase A. Nature. 1995;373:705–709. doi: 10.1038/373705a0. [DOI] [PubMed] [Google Scholar]
- Tabata T, Kornberg TB. Hedgehog is a signaling protein with a key role in patterning Drosophila imaginal discs. Cell. 1994;76:89–102. doi: 10.1016/0092-8674(94)90175-9. [DOI] [PubMed] [Google Scholar]
- Theodosiou NA, Zhang S, Wang WY, Xu T. slimb coordinates wg and dpp expression in the dorsal-ventral and anterior-posterior axes during limb development. Development. 1998;125:3411–3416. doi: 10.1242/dev.125.17.3411. [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Zecca M, Basler K, Struhl G. Sequential organizing activities of engrailed, hedgehog and decapentaplegic in the Drosophila wing. Development. 1995;121:2265–2278. doi: 10.1242/dev.121.8.2265. [DOI] [PubMed] [Google Scholar]