

Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) are extensively used to relieve pain and inflammation in humans via cyclooxygenase inhibition. Our recent research suggests that certain NSAIDs including ibuprofen suppress intracellular RhoA signal and improve significant axonal growth and functional recovery following axonal injury in the CNS. Several NSAIDs have been shown to reduce generation of amyloid-beta42 peptide via inactivation of RhoA signal, supporting potent RhoA-repressing function of selected NSAIDs. In this report, we demonstrate that RhoA-inhibiting NSAIDs ibuprofen and indomethacin dramatically reduce cell death of oligodendrocytes in cultures or along the white matter tracts in rats with a spinal cord injury. More importantly, we demonstrate that treatments with the RhoA-inhibiting NSAIDs significantly increase axonal myelination along the white matter tracts following a traumatic contusion spinal cord injury. In contrast, non-RhoA-inhibiting NSAID naproxen does not have such an effect. Thus, our results suggest that RhoA inactivation with certain NSAIDs benefits recovery of injured CNS axons not only by promoting axonal elongation, but by enhancing glial survival and axonal myelination along the disrupted axonal tracts. This study, together with previous reports, supports that RhoA signal is an important therapeutic target for promoting recovery of injured CNS and that RhoA-inhibiting NSAIDs provide great therapeutic potential for CNS axonal injuries in adult mammals.
Keywords: Spinal cord injury, axon injury, ibuprofen, NSAIDs, RhoA, apoptosis, myelination
Introduction
Axon disconnection in the central nervous system (CNS) usually results in signal conduction failure and persistent functional deficits in many patients, such as white matter stroke, progressive multiple sclerosis, traumatic brain injury as well as spinal cord injury (SCI). Medical treatments for recovering signal conduction along the injured axons are currently not available due to complexity of injury mechanisms and neuronal growth failure. Although a number of compounds are in clinical trials, the only approved drug for SCI is methylprednisolone, an acute neuroprotective therapy administered only shortly after trauma (Bracken et al. 1997). Following CNS injuries, activation of RhoA is a convergent intracellular pathway for numerous extracellular molecules that restrict axonal growth, including CNS myelin, glial scar source of chondroitin sulfate proteoglycans and possibly a few axonal guidance cues during development (Huber et al. 2003; McGee and Strittmatter 2003). RhoA signal activation leads to growth cone collapse and neurite growth inhibition in cultured primary neurons (Kozma et al. 1997; Kranenburg et al. 1999). Inactivation of Rho with C3 transferase or its downstream with Y-27632 (Fournier et al. 2003) promotes neurite outgrowth on myelin or glial scar inhibitory substrates. Accordingly, Rho signal suppression has been shown to be a feasible approach to promote axonal regeneration in rodents with transection and contusion SCI or optic nerve crush injury (Bertrand et al. 2005; Dergham et al. 2002; Dubreuil et al. 2003; Fischer et al. 2004; Fournier et al. 2003; Hara et al. 2000; Nishio et al. 2006; Sung et al. 2003). Thus, Rho signaling pathway is an important therapeutic target for promoting functional recovery in adult mammals with CNS axonal injuries (Dergham et al. 2002; Mueller et al. 2005).
Recently, we have reported that NSAID ibuprofen and indomethacin overcome neuronal growth suppression from various axonal growth inhibitors via potently repressing activity of intracellular RhoA signal (Fu et al. 2007). Especially, we have demonstrated that RhoA silence with ibuprofen is able to stimulate significant axonal regrowth of descending fiber tracts in the distal spinal cord following a dorsal transection or contusion trauma (Fu et al. 2007). In addition, ibuprofen promotes remarkable locomotor functional recovery in SCI rodents, even when applied one week after injury. Our findings have been independently replicated in other spinal cord injury models (Wang et al. 2009b). Given the axon growth-promoting effect of ibuprofen and frequent use of this drug in humans, it may become an effective treatment for clinical conditions characterized by white matter damages. Most recently, our further research work has illustrated that the transcription factor peroxisome proliferator-activated receptor γ (PPARγ) is essential for coupling ibuprofen to RhoA inhibition and subsequent promotion of neurite growth in neurons (Dill et al. 2010), suggesting that PPARγ may be an additional therapeutic target for the disorders characterized by RhoA activation.
On the other hand, a number of glial cells including oligodendrocytes (OLGs) along the white matter tracts undergo apoptosis, and OLG loss appears to be responsible for demyelination and axonal dysfunction following CNS axonal injuries (Jensen et al. 1999; Tamura et al. 2005; Warden et al. 2001). Several extracellular molecules contribute to the post-injury apoptosis (Beattie et al. 2002; Demjen et al. 2004; Kim et al. 2001; Sanchez-Gomez et al. 2003) and intracellular activation of RhoA appears to be important for apoptotic glial cell death following SCI because RhoA inactivation with membrane-permeable C3 transferase reverses apoptotic cell loss (Dubreuil et al. 2003). Thus, it is very likely that the RhoA-inhibiting NSAIDs improve functional recovery following CNS injuries not only by stimulating axonal regrowth, but also by preventing glial cell loss, consequently reducing demyelination and improving remyelination of spared and/or regenerating CNS axons. In this study, we report that ibuprofen and indomethacin applied at the doses to inhibit RhoA activity dramatically reduced OLG cell death in cultures or along the white matter tracts in SCI rodents. Furthermore, we demonstrate that treatments with RhoA-inhibiting NSAIDs significantly increase myelination of CNS axons following a contusion SCI in rats. Together with previous reports, the present study supports that RhoA signal is an important therapeutic target for promoting functional recovery in the injured CNS and that RhoA-inhibiting NSAIDs provide great therapeutic potential for axonal injuries in adult mammals.
Materials and methods
CG4 cell cultures and treatments
CG4 cell line was cultured on plastic coverslips coated with poly-L-lysine in 24-well plates in DMEM containing 10% fetal bovine serum and N2 supplements. After CG4 cells became confluent (usually two days after growth), they were cultured in serum-free medium containing N1 (including insulin) and N2 supplements for 3 days to stimulate cell maturation. CG4 cells develop many processes during maturation 2 days after growth in the second culture medium and the majority of CG4 cells are positive for galactocerebroside C (GalC) staining (Nicholas et al. 2002). Three days after differentiation in the second medium, the differentiated cells were incubated with vehicle (Veh) saline, naproxen (Nap, 400 μM), ibuprofen (Ibu, 400 μM), indomethacin (Ind, 20 μM) or Rho inhibitor C3 transferase (4 μg/ml) for 1hr. Then, cells were stimulated with tumor necrosis factor α (TNF-α, 1.2 μg/ml) for 8 hrs in serum-free medium to induce cell death in the presence of above drugs. Eight hrs following TNF-α incubation, the survival and dead cells on coverslips were stained with fluorescent dyes calcein and ethidium homodimer-1 (EthD1), respectively, via incubation for 30 min in the medium. Cells were fixed with 4% formalin before quantification. The ratio of the dead cells to the total cells was quantified from multiple coverslips in each group. A total of 7,268 -10,782 cells were counted in each group. In selected experiments, differentiated CG4 cells were treated with drugs for 9 hrs in the absence of TNF-α.
Animals and contusion spinal cord lesion
In SCI model, female Sprague-Dawley rats (weight 180–250gram) were deeply anesthetized with ketamine (70 mg/kg) and xylazine (8 mg/kg). For the contusion SCI, a laminectomy was performed at T7-8 and a moderate lesion was made at T8 with the NYU Impactor as we reported previously (Dill et al. 2008; Fu et al. 2007). We performed 4 batches of rat experiments with moderate contusion SCI. 1) A total of 34 rats were used for the apoptotic cell analysis in the SCI rats 5 days following the lesion (Fig. 2 and 3), 8 in each of the vehicle, Nap and Ibu groups, and 10 in the Ind group. The drug delivery was started 1 hr after the injury until 5 days post-trauma. 2) To confirm the role of Ibu in reducing apoptosis, we performed the second set of SCI rats and analyzed cell apoptosis 7 days after a contusion SCI: 11 and 9 rats in the vehicle and Ibu groups, respectively (Fig. 4). In these 20 rats, vehicle or drugs were initiated 1 hr after the injury and terminated 7 days after trauma. 3) For myelination studies at light microscopic level (Fig. 5–6), a total of 34 rats were employed following a moderate contusion injury, 8 in each of the vehicle and Ind groups, 9 in each of the Nap and Ibu groups. These rats started receiving vehicle or drug treatments 1 hr after injury, but the delivery persisted for 28 days post-trauma. These SCI rats were perfused for myelination histology 6 weeks after SCI. 4) Twenty SCI rats were used for myelin ultrastructural and Western blotting assays (Fig. 7 and 8), 5 in each of vehicle, Nap, Ibu and Ind groups. These SCI rats received the same drug treatments as in batch 3 and were perfused for histological and biochemical assays 4 weeks after injury. In all those rat experiments, drugs were delivered via daily subcutaneous injections with syringes at the following doses: Nap: 40 mg/kg/day; Ibu: 60 mg/kg/day; Ind: 4 mg/kg/day. The vehicle-treated animals received the same volume of saline. After SCI, a few rats died during the recovery due to general poor condition, and were consequently excluded from the study. In addition, a few rats without SCI were used as uninjured controls for spinal cord histology or Western blot.
Fig. 2. Systemic treatments with ibuprofen and indomethacin reduce apoptotic cells in the spinal cord 5 days after a contusion lesion.

A. The representative transverse sections of the spinal cord 6–10 mm rostral to the lesion site indicate apoptotic cells in the spinal cord of SCI rats from different groups. The TUNEL-labeled apoptotic cells were mostly distributed to the white matter areas in the dorsal, lateral and ventral columns. B, C, The number of TUNEL-positive cells in different areas of the spinal cord was quantified from transverse sections of the spinal cord 6–10 mm rostral to (B) and caudal to (C) the SCI center. Means ± SEM are reported (n= 8–10 rats/group). The values from ibuprofen (Ibu) and indomethacin (Ind) are statistically different from vehicle-treated group where indicated. Nap: naproxen. DS: dorsal; LT: lateral; VT: ventral; WM: white matter; GM: gray matter. *p<0.05; **p<0.01, Student’s t-test.
Fig. 3. Ibuprofen and indomethacin treatments increase OLGs in the spinal cord 5 days after SCI.

A. Transverse sections of the spinal cord 6–10 mm rostral to the lesion show CC1-labeled OLGs in different white matter areas of the dorsal, lateral and ventral columns in each group. Scale: 50 μm. B, C, The number of CC1-positive cells in different areas of the spinal cord white matter was quantified from transverse sections 6–10 mm rostral to (B) and caudal to (C) the lesion center. Means ± SEM are reported (n= 6–8 rats/group). The values from ibuprofen (Ibu) and indomethacin (Ind) are statistically different from vehicle-treated group where indicated. *p<0.05; **p<0.01, Student’s t-test.
Fig. 4. Ibuprofen treatment attenuates apoptotic cells in the spinal cord 7 days after a contusion lesion and the apoptotic cells are localized to OLGs.

A–L, Transverse sections of the spinal cord 9–11 mm rostral or caudal to the lesion indicate apoptotic cells in SCI rats treated with vehicle or ibuprofen. Scale: 500 μm (A, D, G, J) and 100 μm (B, C, E, F, H, I, K, L). M, N, The number of TUNEL-positive cells was quantified from different areas of the spinal cord 9–11 mm rostral (M) and caudal to (N) the SCI center. Means ± SEM are reported (n= 7–9 rats/group). The values from Ibu group are statistically different from vehicle-treated group where indicated. *p<0.05; **p<0.01, Student’s t-test. O, Longitudinal sections of the spinal cord 15–20 mm rostral to the lesion and transverse sections of the spinal cord 5–10 mm rostral to the lesion were doubly stained with OLG marker CC1 (green) and active caspase-3 (Casp3, red). Active caspase-3 was frequently observed in the nuclei of CC1-positive cells along spinal cord white matter tracts. Scale:10 μm (O).
Fig. 5. Systemic treatments with ibuprofen and indomethacin enhance myelination in the caudal spinal cord 6 weeks after a contusion lesion.

A, Images indicate the representative transverse sections of the spinal cord 3–4 mm caudal to the lesion from different groups. Transverse sections were processed for Luxol fast blue (LFB) staining in Veh, Nap, Ibu or Ind-treated rats 6 weeks after a contusion SCI. The images were collected from different white matter areas (arrowheads at low power, arrows at high power), including dorsal, lateral and ventral columns. In all the sections, dorsal is up. Scale bars: 100μm (low magnification) and 25μm (high magnification). B and C, The myelin signals stained by LFB in different areas were quantified from transverse sections of the spinal cord 3–4 mm (B) or 6–8 mm (C) caudal to the SCI in a blind manner. D, Transverse sections of the normal or injured spinal cord (3–4 mm caudal to the lesion) from different groups were immunostained for MBP (arrowheads) 6 weeks after a contusion SCI. In all the sections, dorsal is up. Scale bars: 100μm. E and F, MBP-stained myelin signals in different areas were quantified from transverse sections of the spinal cord 3–4 mm (E) or 6–8 mm (F) caudal to the SCI. Means ± SEM are reported (n=7–9 rats/group). In, B, C, E and F, the values from Ibu and Ind are statistically different from vehicle-treated group where indicated. *p<0.05; **p<0.01, Student’s t-test.
Fig. 6. Systemic treatments with ibuprofen and indomethacin enhance myelination in the rostral spinal cord 6 weeks after SCI.

A, Images indicate the representative transverse sections of the spinal cord 3–4 mm rostral to the lesion from each group. Transverse sections were processed for LFB staining in Veh, Nap, Ibu or Ind-treated rats 6 weeks after a contusion SCI. The images were collected from different white matter areas (arrowheads at low power, arrows at high power), including dorsal, lateral and ventral columns. In all the sections, dorsal is up. Scale bars: 100μm (low magnification) and 25μm (high magnification). B and C, The LFB-stained myelin signals in different areas were quantified from transverse sections of the spinal cord 3–4 mm (B) or 6–8 mm (C) rostral to the SCI in a blind manner. D, Transverse sections of the normal or injured spinal cord (3–4 mm rostral to the lesion) from different groups were immunostained for MBP (arrowheads) 6 weeks after a contusion SCI. Scale bars: 100μm. E and F, MBP-stained myelin signals in different areas were quantified from transverse sections of the spinal cord 3–4 mm (E) or 6–8 mm (F) rostral to the SCI. Means ± SEM are reported (n=7–9 rats/group). In, B, C, E and F, the values from Ibu and Ind are statistically different from vehicle-treated group where indicated. *p<0.05; **p<0.01, Student’s t-test.
Fig. 7. Treatments with ibuprofen and indomethacin enhance axon myelination in the lesioned spinal cord 4 weeks after SCI.

Low- (left column) and high-magnification electron microscopic imaging (middle and right columns) of transverse sections from the normal or injured spinal cord at the lesion epicenter revealed the ultrastructures of myelinated axons in different groups. The SCI rats treated with Ibu and Ind displayed higher density of myelinated axons and more intact myelin structures than those treated with Veh or Nap. Scale bars: 5 μm in left column and 0.5 μm in middle and right columns.
Fig. 8. Ibuprofen and indomethacin treatments increase MBP levels in the lesioned spinal cord 4 weeks after injury.
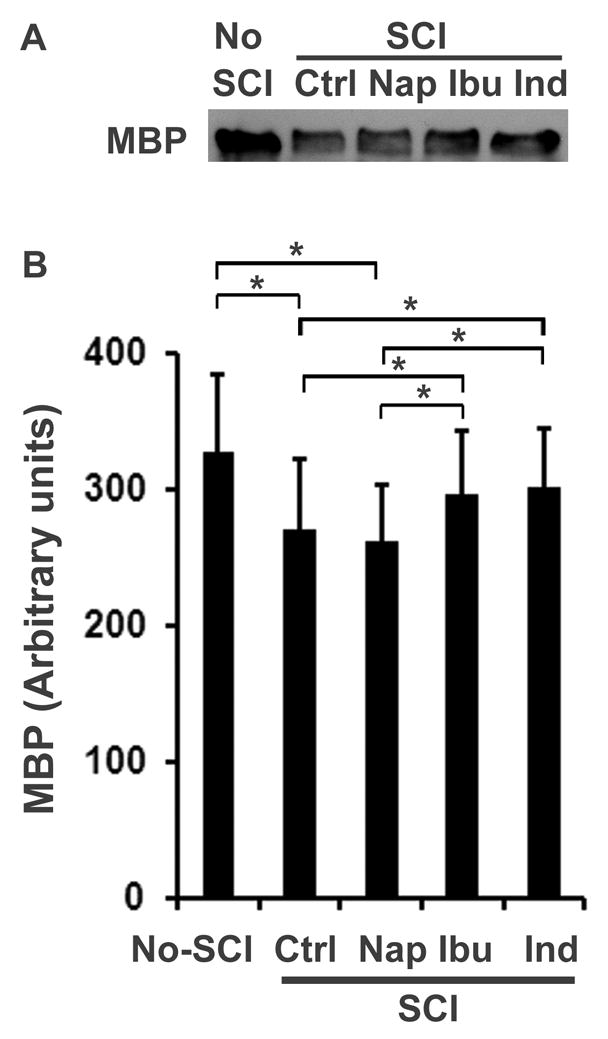
MBP was analyzed by Western blotting from tissue lysates of the spinal cord 3 mm rostral to and caudal to a contusion lesion at T8 (or the same level of the spinal cord from uninjured controls) (A). Bar graph below the bands indicates densitometric analysis of MBP levels in the spinal cord (B). Means ± SEM are reported (n=3 from 3 rats in each group, *p<0.05 compared to the indicated groups, Student’s t test).
Terminal deoxynucleotidyl-transferase-mediated dUTP nick end-labeling (TUNEL) staining and immunohistochemistry for OLGs in the spinal cord
Spinal cord samples at the indicated locations rostral to and caudal to the lesion were transversely cut (40 μm) for apoptotic cell staining via TUNEL method. Following dehydration and rehydration in a gradient of ethanol and xylene, TUNEL labeling was performed to detect DNA fragmentation in the apoptotic cells via a diaminobenzidine-based color reaction or a fluorescent dye using ApopTag® Peroxidase In Situ Apoptosis Detection Kit (Millipore, S7101). The sections were co-stained with Hoechst 33342 to label cell nuclei (Sigma-Aldrich). For the quantification of apoptotic cell number, TUNEL-positive cells were counted from multiple sections at a given level in each animal in a blind manner. To co-localize apoptotic cells to OLG cells, we performed double staining for active caspase 3 and OLG marker CC1 (adenomatous polyposis coli 7, Calbiochem, La Jolla, CA) in selected transverse or parasagittal sections of the spinal cord.
To verify that RhoA inhibition with certain NSAIDs increases numbers of OLGs around the lesioned spinal cord, in some transverse sections rostral to or caudal to the lesion, we immunostained OLGs with an antibody against CC1. The CC1 staining signals were visualized via an Alexa488 anti-mouse secondary antibody. The CC1-labeled individual OLGs in the dorsal, lateral and ventral white matter areas of the spinal cord were counted manually from five random transverse sections at a given level in each rat.
Histology for myelination in the spinal cord
Following animal perfusion with 4% paraformaldehyde, the spinal cords containing the lesion area were dissected out. The spinal cord blocks were transversely cut (10 μm) at the indicated levels rostral to or caudal to the lesion. Myelination of the spinal cord was evaluated via Luxol fast blue (LFB) staining and immunostaining for myelin basic protein. For LFB staining, spinal cord sections were dehydrated in a gradient of ethanol and stained in 0.1% solvent blue 38 (Sigma) in acidified 95% ethanol overnight at 60°C. After rinsing with 95% ethanol and distilled water, sections were then differentiated with 0.05% Li2CO3 and 35% ethanol several times until the contrast between the gray matter and white matter were clearly detected. For myelin basic protein (MBP) immunostaining, spinal cord sections were incubated with a mouse anti-MBP antibody (1:1000, Sternberger Monoclonals Inc., SMI-99) overnight at room temperature. After 5 washes with 0.1% Triton TX100 in tris-buffered saline, sections were incubated in Alexa488-conjugated anti-mouse IgG antibody (1:200) for 2 hrs at room temperature.
For myelin quantification, images were captured with a Nikon digital image-collecting system from multiple spinal cord sections. The measured fields (250 x 360 μm) in each section were randomly selected from three different white mater areas, including dorsal, lateral and ventral columns. The intensity of myelin signal in selected fields was analyzed automatically with ImageJ software. Specifically, after inversion of the loaded photos and conversion of them into binary images, the levels of threshold were adjusted until the clear myelin dots and circles were achieved. Then, myelin signal was automatically calculated as a ratio of myelin signal to the measured areas. To avoid measuring any non-specific background staining signals, the myelin dots <1 μm in diameter were excluded from myelin quantification. The average of 8 random sections was collected at a given level of the spinal cord in each animal. During histological processing and myelin measurements, the researcher was unaware of the drug treatment in each SCI rat.
Electron microscopy
Two SCI rats per group from batch 4 in vivo experiments were employed for axon ultrastructural analysis around the lesion site. Following deep anesthetization, rats were transcardially perfused at 28 days after SCI with 100 ml phosphate buffer saline and then with 300 ml of 4% paraformaldehyde and 2% glutaraldehyde. Two age and sex-matched uninjured rats were used as normal controls. One millimeter tissue slices was dissected from lateral column of the spinal cord at the lesion epicenter, and immersed in 1% paraformaldehyde and 2.5% glutaraldehyde in 0.1M cacodylate buffer for two days. Tissues were rinsed in buffer, dehydrated in graded ethanol and propylene oxide, and embedded in Embed812 epoxy resin. Ultrathin sections were prepared and stained with uranyl acetate and lead citrate and examined an electron microscope.
Western blot for spinal cord tissue
For MBP level assay in the lesioned spinal cord, 3 rats in each group from batch 4 in vivo experiments were used for Western blotting. Four weeks after injury (or age and sex-matched uninjured controls), rats were perfused through heart with cold phosphate buffer saline for 5 min. Tissue blocks of the fresh spinal cord 3 mm rostral and caudal to the lesion epicenter (6 mm long containing the lesion site) were collected to dry ice and stored at −80°C. Then, each of the spinal cord blocks was prepared in 1 ml lysis buffer with protease inhibitors and clarified by centrifugation at 15,000 g for 10 min at 4°C. Following total protein quantification in lysates, samples containing same amount of protein were prepared for Western blot using an antibody against MBP (Covence). Proteins were transferred to nitrocellulose membrane and bands were visualized with enhanced chemiluminescence reagents. For blot densitometry assay, the images of MBP bands were captured with a Bio-Rad Gel Doc XR documentation system and the band density was determined using Quantity One software (Fu et al. 2007).
Results
RhoA-inhibiting NSAIDs protect cultured CG-4 cells from cell death induced by TNF-α
After axonal lesioning in the CNS, OLGs along axotomized axons are usually exposed to various extracellular molecules including cytokines, which contribute to OLG cell death. Predominantly expressed by various cells including OLGs after SCI (Conrad et al. 2005), RhoA signal appears to regulate the apoptosis of OLGs following axonal lesion (Dubreuil et al. 2003). To characterize if RhoA-suppressing NSAIDs reduce OLG loss in vitro, we evaluated treatments with ibuprofen or indomethacin on cell death in differentiated CG-4 cell cultures exposed to a potent cytokine TNF-α. CG-4 cells are rat CNS glia from primary cultures of bipotential oligodendrocyte-type 2-astrocyte (O-2A) progenitors. They could differentiate into OLGs and type 2 astrocytes under certain culture conditions or following transplantation into the CNS (Franklin et al. 1995; Louis et al. 1992; Tontsch et al. 1994). Thus, differentiated CG4 cells possess the properties of OLGs and have been frequently employed to study the functions of OLGs in vitro (Nicholas et al. 2002; Wang et al. 2009a). After two day growth in serum-containing medium, CG4 cells were switched to serum-free medium containing N1 (including insulin) to stimulate maturation. Two-three days after growth in the second medium, the majority of CG4 cells exhibited multiple processes (Fig 1) and became positive to GalC, marking their differentiation (Nicholas et al. 2002). To induce cell death in differentiated CG4 cells, we applied TNF-α (1.2 μg/ml) to cultured cells for 8 hours. Cytokine TNF-α induces RhoA activation (Neumann et al. 2002) and cell apoptosis (Kim et al. 2001; Louis et al. 1993; Selmaj and Raine 1988; Yune et al. 2003). We labeled the survived and dead cells with fluorescent dyes calcein and EthD1, respectively. As expected, application of TNF-α remarkably increased the number of dead CG4 cells (Fig. 1). However, treatments with RhoA-inhibiting ibuprofen (500 μM), indomethacin (20 μM), or selective Rho inhibitor C3 transferase (4 μg/ml) (Fournier et al. 2003; Fu et al. 2007), significantly attenuated EthD1-positive cells induced due to TNF-α exposure. In contrast, naproxen, a non-RhoA inhibiting NSAID failed to protect cells from death. To exclude the possibility that the decreased number of EthD1-positive cells was caused by increased proliferation of CG4 cells, we evaluated effects of these drugs on the density of differentiated CG4 cells in the absence of TNF-α. Treatments with Ibu, Ind or C3 for 9 hrs did not significantly increase CG4 cell number (Fig. 1C). These experiments suggest that RhoA inhibition with NSAIDs or selective inhibitor effectively prevents cultured OLGs from cell death due to application of TNF-α, an upstream signal of RhoA.
Fig. 1. RhoA-inhibiting NSAIDs dramatically protect differentiated CG4 cells from death induced by TNF-α.

A. The representative examples of differentiated CG4 cells were labeled with calcein (green, survival cell bodies and processes, arrowheads) and EthD1 (red, dead cell nuclei, arrows) from different groups treated with vehicle or drugs. In the absence of cytokine TNF-α, only a small number of dead cells were detected. Incubation with TNF-α for 8 hrs remarkably increased the number of dead cells in differentiated CG4 cells. However, treatments with Ibu, Ind or C3 transferase principally prevented cell death labeled by EthD1. In contrast, Nap did not have such a protective effect. Scale bar: 50 μm. B, Bar graph indicates percentage of dead CG4 cells labeled with EthD1 eight hours after TNF-α application. Treatments with Ibu, Ind or C3 transferase significantly protected CG4 cells from death induced due to TNF-α incubation. **P < 0.001, compared to the controls treated with vehicle or Nap (n=4 coverslips in each group). C, Graph shows effects of drug treatments for 9 hrs on the number of CG4 cells in the absence of TNF-α. Treatments with Ibu, Ind or C3 did not significantly alter the number of cultured CG4 cells.
RhoA-inhibiting NSAIDs protect oligodendrocytes from apoptosis in rats with a contusion spinal cord injury
After SCI, numerous cells die through apoptosis along the fiber tracts undergoing Wallerian degeneration (Shuman et al. 1997) and this glial apoptosis contributes to chronic demyelination and axonal dysfunction (Jensen et al. 1999; Tamura et al. 2005; Warden et al. 2001). RhoA activation has been reported to play an essential role in inducing apoptotic cell death (Dubreuil et al. 2003). To detect if inactivating RhoA with NSAIDs protects cells from apoptosis in vivo, we administered RhoA-inhibiting Ibu (60 mg/kg/day) and Ind (4 mg/kg/day) subcutaneously into adult rats with a moderate contusion injury at T8 for 5 continuous days. Those doses have been shown to inhibit RhoA activity following systemic applications (Fu et al. 2007). Five days after SCI, we examined apoptotic positive cells by staining fragmented DNAs using TUNEL. We observed many apoptotic cells in transverse sections of the spinal cord 6–10 mm rostral to and caudal to the lesion in saline- or naproxen-treated controls (Fig. 2). At the indicated range of the spinal cord (6–10 mm from the lesion), we did not detect obvious difference in the number of apoptotic cells. The majority of TUNEL positive cells are present in the lateral, dorsal and ventral white matter areas, and only a few are observed in the gray matter. Consistently, previous studies indicate apoptotic cells on the borders of the lesions and in the vicinity of white matter (Dubreuil et al. 2003; Emery et al. 1998). The distribution of glial apoptosis is probably related to axonal Wallerian degeneration following axotomy (Crowe et al. 1997; Shuman et al. 1997). Double staining for active caspase-3, an apoptotic marker, and for CC1, a marker for OLGs, indicates that the majority of apoptotic cells are localized to OLG glia (Fig. 4O). The other apoptotic cells in the white matter might be microglia. Notably, the number of TUNEL positive cells in both rostral and caudal spinal cord was reduced by 40~50% in SCI rats treated with Ibu or Ind (Fig. 2). Constantly, Rho inhibition with selective inhibitor C3 transferase could reduce the number of TUNEL-labeled cells by ~50% in SCI rodents (Dubreuil et al. 2003).
RhoA-inhibiting NSAIDs attenuated apoptotic cell loss in SCI rats following 5 day treatments, suggesting that RhoA inhibition with Ibu and Ind may increase the numbers of OLGs along the white matter tracts after SCI. To confirm this hypothesis, we counted CC1-labeled OLGs from randomly selected transverse sections of the spinal cord 6–10 mm rostral or caudal to the lesion. Consistently, we detected moderately enhanced number of CC1-positve cells in white matter areas of the spinal cord in SCI rats treated with Ibu and Ind, but not with non-RhoA-inhibiting Nap (Fig. 3). The limited enhancement of OLG numbers after Ibu and Ind treatments was probably due to a small degree of OLG reduction five days following SCI in rodents (Beattie et al. 2002).
To confirm the protective effect of Ibu for OLGs, in another set of experiments, we delivered Ibu or vehicle saline to contusion SCI rats for 7 days and examined apoptosis in the injured spinal cord. At this time point, we detected more apoptotic cells in the rostral spinal cord than in the caudal regions (75 vs. 22/section). However, in the Ibu-treated rats, we also detected a significant reduction of TUNEL-positive cells in the spinal cord both rostral to and caudal to the lesion (Fig. 4A–N). Together, these findings support that RhoA inactivation with selected NSAIDs remarkably attenuates apoptotic cell loss after SCI.
Rho-inhibiting NSAIDs enhance axonal myelination caudal to the lesion in SCI rats
The above experiments demonstrate that RhoA-inhibiting NSAIDs significantly protect OLGs from apoptotic cell death several days after a contusion SCI. Loss of OLGs may directly leads to axonal demyelination and protection to these glial cells should preserve myelin structure around the injury. We next determined whether RhoA inhibition with NSAIDs improved myelination of spinal cord axons around the lesion 6 weeks after a contusion SCI. After SCI, axonal demyelination occurs initially at the lesion epicenter, but it progresses gradually along the white matter tracts of the spinal cord (Wu and Ren 2009). Thus, we examined the integrity of myelin structures several mm from the lesion site in SCI rats 6 weeks after the injury. In particular, we visualized myelinated regions in transverse sections of the spinal cord 3–4 mm or 6–8 mm caudal to the lesion center with Luxol fast blue staining (Fig. 5A–C). For LFB staining, blue areas indicate myelinated axons whereas pale areas in the white matter indicate demyelination of axons. Compared to uninjured spinal cord at same level (not shown), we detected obvious myelin loss along the white matter tracts in SCI rats treated with vehicle. However, we observed significantly increased LFB staining signals at different white matter areas of the spinal cord in SCI rats treated with Ibu or Ind, particularly with the former drug. In contrast, non-RhoA-inhibiting NSAID naproxen did not increase LFB staining signal in the spinal cord.
To confirm the increased myelination of axons in SCI rats treated with Ibu and Ind, we also performed immunohistochemistry for MBP, a widely used marker for myelin structure in the nerve system. Comparing to sections from uninjured rats (Fig. 5D), we frequently observed the regions devoid of myelinated axons in the white matter areas of SCI rats treated with vehicle or naproxen. Nevertheless, the SCI rats treated with Ibu or Ind exhibit significantly higher MBP-staining signals in the spinal cord 3–4 or 6–8 mm caudal to the lesion than SCI rats treated with vehicle or naproxen (Fig. 5D–F). These results suggest that RhoA suppression with Ibu and Ind significantly increases the number of myelinated axons caudal to the lesion probably by preventing demyelination and/or enhancing remyelination of spinal cord axons around the lesion.
RhoA-inhibiting NSAIDs increased axonal myelination rostral to the lesion in SCI rats
Ibu and Ind protect OLGs from apoptotic cell death in the spinal cord both caudal and rostral to the contusion SCI (Fig. 2–4). If protection to OLGs principally contributes to the enhanced myelination in the caudal spinal cord, increased survival of OLGs in the rostral spinal cord should also promote myelination at the corresponding areas. Thus, we also measured myelination of the spinal cord 6–8 or 3–4 mm rostral to the lesion from these SCI rats. Comparing to uninjured spinal cord at the same levels (Fig. 6D, not shown for LFB), we detected remarkably reduced LFB- and MBP-staining signals at the rostral spinal cord in SCI rats treated with saline or naproxen. As expected, we found that treatments with Ibu and Ind significantly increased LFB- and MBP-staining signals at the rostral spinal cord compared with SCI controls (Fig. 6). We observed the increased myelination at different white matter regions including dorsal, lateral and ventral spinal cord white matter columns. Therefore, our experiments support that RhoA inhibition with selected NSAIDs promotes myelination of spinal cord axons both rostral to and caudal to a contusion lesion. Of note, our behavioral tests support that Ibu and Ind enhance locomotor recovery in SCI rats (not shown) (Fu et al. 2007). In addition to improved axonal growth that we reported previously (Fu et al. 2007), increased axonal myelination around the lesion may also contribute to the enhanced functional recovery in SCI rodents treated with RhoA-inhibiting NSAIDs.
Ibu and Ind increased axonal myelination at the lesion site in SCI rats
Electron microscope was utilized to determine axon myelination at the lesion site 28 days after a contusion SCI. Contusion injury usually results in cavitation and loss of gross tissue structure within the central region of the spinal cord at the injury epicenter, especially in the regions close to the dorsal spinal cord (Fu et al. 2007; Siegenthaler et al. 2007). To compare the ultrastructures of myelinated axons at the lesion site, we collected one mm of lateral columns of the spinal cord at the lesion epicenter from different groups of animals. In contrast to uninjured controls, we detected obviously reduced density of axons in animals treated with Veh or Nap (Fig. 7), indicating some spared axons in the lateral columns following a moderate contusion SCI. We frequently detected myelinated axons at the injury site in SCI controls, but some axons were demyelinated (arrows) and most myelinated axons exhibited damaged myelin structures, including less compact myelin and partial myelin loss (arrows). However, in animals treated with Ibu or Ind, we observed higher density of myelinated axons, suggesting that RhoA inhibition with these drugs promotes axonal regeneration or sprouting at the injury site (Fu et al. 2007; Wang et al. 2009b). Although we also found some loose myelin in SCI rats treated with Ibu and Ind, most myelinted axons exhibited intact and compact myelin structures in these rodents. Moreover, comparing to normally myelinated axons (arrowheads), we noticed some thin myelin sheaths (thin arrows) in SCI rats, including those treated with Ibu and Ind, suggesting remyelination of some axons after SCI (Siegenthaler et al. 2007).
To confirm the increased axon myelination following RhoA suppression with NSAIDs, we examined the levels of MBP in lysates of the spinal cord at the lesion site 4 weeks after a contusion SCI at T8 in rats. In comparison to the basic levels in the normal spinal cord at T7-9, MBP levels in the injured spinal cord (3 mm rostral to and caudal to the lesion) were moderately decreased in Veh and Nap-treated rats (Fig. 8). Nevertheless, the SCI rats treated with Ibu or Ind exhibited significantly higher MBP levels at the same spinal cord region than those treated with Veh or Nap.
Discussion
Following CNS axonal injuries, OLG cell loss and demyelination persist along the white matter tracts and significantly contribute to pathophysiology and dysfunction in several neurologic disorders including SCI (Jensen et al. 1999; Tamura et al. 2005; Warden et al. 2001). The signal conduction deficits along demyelinated axons continue chronically although a limited degree of spontaneous remyelination occurs by endogenous OLGs and invading peripheral Schwann cells (Totoiu and Keirstead 2005). A number of factors are responsible for the failure of endogenous remyelination, including limited number of precursor cells, presence of inhibitory factors for OLG differentiation, and lack of growth factors for OLG survival and myelination in the injured CNS. As a result, demyelination is progressive in the chronic stage and the number of demyelinated axons increases even one year post-trauma (Totoiu and Keirstead 2005). Thus, preventing axonal demyelination and enhancing remyelination have become attractive therapies for repairing the injured CNS. By targeting individual factors, several strategies have been shown to regain function of demyelinated axons in adult CNS via remyelination (Butzkueven et al. 2002; Cao et al.; Groves et al. 1993). Obviously, approaches targeting multiple extracellular factors may become more effective for stimulating axonal remyelination. In this study, we find that blockade of RhoA, the convergent signal for numerous extracellular molecules, with NSAIDs Ibu and Ind, remarkably prevents OLG loss and increases myelination along the white matter tracts following traumatic SCI. Our findings demonstrate great therapeutic potential of the selected NSAIDs to the neurological disorders characterized by axonal damages.
OLG loss and axonal demyelination following CNS injuries
OLG cell death and subsequent demyelination of surviving axons substantially contribute to deterioration of neural function after CNS axonal trauma (Beattie et al. 2002; Crowe et al. 1997). Injury to white matter fibers usually results in axonal degeneration and intrafascicular OLG apoptosis. OLG death starts as early as one day after injury and persists for at least 3 weeks (Beattie et al. 2000). Cellular apoptosis predominantly presents in the areas of Wallerian degeneration along the white matter tracts after SCI. In this study, we detected a similar pattern of apoptotic cells along the spinal cord fiber tracts after a moderate contusion SCI. Double staining with a marker protein confirms the apoptotic cell loss of OLGs. However, treatments with RhoA-inhibiting Ibu or Ind, but not with NSAID naproxen, remarkably reduced the number of apoptotic cells (~50%). Consistently, selective Rho inhibitor C3 transferase attenuates cell death to a similar degree in vitro (Fig. 1) and in vivo (Dubreuil et al. 2003). Of note, differentiation defect of OLG precursor cells has been recognized as a major cause for the failure of axon remyelination in multiple sclerosis (John et al. 2002; Syed et al. 2011). Although treatments with Ibu and Ind for 9 hours did not alter the number of cultured CG4 cells (Fig. 1C), we could not exclude the possibility that long-term treatments with RhoA-inhibiting NSAIDs increased myelination in SCI rodents partially via promoting proliferation and differentiation of OLG precursors. In fact, RhoA has been shown to be downregulated during differentiation of OLG precursors (Kippert et al. 2007) and to mediate differentiation of OLG precursors and process elongation and myelin formation of OLGs (Wolf et al. 2001).
Importantly, we evaluated RhoA inhibition with NSAIDs on axon structural alterations in the chronic stage 4–6 weeks after SCI and observed improved myelination around the lesioned spinal cord. Although we could not exclude possible protective effects of RhoA-inhibiting NSAIDs to axonal cylinders, our results suggest that increased survival of OLGs following RhoA suppression with Ibu or Ind attenuates demyelination of spinal cord axons and facilitates remyelination of spared and/or regenerated axons around the lesion, thus enhancing axonal myelination around the lesion. Similarly, preventing OLG loss through other approaches, such as treatments with a p38-MAPK inhibitor or minocycline, could improve axonal myelination and neural function (Horiuchi et al. 2003; Yune et al. 2007). Moreover, over-expression of p35, a broad-spectrum caspase inhibitor, in transgenic mice, increased OLG number and diminished demyelination after SCI (Tamura et al. 2005). Thus, our results, together with previous reports, suggest that OLG apoptosis is an important pathologic factor in axonal demyelination following CNS axotomic injuries.
Molecular basis for OLG protection and myelination improvement of certain NSAIDs after CNS axonal injuries
RhoA inactivation with Ibu and Ind is most likely to be the molecular basis for OLG protection and enhanced axonal myelination detected in this report. First, following axonal injury in SCI rodents, RhoA is highly upregulated and substantially activated in different types of cells including OLGs (Conrad et al. 2005; Dubreuil et al. 2003). RhoA inactivation with selective inhibitor C3 transferase efficiently protects OLGs from cell death in cultures (Fig. 1) and SCI rodents (Dubreuil et al. 2003). Second, by using Rho-GTP binding assay with Rhotekin binding domain, we have demonstrated that ibuprofen and indomethacin effectively reverse increased RhoA activity in cultured cells exposed to a Rho agonist or axonal growth inhibitors, or in rats with traumatic SCI (Fu et al. 2007). Correspondingly, we demonstrate that only RhoA-inhibiting NSAIDs Ibu and Ind are able to reduce OLG cell death in vitro and in vivo. In contrast, naproxen, an NSAID that does not have RhoA-suppressing function, failed to protect OLGs (Fig. 1–3) and myelin structure (Fig. 5–8). Furthermore, the SCI rodents treated with RhoA-inhibiting NSAIDS have significant improvement of the hindlimb locomotion (Fu et al. 2007), but not those treated with naproxen. Although we did not measure RhoA activity in individual OLGs, our results suggest that RhoA suppression is essential for the beneficial effects detected here. Therefore, following CNS axonal injuries, over-activation of RhoA plays an important role in glial cell loss (Dubreuil et al. 2003), which consequently induces myelin damage and contributes to signal conduction collapse and permanent functional deficits (Blight 2002).
Recently, by using different in vitro research approaches, we have convincibly demonstrated that the transcription factor PPARγ is essential for coupling ibuprofen to RhoA inhibition and subsequent promotion of neurite growth in neurons (Dill et al. 2010). Given wide expression of PPARs in different types of cells including OLGs (Granneman et al. 1998), PPARs may also mediate RhoA activation in this type of glial cells. In support of this, PPAR agonists have been reported to protect OLG progenitor cells from oxidant damage and promote differentiation of these cells (Bernardo et al. 2009). Treatments with PPAR agonists were able to reduce secondary tissue damage and promote significant functional recovery in SCI rodents (McTigue et al. 2007; Park et al. 2007). Thus, it will be interesting to examine the potential role of PPARs in regulating OLG loss and demyelination following CNS axonal injuries.
Upstream factors mediating glial RhoA activation following CNS axonal injuries
Combined effects of many different Rho-activating factors may regulate massive Rho activation in glial cells following axonal lesions. After SCI, generation of several extracellular inflammatory mediators and cytokines, such as TNF-α, interferon-γ and FAS, contributes to apoptotic cell death of glial cells in the injured spinal cord (Beattie et al. 2002; Demjen et al. 2004; Dubreuil et al. 2003; Kim et al. 2001). ProNGF, the precursor form of nerve growth factor, plays an important role in SCI-induced OLG apoptosis mediated via P75 receptor (Beattie et al. 2002; Dubreuil et al. 2003). Lack of trophic support after axonal damage may be partly responsible for OLG death because a few trophic factors, including insulin-like growth factors, neurotrophin-3, ciliary-neurotrophic factor, leukemia inhibitory factor, interleukin-6 and neuregulins, have been shown to be essential for OLG survival (Barres et al. 1993). Also, AMPA receptor activation via increased extracellular glutamate may initiate OLG injury (Li et al. 1999; Li and Stys 2000; McDonald et al. 1998) and cause apoptosis of this glial cell following white matter injury (Sanchez-Gomez et al. 2003). Then, activation of membrane receptors by these apoptosis-inducing factors, such as TNF receptor by TNF-α (Neumann et al. 2002), P75 receptor (Huang and Reichardt 2003) by proNGF and lack of neurotrophic factors (Yamashita et al. 1999), and AMPA receptor (Kim et al. 2004) by glutamate, may increase the activity of intracellular RhoA signal, which is linked to apoptotic cell death after SCI (Dubreuil et al. 2003).
Glial cell death via apoptosis, particularly loss of myelinating OLGs, may result in consequent axonal demyelination (Jensen et al. 1999; Tamura et al. 2005; Warden et al. 2001). In support this hypothesis, remyelination of the spinal cord axons via cell transplantation (Akiyama et al. 2002; Groves et al. 1993; Keirstead et al. 2005) or transplants combined with cAMP elevation or neurotrophic factors (Cao et al. 2005; Pearse et al. 2004) have been shown to improve functional recovery in SCI rodents significantly. In addition, RhoA might negatively regulate myelin formation in the CNS because RhoA activation by Lingo1, a protein present in OLGs, appears to suppress myelination of CNS axons (Mi et al. 2005; Mi et al. 2009). Together, our results suggest that silence of RhoA activity with Ibu and Ind prevents OLG loss and promotes myelination of CNS axons following a trauma by attenuating myelin damage and/or enhancing axonal remyelination.
Therapeutic potential of RhoA-inhibiting NSAIDs to patients with white matter injuries
Different pathophysiological pathways are activated following traumatic axonal lesions. The treatments targeting different factors of neural damages should be more effective than targeting an individual component. As the downstream signal for multiple extracellular molecules, RhoA activation plays a critical role in limiting axonal regrowth (Fu et al. 2007) and inducing glial apoptosis after CNS axonal injury (Fig. 2–4), suggesting that RhoA signal is an important therapeutic target for repairing injured CNS neurons. The CNS lesions upregulate RhoA protein in both rodents (Conrad et al. 2005; Schwab et al. 2004) and patients (Brabeck et al. 2004). The increased RhoA activation usually starts shortly after injury and persists for at least several weeks (Brabeck et al. 2004; Conrad et al. 2005), indicating a wide time-window for therapeutic intervention after axonal injuries. Inhibition of Rho signal with C3 transferase or its downstream Rock with Y-27632, fasudil or peptide p21CIP1/WAF1 has been reported to improve recovery in SCI rodents (Dergham et al. 2002; Fournier et al. 2003; Tanaka et al. 2004), but none of these compounds have been approved for use in humans. Interestingly, several NSAIDs, a group of widely used drugs as cyclooxygenase selective and nonselective inhibitors, have been found to repress RhoA activity strongly. Our new findings in this project suggest that RhoA-inhibiting NSAIDs benefit recovery of injured CNS axons not only by promoting axonal elongation (Fu et al. 2007), but by enhancing OLG survival and axonal myelination along the disrupted white matter tracts.
In recent years, a number of researchers have reported progress in developing intervention strategies in animal SCI models via limiting secondary injury and enhancing fiber sprouting/regeneration and promoting functional recovery. However, few basic science discoveries have been applied clinically to improve behavioral outcomes of axonal injury patients. Thus, it is critical that the promising approaches from basic studies are translatable to effective clinical therapies. We have demonstrated that systemic delivery of RhoA-inhibiting NSAID ibuprofen, one of the most widely prescribed drugs for treatment of pain, fever and inflammation in humans, could benefit recovery of axonal injured rodents by promoting regeneration (Fu et al. 2007) and myelination of spinal cord axonal tracts (Fig. 5–8). Although it is difficult to accurately assess the extent to which increased axonal growth and myelination contribute to functional improvement in SCI rodents, our studies should facilitate us to design more rational therapies for traumatic CNS injuries and neurological disorders characterized by axonal damages.
Research highlights.
RhoA-inhibiting ibuprofen and indomethacin reduce oligodendrocyte cell death.
Ibuprofen and indomethacin increase axonal myelination in SCI rodents.
Ibuprofen and indomethacin benefits recovery by promoting axon growth and myelination.
Acknowledgments
We thank John Dill and Dr. Keith Tansy for technical support and the Molecular and Cellular Imaging Center at UT Southwestern Medical Center for electron microscopy. This work was supported by research grants to S.L. from the Paralyzed Veterans of America, NIH (1R21NS066114-01A1), Morton Cure Paralysis Fund and the Christopher and Dana Reeve Foundation.
Abbreviations
- DS
dorsal
- EthD1
ethidium homodimer-1
- GM
gray matter
- Ibu
ibuprofen
- Ind
indomethacin
- LFB
Luxol fast blue
- LT
lateral
- MBP
myelin basic protein
- O-2A
oligodendrocyte-type 2-astrocyte
- PPARγ
peroxisome proliferator-activated receptor γ
- Nap
naproxen
- NSAIDs
nonsteroidal anti-inflammatory drugs
- SCI
spinal cord injury
- TNF-α
tumor necrosis factor α
- TUNEL
terminal deoxynucleotidyl-transferase-mediated dUTP nick end-labeling
- WM
white matter
- VT
ventral
Footnotes
Author Disclosure Statement: No competing financial interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama Y, Radtke C, Kocsis JD. Remyelination of the rat spinal cord by transplantation of identified bone marrow stromal cells. J Neurosci. 2002;22(15):6623–30. doi: 10.1523/JNEUROSCI.22-15-06623.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118(1):283–95. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Farooqui AA, Bresnahan JC. Review of current evidence for apoptosis after spinal cord injury. J Neurotrauma. 2000;17(10):915–25. doi: 10.1089/neu.2000.17.915. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO. ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron. 2002;36(3):375–86. doi: 10.1016/s0896-6273(02)01005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo A, Bianchi D, Magnaghi V, Minghetti L. Peroxisome proliferator-activated receptor-gamma agonists promote differentiation and antioxidant defenses of oligodendrocyte progenitor cells. J Neuropathol Exp Neurol. 2009;68(7):797–808. doi: 10.1097/NEN.0b013e3181aba2c1. [DOI] [PubMed] [Google Scholar]
- Bertrand J, Winton MJ, Rodriguez-Hernandez N, Campenot RB, McKerracher L. Application of rho antagonist to neuronal cell bodies promotes neurite growth in compartmented cultures and regeneration of retinal ganglion cell axons in the optic nerve of adult rats. J Neurosci. 2005;25(5):1113–21. doi: 10.1523/JNEUROSCI.3931-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight AR. Miracles and molecules--progress in spinal cord repair. Nat Neurosci. 2002;5(Suppl):1051–4. doi: 10.1038/nn939. [DOI] [PubMed] [Google Scholar]
- Brabeck C, Beschorner R, Conrad S, Mittelbronn M, Bekure K, Meyermann R, Schluesener HJ, Schwab JM. Lesional expression of RhoA and RhoB following traumatic brain injury in humans. J Neurotrauma. 2004;21(6):697–706. doi: 10.1089/0897715041269597. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M, Fehlings M, Herr DL, Hitchon PW, Marshall LF, et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. Jama. 1997;277(20):1597–604. [PubMed] [Google Scholar]
- Butzkueven H, Zhang JG, Soilu-Hanninen M, Hochrein H, Chionh F, Shipham KA, Emery B, Turnley AM, Petratos S, Ernst M, et al. LIF receptor signaling limits immune-mediated demyelination by enhancing oligodendrocyte survival. Nat Med. 2002;8(6):613–9. doi: 10.1038/nm0602-613. [DOI] [PubMed] [Google Scholar]
- Cao Q, He Q, Wang Y, Cheng X, Howard RM, Zhang Y, DeVries WH, Shields CB, Magnuson DS, Xu XM, et al. Transplantation of ciliary neurotrophic factor-expressing adult oligodendrocyte precursor cells promotes remyelination and functional recovery after spinal cord injury. J Neurosci. 30(8):2989–3001. doi: 10.1523/JNEUROSCI.3174-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Xu XM, Devries WH, Enzmann GU, Ping P, Tsoulfas P, Wood PM, Bunge MB, Whittemore SR. Functional recovery in traumatic spinal cord injury after transplantation of multineurotrophin-expressing glial-restricted precursor cells. J Neurosci. 2005;25(30):6947–57. doi: 10.1523/JNEUROSCI.1065-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad S, Schluesener HJ, Trautmann K, Joannin N, Meyermann R, Schwab JM. Prolonged lesional expression of RhoA and RhoB following spinal cord injury. J Comp Neurol. 2005;487(2):166–75. doi: 10.1002/cne.20561. [DOI] [PubMed] [Google Scholar]
- Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3(1):73–6. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- Demjen D, Klussmann S, Kleber S, Zuliani C, Stieltjes B, Metzger C, Hirt UA, Walczak H, Falk W, Essig M, et al. Neutralization of CD95 ligand promotes regeneration and functional recovery after spinal cord injury. Nat Med. 2004;10(4):389–95. doi: 10.1038/nm1007. [DOI] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22(15):6570–7. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill J, Patel AR, Yang XL, Bachoo R, Powell CM, Li S. A molecular mechanism for ibuprofen-mediated RhoA inhibition in neurons. J Neurosci. 2010;30(3):963–72. doi: 10.1523/JNEUROSCI.5045-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill J, Wang H, Zhou FQ, Li S. Inactivation of glycogen synthase kinase-3 promotes axonal growth and recovery in the CNS. J Neurosci. 2008;28(36):8914–28. doi: 10.1523/JNEUROSCI.1178-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil CI, Winton MJ, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162(2):233–43. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery E, Aldana P, Bunge MB, Puckett W, Srinivasan A, Keane RW, Bethea J, Levi AD. Apoptosis after traumatic human spinal cord injury. J Neurosurg. 1998;89(6):911–20. doi: 10.3171/jns.1998.89.6.0911. [DOI] [PubMed] [Google Scholar]
- Fischer D, Petkova V, Thanos S, Benowitz LI. Switching mature retinal ganglion cells to a robust growth state in vivo: gene expression and synergy with RhoA inactivation. J Neurosci. 2004;24(40):8726–40. doi: 10.1523/JNEUROSCI.2774-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, Takizawa BT, Strittmatter SM. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci. 2003;23(4):1416–23. doi: 10.1523/JNEUROSCI.23-04-01416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ, Bayley SA, Milner R, Ffrench-Constant C, Blakemore WF. Differentiation of the O-2A progenitor cell line CG-4 into oligodendrocytes and astrocytes following transplantation into glia-deficient areas of CNS white matter. Glia. 1995;13(1):39–44. doi: 10.1002/glia.440130105. [DOI] [PubMed] [Google Scholar]
- Fu Q, Hue J, Li S. Nonsteroidal anti-inflammatory drugs promote axon regeneration via RhoA inhibition. J Neurosci. 2007;27(15):4154–64. doi: 10.1523/JNEUROSCI.4353-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman J, Skoff R, Yang X. Member of the peroxisome proliferator-activated receptor family of transcription factors is differentially expressed by oligodendrocytes. J Neurosci Res. 1998;51(5):563–73. doi: 10.1002/(SICI)1097-4547(19980301)51:5<563::AID-JNR3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Groves AK, Barnett SC, Franklin RJ, Crang AJ, Mayer M, Blakemore WF, Noble M. Repair of demyelinated lesions by transplantation of purified O-2A progenitor cells. Nature. 1993;362(6419):453–5. doi: 10.1038/362453a0. [DOI] [PubMed] [Google Scholar]
- Hara M, Takayasu M, Watanabe K, Noda A, Takagi T, Suzuki Y, Yoshida J. Protein kinase inhibition by fasudil hydrochloride promotes neurological recovery after spinal cord injury in rats. J Neurosurg. 2000;93(1 Suppl):94–101. doi: 10.3171/spi.2000.93.1.0094. [DOI] [PubMed] [Google Scholar]
- Horiuchi H, Ogata T, Morino T, Chuai M, Yamamoto H. Continuous intrathecal infusion of SB203580, a selective inhibitor of p38 mitogen-activated protein kinase, reduces the damage of hind-limb function after thoracic spinal cord injury in rat. Neurosci Res. 2003;47(2):209–17. doi: 10.1016/s0168-0102(03)00216-5. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–42. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Huber AB, Kolodkin AL, Ginty DD, Cloutier JF. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu Rev Neurosci. 2003;26:509–63. doi: 10.1146/annurev.neuro.26.010302.081139. [DOI] [PubMed] [Google Scholar]
- Jensen NA, West MJ, Celis JE. Oligodendrocyte programmed cell death and central myelination deficiency induced in transgenic mice by synergism between c-Myc and Oct-6. J Biol Chem. 1999;274(42):29921–6. doi: 10.1074/jbc.274.42.29921. [DOI] [PubMed] [Google Scholar]
- John GR, Shankar SL, Shafit-Zagardo B, Massimi A, Lee SC, Raine CS, Brosnan CF. Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med. 2002;8(10):1115–21. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- Keirstead HS, Nistor G, Bernal G, Totoiu M, Cloutier F, Sharp K, Steward O. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci. 2005;25(19):4694–705. doi: 10.1523/JNEUROSCI.0311-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GM, Xu J, Xu J, Song SK, Yan P, Ku G, Xu XM, Hsu CY. Tumor necrosis factor receptor deletion reduces nuclear factor-kappaB activation, cellular inhibitor of apoptosis protein 2 expression, and functional recovery after traumatic spinal cord injury. J Neurosci. 2001;21(17):6617–25. doi: 10.1523/JNEUROSCI.21-17-06617.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Jeon S, Shin EY, Kim EG, Park J, Bae CD. AMPA, not NMDA, activates RhoA GTPases and subsequently phosphorylates moesin. Exp Mol Med. 2004;36(1):98–102. doi: 10.1038/emm.2004.14. [DOI] [PubMed] [Google Scholar]
- Kippert A, Trajkovic K, Rajendran L, Ries J, Simons M. Rho regulates membrane transport in the endocytic pathway to control plasma membrane specialization in oligodendroglial cells. J Neurosci. 2007;27(13):3560–70. doi: 10.1523/JNEUROSCI.4926-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R, Sarner S, Ahmed S, Lim L. Rho family GTPases and neuronal growth cone remodelling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol Cell Biol. 1997;17(3):1201–11. doi: 10.1128/mcb.17.3.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranenburg O, Poland M, van Horck FP, Drechsel D, Hall A, Moolenaar WH. Activation of RhoA by lysophosphatidic acid and Galpha12/13 subunits in neuronal cells: induction of neurite retraction. Mol Biol Cell. 1999;10(6):1851–7. doi: 10.1091/mbc.10.6.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mealing GA, Morley P, Stys PK. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+-dependent glutamate transport. J Neurosci. 1999;19(14):RC16. doi: 10.1523/JNEUROSCI.19-14-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Stys PK. Mechanisms of ionotropic glutamate receptor-mediated excitotoxicity in isolated spinal cord white matter. J Neurosci. 2000;20(3):1190–8. doi: 10.1523/JNEUROSCI.20-03-01190.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis JC, Magal E, Muir D, Manthorpe M, Varon S. CG-4, a new bipotential glial cell line from rat brain, is capable of differentiating in vitro into either mature oligodendrocytes or type-2 astrocytes. J Neurosci Res. 1992;31(1):193–204. doi: 10.1002/jnr.490310125. [DOI] [PubMed] [Google Scholar]
- Louis JC, Magal E, Takayama S, Varon S. CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science. 1993;259(5095):689–92. doi: 10.1126/science.8430320. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4(3):291–7. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- McGee AW, Strittmatter SM. The Nogo-66 receptor: focusing myelin inhibition of axon regeneration. Trends Neurosci. 2003;26(4):193–8. doi: 10.1016/S0166-2236(03)00062-6. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Tripathi R, Wei P, Lash AT. The PPAR gamma agonist Pioglitazone improves anatomical and locomotor recovery after rodent spinal cord injury. Exp Neurol. 2007;205(2):396–406. doi: 10.1016/j.expneurol.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, Chang J, Thill G, Levesque M, Zhang M, et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8(6):745–51. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- Mi S, Miller RH, Tang W, Lee X, Hu B, Wu W, Zhang Y, Shields CB, Miklasz S, Shea D, et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol. 2009;65(3):304–15. doi: 10.1002/ana.21581. [DOI] [PubMed] [Google Scholar]
- Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005;4(5):387–98. doi: 10.1038/nrd1719. [DOI] [PubMed] [Google Scholar]
- Neumann H, Schweigreiter R, Yamashita T, Rosenkranz K, Wekerle H, Barde YA. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J Neurosci. 2002;22(3):854–62. doi: 10.1523/JNEUROSCI.22-03-00854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas RS, Stevens S, Wing MG, Compston DA. Microglia-derived IGF-2 prevents TNFalpha induced death of mature oligodendrocytes in vitro. J Neuroimmunol. 2002;124(1–2):36–44. doi: 10.1016/s0165-5728(02)00011-5. [DOI] [PubMed] [Google Scholar]
- Nishio Y, Koda M, Kitajo K, Seto M, Hata K, Taniguchi J, Moriya H, Fujitani M, Kubo T, Yamashita T. Delayed treatment with Rho-kinase inhibitor does not enhance axonal regeneration or functional recovery after spinal cord injury in rats. Exp Neurol. 2006;200(2):392–7. doi: 10.1016/j.expneurol.2006.02.123. [DOI] [PubMed] [Google Scholar]
- Park SW, Yi JH, Miranpuri G, Satriotomo I, Bowen K, Resnick DK, Vemuganti R. Thiazolidinedione class of peroxisome proliferator-activated receptor gamma agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. J Pharmacol Exp Ther. 2007;320(3):1002–12. doi: 10.1124/jpet.106.113472. [DOI] [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10(6):610–6. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- Sanchez-Gomez MV, Alberdi E, Ibarretxe G, Torre I, Matute C. Caspase-dependent and caspase-independent oligodendrocyte death mediated by AMPA and kainate receptors. J Neurosci. 2003;23(29):9519–28. doi: 10.1523/JNEUROSCI.23-29-09519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab JM, Conrad S, Elbert T, Trautmann K, Meyermann R, Schluesener HJ. Lesional RhoA+ cell numbers are suppressed by anti-inflammatory, cyclooxygenase-inhibiting treatment following subacute spinal cord injury. Glia. 2004;47(4):377–86. doi: 10.1002/glia.20031. [DOI] [PubMed] [Google Scholar]
- Selmaj K, Raine CS. Tumor necrosis factor mediates myelin damage in organotypic cultures of nervous tissue. Ann N Y Acad Sci. 1988;540:568–70. doi: 10.1111/j.1749-6632.1988.tb27175.x. [DOI] [PubMed] [Google Scholar]
- Shuman SL, Bresnahan JC, Beattie MS. Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res. 1997;50(5):798–808. doi: 10.1002/(SICI)1097-4547(19971201)50:5<798::AID-JNR16>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Siegenthaler MM, Tu MK, Keirstead HS. The extent of myelin pathology differs following contusion and transection spinal cord injury. J Neurotrauma. 2007;24(10):1631–46. doi: 10.1089/neu.2007.0302. [DOI] [PubMed] [Google Scholar]
- Sung JK, Miao L, Calvert JW, Huang L, Louis Harkey H, Zhang JH. A possible role of RhoA/Rho-kinase in experimental spinal cord injury in rat. Brain Res. 2003;959(1):29–38. doi: 10.1016/s0006-8993(02)03717-4. [DOI] [PubMed] [Google Scholar]
- Syed YA, Hand E, Mobius W, Zhao C, Hofer M, Nave KA, Kotter MR. Inhibition of CNS remyelination by the presence of semaphorin 3A. J Neurosci. 2011;31(10):3719–28. doi: 10.1523/JNEUROSCI.4930-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Nakamura M, Ogawa Y, Toyama Y, Miura M, Okano H. Targeted expression of anti-apoptotic protein p35 in oligodendrocytes reduces delayed demyelination and functional impairment after spinal cord injury. Glia. 2005;51(4):312–21. doi: 10.1002/glia.20212. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Yamashita T, Yachi K, Fujiwara T, Yoshikawa H, Tohyama M. Cytoplasmic p21(Cip1/WAF1) enhances axonal regeneration and functional recovery after spinal cord injury in rats. Neuroscience. 2004;127(1):155–64. doi: 10.1016/j.neuroscience.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Tontsch U, Archer DR, Dubois-Dalcq M, Duncan ID. Transplantation of an oligodendrocyte cell line leading to extensive myelination. Proc Natl Acad Sci U S A. 1994;91(24):11616–20. doi: 10.1073/pnas.91.24.11616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totoiu MO, Keirstead HS. Spinal cord injury is accompanied by chronic progressive demyelination. J Comp Neurol. 2005;486(4):373–83. doi: 10.1002/cne.20517. [DOI] [PubMed] [Google Scholar]
- Wang PS, Wang J, Xiao ZC, Pallen CJ. Protein-tyrosine phosphatase alpha acts as an upstream regulator of Fyn signaling to promote oligodendrocyte differentiation and myelination. J Biol Chem. 2009a;284(48):33692–702. doi: 10.1074/jbc.M109.061770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Budel S, Baughman K, Gould G, Song KH, Strittmatter SM. Ibuprofen Enhances Recovery from Spinal Cord Injury by Limiting Tissue Loss and Stimulating Axonal Growth. J Neurotrauma. 2009b doi: 10.1089/neu.2007.0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden P, Bamber NI, Li H, Esposito A, Ahmad KA, Hsu CY, Xu XM. Delayed glial cell death following wallerian degeneration in white matter tracts after spinal cord dorsal column cordotomy in adult rats. Exp Neurol. 2001;168(2):213–24. doi: 10.1006/exnr.2000.7622. [DOI] [PubMed] [Google Scholar]
- Wolf RM, Wilkes JJ, Chao MV, Resh MD. Tyrosine phosphorylation of p190 RhoGAP by Fyn regulates oligodendrocyte differentiation. J Neurobiol. 2001;49(1):62–78. doi: 10.1002/neu.1066. [DOI] [PubMed] [Google Scholar]
- Wu B, Ren X. Promoting axonal myelination for improving neurological recovery in spinal cord injury. J Neurotrauma. 2009;26(10):1847–56. doi: 10.1089/neu.2008.0551. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tucker KL, Barde YA. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron. 1999;24(3):585–93. doi: 10.1016/s0896-6273(00)81114-9. [DOI] [PubMed] [Google Scholar]
- Yune TY, Chang MJ, Kim SJ, Lee YB, Shin SW, Rhim H, Kim YC, Shin ML, Oh YJ, Han CT, et al. Increased production of tumor necrosis factor-alpha induces apoptosis after traumatic spinal cord injury in rats. J Neurotrauma. 2003;20(2):207–19. doi: 10.1089/08977150360547116. [DOI] [PubMed] [Google Scholar]
- Yune TY, Lee JY, Jung GY, Kim SJ, Jiang MH, Kim YC, Oh YJ, Markelonis GJ, Oh TH. Minocycline alleviates death of oligodendrocytes by inhibiting pro-nerve growth factor production in microglia after spinal cord injury. J Neurosci. 2007;27(29):7751–61. doi: 10.1523/JNEUROSCI.1661-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]