

Abstract
The p16INK4a tumor suppressor inhibits cyclin-dependent kinases (CDK4 and CDK6). Here we report the isolation of a novel gene, SEI-1, whose product (p34SEI-1) appears to antagonize the function of p16INK4a. Addition of p34SEI-1 to cyclin D1–CDK4 renders the complex resistant to inhibition by p16INK4a. Expression of SEI-1 is rapidly induced on addition of serum to quiescent fibroblasts, and ectopic expression of p34SEI-1 enables fibroblasts to proliferate even in low serum concentrations. p34SEI-1 seems to act as a growth factor sensor and may facilitate the formation and activation of cyclin D–CDK complexes in the face of inhibitory levels of INK4 proteins.
Keywords: Cyclin D1, CDK4, p16INK4a
In mammalian cells, the cyclin-dependent kinases CDK4 and CDK6 in conjunction with D-type cyclins, regulate entry into the cell cycle and passage through the restriction point in late G1 (Sherr 1993). As critical integrators of mitogenic-signaling pathways, their activities are regulated at multiple levels, including the synthesis and stability of individual components, the assembly of functional complexes, and post-translational modification (Sherr and Roberts 1999). CDK4/CDK6 are further regulated by CDK inhibitors (CKIs), which induce cell cycle arrest in response to different signals (Sherr and Roberts 1999). There are at present two different classes of CKI, the KIP/CIP family (p21Cip1, p27Kip1, and p57KipII) and the INK4 family (p16INK4a, p15INK4b, p18INK4c, and p19INK4d). In contrast to the KIP/CIP family, which inhibits a broad range of CDKs and has been also shown to activate cyclin D-dependent kinases (Zhang et al. 1994; LaBaer et al. 1997; Cheng et al. 1999; Parry et al. 1999), the INK4 family protein specifically binds to and inactivates CDK4/CDK6 (Serrano et al. 1993; Koh et al. 1995; Lukas et al. 1995).
Recent observations strongly suggest that cyclin D kinases contribute to cyclin E–CDK2 activation and S-phase entry through both pRB phosphorylation and sequestration of the KIP/CIP family CKIs (Geng et al. 1999; Sherr and Roberts 1999). This evidence suggests that the activity of cyclin D–CDK4/6 kinase is crucial and is tightly regulated by a number of different factors. To help understand the regulation of cyclin D-dependent kinases, we have identified and characterized a novel CDK4 regulator that prevents p16INK4a from inhibiting the formation of cyclin D1–CDK4 complexes.
Results and Discussion
Because the only known binding partners for INK4 proteins are CDK4 and CDK6, we performed yeast two-hybrid screening using full-length human p16INK4a as bait (Durfee et al. 1993). From a total of ∼1.5 × 106 yeast colonies screened with HeLa cell cDNA library, three were recovered that fulfilled the criteria for a positive interaction. One cDNA encoded CDK4, confirming the reliability of the screening procedure, whereas the other two contained the same novel sequence, which we called SEI-1 because it was selected with INK4A as bait.
The SEI-1 cDNA encodes a 34-kD protein (p34SEI-1) whose predicted amino acid sequence (GenBank accession no. AF117959) shows no obvious similarities to any other proteins in the database. The presence of a typical nuclear localization signal (KRKR) near the amino terminus suggested that p34SEI-1 may be a nuclear protein, this was confirmed by immunodetection of epitope-tagged protein in COS-7 cells (data not shown).
SEI-1 cDNA was found to hybridize to a single 1.35-kb mRNA transcript that is widely expressed in various human tissues, including spleen, prostate, testis, ovary, small intestine, colon, and leukocyte, with the exception of thymus (data not shown). To examine the expression pattern of p34SEI-1 in the cell cycle, human primary fibroblasts, TIG-3 cultures, were rendered quiescent by serum withdrawal and stimulated to re-enter the cell cycle. At various times after serum addition, samples were analyzed for the expression of p34SEI-1 and other cell cycle regulators. Flow cytometry (data not shown) confirmed that the majority of the cells began to enter S phase at ∼16 hr after serum addition, coinciding with the first appearance of cyclin A protein (Fig. 1A). p34SEI-1 expression was very weak in quiescent TIG-3 cells, but the RNA (data not shown) and protein were rapidly induced, reaching peak levels 2 hr after serum addition (Fig. 1A). A similar SEI-1 RNA expression pattern was also observed in rat fibroblasts, 3Y1 (data not shown). Immunoblotting was also used to monitor the expression of cyclin D1, CDK4, and p16INK4a proteins. As reported previously (Li et al. 1994), the levels of cyclin D1 and CDK4 remain essentially constant in serum-stimulated TIG-3 cells, whereas p16INK4a expression was increased by serum stimulation (Tam et al. 1994; Hara et al. 1996; Fig. 1A). Different patterns have been reported in other cell systems in which cyclin D expression can be growth-factor dependent (Matsuhime et al. 1994).
Figure 1.



Cell cycle-dependent expression of SEI-1 and its influence over p16INK4a activity. (A) Early passage (35 PDL) TIG-3 cells were rendered quiescent in medium containing 0.2% serum for 4 days, then stimulated to proliferate by addition of 20% FCS. Total cell lysates were prepared at various time points and 150 μg of protein were subjected to Western blotting with antibodies indicated at right. (B) HPLC-purified cyclin D1–CDK4 complexes, prepared from baculovirus-infected Sf9 cells, were preincubated with His–p16INK4a, with or without His–p34SEI-1 or His–Id1 as indicated, and then assayed for kinase activity toward GST–Rb. (C) 35S-Labeled in vitro-translated p16INK4a, CDK4, CDK6, and p53 proteins (top) were mixed with 10 ng of His–p34SEI-1 protein (bottom). The samples were analyzed directly by SDS-PAGE (top) or after immunoprecipitation with an antibody against p34SEI-1 (bottom).
Interaction of p16INK4a with p34SEI-1 in a yeast two-hybrid screen suggested that p34SEI-1 might have some influence over the CDK inhibitory activity of p16INK4a. Highly purified cyclin D1–CDK4 complex, produced in a baculovirus-based expression system, phosphorylates the retinoblastoma gene product, pRb, on specific residues (Kato et al. 1993). As shown in Figure 1B, the activity of such a complex, assayed by its ability to phosphorylate the carboxy-terminal domain of pRb expressed as a GST fusion protein, can be completely abolished by addition of sufficient amounts (100 or 200 ng) of bacterially produced histidine-tagged p16INK4a protein (His–p16INK4a) (Fig. 1B, lanes 3,4). However, addition of 1 μg of bacterially produced histidine-tagged p34SEI-1 (His–p34SEI-1) rendered the complex resistant to inhibition by p16INK4a (Fig. 1B, lanes 5,6,7), whereas histidine-tagged Id1 protein (His–Id1) (Hara et al. 1997), used here as a nonspecific control, had no effect on the inhibitory properties of p16INK4a (Fig. 1B, lanes 8,9,10). These effects were more significant when the cyclin D1–CDK4 complex was preincubated with His–p34SEI-1 prior to the addition of His–p16INK4a (data not shown). These results are reminiscent of an observation that viral cyclins encoded by Kaposi's sarcoma virus form active kinase complexes with CDK6 that are resistant to inhibition by p16INK4a (Swanton et al. 1997).
To validate the interaction between p34SEI-1 and p16INK4a, His–p16INK4a was tested for its ability to bind in vitro to [35S]methionine-labeled p34SEI-1 synthesized in reticulocyte lysates. Although binding of p34SEI-1 was observed, these interactions appeared much weaker than that between CDK4 and INK4 proteins (data not shown). However, the results in Figure 1B strongly suggest that p34SEI-1 has some influence over the p16INK4a activity. Therefore, we next examined the possibility that p34SEI-1 might bind directly to CDK4. 35S-Labeled CDK4 was coprecipitated with 10 ng of His–p34SEI-1, whereas 35S-labeled p16INK4a was not coprecipitated under similar binding conditions (Fig. 1C, cf. lane 2 and lane 1). CDK6 protein was not coprecipitated with His–p34SEI-1 protein despite its high homology with CDK4 (Fig. 1C, lane 3). We did not observe any interaction between p34SEI-1 and cyclin D1 (data not shown). These data indicate that CDK4 may be a genuine binding target of p34SEI-1.
To identify the region important for SEI-1/CDK4 binding, histidine-tagged CDK4 (His–CDK4) was mixed with 35S-labeled in vitro-translated proteins from a series of p34SEI-1 truncation mutants (Fig. 2A), and the complexes were recovered by immunoprecipitation of His–CDK4. All truncated SEI-1 proteins were of the expected molecular size as determined by SDS-PAGE (Fig. 2B, top). SEI-1 proteins that were progressively truncated from the amino terminus retained the ability to bind CDK4 up until removal of amino acids 44–99, which abrogated this binding (Fig. 2B, bottom, lanes 3–8). Similarly, carboxy-terminal mutants also possessed CDK4-binding activity, but deletion of amino acids 102–161 prevented this binding (Fig. 2B, bottom, lanes 9–12). This result indicates that the region between 44 and 161, which contains a proline-rich sequence, is required for CDK4 binding. Similar analyses were performed with His–p34SEI-1 and 35S-labeled proteins from a series of CDK4 truncation mutants (Fig. 2C). Although none of the truncation mutants of CDK4 were able to bind to p16INK4a (Fig. 2D, middle, lanes 2–5), neither amino-terminal nor carboxy-terminal deletions abrogated the binding to p34SEI-1 (Fig. 2D, bottom, lanes 2–4). The most carboxy-terminal part of CDK4, amino acids 245–303, did not bind to p34SEI-1 (Fig. 2D, bottom, lane 5). This indicates the region lying between 79–245 of CDK4 is required for p34SEI-1 binding.
Figure 2.
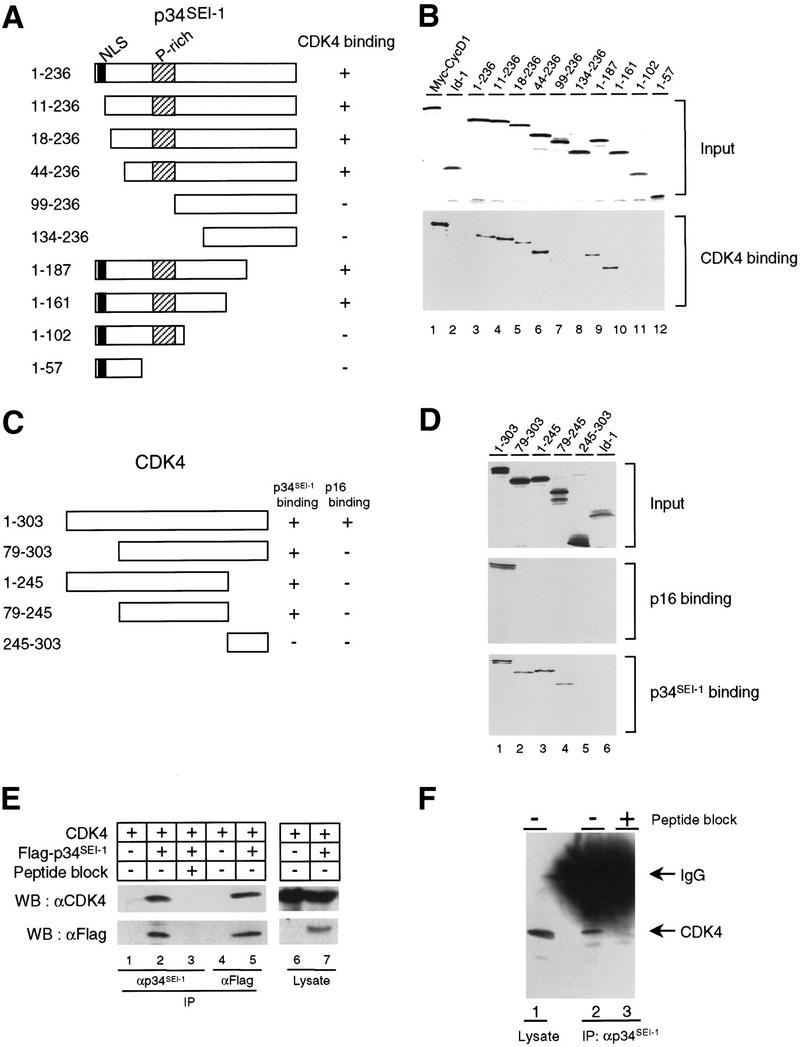
Interaction of p34SEI-1 with CDK4. (A) Schematic representation of p34SEI-1 and its interaction activity. (B) A series of truncation mutants of 35S-labeled p34SEI-1 proteins (top) were incubated with His–CDK4 protein. Bound 35S-labeled p34SEI-1 proteins were detected by autoradiography (bottom). (C) Schematic representation of CDK4 and its interaction activity. (D) A series of truncation mutants of 35S-labeled CDK4 proteins (top) were incubated with either His–p16INK4a (middle) or His–p34SEI-1 protein (bottom). Bound 35S-labeled CDK4 proteins were detected by autoradiography (middle, bottom). (E) Cos7 cells were transfected with expression vectors for Flag-tagged p34SEI-1 and/or CDK4 as indicated (top) and collected 48 hr later. Specific interactions were analyzed by immunoblotting with antibodies indicated at left after immunorecipitation with antibodies indicated at bottom (lanes 1–5). (F) p34SEI-1 immunoprecipitates prepared from C33A cells in the absence (lane 2) or presence (lane 3) of competing SEI-1 peptide were subjected to Western blotting with anti-CDK4 antibody. 1/100 volume of total cell lysate from C33A cells was directly immunoblotted as a postivie control (lane 1).
To confirm the interaction between p34SEI-1 and CDK4 in vivo, Cos 7 cells were transfected with a CDK4 expression vector in the presence or absence of a second expression vector containing Flag-tagged p34SEI-1. Cell lysates were immunoprecipitated with anti-p34SEI-1 antibody, and the immune complexes were subjected to Western blotting with an anti-CDK4 antibody. We could detect CDK4 among the coprecipitated proteins (Fig. 2E, lane 2), but no CDK4 was detected when the immunoprecipitation was competed with excess SEI-1 peptide antigen (Fig. 2E, lane 3). Similar results were obtained when anti-Flag antibody-conjugated agarose beads were used for immunoprecipitation. CDK4 protein was only detected in the cells transfected with both CDK4 and Flag-tagged p34SEI-1 expression vectors (Fig. 2E, lanes 4,5). These results suggest that p34SEI-1 binds to CDK4 in Cos7 cells. We have further shown an interaction between endogenous p34SEI-1 and CDK4 proteins in the cervical carcinoma cell line, C33A. CDK4 was detectable in the p34SEI-1-immunoprecipitated complexes (Fig. 2F, lane 2); whereas no CDK4 was evident when the immunoprecipitation was competed with excess SEI-1 peptide antigen (Fig. 2F, lane 3). There are two possibilities to explain why we obtained p34SEI-1 by two-hybrid screening using p16INK4a as a bait. Cdc28 might be working as a bridging molecule between p34SEI-1 and p16INK4a in yeast. Alternatively, although the binding between p34SEI-1 and p16INK4a is very weak, these proteins may bind each other more efficiently when they are highly overexpressed in yeast.
Because p16INK4a is believed to inhibit the CDK4 kinase activity by binding in competition with D-type cyclin, we next asked what influence p34SEI-1 might have on the competitive binding of cyclin D1 and p16INK4a to CDK4. A Myc-epitope tag was added to the cyclin D1 sequence to aid discrimination of the in vitro-translated proteins, and the complexes were recovered by immunoprecipitation of CDK4. Addition of 1 μg of p34SEI-1 had a small stimulatory effect on cyclin D1–CDK4 binding, whereas increasing amounts of p16INK4a effectively blocked the cyclin D1–CDK4 interaction (Fig. 3A, cf. lanes 1,2 and 4,7) as described previously (Parry et al. 1995; Guan et al. 1996). When added together, p34SEI-1 prevented the disruption of the cyclin D1–CDK4 interaction by p16INK4a (Fig. 3A, lanes 5,8), and Id1 was unable to counteract p16INK4a (Fig. 3A, lanes 6,9). A similar analysis was performed with CDK6 instead of CDK4. Interestingly, p34SEI-1 did not prevent the disruption of the cyclin D1–CDK6 interaction by p16INK4a (Fig. 3B). This correlates well with the observation that p34SEI-1 did not bind to Cdk6 in vitro (Fig. 1C, lane 3). These results suggest that an interaction between p34SEI-1 and CDK4 might be important for rendering the cyclin D1–CDK4 complex resistant to inhibition by p16INK4a.
Figure 3.
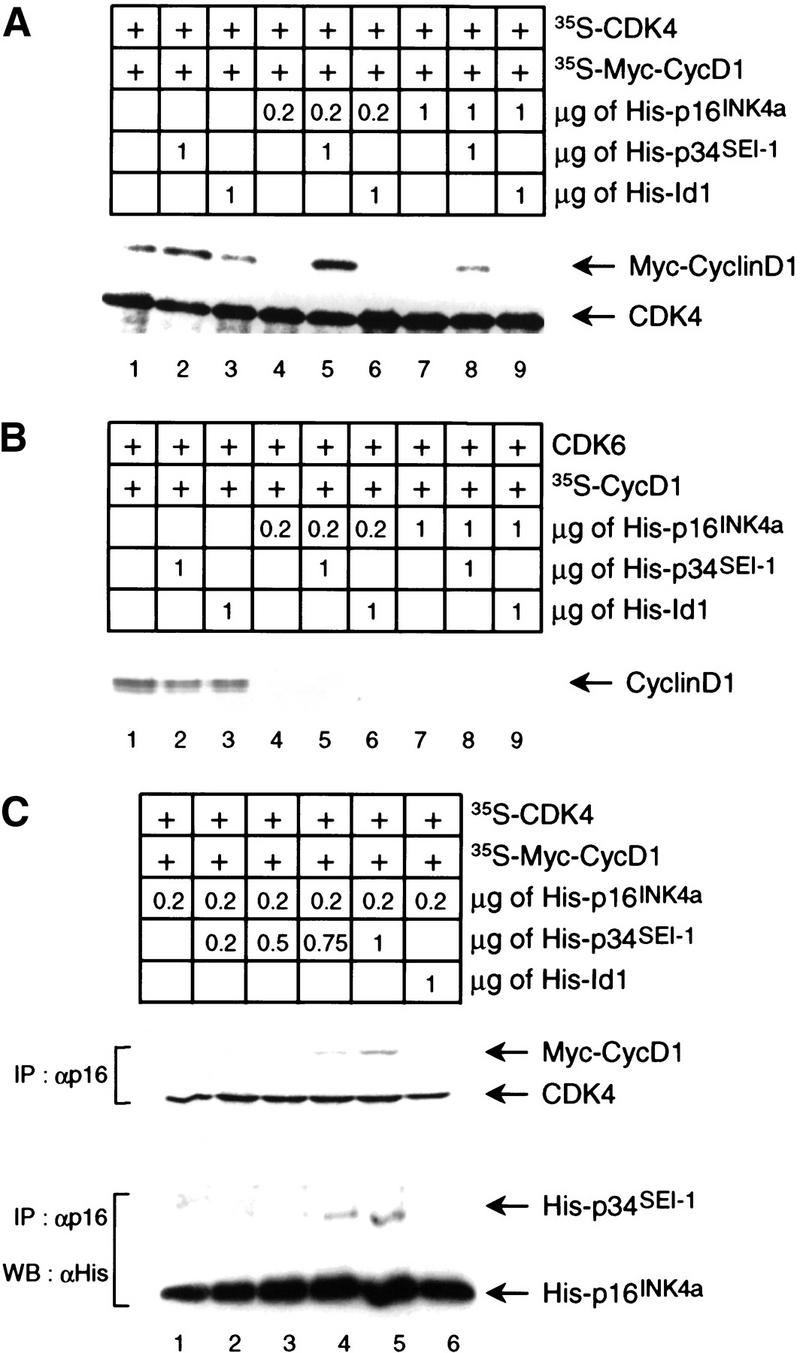
Protection of cyclin D1–CDK4 complex from p16INK4a by p34SEI-1. Both in vitro-translated 35S-labeled Myc-tagged cyclin D1 and CDK4 (A) or unlabeled CDK6 (B) were mixed with His–p16INK4a, p34SEI-1, or Id1 as indicated. The mixtures were immunoprecipitated with antisera to CDK4 (A) or CDK6 (B) and analyzed by SDS-PAGE. Labeled D1 proteins were detected by autoradiography. (B) (C) In vitro-translated 35S-labeled Myc-tagged-cyclin D1 and Cdk4 were mixed with the proteins as indicated, immunoprecipitated with anti-p16 antibody, and analyzed by SDS-PAGE. Labeled proteins were detected by autoradiography (top). These immunoprecipitated samples were also subjected to Western blotting with anti-His-tag antibody (bottom).
To examine the mechanism of p34SEI-1 action in further detail, we checked first whether or not p34SEI-1 could inhibit the interaction between p16INK4a and CDK4. Results indicated that p34SEI-1 did not have any effect on the interaction between p16INK4a and CDK4 (data not shown). This suggests a major difference between p34SEI-1 and another CDK4-binding protein, Cdc37, which can compete with p16INK4a for binding to CDK4 (Dai et al. 1996; Stepanova et al. 1996; Lamphere et al. 1997) Next, we examined the possibility that p34SEI-1 might enable CDK4 to bind cyclin D1 even in the presence of p16INK4a. 35S-Labeled in vitro-translated CDK4 and cyclin D1 proteins were mixed with His–p16INK4a in the presence or absence of His–p34SEI-1. Immunoprecipitated complexes with an anti-p16INK4a antibody were analyzed by autoradiography or by immunoblotting with anti-His-tag antibody. Cyclin D1 was coprecipitated with p16INK4a following the addition of increasing amounts of p34SEI-1 (Fig. 3C, lanes 4,5, top). Cyclin D1, CDK4, p16INK4a, and p34SEI-1 were detected in the same p16INK4a immunoprecipitates (Fig. 3C, lanes 4,5). The most likely explanation for this result is that the binding of p34SEI-1 to CDK4 renders the cyclin D1–CDK4 complex resistant to the action of p16INK4a, and results in the formation of quartenary complexes of p34SEI-1–p16INK4a–cyclin D1–CDK4. This evidence correlates with reports suggesting that INK4 proteins associated with the cyclin D–CDK4 complexes (Hirai et al. 1995; Reynisdottir and Massague 1997). Although the exact mechanism is still not fully understood, these data indicate that p34SEI-1 may be a positive regulator of the cyclin D1–CDK4 complex by counteracting the INK4 proteins. Recent crystallographic studies of the CDK6–INK4 structure indicate that the conformational changes induced by INK4 proteins inhibit both productive binding of ATP and cyclin-induced rearrangement of the kinase from an inactive to an active conformation (Brotherton et al. 1998; Russo et al. 1998). Therefore, although p34SEI-1 does not bind to CDK6, p34SEI-1 binding to CDK4 might induce structural changes that restore the ATP binding and cyclin D binding in the presence of INK4.
To examine the biological significance of p34SEI-1 in cell growth, 3Y1 cells were transfected with SEI-1 cDNA and stable clones were established. Two clones, F-2 and F-11, were chosen for further analysis because they expressed different amounts of exogenous p34SEI-1 (Fig. 4A). No exogenous p34SEI-1 protein could be detected in the control clone, N-2, transfected with empty vector. All three of these clones grew well in normal medium containing 10% serum (Fig. 4B, left). However, F-11 cells, which express the highest levels of exogenous p34SEI-1, continued to grow well in 3% serum, whereas growth of the N-2 control cells was severely impaired (Fig. 4B, middle). The F-2 clone, expressing modest levels of p34SEI-1, had intermediate properties. The effects were similar but even more pronounced in 1% serum (Fig. 4B, right). Similar results were obtained in other cell clones (data not shown). We repeated the same growth assay using a pool of transfected cells rather than cell clones (Fig. 4D,E). The cells transfected with wild-type p34SEI-1 expression plasmid grew better than cells transfected with the CDK4-binding-deficient mutant of p34SEI-1 (amino acids 134–236; Fig. 2B, lane 8) or vector alone in low serum conditions (Fig. 4E). Thus, exogenous SEI-1 was able to sustain cell proliferation even in low-serum concentrations and this activity was dependent on its binding to CDK4.
Figure 4.

Effects of p34SEI-1 on cell growth. (A) Clones of rat 3Y1 fibroblasts stably transfected with a p34SEI-1 expression vector (F-2 and F-11) or empty vector (N-2) were monitored for p34SEI-1 expression by immunoblotting with an antiserum (TH1) against the carboxyl terminus of p34SEI-1. (B) The clones were placed in either 10% serum (left), 3% serum (middle), or 1% serum (right), and relative cell numbers were compared at 12-hr intervals. (□) N-2; (●) F-2; (○) F-11. (C) N-2 and F-11 cells were maintained in medium containing 1% serum for 4 days. Cell lysates were then immunoprecipitated with either CDK4 antiserum or NRS. The precipitates were tested for their ability to phosphorylate GST–Rb protein (top) and were also subjected to Western blotting with anti-CDK4 antibody (bottom). (D) 3Y1 cells transfected with expression plasmids encoding wild-type (WT) or CDK4-binding-deficient mutant; amino acids 134–236 (Mut) of p34SEI-1 or empty vector (Vec), pcDNA3 (Invitogen), were selected against G418 for 2 weeks and pooled. This pooled population was monitored for p34SEI-1 expression by immunoblotting with an antiserum (TH1). (NS) Nonspecific association to TH1 antiserum in 3Y1 cell lysate. (E) The pooled cells were placed in the medium containing 1% serum and relative cell numbers were compared at 12-hr intervals. (□) Vec; (●) Mut; (○) Wt.
Because the working hypothesis is that SEI-1 substitutes for serum factors in activating cyclin D–CDK4 complexes, we compared the CDK4-associated kinase activity in F-11 and N-2 cells that had been maintained in 1% serum for 4 days. Cell lysates were immunoprecipitated with either CDK4 antiserum or normal rabbit serum (NRS) and the complexes were tested for their ability to phosphorylate GST–Rb (Kitagawa et al. 1996). Although equal amounts of CDK4 proteins were used for the kinase assay, only F-11 cells contained significant levels of CDK4 kinase activity, even in 1% serum (Fig. 4C, lanes 2,4). Although we were unable to detect p16INK4a, p18INK4c was readily detected and both CDK4 and cyclin D1 levels were relatively constant among these cell lines (data not shown). Therefore, p34SEI-1 might facilitate the formation and activation of cyclin D1–CDK4 complexes in the face of inhibitory levels of p18INK4c in 3Y1 cells.
Next, we examined whether overexpression of p34SEI-1 can force assembly of cyclin D1–CDK4 complexes in cells expressing cyclin D1 and CDK4 under serum starvation conditions. The expression levels of both cyclin D1 and CDK4 protein are high in TIG-3 cells under serum starvation conditions, but there was no detectable cyclin D1–CDK4 complex (Fig. 5A, lane 1; data not shown). However, forced expression of p34SEI-1 protein enables formation of cyclin D1–CDK4 complex in low-serum condition (Fig. 5A, lane 2). Furthermore, treatment of the TIG-3 cells with antisense oligomers against the SEI-1 gene significantly reduced both SEI-1 RNA and protein levels (Fig. 5B, lane 2, and C, lane 3), attenuated the cyclin D1–CDK4 complex formation (Fig. 5D, lane 3) and inhibited S-phase entry (Fig. 5E, lane 2). Taken together, these data suggest that SEI-1 is a potential mediator of the growth factor-dependent activation of cyclin D–CDK4 that occurs as cells enter the proliferative cycle. A recent report implicated that the MEK/ERK-signaling pathway is involved in this process (Cheng et al. 1998), but endogenous SEI-1 expression was found to be unaffected in a cell line engineered for inducible expression of MEK (data not shown). Moreover, treatment of fibroblasts with PD98059, a MEK1 inhibitor, had no effect on induction of p34SEI-1 expression by serum stimulation (data not shown). Although this argues against SEI-1 being the assembly factor, the biochemical evidence clearly indicates that p34SEI-1 enhances the cyclin D1–CDK4 interaction and renders the complex resistant to inhibition by the INK4 family of CDK inhibitors. Because the abundance of p34SEI-1 may therefore modulate the CDK4 kinase activity, overexpression of SEI-1 could contribute to the deregulated growth of tumor cells. In this context, it is interesting to note that SEI-1 maps to a region of chromosome 19q13.1–q13.2 (data not shown) that is amplified in a number of human cancers (Thompson et al. 1996). Although we have yet to define the precise biological function of p34SEI-1, the data presented here strongly suggest that SEI-1 encodes a new type of cell cycle regulator that is induced by growth factors and counteracts the INK4 family of CDK inhibitors.
Figure 5.

Effects of p34SEI-1 on cyclin D1–CDK4 complex formation in vivo. (A) Early passage TIG-3 cells were infected with retrovirus that encodes SEI-1 (lane 2) or empty vector (lane 1) and selected by puromycin. Selected cells were pooled and were incubated with a medium containing 1% serum for 4 days, then lysates were prepared and immunoprecipitated with anti-cyclin D1 antibody and immunoblotted with either anti-CDK4 (middle) or anti-cyclin D1 (bottom) antibody. Expression level of p34SEI-1 was confirmed by Western blot with anti-SEI-1 antiserum (top). (B,C,D,E) Early passage TIG-3 cells were cultured in a low serum (0.2% FCS) medium for 3 days, then treated with either antisense oligomer against the SEI-1 gene or a control oligomer for 24 hr. (B) Total RNAs were prepared from both control (lane 1) and antisense oligomer (lane 2)-treated cells that had been incubated with high-serum (20% FCS) medium for 2 hr and were subjected to Northern blotting with either SEI-1 (top) or β-actin (bottom) cDNA probes. (C) Cell lysate were prepared from both control (lanes 1,2) and antisense oligomer (lane 3)-treated cells incubated with high serum medium for 2 hr in the presence of [35S]methionine. Cell lysates were then immunoprecipitated with antisera to SEI-1 in the absence (lanes 2,3) or presence (lane 1) of competing SEI-1 peptide and analyzed by SDS-PAGE. (D) Cell lysates were prepared from both control (lane 2) and antisense oligomer (lane 3)-treated cells that were incubated with high serum medium for 8 hr and were immunoprecipitated with anti-cyclin D1 antibody and immunoblotted with either anti-CDK4 (top) or anti-cyclin D1 (bottom) antibody. 1/100 volume of total cell lysate from TIG-3 cells was directly immunoblotted as a positive control (lane 1). (E) Cells were incubated with high-serum medium for 20 hr in the presence of [3H]thymidine, then fixed and developed. The percentages of [3H]thymidine-incorporated cells were measured in both control (lane 1) and antisense oligomer (lane 2) treated cells.
Materials and methods
Cell culture
C33A, COS-7, TIG-3 and 3Y1 cells were cultured in DMEM supplemented with 10% FCS. Cell transfection was carried out with the FuGENE6 reagent (Boehringer Mannheim). Retroviral-mediated gene transfer was performed as described previously (Serrano et al. 1997).
Antisense oligomer treatment
Antisense oligomers were synthesized with phosphorothioate and purified by HPLC. The oligomers were complementary to 16 nucleotides overlapping the translation initiation codon (ATG) of human SEI-1 as follows: antisense 1 (5′-GCTCAGCATCTTGCTC-3′), antisense 2 (5′-CTTGCTCAGCATCTTG-3′), and control oligomer (5′-ACGGCATAGAGCGATG-3′). TIG-3 cells were incubated with DMEM containing 0.4% DMRIE-C (GIBCO-BRL), and either a mixture of 125 nm of both antisense 1 and antisense 2 oligonucleotides or 250 nm of control oligo for 5 hr. The medium was then replaced with DMEM.
Protein analysis
Immunoblotting and immunoprecipitation were performed as described previously (Stott et al. 1998). The primary antibodies used in these studies were against p16INK4a (DCS50), p34SEI-1 [TH1 (this paper)], cyclin A (Santa Cruz H432), cyclin D1 (Santa Cruz R124), CDK4 (Santa Cruz H303, C-22), His (Qiagen 34610), p18INK4c (Santa Cruz N-20), and Flag M2 (Sigma A-1205).
Acknowledgments
We thank Drs. M.F. Roussel and C.J. Sherr for providing us with the MEK-1-inducible cell line. We are grateful to Drs. G. Peters, N. Jones, J.-Y. Kato, H. Hirai, T. Okuda, and J. Campisi for valuable suggestions, and to T. Huot for help in generating the antiserum (TH1). This work was supported by United Kingdom Cancer Research Campaign and Sumitomo Electric Industries.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Ehara@picr.man.ac.uk; FAX +44-161-446-3109.
References
- Brotherton DH, Dhanaraj V, Wick S, Brizuela L, Domaille PJ, Volyanik E, Xu X, Parisini E, Smith BO, Archer SJ, et al. Crystal structure of the complex of the cyclin D-dependent kinase Cdk6 bound to the cell-cycle inhibitor p19INK4d. Nature. 1998;395:244–250. doi: 10.1038/26164. [DOI] [PubMed] [Google Scholar]
- Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclinD-dependent kinase and titration of p27KIP1 regulated by mitogen-activated protein kinase kinase (MEK1) Proc Natl Acad Sci. 1998;95:1091–1096. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21Cip1 and p27Kip1 CDK “inhibitors” are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai K, Kobayashi R, Beach D. Physical interaction of mammalian CDC37 with CDK4. J Biol Chem. 1996;271:22030–22034. doi: 10.1074/jbc.271.36.22030. [DOI] [PubMed] [Google Scholar]
- Durfee T, Becherer K, Cheng P-L, Yeh S-H, Yang Y, Kilburn AE, Lee W-H, Elledge SJ. The retinoblastoma protein associates with the protein phosphatase type I catalytic subunit. Genes & Dev. 1993;7:555–569. doi: 10.1101/gad.7.4.555. [DOI] [PubMed] [Google Scholar]
- Geng Y, Whoriskey W, Park MY, Bronson RT, Meclema RH, Tiansen L, Weinberg RA, Sicinski P. Rescue of cyclinD1 deficiency by knockin cyclinE. Cell. 1999;97:767–777. doi: 10.1016/s0092-8674(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Guan K-L, Jenkins CW, Li Y, O'Keefe CL, Noh S, Wu X, Zariwala M, Matera AG, Xiong Y. Isolation and characterization of p19INK4d, a p16-related inhibitor specific to CDK6 and CDK4. Mol Biol Cell. 1996;7:57–70. doi: 10.1091/mbc.7.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–867. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara E, Hall M, Peters G. Cdk2-dependent phosphorylation of Id2 modulates activity of E2A-related transcription factors. EMBO J. 1997;16:332–342. doi: 10.1093/emboj/16.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Roussel MF, Kato J-Y, Ashmun RA, Sherr CJ. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclinD-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15:2672–2681. doi: 10.1128/mcb.15.5.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J-Y, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes & Dev. 1993;7:331–342. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Higashi H, Jung H-K, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J-Y, Segawa K, Yoshida E, Nishimura S, Taya Y. The consensus motif for phosphorylation by cyclinD1-Cdk4 is different from that for phosphorylation by cyclinA/E-Cdk2. EMBO J. 1996;15:7060–7069. [PMC free article] [PubMed] [Google Scholar]
- Koh J, Enders GH, Dynlacht BD, Harlow E. Tumor-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature. 1995;375:506–510. doi: 10.1038/375506a0. [DOI] [PubMed] [Google Scholar]
- LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for p21 family of CDK inhibitors. Genes & Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Lamphere L, Fiore F, Xu X, Brizuela L, Keezer S, Sardet C, Draetta GF, Gyuris J. Interaction between Cdc37 and Cdk4 in human cells. Oncogene. 1997;14:1999–2004. doi: 10.1038/sj.onc.1201036. [DOI] [PubMed] [Google Scholar]
- Li Y, Jenkins CW, Nichols MA, Xiong Y. Cell cycle expression and p53 regulation of the cyclin-dependent kinase inhibitor p21. Oncogene. 1994;9:2261–2268. [PubMed] [Google Scholar]
- Lukas J, Parry D, Aagaard L, Mann D, Bartkova J, Strauss M, Peters G, Bartek J. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumor suppressor p16. Nature. 1995;375:503–506. doi: 10.1038/375503a0. [DOI] [PubMed] [Google Scholar]
- Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato J-Y. D-type cyclin dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry D, Bates S, Mann DJ, Peters G. Lack of cyclin D-Cdk complexes in Rb-negative cells correlates with high levels of p16INK4/MTS1 tumor suppressor gene product. EMBO J. 1995;14:503–511. doi: 10.1002/j.1460-2075.1995.tb07026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry D, Mahony D, Wills K, Lees E. CyclinD-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol Cell Biol. 1999;19:1775–1783. doi: 10.1128/mcb.19.3.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisdottir I, Massague J. The subcellular localization of p15INK4b and p27KIP1 coordinate their inhibitory interactions with Cdk4 and Cdk2. Genes & Dev. 1997;11:492–503. doi: 10.1101/gad.11.4.492. [DOI] [PubMed] [Google Scholar]
- Russo AA, Tong L, Lee J-O, Jeffrey PD, Pavletich NP. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumor suppressor p16INK4a. Nature. 1998;395:237–343. doi: 10.1038/26155. [DOI] [PubMed] [Google Scholar]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell cycle control causing specific inhibition of cyclinD/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes & Dev. 1999;13:1504–1412. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Stepanova L, Leng X, Parker SB, Harper JW. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes & Dev. 1996;10:1491–1502. doi: 10.1101/gad.10.12.1491. [DOI] [PubMed] [Google Scholar]
- Stott F, Stewart B, James MC, McConnell BB, Starborg M, Brookes S, Palmero I, Ryan K, Hara E, Vousden KH, Peters G. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17:5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton C, Mann DJ, Fleckenstein B, Neipel F, Peters G, Jones N. Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature. 1997;390:184–187. doi: 10.1038/36606. [DOI] [PubMed] [Google Scholar]
- Tam SW, Theodoras AM, Shay JW, Draetta GF, Pagano M. Differential expression and regulation of Cyclin D1 protein in normal and tumor human cells: Association with Cdk4 is required for Cyclin D1 function in G1 progression. Oncogene. 1994;9:2663–2674. [PubMed] [Google Scholar]
- Thompson FH, Nelson MA, Trent JM, Guan XY, Liu Y, Yang JM, Emerson J, Adair L, Wymer J, Balfour C, et al. Amplification of 19q13.1-q13.2 sequences in ovarian cancer. G-band, FISH, and molecular studies. Cancer Genet Cytogenet. 1996;87:55–62. doi: 10.1016/0165-4608(95)00248-0. [DOI] [PubMed] [Google Scholar]
- Zhang H, Hannon GJ, Beach D. p21-containing cyclin kinases exist in both active and inactive states. Genes & Dev. 1994;8:1750–1758. doi: 10.1101/gad.8.15.1750. [DOI] [PubMed] [Google Scholar]