

Abstract
The bacteriophage λ repressor and its relatives bind cooperatively to adjacent as well as artificially separated operator sites. This cooperativity is mediated by a protein–protein interaction between the DNA-bound dimers. Here we use a genetic approach to identify two pairs of amino acids that interact at the dimer–dimer interface. One of these pairs is nonconserved in the aligned sequences of the λ and P22 repressors; we show that a λ repressor variant bearing the P22 residues at these two positions interacts specifically with the P22 repressor. The other pair consists of a conserved ion pair; we reverse the charges at these two positions and demonstrate that, whereas the individual substitutions abolish the interaction of the DNA-bound dimers, these changes in combination restore the interaction of both λcI and P22c2 dimers.
Keywords: Charge reversal mutants, cooperativity, λ repressor, P22 repressor, mutant-suppressor pairs, protein–protein interactions
Transcriptional regulatory proteins that bind to DNA participate in a variety of protein–protein interactions, both strong and weak, to carry out their functions. They interact with the transcription apparatus and also with one another in homologous and heterologous combinations. The strong interactions occur in solution prior to DNA binding, whereas, under physiological conditions at least, the weaker interactions result in the formation of stable complexes only when the participants are appropriately positioned on the DNA. Thus, DNA-binding regulators that interact weakly with one another exhibit cooperative binding to the DNA, with the result that relatively modest changes in protein concentrations can produce large changes in the fractional occupancy of the relevant binding sites.
There are only a few cases in which the interactions of cooperatively bound DNA-binding proteins have been characterized in chemical or structural detail (e.g., see Hughes et al. 1990; Somers and Phillips 1992; Raumann et al. 1994). The bacteriophage λ cI protein (the λ repressor, or λcI) provides an example of cooperative binding to DNA that has been well characterized biochemically and genetically (for review, see Sauer et al. 1990; Hochschild and Ptashne 1988; Beckett et al. 1993; Benson et al. 1994; Burz and Ackers 1994; Whipple et al. 1994). λcI, which functions as both a repressor and an activator of transcription, is a two-domain protein that binds each of its operator sites as a dimer (Ptashne 1992). Pairs of dimers can also interact, binding cooperatively to adjacent operator sites on the phage chromosome (Johnson et al. 1979) and also to artificially separated operators (Hochschild and Ptashne 1986). Figure 1A depicts the arrangement of λcI molecules within the phage’s right operator region (OR) in a λ lysogen. A λcI dimer bound at the high-affinity operator OR1 interacts with a second dimer, stabilizing its binding to the lower affinity site OR2, and that dimer in turn interacts with RNA polymerase (RNAP) to activate transcription from promoter PRM (for review, see Ptashne 1992). This pattern of site occupancy simultaneously represses transcription from promoter PR (by promoter occlusion). The amino-terminal domain (NTD) of λcI is the DNA-binding domain and also contains the residues involved in activation (Pabo and Lewis 1982; Guarente et al. 1982; Hochschild et al. 1983; Bushman et al. 1989), whereas the carboxy-terminal domain (CTD) mediates both dimerization and cooperativity (Pabo et al. 1979), using distinct surfaces to do so (see below).
Figure 1.

(A) The arrangement of molecules at the right operator (OR) region of a λ lysogen. Dimers of λcI protein (λ repressor) bind cooperatively to the strong OR1 and the weaker OR2 operator sites, repressing promoter PR and activating promoter PRM. Activation is mediated specifically by the dimer bound at OR2, which interacts with RNAP. (B) The identities of selected amino acids in λcI and their counterparts in P22c2.
The related lambdoid phage P22 also encodes a repressor (P22c2) that binds cooperatively to adjacent and artificially separated operators (Poteete and Ptashne 1982; Valenzuela and Ptashne 1989). Like λcI, P22c2 has two domains, and the NTD mediates DNA binding whereas the CTD mediates dimerization and cooperativity (Poteete and Ptashne 1982). Both λcI and P22c2 mutants that are specifically defective for cooperativity have been identified (Hochschild and Ptashne 1988; Valenzuela and Ptashne 1989; Beckett et al. 1993; Benson et al. 1994; Burz and Ackers 1994; Whipple et al. 1994); most of these mutants, which bear amino acid substitutions in the CTD, manifest no defects in dimerization. Although λcI and P22c2 exhibit considerable amino acid sequence homology in their CTDs (Sauer et al. 1982), heterodimers do not form nor do the respective homodimers interact with one another. Nevertheless, previous work suggests that the two proteins use homologous surfaces to mediate cooperativity. In particular, a number of conserved residues have been identified that are apparently critical for both λcI and P22c2 cooperativity (Valenzuela and Ptashne 1989; Whipple et al. 1994). Furthermore, we have shown previously that the replacement of six nonconserved residues in λcI with their P22c2 counterparts permits the resulting λcI variant to form dimers that interact specifically with P22c2 dimers (Whipple et al. 1994).
Here, we test the hypothesis that the specificity of these protein–protein interactions derives, at least in part, from the interactions of specific pairs of amino acids at the dimer–dimer interface. We first identify a pair of nonconserved residues whose side chains interact at the dimer–dimer interfaces of both λcI and P22c2. This pair comprises two of the six residues altered in the previously constructed switched specificity variant of λcI. We show that the identity of this residue pair alone accounts fully for the abilities of λcI and P22c2 dimers to discriminate between one another. We then identify a pair of conserved residues whose side chains interact at both the λcI and the P22c2 dimer–dimer interfaces. These residues are oppositely charged, forming an ion pair. We construct both λcI and P22c2 mutants bearing amino acid substitutions that reverse the charges at these two positions. In each case, dimers bearing both substitutions can interact, whereas dimers bearing either one or the other cannot, indicating that the complementarity of these side chains is a critical determinant of the dimer–dimer interaction.
Results
Identification of a nonconserved pair of interacting residues
To identify pairs of amino acids that interact at the λcI dimer–dimer interface, we sought to isolate specific suppressors of selected cooperativity mutants. We first focused on a nonconserved residue that had previously been implicated in both λcI and P22c2 cooperativity, asparagine (N) 148 in λcI and the corresponding aspartic acid (D) 131 in P22c2 (see Fig. 1B). Replacement of each of these residues with its counterpart (i.e., λcI N148D and P22c2 D131N) weakens cooperativity (Valenzuela and Ptashne 1989; Whipple et al. 1994). Furthermore, the N148D replacement is necessary (but not sufficient) to switch the specificity of λcI dimers so that they can interact specifically with P22c2 dimers (Whipple et al. 1994). We sought to identify the interacting partner for residue 148 by screening for a repressor mutant that could interact efficiently with λcI mutant N148D (λcI–N148D).
Our strategy utilized a hybrid repressor that bears the DNA-binding domain of P22c2 and the CTD of λcI. This hybrid repressor forms dimers that bind specifically to P22 operators and can interact with a λcI dimer if the latter is bound nearby on the same side of the DNA helix (Whipple et al. 1994). The use of this hybrid repressor permitted us to direct the binding of a preselected cooperativity mutant to a λ operator and potential suppressor mutants to a P22 operator. To detect the interaction of the DNA-bound repressors, we used a reporter system in which the lacZYA operon is controlled by an artificial promoter that is flanked by λ and P22 operator sites (Fig. 2). These operators are positioned such that transcription of the linked lacZYA operon is repressed only if the DNA-bound repressors interact (i.e., not by site occupancy per se). Therefore, the assay can be done under conditions of full site occupancy so that increased repression cannot result from increased site occupancy, but instead reflects a stronger interaction between the DNA-bound dimers.
Figure 2.

Interactions of hybrid repressor derivatives with λcI derivatives. Strain FW89B (which contains the depicted reporter construct) was cotransformed with versions of plasmids pFW7-280Δ and pA3B2 that encode the hybrid repressor and λcI, respectively. Each repressor protein bears either a wild-type λcI CTD or a mutant derivative thereof, as listed. β-Galactosidase activities of cultures grown in the presence of 300 μm IPTG were measured. Results are averages of duplicate assays, which varied by less than ±3% from the mean.
A strain harboring this reporter on an F′ episome produces red colonies on lactose tetrazolium indicator plates when transformed with two compatible plasmids that express wild-type λcI and the unmutated hybrid repressor, respectively. Introduction of a cooperativity mutation into the CTD of either repressor disrupts repression resulting in the formation of pink or white colonies. Thus, we reasoned that compensatory mutations could be identified because transformants containing mutant-suppressor pairs would form red colonies. We therefore mutagenized the CTD-encoding portion of a plasmid that directs expression of the hybrid repressor and screened for variants that resulted in the production of red colonies when introduced into indicator strain cells already containing the λcI–N148D mutant.
We isolated three hybrid repressor mutants on the basis of both colony color and quantitative β-galactosidase assays. Two of them exhibited increased repression when tested in combination with wild-type λcI; these were not studied further. However, one of the mutants, bearing the substitution glutamine 204 to arginine (Q204R), specifically compensated for the defect of λcI–N148D. When tested together with either wild-type λcI or another arbitrarily chosen cooperativity mutant (λcI–R196G; Whipple et al. 1994), it manifested decreased repression (see Fig. 2).
The replacement of Q204 with a basic residue in the suppressor mutant suggested that the presence of a pair of oppositely charged residues at positions 148 and 204 can function in place of the pair of hydrophilic residues present in wild-type λcI. Noting that the corresponding positions in P22c2 also comprise a charged pair (aspartic acid and lysine; see Fig. 1B), we constructed a hybrid repressor variant bearing lysine at position 204 and tested it together with λcI–N148D. Like the Q204R change, the Q204K change decreased the ability of the hybrid repressor to repress transcription in the presence of wild-type λcI, but increased its ability to repress transcription in the presence of λcI–N148D (Fig. 2). In fact, both mutant-suppressor pairs (the hybrid repressor bearing either the Q204R or the Q204K change together with λcI–N148D) repressed transcription as efficiently as the unmodified parent proteins (cf. lines 6 and 9 with line 2), suggesting that each pair interacted as strongly as the parent proteins.
Then, we tested whether removal of the hydrophilic side chain at position 204 would suffice to suppress the defect of λcI–N148D. We found that introduction of an alanine (A) at position 204 in the hybrid repressor did not significantly increase its ability to repress transcription in the presence of λcI–N148D (Fig. 2, cf. lines 3 and 11), consistent with the hypothesis that a basic side chain at position 204 is required to suppress the defect caused by the introduction of an acidic side chain at position 148 in λcI. Strikingly, the Q204A substitution did not decrease the ability of the hybrid repressor to repress transcription in the presence of wild-type λcI (Fig. 2, cf. lines 2 and 10). This suggests that, in wild-type λcI, any interaction between the amide side chains of residues 148 and 204 does not contribute in a significant way to the overall interaction energy of the cooperatively bound dimers (see Discussion).
A λcI variant bearing substitutions at both positions 148 and 204
On the basis of the results of Figure 2, we predicted that λcI dimers bearing both the N148D and the Q204K substitutions would interact as efficiently as wild-type λcI dimers. Therefore, we constructed a λcI variant bearing both of these substitutions and tested it using a reporter system in which the lacZ gene is controlled by the λ PRM promoter modified so that OR1 is centered either an integral or a nonintegral number of turns of the DNA helix away from OR2 (either 5.9 or 5.5 turns; Fig. 3; Hochschild and Ptashne 1988; Whipple et al. 1994). Ordinarily, a λcI dimer bound at OR2 activates transcription from PRM. On the in-phase template, however, the interaction of nonadjacently bound dimers inhibits this activation, whereas on the out-of-phase template, the dimers do not interact and, therefore, full stimulation is observed provided λcI is supplied at a concentration sufficient to ensure full occupancy of OR2 (the assay conditions used).
Figure 3.

Activation of PRM by wild-type and mutant λcI dimers bound at nonadjacent operator sites. Strains AH-5.9 (in-phase reporter) and AH-5.5 (out-of-phase reporter) were transformed with derivatives of plasmid pA3B2 (A) or pLR2 (B) encoding wild-type or mutant derivatives of λcI. Fold activation was determined by calculation of the ratio of β-galactosidase activity observed in each case to that observed in cells transformed with a control plasmid that lacks the λcI gene. β-Galactosidase activities in the control cultures were between 1.6 and 2.5 Miller units in all cases. Results are averages of duplicate assays, which varied by less than ±6.5% from the mean.
Figure 3A shows the results of β-galactosidase assays performed to quantify the effects of the single and double amino acid substitutions at positions 148 and 204 on the abilities of λcI dimers to interact. Plasmids directing the synthesis of each mutant were introduced into the in-phase and the out-of-phase reporter strains. As we showed previously, λcI dimers bearing the N148D substitution interacted more weakly than wild-type λcI dimers (Whipple et al. 1994), and λcI dimers bearing the Q204K substitution failed to interact. However, as expected on the basis of the results shown in Figure 2, dimers bearing both substitutions behaved indistinguishably from wild-type dimers, as did dimers bearing only the Q204A substitution. Dimers bearing the N148A substitution exhibited a detectable, but relatively small defect in their ability to interact.
Two amino acid substitutions suffice to permit λcI dimers to interact specifically with P22c2 dimers
As mentioned above, the residues at positions 148 and 204 are among the six that were altered in the previously constructed λcI variant that forms dimers able to interact specifically with P22c2 dimers (Whipple et al. 1994). This led us to ask whether the λcI variant bearing only the N148D and the Q204K substitutions would interact specifically with P22c2 dimers. To perform this test, we used a pair of strains bearing a lacZ gene controlled by an artificial promoter with a low-affinity P22 operator between its −10 and −35 hexamers and a high-affinity λ operator located either an integral (5.0) or a nonintegral (5.4) number of turns of the helix upstream (Fig. 4). The binding of P22c2 to the low-affinity site represses lacZ transcription, and cooperative binding to the in-phase operators on the 5.0-turn template results in enhanced occupancy of this site, and hence, enhanced repression (Whipple et al. 1994). Therefore, in this case, the assay conditions were chosen to permit P22c2 to occupy partially the low-affinity P22 operator on the out-of-phase reporter.
Figure 4.

Cooperative binding of λcI with P22c2 or with the P22c2–λcI hybrid repressor. Strains FW40 (in-phase reporter) and FW42 (out-of-phase reporter) were cotransformed with a plasmid encoding the listed λcI variant (pA3B2 or a derivative thereof) and a second plasmid encoding either P22c2 (A; pPR2) or the P22c2–λcI hybrid repressor (B; pFW7-280Δ). β-Galactosidase activities were measured, and the degree of repression was calculated relative to cultures of the same strains transformed with a pair of control plasmids neither of which encodes a repressor. All cultures were grown in medium containing 300 μm IPTG. Results are averages of duplicate assays, which varied by less than ±8% from the mean. β-Galactosidase activities in the absence of any repressor in strains FW40 and FW42 were 3487 and 2916 Miller units, respectively, in the experiment of A, and 3790 and 3971 Miller units, respectively, in the experiment of B.
The experiment of Figure 4A shows that substitutions N148D and Q204K suffice to confer on λcI dimers the ability to interact with P22c2 dimers. In the presence of P22c2 only, lacZ transcription in either the strain bearing the in-phase or the out-of-phase reporter was repressed approximately fivefold relative to the level measured in the absence of any repressor. This repression was enhanced marginally when wild-type λcI was introduced together with P22c2 in the in-phase reporter strain only. However, when either the previously described switched-specificity variant λcIv1–5;148 (Whipple et al. 1994) or the λcI–N148D;Q204K double mutant was introduced together with P22c2, the magnitude of the repression in the in-phase reporter strain was increased substantially (to between 22- and 27-fold), indicating that both of these λcI variants can interact efficiently with wild-type P22c2.
To confirm that the λcI–N148D;Q204K double mutant is a switched-specificity mutant and not simply relaxed in its specificity, we also tested its ability to interact with wild-type λcI (i.e., its CTD). To do this, we used the same pair of reporter strains and replaced P22c2 with the P22c2–λcI hybrid repressor used in the experiment of Figure 2. Whereas wild-type λcI was able to interact with the hybrid repressor, both variants were unable to do so (Fig. 4B).
The results presented so far strongly suggest that λcI residues 148 and 204 approach each other at the dimer–dimer interface. We first identified the substitutions Q204R and Q204K as specific extragenic suppressors of the cooperativity defect caused by the N148D substitution. Then, we showed that the dimer–dimer interaction is not impaired when the wild-type amide side chains at these two positions of λcI are replaced with oppositely charged side chains, as are found at the corresponding positions of P22c2. Finally, we showed that these two changes are sufficient to confer on λcI dimers a switched specificity such that they can interact specifically with P22c2 dimers.
Identification of a conserved pair of interacting residues
Next, we sought to identify amino acids conserved in λcI and P22c2 that interact at the dimer–dimer interface. We had suggested previously that D197 in λcI and the corresponding D179 in P22c2 play essential roles in cooperativity on the basis of the fact that replacement of each of these aspartic acids with a glycine abolished cooperativity (Whipple et al. 1994; see also Benson et al. 1994). We hypothesized that the aspartic acid side chain might participate in an electrostatic bond at the dimer–dimer interface, and therefore, we sought a conserved basic residue as a candidate partner for D197. Inspection of the amino acid sequences of the two repressors revealed only one conserved basic residue previously implicated in cooperativity, K192 in λcI (corresponding to K174 in P22c2; see Fig. 1B). Therefore, we tested whether residues K192 and D197 are partner residues that interact specifically at the λcI dimer–dimer interface.
In this case, we used a recently developed method for detecting the interaction of protein domains (Dove et al. 1997; Dove and Hochschild 1998). This system is based on the observation that contact between any protein domain (Y) fused to a DNA-bound protein and a partner domain (X) fused to a subunit of RNA polymerase (RNAP) can activate transcription from an appropriately engineered test promoter. In particular, the interaction of a DNA-bound λcI dimer with the wild-type λcI CTD fused to the α subunit of RNAP (hereafter referred to as the wild-type α–cI chimera) activates transcription from a modified lac promoter bearing a λ operator in its upstream region (see Fig. 5). This activation is abolished when either λcI or the α–cI chimera bears an amino acid substitution that normally disrupts cooperativity (Dove et al. 1997). We first verified that variants bearing either the substitution Q204K (introduced into λcI) or N148D (introduced into the α–cI chimera) caused a reduction in activation when tested with a wild-type partner, but resulted in full activation when tested together (data not shown).
Figure 5.

Effects of alanine substitutions at positions 192 and 197 on interaction of λcI CTDs. Depicted above the graph is the reporter construct present in strain KS1. Interaction between the CTDs of the DNA-bound λcI dimer and the CTDs fused to the α-NTD results in transcriptional activation from this reporter. The graph shows the results of β-galactosidase assays performed on cultures of KS1 cells containing plasmids pACλcI and pBRα–cI (○), pACλcI and pBRα–cI–D197A (▾), or pACλcI–K192A and pBRα–cI (•) grown in medium containing IPTG at concentrations of 0, 5, and 20 μm. Graph labels list the λcI allele first and the α–cI allele second. Previous experiments with this system have shown that maximum β-galactosidase values are achieved at IPTG concentrations between 10 and 20 μm (Dove et al. 1997). Control experiments indicate that, at these IPTG concentrations, the λ operator is saturated and the fraction of RNAP molecules containing the α–cI chimera approaches a maximum (S.L. Dove and A. Hochschild, unpubl.).
To confirm that the side chains of both D197 and K192 are essential for the dimer–dimer interaction, we introduced alanine residues at positions 192 (in λcI) and 197 (in the α–cI chimera) and measured β-galactosidase activities in reporter strain cells bearing compatible plasmids that direct the expression of λcI and the α–cI chimera under the control of IPTG-inducible promoters. As expected, neither alanine-substituted variant was able to interact with a wild-type partner molecule (Fig. 5).
Then we sought to obtain evidence that the side chains of K192 and D197 interact directly at the dimer–dimer interface by identifying mutant-suppressor pairs. We first tried exchanging the amino acids at these positions. That is, we replaced K192 with asparatic acid in the λcI molecule and D197 with lysine in the α–cI chimera. Whereas neither substituted variant interacted with a wild-type partner, the two mutants interacted with one another, resulting in a partial restoration of transcriptional activation (Fig. 6A).
Figure 6.
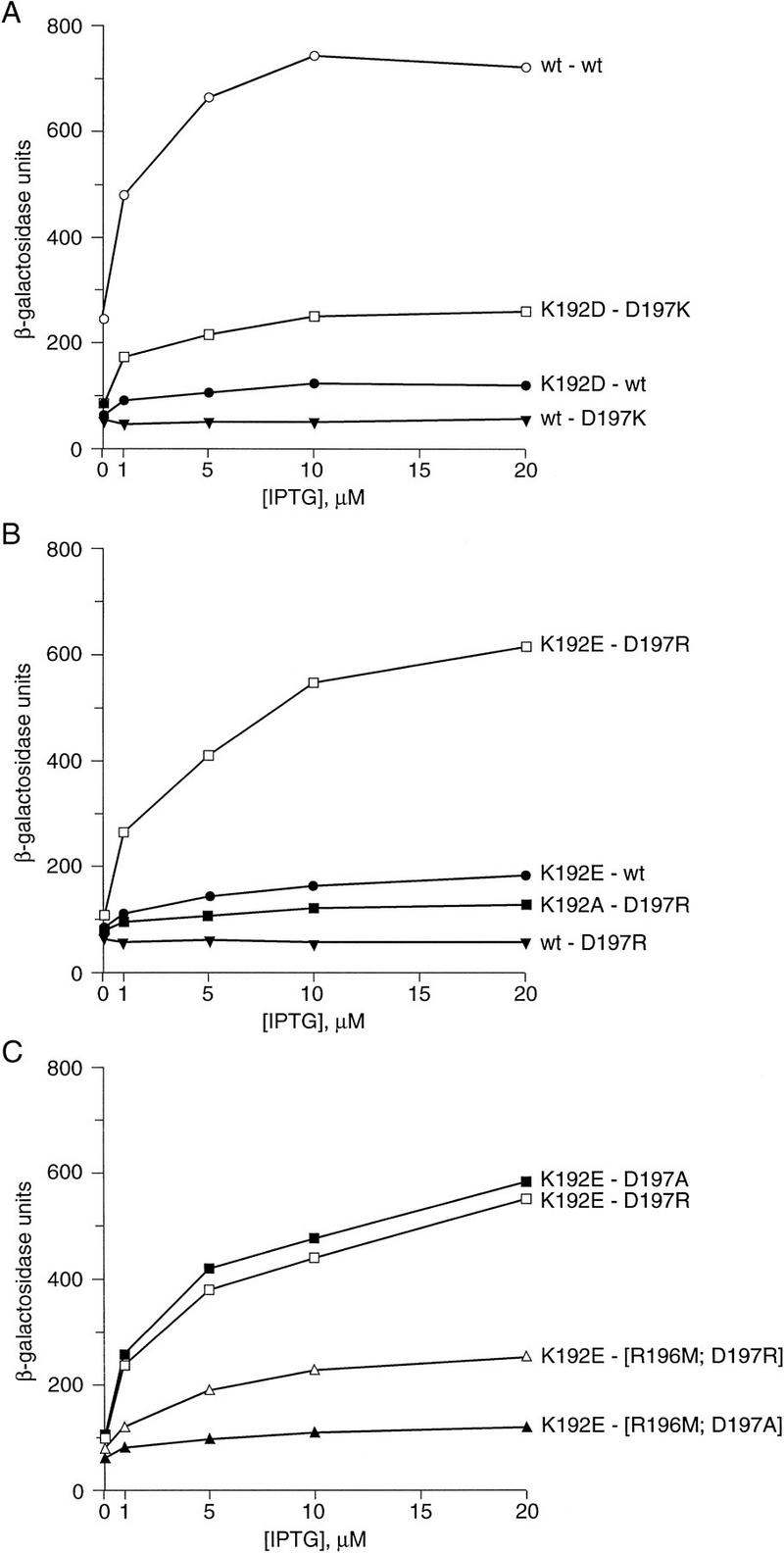
Effects of substitutions at positions 192 and 197 on interaction of λcI CTDs. Strain KS1 was cotransformed with a derivative of plasmid pACλcI encoding λcI with the wild-type or a mutant amino acid at position 192 and a derivative of plasmid pBRα–cI encoding the α–cI chimera with the wild-type or a mutant amino acid at position 197. For the experiment in C, the α–cI chimeras tested bore a mutant residue at position 197 and either the wild-type or a mutant residue at position 196. Graph labels list the λcI first and the α–cI allele second. β-Galactosidase activities were measured in cultures grown in the presence of 0, 1, 5, 10, and 20 μm IPTG. Each panel presents results of an individual representative experiment. Similar experiments performed on separate days yielded the same relative patterns of activation.
Seeking to identify a more strongly interacting mutant-suppressor pair, we assayed another combination of amino acids at these two positions, again reversing the charges of the wild-type residues. Comparison of Figure 6, A and B, shows that the mutants λcI–K192E and α–cI–D197R interacted more strongly than did λcI–K192D and α–cI–D197K, resulting in a level of transcriptional activation approaching that observed with wild-type λcI and the wild-type α–cI chimera. Again, neither mutant interacted with a wild-type partner protein.
To test the specific requirement for an oppositely charged residue to suppress the defects caused by the K192E and D197R changes, we tested each charge reversal mutant in combination with a partner molecule bearing an alanine substitution. As expected, λcI–K192A and α–cI–D197R did not interact with each other (Fig. 6B). Unexpectedly, however, λcI–K192E and α–cI–D197A did interact; in fact λcI–K192E activated transcription as efficiently when tested with α–cI–D197A as with α–cI–D197R (Fig. 6C). Noting the presence of a basic residue (R196) adjacent to residue 197 (see Fig. 1B), we considered the possibility that the mutant glutamic acid residue at position 192 might interact productively with a basic residue located either at position 197 or, if the acidic side chain of residue 197 is removed, at position 196.
We tested this idea by removing the basic side chain at position 196 of the α–cI chimera. We introduced the previously characterized cooperativity mutation R196M (Whipple et al. 1994) into α–cI chimeras bearing either the D197R or the D197A substitution and tested the ability of λcI–K192E to interact with the resulting double mutants. The R196M substitution weakened the interaction in both cases (Fig. 6C), as expected on the basis of the fact that it causes a moderate cooperativity defect when introduced into an otherwise wild-type λcI molecule. However, this substitution had a more severe effect in the context of the alanine residue at position 197, as would be expected if, in the absence of a charged residue at position 197, the basic side chain of residue 196 paired with the acidic side chain at position 192 in the λcI–K192E mutant.
We performed additional tests to verify that the activation observed with the K192E–D197R and K192E–D197A mutant combinations could not be achieved by the introduction of non-native acidic or basic residues at arbitrary positions on the cooperativity interface. Specifically, cooperativity mutant λcI–G199D (Whipple et al. 1994) could not substitute for λcI–K192E when tested with α–cI chimeras bearing either the D197R or the D197A substitution. Similarly, a chimera bearing the substitution E188K (Benson et al. 1994; Whipple et al. 1994) could not substitute for α–cI–D197R when tested with λcI–K192E. In each of these three tests, the level of lacZ expression was <10% of that observed in the wild type–wild type case (data not shown).
A λcI variant bearing substitutions at both positions 197 and 192
The proposal that residues 192 and 197 interact at the dimer–dimer interface, and, in particular, that a mutant glutamic acid residue at position 192 can interact with a mutant arginine residue at position 197 suggests that a double mutant bearing both the K192E and the D197R substitutions should bind cooperatively to pairs of λ operator sites. To test this prediction, we compared the abilities of λcI mutants bearing either one or both of these substitutions to interact when bound at nonadjacent operators using the PRM–lacZ reporter system described above (see Fig. 3). Figure 3B shows that, whereas dimers of each single mutant (either K192E or D197R) were unable to interact when bound at nonadjacent operator sites, dimers of the double mutant interacted relatively efficiently, as reflected in the inhibition of transcriptional activation in the in-phase reporter strain.
P22c2 mutants bearing substitutions at the positions corresponding to λcI positions 197 and 192
We sought to determine whether the P22c2 residues corresponding to λcI residues K192 and D197 interact at the P22c2 dimer–dimer interface. To do this, we constructed P22c2 mutants bearing either the amino acid substitution corresponding to K192E (K174E) or that corresponding to D197R (D179R), or both changes. We tested the abilities of these P22c2 mutants to interact when bound at nonadjacent P22 operators using a previously described reporter system similar to the one depicted in Figure 4 (Valenzuela and Ptashne 1989). In this system, the test strains bear a lacZ gene controlled by an artificial promoter with the low-affinity P22 operator between its −10 and −35 hexamers with or without a high-affinity P22 operator located an integral (5.0) number of turns of the DNA helix upstream (see Fig. 7).
Figure 7.

Cooperative binding of wild-type and mutant variants of P22c2. Strains DV59 (single-site reporter) and DV72 (two-site reporter) were transformed with wild-type or mutant versions of plasmid pPR4 encoding P22c2. Fold repression was determined by calculation of the ratio of β-galactosidase activity observed in each case to that observed in cells transformed with the control plasmid that lacks the P22c2 gene (4404 and 4231 Miller units in strains DV59 and DV72, respectively). All cultures were grown in medium containing 10 μm IPTG. Results are averages of duplicate assays that varied by less than ±3% from the mean. Repression levels are higher than those of Figure 4A because pPR4 (which resembles the plasmid used in the initial characterization of this system; Valenzuela and Ptashne 1989) directs expression of high levels of P22c2. Note that all of the mutant proteins repressed transcription somewhat less efficiently than the wild-type protein in the single-site reporter strain, reflecting a corresponding decrease in the fractional occupancy of the P22 operator on this reporter template. The double mutant, however, repressed transcription slightly more efficiently than the wild-type protein in the two-site reporter strain (a difference that was reproducible).Therefore, we conclude that the double mutant dimers interact at least as efficiently as the wild-type dimers.
The results shown in Figure 7 indicate that the P22c2 double mutant K174E;D179R formed dimers that interacted at least as efficiently as the wild-type dimers. The data show first that wild-type P22c2 manifests enhanced repression in the strain bearing the two-site template relative to the strain bearing the single-site template, as demonstrated previously (Valenzuela and Ptashne 1989). In contrast, the single mutants behaved indistinguishably in the two strains, indicating that dimers of each single mutant are unable to interact when bound at nonadjacent operators. Finally, the double mutant manifested greatly enhanced repression in the strain bearing the two-site template, the double mutant dimers apparently interacting even more strongly than the wild-type dimers (see Fig. 7, legend). We also tested the effects of replacing either D179 or K174 with an alanine residue. Each of the resulting P22c2 mutants behaved indistinguishably in the two strains, indicating that, as for λcI, removal of the wild-type side chains at either of these two positions abolishes the dimer–dimer interaction.
Discussion
We have identified two pairs of amino acids that interact at the dimer–dimer interface of cooperatively bound dimers of both λcI and P22c2. One of these pairs involves amino acid residues that are conserved at the corresponding positions of λcI and P22c2. These residues, K192 and D197 in λcI, are oppositely charged and evidently form a salt bridge. The other pair involves nonconserved amino acids that are hydrophilic in λcI (N148 and Q204) and oppositely charged in P22c2 (D131 and K186). We have shown that a λcI mutant bearing the P22c2 residues at both of these positions (148 and 204) interacts specifically with wild-type P22c2, indicating that these nonconserved residues are the critical specificity determinants that preclude interaction between wild-type λcI and P22c2 dimers.
The role of specific amino acid–amino acid interactions at the dimer–dimer interface
In the case of the conserved ion pair (D197 and K192), individual alanine replacements (i.e., replacement of either charged side chain with a methyl group) essentially abolished the dimer–dimer interaction for both λcI and P22c2 within the detection limits of our in vivo assays. Although such results are often taken as evidence that the deleted side chain participates in an energetically important interaction at the protein–protein interface, these individual alanine replacements have at least two potential effects: (1) any energetically favorable interaction of the wild-type side chain involving atoms beyond the β carbon is eliminated, and (2) the presence of an unpaired partner residue (here bearing a charged side chain) at the protein–protein interface may destabilize the complex. We have attempted to separate these two potential effects by replacing both of the interacting residues with alanines. We found that activation in the presence of the two alanine-substituted variants (λcI–K192A and α–cI–D197A) was roughly half that observed with the wild type–wild type combination (F. Whipple and A. Hochschild, unpubl.), suggesting that both of these effects are involved. Irrespective of any energetic contribution to the dimer–dimer complex, the presence of charged residues K192 and D197 at the interface is likely to help prevent formation of nonspecific complexes with unrelated proteins as a result of the energetic cost of burying unpaired charges at a protein–protein interface (Fersht 1977).
In the case of nonconserved residues N148 and Q204, our data suggest that interaction of the amide side chains in wild-type λcI is not energetically significant. Specifically, we found that although the N148D and Q204K substitutions each resulted in a cooperativity defect, replacement of either residue with an alanine (i.e., removal of either amide side chain) was tolerated with little or no loss in interaction energy. Thus, relative to the wild-type amide pair, the juxtaposition of an amide side chain with a charged side chain is disfavored, but the juxtaposition of an amide side chain with a methyl group is energetically neutral.
We do not know how many energetically significant amino acid–amino acid interactions occur at the dimer–dimer interface. Although mutations that result in cooperativity defects have been identified at at least sixteen positions in λcI (Hochschild and Ptashne 1988; Beckett et al. 1993; Benson et al. 1994; Burz and Ackers 1994; Whipple et al. 1994), the defects in many cases are likely attributable entirely to the disruptive presence of a mutant side chain. For example, charge reversal mutations at a conserved position (E188K in λcI and E170K in P22c2) result in large cooperativity defects (Valenzuela and Ptashne 1989; Benson et al. 1994; Burz and Ackers 1994; Whipple et al. 1994), but a mutation that simply removes this side chain (E188G in λcI) actually enhances cooperativity slightly (F. Whipple and A. Hochschild, unpubl.). As a second example, the λcI mutation G199D (Whipple et al. 1994), which introduces a new acidic side chain, results in a severe cooperativity defect. However, other mutations at this position result in a much weaker defect (G199S; Whipple et al. 1994) or no defect at all (G199R; N. Kuldell and A. Hochschild, unpubl.). Two aromatic residues have been implicated in cooperativity (F202 and Y210 in λcI; Benson et al. 1994; Burz and Ackers 1994; Whipple et al. 1994), but it is not known whether their side chains make contacts at the dimer–dimer interface. In addition to specific side chain–side chain interactions, interactions involving the polypeptide backbone might also be energetically significant.
A structural model
Although the structure of the λcI CTD is not yet known, the structure of a related Escherichia coli protein, UmuD′, has been determined recently by X-ray crystallography (Peat et al. 1996). UmuD′, which is involved in the cell’s SOS response to DNA damage, is initially synthesized as a larger precursor (UmuD). This precursor undergoes an autocleavage reaction to generate the active product, which corresponds to the CTDs of λcI and its relatives (Perry et al. 1985). This autocleavage reaction is a conserved feature within this family of proteins and depends on an activated form of the bacterial RecA protein, which is generated on exposure of the cell to DNA-damaging agents (for review, see Little 1993). In the case of the phage repressors, the corresponding autocleavage reaction separates the NTD from the CTD and results in prophage induction (for review, see Roberts and Devoret 1983).
The structure of UmuD′ consists of seven β strands preceded by a short helical segment. NMR studies of UmuD′ dimers in solution suggest that the monomers within the dimer interact as shown in the space filling model of Figure 8 (Ferentz et al. 1997). In this stereo diagram, the residues corresponding to λcI residue pairs N148–Q204 and K192–D197 (UmuD residues D59–I108 and K97–R102, dark blue and purple, respectively) are visible on the surface of the dimer. On the right-hand monomer, which is green in Figure 8, residues corresponding to λcI–Q204 and –D197 are visible, whereas residues corresponding to λcI–N148 and –K192 are exposed on the back and hence not visible in this view. Conversely, on the left-hand monomer (pale blue), which is related to its partner by a vertical twofold axis in the plane of the page, residues corresponding to λcI–N148 and –K192 are visible on the front side and residues corresponding to λcI–Q204 and –D197 are exposed on the back.
Figure 8.
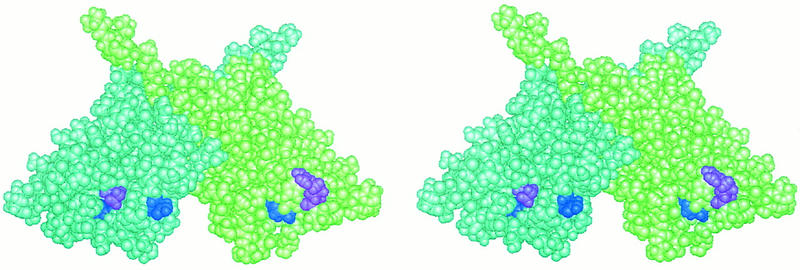
Space filling model of the UmuD′ dimer. In this stereo view of the UmuD′ dimer the left monomer is pale blue and the right monomer is pale green. Pairs of residues that correspond to interacting pairs of λcI amino acids are highlighted. Residues D59 and I108, which correspond to N148 and Q204 in λcI, are shown in dark blue on the left and right monomers, respectively. Residues K97 and R102, which correspond to K192 and D197 in λcI, are shown in purple on the left and right monomers, respectively. Interactions of these residue pairs at the cooperativity interface of λcI can be imagined by treating the two images as separate dimers (rather than as a stereo view) and folding them together along a vertical axis midway between them.
This arrangement of the interacting residues suggests that a pair of dimers might come together in a sandwich-like fashion that can be visualized by viewing the two images depicted in Figure 8 as individual dimers (rather than as a stereo pair) and folding them together along a vertical axis midway between them. The positions of the interacting residues in three-dimensional space are consistent with this model. Specifically, the distance between the α carbons of UmuD′ residues 97 and 59 (corresponding to λcI residues K192 and N148) is roughly the same as the distance between the α carbons of UmuD′ residues 108 and 102 (corresponding to λcI residues 204 and 197), and whereas residues 97 and 108 lie in cavities on the protein surface, residues 59 and 102 protrude from the surface. This model suggests that all four monomers could, in principle, interact at the dimer–dimer interface contrary to the traditional cartoon representations (e.g., see Fig. 1A). However, it is possible that constraints imposed by the DNA when the interacting dimers are DNA bound might limit the interaction to only two monomers. We also note that this model would, in principle, permit the simultaneous interaction of more than two dimers (Liu and Little 1998) because each dimer contributes two potential contact surfaces. Finally, we stress that, although the UmuD′ structure is likely to resemble the structures of the λcI and P22c2 CTDs, UmuD′ dimers have not been observed to form higher order oligomers in solution (Ferentz et al. 1997), and neither UmuD′ residues 59 and 108 nor residues 97 and 102 would be expected to pair. Furthermore, nothing is known about what effect, if any, the loss of the amino-terminal fragments after cleavage may have on the structures of the proteins within this family.
Cooperativity of the HK022 repressor
The repressor of another lambdoid phage, HK022, binds adjacent operators with a much higher degree of cooperativity than do λcI and P22c2 and may participate in long-range cooperative interactions during the natural life cycle of the phage (Carlson and Little 1993). Cooperativity mutations in the HK022 repressor have been identified, some of which are pleiotropic (Mao and Little 1998). Although mutually suppressive pairs have been identified, the amino acids involved are not thought to interact directly (Mao and Little 1998). Mao and Little have pointed out that strong cooperativity mutants were likely to have been eliminated from their screen, and, therefore, it remains to be learned whether or not the residues that mediate HK022 repressor cooperativity correspond with those that mediate λcI and P22c2 cooperativity. In either case, the greater strength of the HK022 dimer–dimer interaction implies that the respective dimer–dimer interfaces must differ to some extent.
Mao and Little (1998) have suggested that the CTD can assume multiple conformations that support different functions of this domain (i.e., cooperativity and autocleavage). During autocleavage a conserved lysine (K192 in λcI) deprotonates a conserved serine (S149 in λcI), activating it to perform a nucleophilic attack on a specific peptide bond within the linker that connects the two domains (for review, see Little 1993). The results presented here indicate that K192 is directly involved in both processes, participating not only in the intramolecular interaction with S149, but also in an intermolecular salt bridge with residue D197 at the dimer–dimer interface. Consistent with the proposal of Mao and Little, it is therefore likely (though not necessarily required) that autocleavage and cooperativity involve different conformations of the CTD that position the lysine side chain differently under conditions that either do or do not favor cleavage.
Mutant-suppressor analysis and charge reversal mutants
Mutant-suppressor analysis is often used to attempt to identify partner proteins that interact with a protein of interest (Jarvik and Botstein 1975; Phizicky and Fields 1995), or to identify interacting residues at protein–DNA or protein–protein interfaces. In the case of sequence-specific DNA-binding proteins, specific amino acid–base pair contacts have been identified by screening for amino acid substitutions that permit the mutant protein to bind a specifically altered recognition site (e.g., see Youderian et al. 1983; Ebright et al. 1984; Wharton and Ptashne 1987). In the case of protein–protein interactions, an analogous approach has been taken by screening for amino acid substitutions that restore an interaction with a specifically altered partner protein (e.g., see Li et al. 1994; Kallipolitis et al. 1997). There are, however, few examples in which mutant-suppressor analysis has resulted in the identification of specific pairs of interacting amino acid side chains; thus, it is not clear from such studies how difficult it is to obtain an amino acid substitution that restores a given protein–protein interaction by virtue of its specific ability to interact with a mutant residue in the partner protein.
Our results demonstrate that individual amino acid–amino acid contacts can function independently at a protein–protein interface and that pairwise alterations can be accommodated. Specifically, the demonstration that oppositely charged residues can be exchanged provides evidence for a degree of plasticity that would not necessarily have been anticipated at a protein–protein interface (for another example of plasticity, see Atwell et al. 1997). Genetic engineering with structurally characterized proteins has provided a number of other examples of ion pair reversals. Several enzyme–substrate systems have been used to evaluate the effects of active-site ion pair reversal on catalysis (Graf et al. 1987; Cronin and Kirsch 1988; Almo et al. 1994). The general conclusion from these studies is that such reversals do not tend to work very well (Hwang and Warshel 1988). However, the optimization of an active site by evolution is likely to be governed by more stringent requirements than the optimization of a simple protein–protein interface, and it may be that ion pair reversal will prove more successful in the latter context. We are aware of two examples in which genetic engineering has been used successfully to restore a protein–protein interaction by ion pair reversal, one involving an enzyme and its specific inhibitor (Jucovic and Hartley 1996), and the other involving the human TATA box-binding protein and the general transcription factor TFIIB (Tansey and Herr 1997). These studies and those described here suggest that manipulation of the specificity of protein–protein interaction may be easier than previously thought.
Materials and methods
Media and stock strains
LB liquid medium and plates were prepared as described by Miller (1972). Strain MC1000 [F−araD139, Δ(ara, leu)7697, Δlac X74, galU, galK, strA; Casadaban and Cohen 1980] was from our laboratory collection. Strains CSH100 [F′lacproA+,B+(lacIq lacPL8)/araD(gpt–lac)5] and CSH142 [F−/araD(gpt–lac)5; Miller 1992] were purchased from Cold Spring Harbor Laboratory.
Indicator strains
Strain FW89, used for isolation of mutants of the P22–λ hybrid repressor, is a derivative of strain MC1000 that contains an F′ episome, which carries a kanamycin resistance gene together with a fusion of the test promoter depicted in Figure 2 to the lacZYA operon. The reporter construct, which was first assembled on a precursor of plasmid pFW11 (Whipple 1998), consists of a modified lacUV5 promoter that bears a consensus P22 operator site (ATTTAAGAATTCTTAAAT) and a λ OR1 operator site (TACCTCTGGCGGTGATA) centered 80.5 bp upstream and 35 bp downstream of the start point of transcription, respectively (a center-to-center spacing of 11.0 turns of the DNA helix). An ATG start codon preceded by a ribosome binding site is located at position +116 of the transcribed region and is followed by the leader peptide and lacZ′ gene fragment of pFW11. This construct was transferred onto the F′ lacpro episome of strain CSH100 and moved into strain MC1000 by the method described in Whipple (1998). Strain FW89B was subsequently constructed for technical reasons unrelated to the work reported here by moving the recombinant F′ into a different background strain, CSH142, via conjugation. The quantitative assays reported in Figure 2 were performed with strain FW89B; similar assays performed with FW89 yielded the same results.
Indicator strains AH-5.9 and AH-5.5 (Hochschild and Ptashne 1988), FW40 and FW42 (Whipple et al. 1994), KS1 (Dove et al. 1997), and DV72 and DV59 (Valenzuela and Ptashne 1989), which were used for the assays of Figures 3, 4, 5 and 6, and 7, respectively, have all been described previously. Although derived from various background strains, all of these strains bear fusions of artificial promoter–operator constructs to the lacZ gene. These fusion constructs are located on prophages that are present in single copy in the bacterial chromosomes. All of these strains except AH-5.9 and AH-5.5 also contain high levels of the lac repressor protein encoded by the lacIq allele. In AH-5.9 and AH-5.5 lacZ is under the control of a modified λ PRM promoter region in which OR3 has been inactivated and OR1 has been moved away from OR2 such that the center-to-center distances correspond to 5.9 and 5.5 turns of the DNA helix, respectively. In FW40 and FW42, lacZ is controlled by an artificial promoter that bears a weak P22 operator (P22 OR2) between the −35 and −10 hexamers and a strong λ operator (λ OR1) positioned 5.0 or 5.4 turns of the DNA helix upstream of P22 OR2, respectively. In strain KS1, lacZ is controlled by a modified lac promoter that bears a single λ operator (OR2) centered 62 bp upstream of the start point of transcription. In strain DV72, the reporter construct is identical to that of strain FW40 except that the upstream operator is a consensus P22 operator. DV59 is identical to DV72 except that the upstream operator has been removed.
Plasmids
Plasmids pLR2, pPR2, and pFW7 (Whipple et al. 1994) have been described. They are ampicillin-resistant derivatives of pBR322 that bear the λcI gene, the P22c2 gene, and the P22c2–λcI hybrid repressor gene, respectively, under the control of a truncated lacUV5 promoter–operator region. Plasmid pFW7-280Δ is a derivative of pFW7 that lacks the DNA between the EcoRI site and the MspI–ClaI junction that was created during the construction of pFW7 (see Whipple et al. 1994). This deletion has no significant effect on expression of the hybrid repressor gene. Both pFW7 and pFW7-280Δ bear an artificial translationally silent XbaI site at nucleotide 232 of the P22c2 gene and bear a deletion of 882 bp of vector DNA between the first and last NgoMI sites located downstream of the hybrid repressor gene.
Plasmid pPR4 is a derivative of pPR2 in which the promoter region (between the EcoRI site and the beginning of the repressor gene) has been replaced with the corresponding region from plasmid pTP15 (Poteete and Roberts 1981), which contains the full lacUV5 promoter. Additionally pPR4 contains artificial translationally silent XbaI and HindIII sites at base pairs 232 and 523 of the P22c2 gene, and bears the same downstream deletion in the vector DNA as do pFW7 and pFW7-280Δ.
Plasmid pBRα–cI (Dove et al. 1997) is a derivative of pBR322 that encodes a chimeric protein consisting of the amino domain and linker region of the α subunit of RNAP fused to the CTD of λcI. This gene is under the control of tandem promoters: a constitutive lpp promoter and the repressible lacUV5 promoter.
Plasmid pA3B2 (Whipple et al. 1994) is a chloramphenicol-resistant derivative of pACYC184 that bears the λcI gene under the control of a relatively weak truncated lacUV5 promoter. pAC-λcI (Dove et al. 1997) is identical to pA3B2 except that the region between the EcoRI–HindIII junction (see Whipple et al. 1994) and the beginning of the λcI gene contains a strong nontruncated lacUV5 promoter derived from plasmid pKB280 (Backman and Ptashne 1978).
Mutagenesis
To perform random mutagenesis of the CTD of the P22c2–λcI hybrid repressor, a pool of pFW7 plasmid DNA was generated by trimming PCR-amplified DNA containing this region with XbaI and BamHI and ligating the resulting fragment into the backbone of pFW7 cleaved with the same enzymes (Zhou et al. 1991). The ligation mix was introduced by electroporation into cells of strain FW89 that already bore plasmid pA3B2-N148D. The cells were then spread on lactose tetrazolium plates containing carbenicillin, chloramphenicol, and kanamycin and incubated overnight. Plasmid DNAs isolated from clones that produced red colonies were purified away from the pA3B2 plasmid by passage through strain MC1000 and then verified by reintroducing them into FW89 together with pA3B2-N148D and reconfirming their in vivo phenotypes. Then, they were analyzed by restriction analysis and DNA sequencing.
Site-directed mutagenesis of plasmid-borne repressor genes was performed using the Bio-Rad Mutagene phagemid mutagenesis kit, or by a PCR technique in which one or both of the PCR primers introduced specific changes near a restriction site, followed by reconstruction of the plasmid by use of the appropriate restriction enzymes. All constructions were confirmed by restriction analysis and by DNA sequencing as appropriate. Whenever the PCR was involved, the DNA sequence of final plasmids was verified over the entire region that was subjected to PCR amplification.
Enzyme assays
β-Galactosidase assays were performed as described by Miller (1972) except that cells were grown in LB medium supplemented with carbenicillin (50 μg/ml) and chloramphenicol (30 μg/ml) as appropriate. With strains FW89B, FW40, FW42, and KS1, which contain kanamycin-resistant F′ episomes, the medium was also supplemented with kanamycin (30 μg/ml). Strains DV72 and DV59 were grown at 30°C to avoid induction of their temperature-sensitive prophages. As indicated in the figure legends, IPTG was added to the medium at various concentrations for all assays except those performed with strains AH-5.9 and AH-5.5.
Acknowledgments
We are very grateful to Ann Ferentz for helpful discussions, and to both Ann Ferentz and Gerhard Wagner for providing the molecular model of the UmuD′ dimer. We thank Ana Pis-Lopez and Vladimir Podolny for technical assistance. We also thank Simon Dove and Mark Ptashne for helpful comments on the manuscript, as well as Richard Ebright and Jim Hu for bringing relevant literature to our attention. This work was supported by National Institutes of Health grant GM44025 (A.H.), by an established investigatorship from the American Heart Association (A.H.), and by a Postdoctoral Cancer Fellowship from the American Cancer Society, Massachusetts Division, Inc. (F.W.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL ahochsch@warren.med.harvard.edu; FAX (617) 738-7664.
References
- Almo SC, Smith DL, Danishefsky AT, Ringe D. The structural basis for the altered substrate specificity of the R292D active site mutant of aspartate aminotransferase from E. coli. Protein Eng. 1994;7:405–412. doi: 10.1093/protein/7.3.405. [DOI] [PubMed] [Google Scholar]
- Atwell S, Ultsch M, De Vos AM, Wells JA. Structural plasticity in a remodeled protein–protein interface. Science. 1997;278:1125–1128. doi: 10.1126/science.278.5340.1125. [DOI] [PubMed] [Google Scholar]
- Backman K, Ptashne M. Maximizing gene expression on a plasmid using recombination in vitro. Cell. 1978;13:65–71. doi: 10.1016/0092-8674(78)90138-1. [DOI] [PubMed] [Google Scholar]
- Beckett D, Burz DS, Ackers GK, Sauer RT. Isolation of λ repressor mutants with defects in cooperative operator binding. Biochemistry. 1993;32:9073–9079. doi: 10.1021/bi00086a012. [DOI] [PubMed] [Google Scholar]
- Benson N, Adams C, Youderian P. Genetic selection for mutations that impair the co-operative binding of lambda repressor. Mol Microbiol. 1994;11:567–579. doi: 10.1111/j.1365-2958.1994.tb00337.x. [DOI] [PubMed] [Google Scholar]
- Burz DS, Ackers G. Single-site mutations in the C-terminal domain of bacteriophage λ cI repressor alter cooperative interactions between dimers adjacently bound to OR. Biochemistry. 1994;33:8406–8416. doi: 10.1021/bi00194a004. [DOI] [PubMed] [Google Scholar]
- Bushman FD, Shang C, Ptashne M. A single glutamic acid residue plays a key role in the transcriptional activation function of lambda repressor. Cell. 1989;58:1163–1171. doi: 10.1016/0092-8674(89)90514-x. [DOI] [PubMed] [Google Scholar]
- Carlson NG, Little JW. Highly cooperative DNA binding by the coliphage HK022 repressor. J Mol Biol. 1993;230:1108–1130. doi: 10.1006/jmbi.1993.1229. [DOI] [PubMed] [Google Scholar]
- Casadaban MJ, Cohen SN. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol. 1980;138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- Cronin CN, Kirsch JF. Role of arginine-292 in the substrate specificity of aspartate aminotransferase as examined by site-directed mutagenesis. Biochemistry. 1988;27:4572–4579. doi: 10.1021/bi00412a052. [DOI] [PubMed] [Google Scholar]
- Dove SL, Hochschild A. Conversion of the Ω subunit of Escherichia coli RNA polymerase into a transcriptional activator or an activation target. Genes & Dev. 1998;12:745–754. doi: 10.1101/gad.12.5.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove SL, Joung JK, Hochschild A. Activation of prokaryotic transcription through arbitrary protein-protein contacts. Nature. 1997;386:627–630. doi: 10.1038/386627a0. [DOI] [PubMed] [Google Scholar]
- Ebright RH, Cossart P, Gicquel-Sanzey B, Beckwith J. Mutations that alter the DNA sequence specificity of the catabolite gene activator protein of E. coli. Nature. 1984;311:232–235. doi: 10.1038/311232a0. [DOI] [PubMed] [Google Scholar]
- Ferentz AE, Opperman T, Walker GC, Wagner G. Dimerization of the UmuD′ protein in solution and its implications for regulation of SOS mutagenesis. Nature Struct Biol. 1997;4:979–983. doi: 10.1038/nsb1297-979. [DOI] [PubMed] [Google Scholar]
- Fersht A. Enzyme structure and mechanism, Chapter 9. San Francisco, CA: W.H. Freeman and Company; 1977. Forces between molecules and enzyme-substrate binding energies; pp. 226–243. [Google Scholar]
- Graf L, Craik CS, Patthy A, Roczniak S, Fletterick RJ, Rutter WJ. Selective alteration of substrate specificity by replacement of aspartic acid-189 with lysine in the binding pocket of trypsin. Biochemistry. 1987;26:2616–2623. doi: 10.1021/bi00383a031. [DOI] [PubMed] [Google Scholar]
- Guarente L, Nye JS, Hochschild A, Ptashne M. Mutant lambda phage repressor with a specific defect in its positive control function. Proc Natl Acad Sci. 1982;79:2236–2239. doi: 10.1073/pnas.79.7.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochschild A, Ptashne M. Cooperative binding of λ repressors to sites separated hy integral turns of the DNA helix. Cell. 1986;44:681–687. doi: 10.1016/0092-8674(86)90833-0. [DOI] [PubMed] [Google Scholar]
- ————— Interaction at a distance between λ repressors disrupts gene activation. Nature. 1988;336:353–357. doi: 10.1038/336353a0. [DOI] [PubMed] [Google Scholar]
- Hochschild A, Irwin N, Ptashne M. Repressor structure and the mechanism of positive control. Cell. 1983;32:319–325. doi: 10.1016/0092-8674(83)90451-8. [DOI] [PubMed] [Google Scholar]
- Hughes RE, Hatful GF, Rice P, Steitz TA, Grindley NDF. Cooperativity mutants of the γδ resolvase identify an essential interdimer interaction. Cell. 1990;63:1331–1338. doi: 10.1016/0092-8674(90)90428-h. [DOI] [PubMed] [Google Scholar]
- Hwang J-K, Warshel A. Why ion pair reversal by protein engineering is unlikely to succeed. Nature. 1988;334:270–272. doi: 10.1038/334270a0. [DOI] [PubMed] [Google Scholar]
- Jarvik J, Botstein D. Conditional-lethal mutations that suppress genetic defects in morphogenesis by altering structural proteins. Proc Natl Acad Sci. 1975;72:2738–2742. doi: 10.1073/pnas.72.7.2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AD, Meyer BJ, Ptashne M. Interactions between DNA-bound repressors govern regulation by the λ phage repressor. Proc Natl Acad Sci. 1979;76:5061–5065. doi: 10.1073/pnas.76.10.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucovic M, Hartley RW. Protein-protein interaction: A genetic selection for compensating mutations at the barnase-barstar interface. Proc Natl Acad Sci. 1996;93:2343–2347. doi: 10.1073/pnas.93.6.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallipolitis BH, Norregaard-Madsen M, Valentin-Hansen P. Protein-protein communication: Structural model of the repression complex formed by CytR and the global regulator CRP. Cell. 1997;89:1101–1109. doi: 10.1016/s0092-8674(00)80297-4. [DOI] [PubMed] [Google Scholar]
- Li M, Moyle H, Susskind MM. Target of the transcriptional activation function of phage λ cI protein. Science. 1994;263:75–77. doi: 10.1126/science.8272867. [DOI] [PubMed] [Google Scholar]
- Little JW. LexA cleavage and other self-processing reactions. J Bacteriol. 1993;175:4943–4950. doi: 10.1128/jb.175.16.4943-4950.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Little JW. The spacing between binding sites controls the mode of cooperative DNA-protein interactions: Implications for evolution of regulatory circuitry. J Mol Biol. 1998;278:331–338. doi: 10.1006/jmbi.1998.1695. [DOI] [PubMed] [Google Scholar]
- Mao C, Little JW. Mutations affecting cooperative DNA binding of phage HK022 CI repressor. J Mol Biol. 1998;279:31–48. doi: 10.1006/jmbi.1998.1725. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- ————— . A short course in bacterial genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- Pabo CO, Lewis M. The operator-binding domain of λ repressor and DNA recognition. Nature. 1982;298:443–447. doi: 10.1038/298443a0. [DOI] [PubMed] [Google Scholar]
- Pabo CO, Sauer RT, Sturtevant JM, Ptashne M. The λ repressor contains two domains. Proc Natl Acad Sci. 1979;76:1608–1612. doi: 10.1073/pnas.76.4.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat TS, Frank EG, McDonald JP, Levine AS, Woodgate R, Hendrickson W. Structure of the UmuD′ protein and its regulation in response to DNA damage. Nature. 1996;380:727–730. doi: 10.1038/380727a0. [DOI] [PubMed] [Google Scholar]
- Perry KL, Elledge SJ, Mitchell BB, Marsh L, Walker GC. umuDC and mucAB operons whose products are required for UV light- and chemical-induced mutagenesis: UmuD, MucA, and LexA proteins share homology. Proc Natl Acad Sci. 1985;82:4331–4335. doi: 10.1073/pnas.82.13.4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Fields S. Protein-protein interactions: Methods for detection and analysis. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteete AR, Ptashne M. Control of transcription by the bacteriophage P22 repressor. J Mol Biol. 1982;157:21–48. doi: 10.1016/0022-2836(82)90511-3. [DOI] [PubMed] [Google Scholar]
- Poteete AR, Roberts TM. Construction of plasmids that produce phage P22 repressor. Gene. 1981;13:153–161. doi: 10.1016/0378-1119(81)90004-4. [DOI] [PubMed] [Google Scholar]
- Ptashne M. A genetic switch. Cambridge, MA: Cell Press and Blackwell Scientific Publications; 1992. [Google Scholar]
- Raumann BE, Rould M, Pabo CO, Sauer RT. DNA recognition by β-sheets in the Arc repressor-operator crystal structure. Nature. 1994;367:754–757. doi: 10.1038/367754a0. [DOI] [PubMed] [Google Scholar]
- Roberts JW, Devoret R. Lysogenic induction. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA, editors. Lambda II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1983. pp. 123–144. [Google Scholar]
- Sauer RT, Yocum RR, Doolittle RF, Lewis M, Pabo CO. Homology among DNA-binding proteins suggests use of a conserved super-secondary structure. Nature. 1982;298:447–451. doi: 10.1038/298447a0. [DOI] [PubMed] [Google Scholar]
- Sauer RT, Jordan SR, Pabo CO. λ Repressor: A model system for understanding protein-DNA interactions and protein stability. Adv Protein Chem. 1990;40:1–61. doi: 10.1016/s0065-3233(08)60286-7. [DOI] [PubMed] [Google Scholar]
- Somers WS, Phillips SEV. Crystal structure of the met repressor-operator complex at 2.8 Å resolution reveals DNA recognition by β strands. Nature. 1992;359:387–393. doi: 10.1038/359387a0. [DOI] [PubMed] [Google Scholar]
- Tansey WP, Herr W. Selective use of TBP and TFIIB revealed by a TATA-TBP-TFIIB array with altered specificity. Science. 1997;275:829–831. doi: 10.1126/science.275.5301.829. [DOI] [PubMed] [Google Scholar]
- Valenzuela D, Ptashne M. P22 repressor mutants deficient in co-operative binding and DNA loop formation. EMBO J. 1989;8:4345–4350. doi: 10.1002/j.1460-2075.1989.tb08621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton RP, Ptashne M. A new-specificity mutant of 434 repressor that defines an amino acid-base pair contact. Nature. 1987;326:888–891. doi: 10.1038/326888a0. [DOI] [PubMed] [Google Scholar]
- Whipple FW, Kuldell NH, Cheatham LA, Hochschild A. Specificity determinants for the interaction of λ repressor and P22 repressor dimers. Genes & Dev. 1994;8:121–1223. doi: 10.1101/gad.8.10.1212. [DOI] [PubMed] [Google Scholar]
- Whipple FW. Genetic analysis of prokaryotic and eukaryotic DNA-binding proteins in E. coli. Nucleic Acids Res. 1998;26:3700–3706. doi: 10.1093/nar/26.16.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youderian P, Vershon AK, Bouvier S, Sauer RT, Susskind MM. Changing the DNA-binding specificity of a repressor. Cell. 1983;35:777–783. doi: 10.1016/0092-8674(83)90110-1. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhang X, Ebright RH. Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase. Nucleic Acids Res. 1991;19:6052. doi: 10.1093/nar/19.21.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]