

Abstract
Estradiol attenuates the ATP-induced increase of intracellular calcium concentration ([Ca2+]i) in rat dorsal root ganglion (DRG) neurons by blocking the L-type voltage gated calcium channel (VGCC). Since ATP is a putative nociceptive signal, this action may indicate a site of estradiol regulation of pain. In other neurons, 17β-estradiol (E2) has been shown to modulate L-type VGCC through a membrane estrogen receptor-group II metabotropic glutamate receptor (mGluR2/3). The present study investigated whether the rapid estradiol attenuation of ATP-induced increase [Ca2+]i requires the mGluR2/3. Previously we showed that DRG (L1-S3) express ERα, P2X3 and mGluR2/3 receptors. DRG were acutely dissociated by enzyme digestion and grown in short-term culture for imaging analysis. DRG neurons were stimulated twice, once with ATP (50 μM) for 5 seconds, and then again in the presence of E2 (100 nM) or E2 (100 nM) + LY341495 (100nM), an mGluR2/3 inhibitor. ATP induced transient increase in [Ca2+]i (216.3 ± 41.2 nM). This transient could be evoked several times in the same DRG neurons if separated by a 5 min washout. Treatment with estradiol significantly attenuated the ATP-induced [Ca2+]i in 60% of the DRG neurons, to 163.3±20.9 nM (p<0.001). Co-application of E2 and the mGluR2/3 inhibitor LY341495 blocked the 17β-estradiol-attenuation of the ATP-induced [Ca2+]i transient (209.1±32.2 nM, p>0.05). These data indicate that the rapid action of E2 in DRG neurons is dependent on the mGluR2/3, and demonstrate that membrane estrogen receptor-α initiated signaling involves tan interaction with mGluRs.
Keywords: metabotropic glutamate receptors, estradiol, ATP, calcium, sensory neurons
Introduction
Gonadal hormones have been implicated in the regulation of pain in animal models as well as in humans (Gintzler and Liu, 2001, Craft et al., 2008). While the ability of estrogens to regulate nociception is now well accepted, the complex nature of this mechanism is poorly understood. Nociceptive sensitivity appears to change when 17β-estradiol (E2) levels are elevated. Indeed in most clinical studies, women report different pain levels and duration of pain than men (Giamberardino, 1999). They are consistent with clinical reports that female pelvic pain can increase through P2X3-receptors activation after reductions in circulatingestrogens (Xu et al., 2008). Moreover, there is a variety of non-genomic effects of estrogen which depend on the presence of classical estrogen receptors, ERα and ERβ. For example, the rapid non-genomic effect of E2 on nociceptive signaling in the mouse dorsal root ganglion neurons is mediated by ERα (Chaban and Micevych, 2005) and it is essential for estradiol effects on nociception (Evrard and Balthazart, 2004, Evrard, 2006). E2-induced alterations in sensory processing may underlie sex-based differences in functional pain symptoms such as irritable bowel syndrome (IBS), fibromyalgia, interstitial cystitis, chronic pelvic pain syndrome and others; however reports of E2 modulation of visceral and somatic nociceptive sensitivity are inconsistent.
Little is known about E2-mediated mechanisms in peripheral nervous system, but the fact that DRG neurons express ERs and respond to E2 treatment suggest that they are a potential target for mediating nociception. Adult DRG neurons in short-term culture retain the expression of receptors which mediate the response to putative nociceptive signals such as ATP (Chaban, 2003). They continue to respond to ER agonists mimicking in vivo activation. Many of the E2-mediated signaling processes have been ascribed as membrane-associated. E2 modulates cellular activity by altering ion channel opening, G-protein signaling, and activation of trophic factor-like signal transduction pathways (Kelly and Levin, 2001). However, the mechanism(s) by which ERs are able to trigger cell signaling when localized at the neuronal membrane surface has yet to be determined. Recent data indicate that membrane-associated ER directly interacts with mGluRs to mediate intracellular signaling (Boulware et al., 2005, Dewing et al., 2007). DRG neurons express mGluR2/3 receptors indicating that glutamate could have a substantial inhibitory effect of primary afferent function, reducing and/or fine-tuning sensory input before transmission to the spinal cord (Carlton and Hargett, 2007). Here we addressed the question of whether ERs interact with group II metabotropic glutamate receptors by using well-established cellular model to study the effect of blockade of mGluR2/3 on E2-mediated ATP-induced Ca2+ signaling in small DRG neurons to help achieve a deeper understanding of the mechanism of gender differences in nociception presented in clinical aspects of clinical symptoms.
Materials and Methods
The isolation procedure and primary culture of rat DRGs has been published in detail (Chaban, 2003). Briefly, lumbosacral adult DRGs (level L1-S3) from ovariectomized Long-Evans rats were collected under sterile technique and placed in ice-cold medium Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma Chemical Co., St. Louis, MO). Adhering fat and connective tissue was removed and each DRG was minced with scissors and placed immediately in a medium consisting of 5 ml of DMEM containing 0.5 mg/ml of trypsin (Sigma, type III), 1 mg/ml of collagenase (Sigma, type IA) and 0.1 mg/ml of DNAase (Sigma, type III) and kept at 37°C for 15 minutes with agitation. After dissociation of the ganglia, soybean trypsin inhibitor (Sigma, type III) terminated the dissociation. The cell suspension was centrifuged for one minute at 1500 rpm and resuspended in DMEM supplemented with 5% fetal bovine serum, 5% horse serum, 2 mM glutamine-penicillin-streptomycin mixture, 1 μg/ml DNAase and 5 ng/ml NGF (Sigma). Cells were plated on Matrigel®-coated 15-mm coverslips (Collaborative Research Co., Bedford, PA) and kept at 37°C in 10% CO2 incubator for 24 hrs, given fresh media and maintained in primary culture until used for experimental procedures.
Ca2+ fluorescence imaging was carried out as previously described (Chaban et al., 2003). DRG neurons were loaded with fluorescent dye Fura-2 AM for one hour at 37°C in HBSS supplemented with 20 mM HEPES, pH 7.4. The coverslips were mounted on a fast-perfusion chamber P-4 (World Precision Instrument) and placed on a stage of Olympus IX51 inverted microscope. Observations were made at room temperature (20–22°C) with 20X UApo/340 objective. A fast superfusion system was used to perfuse the cells with HBSS and rapidly apply E2 and other chemicals. Fluorescence intensity at 505 nm with excitation at 334 nm and 380 nm was captured as digital images (sampling rates of 0.1–2 s). Regions of interest were identified within the soma from which quantitative measurements were made by re-analysis of stored image sequences using Slidebook® Digital Microscopy software. [Ca2+]i was determined by ratiometric method of Fura-2 fluorescence from calibration of series of buffered Ca2+ standards. E2 was acutely applied for 3–5 minutes onto the experimental chamber. Repeated application of drugs was achieved by superfusion in a rapid mixing chamber into individual neurons for specific intervals (100–500 ms). Cells were perfused with experimental media (2 ml/min) using a Rainin® peristaltic pump. The amplitude of [Ca2+]i response represents the difference between baseline concentration and the transient peak response to drug stimulation. Significant differences in response to chemical stimulation were obtained by comparing [Ca2+]i increases during the first stimulation with the second or consequent stimulation. A cell was judged responsive if E2 inhibited the second [Ca2+]i transient by ≥30% of the previous. This criterion was empirically derived in previous experiments (Chaban et al., 2003, 2005). Statistical significance was determined using Student t-test or ANOVA and when appropriate followed by Tukey’s post-hoc comparison (GraphPad Prism software, San Diego, CA). Differences of p<0.05 were considered statistically significa
Results
ATP induced transient increase in [Ca2+]i (Chaban et al., 2003). This transient could be evoked several times in the same DRG neurons if separated by a 5 min washout. We previously showed that E2 inhibited ATP-induced [Ca2+]i but an equimolar amount of the ERα/β-inactive stereoisomer, 17α-E2, did not attenuate the ATP-induced [Ca2+]i influx, establishing that the E2 effect was stereospecific (Chaban et al., 2003). This response is ERα-mediated, since it was missing in the ERα knock-out mouse (Chaban and Micevych, 2005). Moreover, the ER antagonist ICI 182,780 (1μM) blocked the E2 attenuation of ATP-induced [Ca2+], but had no effect on ATP actions in the absence of E2 (Chaban, 2003).
In our experiments we investigated whether the rapid E2 attenuation of ATP-induced increase [Ca2+]i requires the mGluR2/3. DRGs were stimulated twice, once with ATP (50 μM) for 5 seconds, and then again in the presence of E2 (100 nM) or E2 (100 nM) + LY341495 (100nM), a mGluR2/3 inhibitor. Treatment with E2 significantly attenuated the ATP-induced [Ca2+]i in 65 % of the DRG neurons, to 163.3±20.9 nM vs 216.3 ±41.2 nM in control (p<0.001) (Fig 1A). The summary of these experiments (n=6) are summarized in Fig 2A. Co-application of E2 and LY341495 blocked the E2 attenuation of the ATP-induced [Ca2+]i transient (209.1±32.2 nM, p>0.05) (Fig 1B). The summary of these experiments (n=12) presented in Fig 2B. Together, these data indicate that the rapid action of E2 in DRG neurons is dependent on the mGluR2/3, and demonstrate that membrane estrogen receptor initiated signaling involves an interaction with mGluRs.
Fig 1.
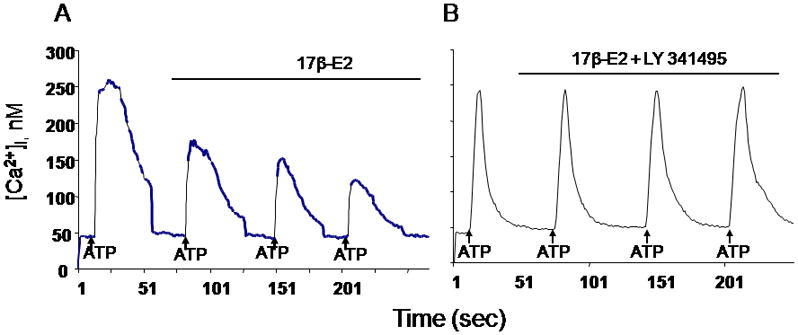
A. Treatment with 17β-estradiol (17β-E2) significantly attenuated the ATP-induced [Ca2+]i of the DRG neurons, from 216.3 ±41.2 nM to 163.3±20.9 nM (p<0.05) in 65% small DRG neurons.
B. Co-application of 17β-estradiol and the mGluR2/3 inhibitor LY341495 blocked the estradiol-attenuation of the ATP-induced [Ca2+]i (209.1±32.2 nM) compared to control (216.3 ±41.2 nM; p>0.05)
Fig. 2.
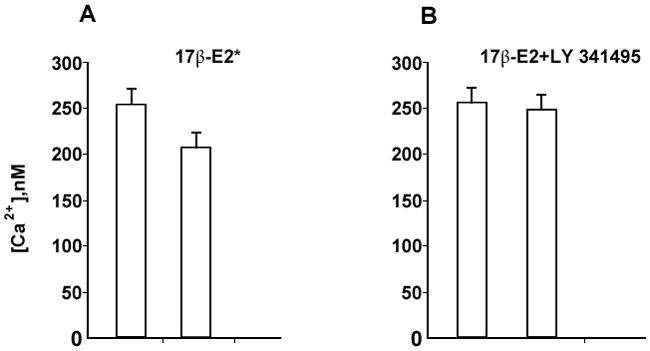
A. Summary of effects of 17β-estradiol (100 nM) on ATP-induced [Ca2+]i increase in DRG neurons (n=6).
B. Summary of the effect of mGluR2/3 inhibitor LY341495 (100 nM) on blocking the 17β-estradiol-attenuation of the ATP-induced [Ca2+]I (n=12).
Discussion
Sex steroids have been suggested as a plausible mechanism of pain regulation and our results provide the evidence that estrogens may directly influence nociception at the level of primary sensory neurons by interacting with group II metabotropic glutamate receptors and suggest that lack of estrogens enhances pro-nociception. Several recent studies have shown that the peripheral nervous system is under substantial influence of gonadal hormones (Xu et al., 2008). In this study we hypothesized that DRG neurons responded to E2 attenuation of ATP-induced [Ca2+]i spikes through interaction with mGluR2/3 (Fig. 3).
Fig. 3.
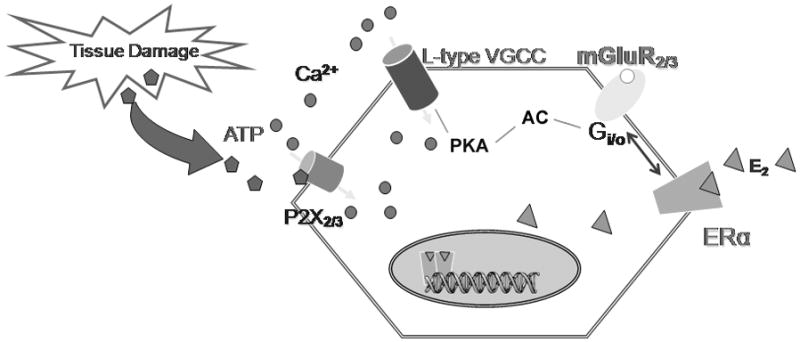
Proposed mechanism of estradiol effect on ATP-induced [Ca2+]i signaling in primary sensory neurons. ATP released by tissue damage acts on P2X3 that activate voltage-gated Ca2+ channels (VGCC)-mediated nociception signaling. 17β-E2 modulates L-type VGCC in DRG neurons via the direct interaction of a membrane ERα with the mGluR2/3 inhibiting adenylyl cyclase activation of L-type VGCC.
ERβ has a significant role in the brain, often counterbalancing that of ERα (Gronemeyer et al., 2004). Recent studies shown that ERβ may mediate part of pro-nociceptive effects of estrogens (Spooner et al., 2007) and that it is crucial for the development of spinal nociceptive system. We had previously demonstrated using ERαKO and βERKO mice that the rapid membrane-initiated signaling in DRGs is through the ERα (Chaban et al., 2005) and therefore did not explore role of ERβ in this study.
The action of estrogens at the level of primary sensory neurons is complex and determined by the characteristics of the target genes and co-regulators, as well as, the regional availability of estradiol. Several authors have suggested that the sex differences in pain sensitivity, and the prevalence of chronic pain disorders may result from a malfunctioning endogenous pain inhibitory response rather than an increase nociceptive activity (Gaumond et al., 2002). The formalin-induced sex hormone modulation of nociception is dependent of the endogenous opioid anti-nociceptive system opioid-mediated, most likely at a spinal level (Gaumond et al., 2007). These results do not preclude an additional mechanism through which estradiol may be acting, such as in the presence of opioids, estrogen acts differently than by itself on nociceptive-mediated pathways.
Many previous studies have established connections between estrogens and the modulation of different nociceptive pathways. In this paper, we show for the first time that an estrogen can inhibit nociceptive stimulus such as ATP through interaction with mGluR2/3. Painful stimuli initiate action potentials in the peripheral terminals of DRG neurons evoking release of excitatory neurotransmitters such as glutamate. Ligand-gated P2X receptors which can be activated by endogenous ATP during induction of action potentials are highly expressed in identified nociceptors (Burnstock, 2001). The data from our experiments suggested that inhibition of mGluR2/3 prevented E2 from inhibiting P2X3 channels suggesting that the rapid action of estradiol in DRG neurons is dependent on the mGluR2/3. Thus, E2-modulated encoding of nociceptive stimuli at the level of primary sensory neurons may contribute to our understanding of complex mechanism of non-genomic effect of gonadal steroids.
Acknowledgments
Supported by NIH DA 013185 to PM and NS 063939 to VC grants.
References
- Boulware MI, Weick JP, Becklund BR, Kuo SP, Groth RD, Mermelstein PG. Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J Neurosci. 2005;25:5066–5078. doi: 10.1523/JNEUROSCI.1427-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Purine-mediated signalling in pain and visceral perception. Trends Pharmacol Sci. 2001;22:182–188. doi: 10.1016/s0165-6147(00)01643-6. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Hargett GL. Colocalization of metabotropic glutamate receptors in rat dorsal root ganglion cells. J Comp Neurol. 2007;501:780–789. doi: 10.1002/cne.21285. [DOI] [PubMed] [Google Scholar]
- Chaban V, Mayer E, Ennes H, Micevych P. Rapid Effect of Estrogen on ATP-induced [Ca2+]i Signaling in Cultured DRG Neurons from Wild Type and ERKO Mice. Gastroenterology. 2003;124:A1. [Google Scholar]
- Chaban VV, Mayer EA, Ennes HS, Micevych PE. Estradiol inhibits ATP-induced intracellular calcium concentration increase in dorsal root ganglia neurons. Neuroscience. 2003;118:941–948. doi: 10.1016/s0306-4522(02)00915-6. [DOI] [PubMed] [Google Scholar]
- Chaban VV, Micevych PE. Estrogen receptor-alpha mediates estradiol attenuation of ATP-induced Ca2+ signaling in mouse dorsal root ganglion neurons. J Neurosci Res. 2005;81:31–37. doi: 10.1002/jnr.20524. [DOI] [PubMed] [Google Scholar]
- Craft RM, Ulibarri C, Leitl MD, Sumner JE. Dose- and time-dependent estradiol modulation of morphine antinociception in adult female rats. Eur J Pain. 2008;12:472–479. doi: 10.1016/j.ejpain.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Dewing P, Boulware MI, Sinchak K, Christensen A, Mermelstein PG, Micevych P. Membrane estrogen receptor-alpha interactions with metabotropic glutamate receptor 1a modulate female sexual receptivity in rats. J Neurosci. 2007;27:9294–9300. doi: 10.1523/JNEUROSCI.0592-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrard HC. Estrogen synthesis in the spinal dorsal horn: a new central mechanism for the hormonal regulation of pain. Am J Physiol Regul Integr Comp Physiol. 2006;291:R291–299. doi: 10.1152/ajpregu.00930.2005. [DOI] [PubMed] [Google Scholar]
- Evrard HC, Balthazart J. Rapid regulation of pain by estrogens synthesized in spinal dorsal horn neurons. J Neurosci. 2004;24:7225–7229. doi: 10.1523/JNEUROSCI.1638-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaumond I, Arsenault P, Marchand S. The role of sex hormones on formalin-induced nociceptive responses. Brain Res. 2002;958:139–145. doi: 10.1016/s0006-8993(02)03661-2. [DOI] [PubMed] [Google Scholar]
- Gaumond I, Spooner MF, Marchand S. Sex differences in opioid-mediated pain inhibitory mechanisms during the interphase in the formalin test. Neuroscience. 2007;146:366–374. doi: 10.1016/j.neuroscience.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Giamberardino MA. Recent and forgotten aspects of visceral pain. Eur J Pain. 1999;3:77–92. doi: 10.1053/eujp.1999.0117. [DOI] [PubMed] [Google Scholar]
- Gintzler AR, Liu NJ. The maternal spinal cord: biochemical and physiological correlates of steroid-activated antinociceptive processes. Prog Brain Res. 2001;133:83–97. doi: 10.1016/s0079-6123(01)33007-8. [DOI] [PubMed] [Google Scholar]
- Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Levin ER. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol Metab. 2001;12:152–156. doi: 10.1016/s1043-2760(01)00377-0. [DOI] [PubMed] [Google Scholar]
- Spooner MF, Robichaud P, Carrier JC, Marchand S. Endogenous pain modulation during the formalin test in estrogen receptor beta knockout mice. Neuroscience. 2007;150:675–680. doi: 10.1016/j.neuroscience.2007.09.037. [DOI] [PubMed] [Google Scholar]
- Xu GY, Shenoy M, Winston JH, Mittal S, Pasricha PJ. P2X receptor-mediated visceral hyperalgesia in a rat model of chronic visceral hypersensitivity. Gut. 2008;57:1230–1237. doi: 10.1136/gut.2007.134221. [DOI] [PubMed] [Google Scholar]