

Abstract
The σ subunit of eubacterial RNA polymerase is required throughout initiation, but how it communicates with core polymerase (α2ββ′) is poorly understood. The present work addresses the location and function of the interface of σ with core. Our studies suggest that this interface is extensive as mutations in six conserved regions of σ70 hinder the ability of σ to bind core. Direct binding of one of these regions to core can be demonstrated using a peptide-based approach. The same regions, and even equivalent residues, in σ32 and σ70 alter core interaction, suggesting that σ70 family members use homologous residues, at least in part, to interact with core. Finally, the regions of σ that we identify perform specialized functions, suggesting that different portions of the interface perform discrete roles during transcription initiation.
Keywords: σ factor, core, RNA polymerase, transcription, initiation, pausing
In prokaryotic cells, the σ subunits of RNA polymerase direct the process of transcription initiation. Bacterial cells utilize one primary σ, called σ70 in Escherichia coli, to direct transcription of housekeeping genes during exponential growth. In addition, most bacteria have several alternative σs, each of which directs transcription of a specific set of genes in response to either environmental or developmental signals. The primary σs and almost all alternative σs constitute a homologous set of proteins, the σ70 family of proteins. E. coli contains six members of this family and one additional σ, σ54, which is unrelated in sequence or mechanism to the σ70 protein family (Lonetto et al. 1992). The conserved regions of the σ70 family are shown in Figure 1D (Helmann and Chamberlin 1988; Lonetto et al. 1992; Wosten 1998).
Figure 1.
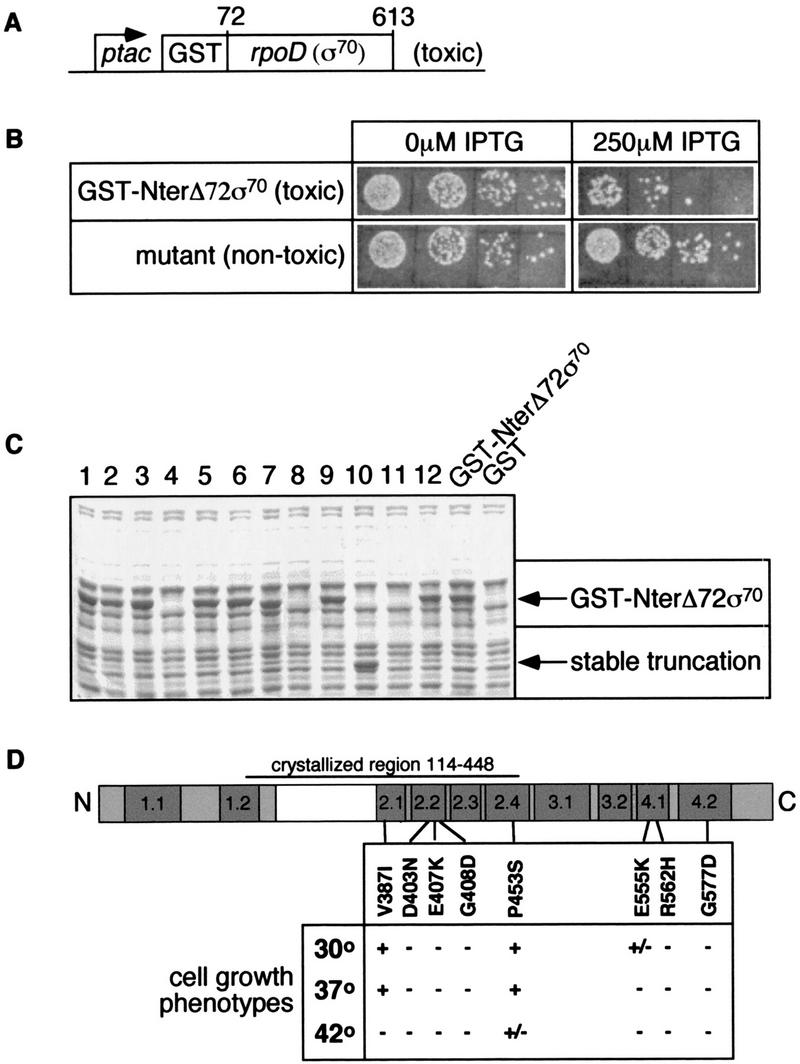
Selection for σ70 mutants defective in σ-core interaction. (A) A schematic of the toxic σ variant mutagenized with hydroxylamine. (B) Serial dilutions of cells with a toxic or nontoxic variant of σ70. DH5α cells containing either the toxic GST–NterΔ72σ70 fragment or a nontoxic hydroxylamine-generated mutant variant were plated with (right) or without (left) expression of the σ variants. (C) SDS gel analysis of hydroxylamine-generated nontoxic variants of GST–NterΔ72σ70. Cells were grown at 30°C in LB to an OD600 = 0.5, induced with 1 mm IPTG for 1 hr, lysed by resuspending in sample loading buffer and boiling for 5 min, and loaded on an 8% SDS Tris-glycine gel. The unstable (lanes 4,8,11) and truncated (lane 10) variants were discarded. (D) Location of mutations in σ70 and their cell-growth phenotypes. A linear diagram of σ indicates the four conserved regions, which have the following functional assignments: Region 1.1, autoinhibition of DNA binding; regions 2.3–2.4, DNA melting and −10 region promoter recognition; region 4.2, −35 region promoter recognition and activator response. Position of mutants is indicated below the linear diagram. Ability of the mutant plasmid-encoded σs to support cell growth was determined by comparing plating efficiency with and without expression of chromosomal σ at three different temperatures.
For σ70-type σs, the process of transcription initiation begins when σ binds to core RNA polymerase (α2ββ′) to form RNA polymerase holoenzyme (α2ββ′σ). As a result of this interaction, both partners undergo major conformational changes and become competent for initiation. For σ, this involves exposure and reorientation of its DNA-binding domains so that it can specifically recognize the two conserved hexamers that constitute the prokaryotic promoter (Dombroski et al. 1992, 1993; Callaci and Heyduk 1998; Callaci et al. 1999). For core RNA polymerase (core), a channel with dimensions large enough to fit duplex DNA that is closed in core opens in the holoenzyme form of the enzyme (Polyakov et al. 1995). Despite the interface's importance in facilitating these conformational changes, little is known about the contacts between σ and core polymerase. In this report, we identify residues in σ70 that are important for interaction with core and discuss their functional significance.
Previous studies have identified conserved region 2.1 of σ as an important determinant of core binding. Deletion derivatives of σ70 and σ32 of E. coli lacking region 2.1 were unable to bind to core, as was a single point mutation in region 2.1 of the σ70 family member σE in Bacillus subtilis (Lesley and Burgess 1989; Lesley et al. 1991; Shuler et al. 1995). In addition, the only short stretch of amino acids in σ54 that seems homologous to σ70 corresponds to a portion of region 2.1. This portion of σ54 is implicated in core binding as well (Tintut and Gralla 1995).
Additional regions of σ have also been suggested to interact with core. A recent genetic analysis of σ32 found residues in four conserved regions (but not region 2.1) that were involved in interaction with core (Joo et al. 1997, 1998). Protein footprinting studies of σ70 and gp55 (a small, divergent, T4 phage-encoded σ) suggested that several regions of these σs are protected on interaction with core (Nagai and Shimamoto 1997; Leonetti and Geiduschek 1998). Finally, genetic analysis suggests that Mftf1p (a distantly related promoter specificity factor in yeast mitochondrial RNA polymerase) uses amino acids in regions homologous to conserved regions 2 and 3 of σ70 to interact with its single subunit core polymerase (Cliften et al. 1997).
We used an unbiased genetic approach as well as directed changes to identify residues in σ70 that are important for interaction with core. Our genetic studies indicate that residues in several conserved regions of σ are important for this interaction, indicating that the interface is extensive. One of these regions is also identified by a peptide inhibition approach. Our results indicate that the same regions and even the same residues in two different σs alter this interaction, suggesting that σ70 family members use homologous residues, at least in part, to interact with core. We show further that the interface of σ with core is functionally specialized, as certain functions are localized to particular regions of the interface.
Results
Selection for σ70 mutants defective in binding core RNA polymerase
GST fused to the amino terminus of a σ70 derivative lacking its first 72 amino acids (GST–NterΔ72σ70; Fig. 1A) is toxic to the cell when overexpressed. RNA polymerase containing this σ fusion protein has a severe defect in open complex formation, suggesting that toxicity results from sequestering core RNA polymerase in a nonproductive holoenzyme complex (Wilson and Dombroski 1997). We reasoned that σ70 mutants defective in binding to core would relieve the toxicity of GST–NterΔ72σ70.
Following hydroxylamine mutagenesis, ∼300 of 120,000 colonies showed decreased toxicity on overexpression of GST–NterΔ72σ70 (Fig. 1B). To screen out mutations that relieved toxicity because they encoded either truncated or misfolded σs, we examined 90 of the mutant σs on SDS–polyacrylamide gels (Fig. 1C). About half of the σs were either unstable, indicating that they were misfolded (Fig. 1C, lanes 4,8,11), or truncated (Fig. 1C, lane 10). These mutants were discarded. Twenty-five of the remaining mutants were cloned back into GST–NterΔ72σ70 to confirm their phenotype. The 20 mutants that relieved toxicity when recloned were sequenced. Ten of these proved to be point mutations in rpoD (σ70). The others either did not have a detectable base change within rpoD (σ70) or produced truncations at the very carboxyl terminus that had escaped our initial screen.
The ten mutations were located in five different conserved regions of σ70; one in region 2.1, three in region 2.2, one in region 2.4, two in region 4.1, and one in region 4.2 (Fig. 1D). Two mutations, D403N and E407K, were isolated twice, implying that these residues were mutational hot spots or that we were approaching saturation in the mutagenesis. The eight remaining σ70 mutants potentially defective in σ-core interaction were cloned into a construct expressing his-tagged full-length σ70.
We used a strain whose chromosomal copy of σ70 is regulated by the trp promoter to assess the ability of the plasmid-encoded mutant σ factors to support cell growth. When chromosomal σ70 was shut off, one of the mutant σs supported growth at all temperatures tested, a second was defective at high temperature only, and the remaining six were unable to support cell growth at any temperature tested (Fig. 1D). Two of the mutations changed glycine residues located at positions 408 and 577. We did not analyze the phenotypes of the glycine mutants further, as such changes are likely to perturb the structure of the protein.
A competitive binding assay demonstrates that the mutant σs are defective in binding to core
We developed a competitive binding assay and then used it to examine the ability of the σ70 mutants to bind core. Increasing amounts of nonradioactive σ70 were mixed with a fixed amount of radioactive wild-type σ70 (σ70*) and allowed to bind to a limiting amount of core. The complexes were separated by immunoprecipitation with an antibody directed against core and then analyzed for the amount of radioactive σ70 present. Order of addition experiments performed over a 10-fold range (10–100 nM σ70*), showed that this assay is in equilibrium (Fig. 2A; data not shown). A titration with increasing amounts of nonradioactive wild-type σ70 indicates that the assay is well behaved over the entire experimental range (Fig. 2B). We can reliably quantify our results in the range of 25%–90% of σ70* competed, thus setting upper and lower limits for the assay.
Figure 2.

A coimmunoprecipitation competition assay measures σ binding to core polymerase. (A) The σ-core-binding assay is in equilibrium. When a twofold excess of nonradioactive wild-type σ70 competitor is added after (lane 2), before (lane 3), or at the same time as (lane 4) 100 nm wild-type σ70 labeled with [γ-32P]ATP, equivalent competition for binding to core is observed (see Materials and Methods for details of the assay). (B) A titration with wild-type σ70 competitor. 100 nm-labeled wild-type σ70* was competed against 1–1000 nm cold σ70 competitor (♦) for binding to core. Experimental data compares well with the theoretical curve. (C) Relative binding of the mutant σs to core determined from the coimmunoprecipitation competition assay. Wild-type σ70* was competed against a twofold (dark gray bars) or fivefold (light gray bars) excess of wild-type or mutant σ70 for binding to core. Competition with a fivefold excess of both mutant and wild-type σ70 is indicated by white open bars. (D) Relative binding of σ70 alanine substitution derivatives. Symbols and protocols as in Fig. 2C.
To determine the binding of each mutant σ relative to wild-type σ, we competed σ70* with a twofold and fivefold excess of nonradioactive mutant σ70. Each of the mutants competes more poorly than wild-type σ70 (Fig. 2C). We excluded two trivial possibilities for lack of competition by mutant σs. First, we show that addition of wild-type σ70 restored competition, eliminating the possibility that the high recovery of σ70* resulted from aggregation of σ70* with the mutant σs (Fig. 2C). Second, we used two independent assays to show that the purified mutant σ70 proteins were approximately as active as wild-type σ70. Both a noncompetitive binding assay performed with core in excess and a transcription assay (see Materials and Methods), indicated that ≥50% of the mutant σ was active (M. Sharp, data not shown). We conclude that the mutants are specifically defective in binding to core.
The binding assay data can be quantified by comparing the concentrations of wild-type and mutant σ70 required for equivalent competition. For example, when 450 nm mutant competes as if it were present at 30 nm wild-type, the mutant has a 15-fold defect in core binding. Experimental limits prevent us from quantifying defects >15-fold. Five of the six σ70 mutant proteins exhibited defects ranging from 7-fold to >15-fold in binding core RNA polymerase (Table 1). The last mutant, V387I had little or no binding defect.
Table 1.
Estimation of the core binding defects of σ70 mutants
| R374A | 1.0 | L598A | 13.0 |
| K392A | 1.0 | N409D | 15.0 |
| V387I | 2.0 | L384A | >15 |
| I405A | 2.0 | V387A | >15 |
| I388A | 3.0 | D403A | >15 |
| F401A | 3.0 | Q406A | >15 |
| P453S | 7.0 | E407A | >15 |
| P504A | 8.0 | M413T | >15 |
| D403N | 9.0 | E555K | >15 |
| R562H | 9.0 | E555A | >15 |
| L402F | 10.0 | R562A | >15 |
| E407K | 11.0 | I565A | >15 |
Data generated from competition with a 5-fold excess of mutant σ was compared to a standard curve generated with increasing concentrations of wild-type σ (Fig. 2B). We express the defect in terms of wild-type σ equivalents, that is, how much more mutant than wild-type protein is required to achieve the same amount of competition. The binding defects calculated from a two-fold excess of mutant σ were similar to those reported above. Binding defects 15-fold or greater cannot be reliably distinguished (see text).
Our mutants could directly disrupt the interaction between σ and core by removing a favorable interaction or by creating a steric clash. We eliminated steric clashes by changing the mutated residues to alanine. Alanine substitutions are also less likely to perturb the hydrophobic core, although in our case, the crystal structure of a fragment of σ70 indicates that two of the affected residues (D403 and E407) are surface exposed. Two additional mutants alter charged residues, suggesting that they are also surface exposed. Only one residue, V387, is partially buried.
All of the alanine substitution mutant proteins (V387A, D403A, E407A, E555A, and R562A) had binding defects equal to or greater than that of the original mutants (Fig. 2D; Table 1), and had activities within twofold of wild-type σ70 in a transcription assay (see Materials and Methods), suggesting that the original mutants removed favorable interactions with residues in core. Alanine substitutions of putative surface-exposed residues may remove favorable interactions, present in the original mutant changes, thus explaining their increased severity. For example, the histidine substitution at R562 might maintain some of the interactions normally provided by an arginine side chain, whereas the alanine substitution would have none of these contacts.
Regions of σ70 and σ32 that affect binding to core are highly conserved
Using a genetic approach, Joo and Calendar had previously identified residues in σ32 important for interaction with core (Joo et al. 1997, 1998). For both σ70 and σ32, multiple residues in both regions 2.2 and 4.1 were implicated in binding to core, indicating that both σs share some similarities in their core binding surfaces. However, the σ32 selection and our σ70 selection had also identified different regions. For σ70, residues in regions 2.1 and 2.4 have a core-binding defect, although the V387A change could be perturbing the structure of the protein. For σ32, residues in regions 3.1, 4.2, and a region unique to σ32 (the RpoH box) have core-binding defects (Fig. 3A). We chose one residue implicated in core binding from each of the four regions in σ32 that are conserved in σ70, mutated the comparable residue in σ70 to alanine, and tested core binding. Q406A, P504A, I565A, and L598A are all defective (Fig. 3B; Table 1). Therefore, these residues are important for both σ70 and σ32 interaction with core RNA polymerase.
Figure 3.


Derivation and binding properties of a second set of σ70 alanine substitution mutations. (A) Comparison of the residues in σ70 and σ32 shown to be important for interaction with core polymerase. The σ32 mutant changes reconstructed as alanine substitutions in equivalent σ70 residues are indicated in boldface. (B) Relative binding of mutant σs to core determined from the coimmunoprecipitation competition assay. Symbols and protocols as in Fig. 2C.
Q406N and E407K abolish the inhibitory effect of a σ-derived peptide on transcription
Region 2.2 of σ70 had been independently implicated in binding to core by S. Roychoudhury, F. Wang, E.W. Merritt, E.P. Rennells, C. Kasibhatla, and C.N. Parker (unpubl.), who showed that a small peptide spanning residues 390–407 of σ70 (SF2.4) inhibited transcription by holoenzyme. This includes part of regions 2.1 and 2.2 and three (D403, Q406, and E407) of the residues identified in our mutational study as important for core binding. We made three peptides, each having a different mutational change, and determined whether they could relieve inhibition. As measured by abortive initiation, two of the three peptides, SF2.4-Q406N and SF2.4-E407K inhibited formation of the abortive trimer, CpApU, less than the wild-type peptide (Fig. 4A, cf. lanes 8, 9, 11, and 12 with lanes 2 and 3). In each case, excess wild-type σ70 overcomes the inhibitory effect of the peptide (Fig. 4A). Together, these results support the view that this peptide inhibits transcription by competing for authentic σ-binding sites on core. Consistent with this idea, a gel-shift assay carried out at 0°C shows that the inhibitory peptide worked at an early step of transcription, preventing formation of the closed complex (Fig. 4B). As expected, both excess σ and attenuation by mutational alteration relieves inhibition (Fig. 4B).
Figure 4.
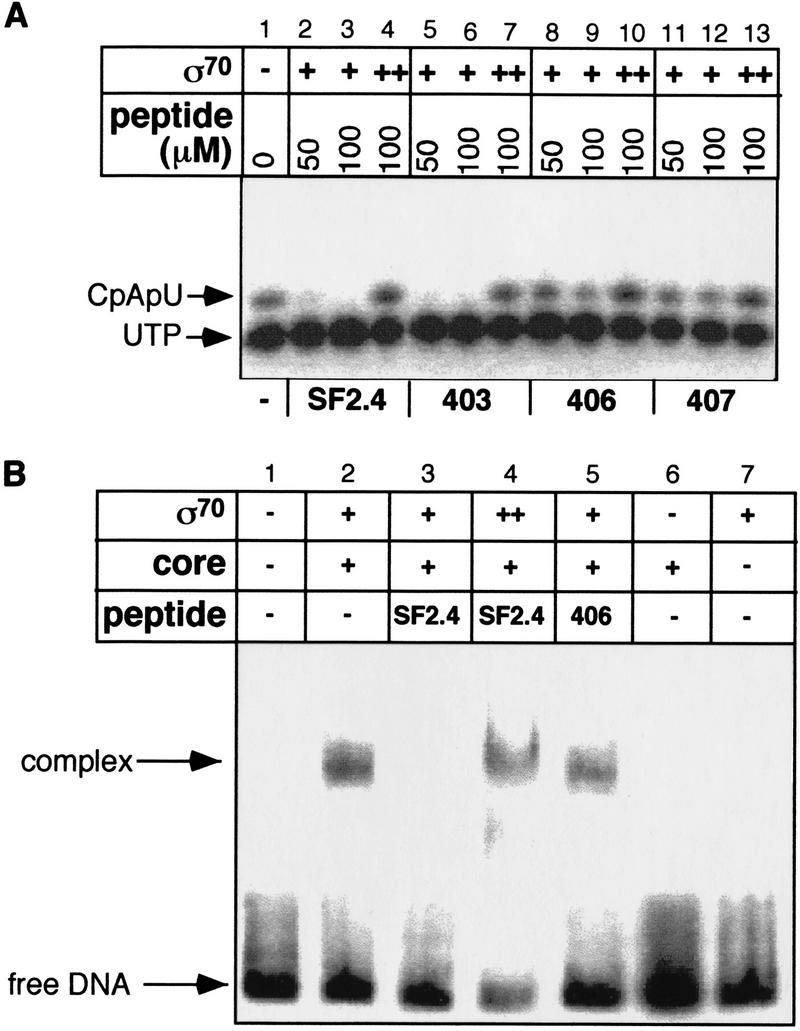
Inhibition of σ-core interactions by synthetic peptides spanning 390–407 of σ70. (A) The effect of synthetic σ factor peptide, SF2.4 (amino acids 390–407 of σ70), on σ70-dependent abortive initiation. SF 2.4 peptide containing either the wild-type sequence (lanes 2–4), the D403N substitution (lanes 5–7), the Q406A substitution (lanes 8–10), or the E407K substitution (lanes 11–13) were combined with 0.2 pmole RNAP core enzyme and 0.1 pmole σ70 (lane 1) and assessed for their ability to make a CpApU abortive transcript from 20 ng of a 130-bp DNA fragment containing T7A1 promoter (see Materials and Methods). The first two lanes in each set of three show the effect of increasing concentration of peptide on abortive initiation; the third lane assesses the effect of a fivefold molar excess of σ relative to core on peptide inhibition. The SF2.4 peptide inhibits transcription (lanes 2,3); this inhibitory effect is ameliorated by the Q406A and E407K substitutions (lanes 8,9,11,12) but not by the D403N substitution (lane 5,6). Excess σ overcomes inhibition (lanes 4,7,10,13). (B) SF2.4 peptide prevents the formation of the closed promoter complex. RNAP core enzyme was incubated with SF2.4 peptide and σ70 as in A, transferred to ice, and incubated with a 32P-labeled 130-bp DNA containing the T7A1 promoter for 30 min. Promoter–RNA polymerase complexes were resolved on nondenaturing polyacrylamide gels and visualized (see Materials and Methods).
Mutants in regions 2.1 and 2.2 of σ70reduce a promoter proximal pause required for λQ function
We had described previously four σ70 mutants (L402F, Q406A, N409D, and M413T) that are specifically defective in λQ-mediated antitermination because they failed to establish the pause required for Q to function (Ko et al. 1998). These mutants were located very close to the mutants we isolated in regions 2.1 and 2.2, and an in vivo assay had suggested that two of these mutants were defective in core interaction. All of these mutants have severe core-binding defects in our in vitro assay (Fig. 5A; Table 1), which are of greater magnitude, but the same rank order as those estimated from the in vivo assay (Ko et al. 1998). The less severe binding defects measured in vivo could result from the indirect nature of the in vivo assay, or from the many differences in conditions between the two assays. In particular, in the rapid initiation conditions present in the cell, nucleotide addition may be faster than dissociation, so that only the forward rates of σ binding to core contribute to competition.
Figure 5.


Phenotypes of σ mutants. (A) Relative core binding of σ70 mutants defective in promoter-proximal pausing. Symbols and protocols as in Fig. 2C. (B) Effects of σ mutants on promoter proximal pausing. Each mutant σ was assessed in vitro for the ability to facilitate the pause in transcription that occurs at +16 after initiation at the λPRpromoter. Transcription reactions were sampled at 0.5, 1, 2, and 5 min.
We wondered whether a defect in producing paused transcripts from the pR′ promoter was characteristic of all mutants with reduced binding to core or limited to σ70 mutants in regions 2.1 and 2.2. The latter proved to be the case. Only the region 2.1 and 2.2 mutants (V387I, D403N, and E407K) were significantly defective, reducing both the fraction of RNA polymerase that pauses and the pause half-life (Fig. 5B). Our region 2.4 mutant (P453S) and the two region 4.1 mutants (E555K and R562H) had little or no effect on pausing at λpR′.
Another potential binding surface in σ70 has little or no effect on holoenzyme formation
On the basis of genetic analysis that included a single-point mutant in σE in B. subtilis at a position homologous to R385 of σ70 and deletions in region 2.1 that altered the ability of both σ70 and σ32 to bind core, it was thought that region 2.1 of σ70 was a primary determinant for binding to core (Lesley and Burgess 1989; Lesley et al. 1991; Shuler et al. 1995). On analysis of the crystal structure of a fragment of σ70, Darst and colleagues proposed that five highly conserved exposed hydrophobic residues and two highly conserved charged residues in regions 2.1 and 2.2 might interact with core (Malhotra et al. 1996). Thus, we wondered if these other solvent-exposed residues of σ70 interacted with core. We made alanine substitutions at four highly conserved hydrophobic residues (L384A, I388A, F401A, and I405A) and at the two highly conserved charged residues (R374A and K392A) proposed to interact with core (Malhotra et al. 1996). With the exception of L384A, these mutants had little or no defect in core binding (Fig. 6; Table 1) and therefore, at least under the conditions of our assay, interactions with these residues are not required for holoenzyme formation. The fifth highly conserved hydrophobic residue had already been identified in our selection for mutants defective in λQ antitermination. This mutant is also defective in core binding as shown in Figure 5. Interestingly, the two mutants with core-binding defects (L384 and L402) are both located within the cluster of mutations that we have defined (see Fig. 7A).
Figure 6.

Relative core binding properties of alanine substitution σ70 mutants located in region 2.1. Symbols and protocols as in Fig. 2C.
Figure 7.
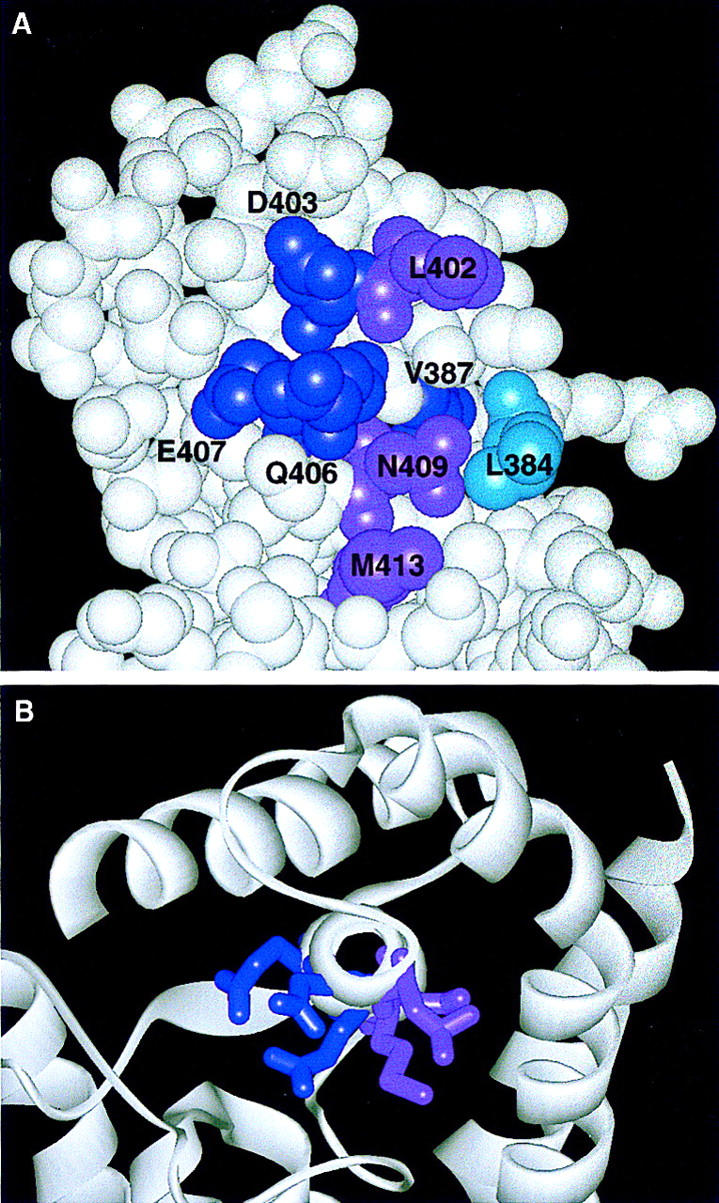
Location on the crystal structure of the σ70 residues implicated in core binding. (A) A space-filling model indicates residues from regions 2.1 and 2.2 implicated in core binding. Residues in dark purple were identified by genetic selection for relief of toxicity (V387, D403, and E407) or by a comparable σ32 selection (Q406); those in light purple were identified because of their defect in Q-mediated antitermination (L402, N409, and M413); the residue in cyan was proposed from the crystal structure (L384) (Malhotra et al. 1996). (B) A view of the σ70 crystal structure looking down or through helix 13 (region 2.2) showing that the region 2.2 mutants from the two different selections (indicated by light and dark purple) are localized on two different faces of the helix.
Discussion
Two complementary genetic approaches allowed us to identify residues in σ70 that are important for interaction with core. An unbiased genetic selection (Fig. 1) as well as site-directed mutagenesis of residues implicated in core binding identified a patch of amino acids composed almost entirely of residues from region 2.2 as well as other residues located in regions 2.4, 3.1, 4.1, and 4.2 that are important for interaction of σ70 with core RNA polymerase. Also, transcription inhibition by a small peptide from region 2.2 is relieved by two of our mutational changes that reduced core binding. It is possible that some of the amino acid substitutions indirectly affect σ binding to core, for example, by stabilizing a protein conformation unfavorable for binding to core. However, our genetic experiments, in combination with other data, provide strong reason to believe that the regions we have identified directly contact core. These same amino acid substitutions alter interaction of σ with selected fragments of core in a yeast two-hybrid analysis (T. Gruber and C. Gross, unpubl.). Moreover, each residue we have identified is located in a region of σ70 that is protected from cleavage when bound to core (Nagai and Shimamoto 1997). We suggest that these regions (and possibly others) constitute the interface used by the σ70 family of proteins to interact with core RNA polymerase and modulate initiation. Our experiments indicate that this interface is functionally specialized.
Much of the σ70and σ32 interface with core is highly conserved
The six members of the E. coli σ70 family of proteins all bind to the same core RNA polymerase with different affinities. As core is limiting in the cell, competition among σ factors could exert global control over gene expression (Osawa and Yura 1981; Ingraham et al. 1983; Ishihama 1991; Zhou and Gross 1992). To understand how different σs bind core, we asked whether residues implicated in σ32 binding to core were also important for σ70 binding. In each of the four cases tested (one residue from each conserved region of σ32 implicated in binding), the σ70 mutants were also very defective in core binding. These results suggest that all σ70 family members use homologous regions and possibly, in some cases, homologous residues when binding to core. However, σs also use nonhomologous regions for this interaction. Both the RpoH box and region 1.1, unique to σ32 and σ70 respectively, contain core-binding determinants (Joo et al. 1998; T. Gruber and C. Gross, unpubl.). The combination of conserved and nonconserved surfaces for interaction may contribute to the functional specialization of each σ factor.
Mutational analysis suggests that the σ70 interface with core is extensive
The core-interaction site that we have defined most completely is a patch of residues on the crystal structure of a fragment of σ70 consisting almost entirely of residues from region 2.2 with contributions from two nearby residues in region 2.1. Among the residues identified, only one (V387) is partially buried in the hydrophobic core; all others, including three hydrophobic residues (L402 and M413 in region 2.2 and L384 in region 2.1), are surface exposed and available to interact with core (Fig. 7). A small peptide (residues 390–407) that includes four of these residues (L402, D403, Q406, and E407) inhibits formation of the closed complex and abortive initiation by binding to core competitively with σ. The simplest interpretation of this result is that inhibition results from occlusion of sites on core that bind σ. The fact that peptides with a Q406N or E407K substitution show reduced inhibition validates this idea, thereby providing direct evidence that Q406 and E407, and by extension, this entire patch of residues directly contact core. The third substitution, D403N, showed severe core-binding defects only in intact σ70. This mutation may inhibit core binding by destabilizing the entire cluster, an effect that would be unlikely to occur in the small peptide that lacks the structural constraints of the intact molecule.
The region 2.2 interaction patch includes mutants obtained from two different types of genetic selections, one on the basis of defects in Q-mediated antitermination (L402F, N409D, and M413T) and the other on the basis of the ability of a toxic fragment of σ to compete with authentic σ for forming productive holoenzyme (D403, E407, and Q406). Both classes of mutants are comparable in their defects in core binding in vitro, but only the former class is viable. Interestingly, an axial view of helix 13, which comprises most of region 2.2, reveals that each mutant class is in a slightly different location, with the Q-defective class slightly to the right of the other class (Fig. 7B). Further examination of the two groups of mutants may reveal additional differences in their function.
In addition to this well-defined patch, residues in regions 2.4, 3.1, 4.1, and 4.2 also reduce interaction of σ with core. When the residues shown to be important for both σ70 and σ32 binding to core are included, the mutationally defined core-interacting surface of region 4.1 is defined by four closely spaced residues (E555, R562, F563, and I565) and that of 4.2 is defined by two residues (I590 and L598). By use of this same criterion, and including as well a substitution in σ70 that has been shown by glycerol gradient sedimentation to have a decreased affinity for core, the mutationally defined core interaction surface of region 3.1 includes three residues (M487, P504, and S506) (Hernandez and Cashel 1995; Joo et al. 1998). Because all of the alanine-substitution mutants we tested retain the core-binding defects of the original mutations, we favor the idea that the originally selected mutations disrupted existing interactions rather than creating unfavorable interactions with residues in core.
Other data suggests that additional regions of σ70 interact with core. The fusion protein used to select mutations lacked the first 71 amino acids of σ70, and could not be used to assess the role of region 1.1 in core binding. Evidence from an independent approach that relies on a yeast two-hybrid analysis indicates that region 1.1 does interact with core (T. Gruber and C. Gross, unpubl.). Consistent with this idea, protein footprinting data suggests burial of region 1.1 on holoenzyme formation (Nagai and Shimamoto 1997). Furthermore, neither the selection for mutants in σ32 nor our selection for mutants in σ70 were saturating. Because hydroxylamine only makes guanine/cytosine to adenine/thymine transitions, it is likely that we were unable to identify several residues important for core interaction. Therefore, conserved regions other than those identified could be important for binding core.
Finally, as holoenzyme undergoes major conformational changes during transcription initiation, it is likely that contacts between σ and core change during initiation. Because our binding assay identifies only those mutants defective in forming the initial interface, other in vitro techniques will be required to identify such contacts. On the basis of genetic analysis, region 2.1 had been previously considered to be a major core-binding determinant (Lesley and Burgess 1989; Shuler et al. 1995). Yet the point mutations we have tested have only little or no binding defects when tested under the conditions of our competitive binding assay. Region 2.1 is an excellent candidate for a core contact surface utilized later in initiation, as mutants in this region do manifest a variety of cell growth phenotypes and transcriptional defects. For example, F401A has <5% of the transcriptional activity of wild type in a single-round transcription experiment (M. Sharp, data not shown), but has little or no defect in binding core. Although these residues have no obvious role in forming the initial interface, they could quite possibly contact core during later steps of initiation.
Comparison of the residues of the B. subtilis σ factor, σF, required for binding to its cognate anti-σ, SpoIIAB, with residues implicated in binding to core, further supports the idea that the σ interface with core is extensive. The simplest way for an anti-σ to disable σ is to occlude its interaction with core. In that case, anti-σ-binding sites should be located in our proposed core interaction regions. This proves to be the case for the three contact sites identified in σF, as the equivalent residues in σ70 (V385; region 2.1, P479; region 3.1 and M561; region 4.1) (Decatur and Losick 1996) are located in close proximity to residues identified here because of their defects in binding to core (V387, P504, and I565).
The mutationally defined σ70 interface with core is functionally specialized
σ mutants in three of the regions implicated in binding to core have specific additional phenotypes. Each of these phenotypes appear to be exclusively associated with that particular region of the interface. Mutations in the core interaction patch in region 2.2 also disrupt λQ-mediated anti-termination by preventing a pause very early after transcription initiation from the λpR′ promoter (Fig. 5B) (Ko et al. 1998). This phenotype is exhibited only by mutants in this region of the interface. An exhaustive search for mutations allowing cells lacking ppGpp to grow on minimal medium identified only a small cluster of residues including the very residue (P504) in region 3.1 implicated in core binding, indicating that this region of the interface has a unique function in transcription. Finally, after extensive searches, only two regions of σ, located upstream and downstream of the helix-turn-helix in region 4.2, have been implicated in activator response. Our region 4.2 core-interaction patch is located within the downstream region. One of the two mutants in this region, L598, has a defect in FNR activation (Lonetto et al. 1998). The coincidence of residues conferring specific phenotypes with particular portions of the σ interface suggests that different portions of the σ-core interface perform different, specialized functions.
The extensive information concerning recognition of the −10 region of the promoter allows us to speculate how a defect in the region 2.2 portion of the σ-core interface could lead to pausing defects. Region 2.2 is primarily composed of a helix positioned immediately behind the −10 recognition helix (Malhotra et al. 1996). The pause mediated by σ is a consequence of its interaction with the nontemplate strand at a reiterated −10 region present early in the pR′ transcript. Because core is required for the ability of σ to discriminate between the template and nontemplate strands, the interaction of region 2.2 with core could mediate the core-induced conformational change necessary for discrimination (Marr and Roberts 1997; Callaci and Heyduk 1998). More generally, it is interesting to note that information from the nontemplate strand is utilized uniquely during the initiation process (Ring and Roberts 1994; Ring et al. 1996; Roberts and Roberts 1996). It would not be surprising to find that this portion of the σ-core interface is necessary to properly orient the nontemplate strand, so that the sequence-specific information present in this strand can be decoded.
The junctions between σ and core are ideally positioned to communicate information concerning initiation and DNA binding from σ to core and back. One region (3.1) is in close proximity to the initiating nucleotide and four regions (2.2, 2.4, 4.1, and 4.2) are in close proximity to the two DNA-binding determinants in σ70 that recognize the conserved −10 and −35 regions of the promoter (Gardella et al. 1989; Siegele et al. 1989; Severinov et al. 1994). The distance between the two DNA-binding domains of σ increases on binding to core RNA polymerase (Callaci et al. 1999), and this may be one function of the regions of the interface adjoining the DNA-binding domains.
Materials and methods
Plasmids and DNA manipulations
Standard methods were used in plasmid construction and other DNA manipulations. Mutations in rpoD were identified with Applied Biosystems BigDye Terminator Cycle Sequencing Ready Reaction Kit following the manufacturer's instructions.
The parent plasmid for fusions of GST and σ70 were variations of pGEXσ[8] and pGEXσ[72] (Dombroski et al. 1992). To facilitate later cloning steps, a HindIII site following the rpoD gene was added to these plasmids by cloning a BglII–HindIII fragment from pJH76 into pUC21 (Vieira and Messing 1991) and then cloning the SphI–AatII fragment from the resulting plasmid (pCL357) between the same two sites in either pGEXσ[8] or pGEXσ[72] generating pCL359 and pCL361, respectively.
pCL360 was constructed by ligating an EcoRI–HindIII fragment from pCL359 between the same two sites in pET28c generating a hexahistidine-tagged variant of σ70 (Novagen). pCL390 was constructed by ligating a NcoI–PstI fragment from pCL360 between the same two sites in ptrc99a (Pharmacia). A PstI–HindIII fragment from pCL360 was ligated between the same two sites in pCL390. The resulting plasmid (pCL391) carries the rpoD gene and the expressed fragment includes a hexahistidine tag, a T7 tag, a thrombin cleavage site, and amino acids 8–613 of σ70.
To generate mutants that relieve the toxicity of an overexpressed amino-terminal–GST fusion of rpoD, which lacks the first 71 amino acids of σ70, 10 ng of pCL361 (expresses the toxic fusion protein, GST–NterΔ72σ70) in 500 μl was treated with 400 mm NH2OH in 100 mm KH2PO4 (pH 6), 1 mm EDTA, for 1 hr at 65°C. Hydroxylamine was removed with the QIAquick PCR Purification Kit (Qiagen), and pCL361 was transformed into DH5α via electroporation. Transformants were plated on LB containing 250 μm IPTG at 37°C. Transformants that grew on IPTG were retained for further analysis.
The QuikChange Site-Directed Mutagenesis Kit was used to make alanine substitutions in σ70 following the manufacturer's instructions (Stratagene). Candidate mutants were screened for the appropriate alanine change by dideoxy sequencing.
Screening of σ70 mutants by SDS-PAGE
Hydroxylamine mutants were run on denaturing gels to screen out unstable and truncated mutant sigmas. Cells were grown in LB to an O.D. of 0.5 and induced for 1 hr with 1 mm IPTG. A total of 500 μl of cells were spun down, the supernatant removed, the pellet resuspended in 50 μl of SDS loading dye, and boiled 3–5 min. Boiled samples (10 μl) were loaded on an 8% Tris-glycine SDS–polyacrylamide gel.
Ability of σ70 to support cell growth
Candidate σ70-binding mutants were cloned into pCL391 (see above) and transformed into CAG20176 in which rpoD is under the control of the trp promoter (Lonetto et al. 1998). Growth of CAG20176 is inhibited by tryptophan and requires indole acrylic acid (IAA). The ability of the mutants to support cell growth was determined by plating dilutions of an overnight culture grown at 37°C in LB containing 200 μm IAA, on plates with or without IAA at 30°C, 37°C, and 42°C.
Source of σ and core enzyme
Source of σ and core are described for all experiments except those in Figure 4. Core polymerase was purified as described by Burgess and Jendrisak (1975) with modifications as described in Hager et al. (1990).
σ70 wild-type and mutant protein was isolated from CAG20176 transformed with mutant derivatives of pCL391. Expression of the mutants was induced for 2 hr by adding 100 μm IPTG to cultures growing exponentially at 37°C in LB containing 200 μm IAA. Cells were harvested by centrifugation, resuspended in binding buffer [10 mm Tris-HCl, (pH 8), 5% glycerol, 10 mm β-mercaptoethanol, 0.5 m NaCl], lysed with 150 μg lysozyme /ml and sonicated. The supernatant was collected by centrifugation for 60 min at 25,000 rpm and applied to a 1.5-ml Talon Metal Affinity column (Clontech). The column was washed with binding buffer containing 10 mm imidazole and eluted with binding buffer containing increasing concentrations of imidazole (20, 40, 80, and 150 mm). Most of the mutants eluted in the 80 and 150 mm imidazole fractions. Fractions containing the mutants were dialyzed against 10 mm Tris-HCl, (pH 8), 50% glycerol, 10 mm β-mercaptoethanol, 0.5 m NaCl, and 0.05% Tween 20 and stored at −80°C.
RNA polymerase preparation
RNA polymerase preparation in Figure 4 is described. RNA polymerase core enzyme was reconstituted from individually overexpressed subunits. The plasmids pT7α, pT7σ, and pMKSe2 expressing RNAP α, σ70, and β subunits, respectively, used for in vitro reconstitution experiments were as described (Zalenskaya et al. 1990; Severinov et al. 1993). The plasmid pCYB2β′ (Nechaev and Severinov 1999) was a source of the wild-type β′ subunit. RNAP reconstitution was performed as described (Borukhov and Goldfarb, 1993; Tang et al. 1995). The enzymes were further purified by gel exclusion chromatography on Superose 6 (Pharmacia) and anion exchange chromatography on Resource Q (Pharmacia). Fractions containing the RNAP core were pooled, concentrated on microcentrifuge filters (Amicon) to ∼1 mg/ml, and stored in 10 mm Tris-HCl, (pH 8), 50% glycerol, 10 mm β-mercaptoethanol, 0.5 m NaCl, and 0.05% Tween 20 at −20°C. Recombinant σ70 subunit was purified from BL21(DE3) cells harboring the pT7σ plasmid (Zalenskaya et al. 1990) as described (Borukhov and Goldfarb 1993).
Labeling σ70 with [γ-32P]ATP (kinase reactions)
σ70 was labeled at a naturally occurring serine protein kinase site located in the nonconserved region of σ70 (RRMSI, residues 363–367) by incubating 25 units of protein kinase from bovine heart (Sigma) with 14 μm σ70 and 10 μm [γ-32P]ATP (30 Ci/pmole) in 20 mm Tris-HCl (pH 8), 100 mm NaCl, 12 mm MgCl2, and 10 mm β-mercaptoethanol at 37°C for 30 min to 1 hr at 37°C. The kinase reaction was stopped by adding a 1/10 volume of stop solution [20 mm NaPO4 (pH 8), 20 mm P2O7, 100 mm imidazole, 0.5% Tween 20, 200 μg of BSA/ml] (Kaelin et al. 1992). Protein kinase was resuspended at 5 U/μl in 20 mm Tris-HCl (pH 8), 50% glycerol, 100 mm NaCl, 12 mm MgCl2, and 100 mm β-mercaptoethanol and stored at −20°C. Free label and kinase were removed by batch binding for 1 hr at 4°C to 100 μl of Ni–NTA agarose (Qiagen) in binding buffer [10 mm Tris-HCl (pH8.8), 0.5 m NaCl, 5% glycerol, 10 mm MgCl2, 10 mm imidazole, 1 mm β-mercaptoethanol, 0.05% Tween 20) and washed once with binding buffer followed by two additional washes with binding buffer plus 0.5 mm EDTA. Labeled σ70 was eluted by incubating two successive 50-μl aliquots of binding buffer plus 100 mm imidazole for 15 min each. The concentration of the labeled protein (σ70*) was determined by removing a sample of the initial kinase reaction, in which the concentration of σ70 is known, and comparing it with that of the two Ni–NTA elutates on an 8% Tris-glycine SDS–polyacrylamide gel with the PhosphorImager (Molecular Dynamics) to quantitate the relative amounts of labeled protein.
σ-core binding assay
σ70 mutants potentially defective in σ-core interaction were tested in a competitive assay in which σ70 mutants compete against labeled wild type σ70 for core binding. The buffer used in all steps of this experiment contained 10 mm Tris-HCl, (pH 8), 5% glycerol, 1 mm β-mercaptoethanol, 0.5 m NaCl, 10 mm MgCl2, and 200 μg/ml BSA. Siliconized Eppendorf tubes soaked overnight in buffer were used throughout this experiment to decrease σ70 binding to the Eppendorf tubes. A total of 150 nm wild-type σ70*, 75 nm core and 0, 300, or 750 nm cold σ70 mutant competitor were incubated at 37°C for 1 hr. A total of 100 μl pre-equilibrated protein A–Sepharose beads (10% volume) coupled to anti-core antibody was added to this mixture and rocked at 4°C for 1 hr. Polyclonal antibodies were raised in rabbits against core RNA polymerase (Animal Pharm Services). The beads were washed with 1 ml of buffer, spun down, and the supernatant removed. A final spin, followed by aspiration, was required to remove all of the liquid from the beads and the sides of the Eppendorf tube. The dry beads were resuspended in 100 μl of water, and 50 μl of the resuspended bead solution was added to 2 ml of scintillation fluid. Results were analyzed via scintillation counter. Coimmunoprecipitation of labeled σ70* is dependent on the presence of core RNA polymerase as anti-core antibodies did not cross react with σ70. All mutants were tested in at least two independent experiments performed in triplicate. Order of addition experiments performed at 0.25 m NaCl and 0.5 m NaCl reveals that the competitive binding assay reaches equilibrium in 20 min at 0.5 m NaCl, but is not at equilbrium at the lower salt concentration (data not shown).
Determining the activity of the mutant σ preps
The activities of the mutant σ preps were determined via a stalled transcription complex assay. Open complexes were assembled on the T7A1 promoter by incubating 100 nm σ70, 100 nm core, and 200 nm DNA fragment from pCL185 (Feng et al. 1994), at 37°C for 15 min in 10 mm Tris-HCl, (pH 8), 5% glycerol, 1 mm β-mercaptoethanol, 0.1 m NaCl, 10 mm MgCl2, 0.1 mm EDTA, 0.05% Tween 20, and 200 μg of BSA/ml. Transcription was initiated and complexes were halted prior to addition of U17 by adding 250 μm ApU dinucleotide; 10 μm ATP, 10 μm GTP, and 10 μm [α-32P]CTP (50 Ci/100 μmoles) were added to the mixture and incubated for an additional 10 min. In the absence of UTP, RNA polymerase will stall at the first position requiring UTP (+17 for T7A1) (Levin et al. 1987). The labeled 16-nucleotide product was analyzed by denaturing-PAGE [7 m urea, 19% acrylamide [acrylamide:bis 19:1 (wt/wt)] and quantitated with the PhosphorImager (Molecular Dynamics). Mutants with core-binding defects had, within twofold, the activity of wild-type σ70.
In addition, some of the mutant σ preps (V387I, D403N, E407K, P453S, and E555K) were tested for activity in a noncompetitive σ core-binding assay performed under low-salt conditions. The buffer used in all steps of this experiment contained 10 mm Tris-HCl, (pH 8), 5% glycerol, 1 mm β-mercaptoethanol, 0.1 m NaCl, 10 mm MgCl2, and 200 μg/ml BSA. Siliconized Eppendorf tubes were used throughout this experiment to decrease σ70 binding to the Eppendorf tubes. A reaction containing 100 nm mutant or wild-type σ70* (labeled as described previously) and 2 μm core was incubated at 37°C for 1 hr, then 100 μl of pre-equilibrated protein A–Sepharose beads (10% volume) coupled to anti-core antibody was added to this mixture and rocked at 4°C for 1hr. Polyclonal antibodies were raised in rabbits against core RNA polymerase (Animal Pharm Services). The beads were washed with 1 ml of buffer, spun down, and the supernatant removed. A final spin, followed by aspiration, was required to remove all of the liquid from the beads and the sides of the Eppendorf tube. The dry beads were resuspended in 100 μl of SDS loading dye, and boiled for 10 min. A total of 20 μl of the resuspended bead solution was run on an 8% Tris-glycine SDS–polyacrylamide gel. Results were analyzed via PhosphorImager (Molecular Dynamics). Coimmunoprecipitation of labeled σ70* is dependent on the presence of core RNA polymerase as anti-core antibodies did not cross react with σ70. All mutants were tested in at least two independent experiments performed in duplicate.
Peptides
Synthetic σ factor (SF) peptides were synthesized by standard automated FMOC chemistry. The peptide sequences were as follows (single letter amino acid code): IAKKYTNRGLQFLDLIQE (SF2.4, corresponds to σ70 amino acids 390–407), IAKKYTNRGLQFLNLIQE (SF2.4-D403N), IAKKYTNRGLQFDLIAE (SF2.4-Q406A), and IAKKYTNRGLQFLDLIQK (SF2.4-E407K). The substitutions are in boldface type in the three peptides. The peptides used were at least 90% pure as judged by reverse-phase HPLC.
Abortive initiation assay
For abortive initiation analysis (used in Fig. 4), 0.2 pmole of core RNA polymerase was incubated with or without SF2.4 peptide for 5 min at 37°C in 20 μl of 20 mm Tris-HCl (pH 8.0), 40 mm KCl, and 10 mm MgCl2, then 0.1 pmole of the purified σ70 was added and incubated for an additional 5 min at 37°C. Abortive initiation reactions were started by adding 20 ng of a 130-bp DNA fragment containing T7A1 promoter (Severinov et al. 1993), 0.5 mm initiating ribodinucleotide CpA, and 10 mCi of [α-32P]UTP (3000 Ci/mmole, New England Nuclear) and were allowed to proceed for 15 min at 37°C. Reactions were terminated by the addition of an equal volume of urea-loading buffer, resolved by urea-PAGE [7 m urea, 20% polyacrylamide, acrylamide to bis 19:1 (wt/wt)], and revealed by PhosphorImager (Molecular Dynamics).
Native polyacrylamide gel shift assay
RNAP core enzyme (0.2 pmole) was combined with 0.1 pmole of σ70 and incubated 5 min at 37°C to allow formation of holoenzyme. Where indicated, SF2.4 was added just prior to σ70. Reactions were transferred to ice and supplemented with ∼5000 cpm of 32P 5′-labeled 130-bp DNA fragment containing T7A1 promoter. After incubation on ice for 30 min, the reactions were loaded on a 0.5 × TBE nondenaturing 4% polyacrylamide gel [acrylamide/bis 39:1 (wt/wt)] kept at 4°C and RNAP-promoter complexes were revealed by PhosphorImager (Molecular Dynamics).
Promoter-proximal pause measured in vitro
The percent of promoter-proximal-paused complexes for σ70 mutants was determined as described in Ko et al. (1998).
Acknowledgments
We thank Robert Landick for helpful discussions and critical reading of the manuscript; Jon Tupy for critical reading of the manuscript, help with figure preparation, and computer modeling; Tracy Cao for critical reading of the manuscript; Dan Joo and Christian N. Parker for helpful discussions; Tanja Gruber for helpful discussions and sharing unpublished results; and Ding Jun for providing anti-core antibody. This work was supported by the National Institutes of Health (NIH) (GM30477). M.M.S. was supported in part by National Science Foundation. C.L.C. was supported by NIH (GM16603). M.T.M. and J.W.R. were supported by NIH (21941). K.S. and S.N. were supported by NIH (GM59295) and a Burroughs Wellcome Fund Career Award.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL cgross@cgl.ucsf.edu; FAX (415) 476-4204.
References
- Borukhov S, Goldfarb A. Recombinant Escherichia coli RNA polymerase: Purification of individually overexpressed subunits and in vitro assembly. Protein Expression Purif. 1993;4:1–8. doi: 10.1006/prep.1993.1066. [DOI] [PubMed] [Google Scholar]
- Burgess R, Jendrisak J. A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving polymin P precipitation and DNA-cellulose chromatography. Biochemistry. 1975;14:4634. doi: 10.1021/bi00692a011. [DOI] [PubMed] [Google Scholar]
- Callaci S, Heyduk T. Conformation and DNA binding properties of a single-stranded DNA binding region of σ70 subunit from Escherichia coli RNA polymerase are modulated by an interaction with the core enzyme. Biochemistry. 1998;37:3312–3320. doi: 10.1021/bi972041m. [DOI] [PubMed] [Google Scholar]
- Callaci S, Heyduk E, Heyduk T. Core RNA polymerase from E. coli induces a major change in the domain arrangement of the σ70 subunit. Mol Cell. 1999;3:229–238. doi: 10.1016/s1097-2765(00)80313-5. [DOI] [PubMed] [Google Scholar]
- Cliften PF, Park J-Y, Davis BP, Jang S-H, Jaehning JA. Identification of three regions essential for interaction between a σ-like factor and core RNA polymerase. Genes & Dev. 1997;11:2897. doi: 10.1101/gad.11.21.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decatur AL, Losick R. Three sites of contact between the Bacillus subtilis transcription factor σF and its anti-σ factor SpoIIAB. Genes & Dev. 1996;10:2348–2358. doi: 10.1101/gad.10.18.2348. [DOI] [PubMed] [Google Scholar]
- Dombroski AJ, Walter WA, Record MT, Jr, Siegele DA, Gross CA. Polypeptides containing highly conserved regions of transcription initiation factor σ70 exhibit specificity of binding to promoter DNA. Cell. 1992;70:501–512. doi: 10.1016/0092-8674(92)90174-b. [DOI] [PubMed] [Google Scholar]
- Dombroski AJ, Walter WA, Gross CA. Amino-terminal amino acids modulate σ-factor DNA-binding activity. Genes & Dev. 1993;7:2446–2455. doi: 10.1101/gad.7.12a.2446. [DOI] [PubMed] [Google Scholar]
- Feng G, Lee DN, Wang D, Chan CL, Landick R. GreA-induced transcript cleavage in transcription complexes containing Escherichia coli RNA polymerase is controlled by multiple factors, including nascent transcript location and structure. J Biol Chem. 1994;269:22282–22294. [PubMed] [Google Scholar]
- Gardella T, Moyle H, Susskind M. A mutant Escherichia coli σ70 subunit of RNA polymerase with altered promoter specificity. J Mol Biol. 1989;206:579–590. doi: 10.1016/0022-2836(89)90567-6. [DOI] [PubMed] [Google Scholar]
- Hager D, Jin D, Kornberg R. Use of Mono Q high-resolution ion-exchange chromatography to obtain highly pure and active Escherichia coli RNA polymerase. Biochemistry. 1990;29:7890–7894. doi: 10.1021/bi00486a016. [DOI] [PubMed] [Google Scholar]
- Helmann JD, Chamberlin MJ. Structure and function of bacterial sigma factors. Annu Rev Biochem. 1988;57:839–872. doi: 10.1146/annurev.bi.57.070188.004203. [DOI] [PubMed] [Google Scholar]
- Hernandez VJ, Cashel M. Changes in conserved region 3 of Escherichia coli σ70 mediate ppGpp-dependent functions in vivo. J Mol Biol. 1995;252:536. doi: 10.1006/jmbi.1995.0518. [DOI] [PubMed] [Google Scholar]
- Ingraham JL, Maaloe O, Neidhardt FC. Growth of the bacterial cell. Sutherland, MA: Sinauer Associated Inc.; 1983. [Google Scholar]
- Ishihama A. Control of cell growth and division. Heidelberg, Germany: Springer; 1991. [Google Scholar]
- Joo DM, Ng N, Calendar R. A σ32 mutant with a single amino acid change in the highly conserved region 2.2 exhibits reduced core RNA polymerase binding specificity. Proc Natl Acad Sci. 1997;94:4907–4912. doi: 10.1073/pnas.94.10.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo DM, Nolte A, Calendar R, Zhou YN, Jin DJ. Multiple regions on the Escherichia coli heat shock transcription factor σ32 determine core RNA polymerase binding specificity. J Bacteriol. 1998;180:1095–1102. doi: 10.1128/jb.180.5.1095-1102.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Krek W, Sellers WR, DeCaprio JA, Ajchembaum F, Fuchs CS, Chittenden T, Li Y, Farnham P, Blanar MA. Expression cloning of a cDNA encoding a retinoblastoma-binding protein with E2F-like properties. Cell. 1992;70:351–364. doi: 10.1016/0092-8674(92)90108-o. [DOI] [PubMed] [Google Scholar]
- Ko DC, Marr MT, Guo J, Roberts JW. A surface of Escherichia coli σ70 required for promoter function and antitermination by phage λQ protein. Genes & Dev. 1998;12:3276–3285. doi: 10.1101/gad.12.20.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti J-P, Geiduschek EP. Core-sigma interaction: Probing the interaction of the bacteriophage T4 gene 55 promoter recognition protein with E. coli RNA polymerase core. EMBO J. 1998;17:1467–1475. doi: 10.1093/emboj/17.5.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesley SA, Burgess RR. Characterization of the Escherichia coli transcription factor sigma 70: Localization of a region involved in the interaction with core RNA polymerase. Biochemistry. 1989;28:7728–7734. doi: 10.1021/bi00445a031. [DOI] [PubMed] [Google Scholar]
- Lesley SA, Brow MAD, Burgess RR. Use of in vitro protein synthesis from polymerase chain reaction-generated templates to study interaction of Escherichia coli transcription factors with core RNA polymerase and for epitope mapping of monoclonal antibodies. J Biol Chem. 1991;266:2632–2638. [PubMed] [Google Scholar]
- Levin JR, Krummel B, Chamberlin MJ. Isolation and properties of transcribing ternary complexes of Escherichia coli RNA polymerase positioned at a single template base. J Mol Biol. 1987;196:85–100. doi: 10.1016/0022-2836(87)90512-2. [DOI] [PubMed] [Google Scholar]
- Lonetto M, Gribskov M, Gross C. The σ70 Family: Sequence conservation and evolutionary relationships. J Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonetto MA, Rhodius V, Lamberg K, Kiley P, Busby S, Gross C. Identification of a contact site for different transcription activators in region 4 of the Escherichia coli RNA polymerase σ70 subunit. J Mol Biol. 1998;284:1353–1365. doi: 10.1006/jmbi.1998.2268. [DOI] [PubMed] [Google Scholar]
- Malhotra A, Severinova E, Darst SA. Crystal structure of a σ70 subunit fragment from E. coli RNA polymerase. Cell. 1996;87:127–136. doi: 10.1016/s0092-8674(00)81329-x. [DOI] [PubMed] [Google Scholar]
- Marr MT, Roberts JW. Promoter recognition as measured by binding of polymerase to nontemplate strand oligonucleotide. Science. 1997;276:1258–1260. doi: 10.1126/science.276.5316.1258. [DOI] [PubMed] [Google Scholar]
- Nagai H, Shimamoto N. Regions of the Escherichia coli primary sigma factor σ70 that are involved in interaction with RNA polymerase core enzyme. Genes Cells. 1997;2:725–734. doi: 10.1046/j.1365-2443.1997.1600357.x. [DOI] [PubMed] [Google Scholar]
- Nechaev S, Severinov K. Inhibition of Escherichia coli RNA polymerase by bacteriophage T7 gene 2 protein. J Mol Biol. 1999;289:815–826. doi: 10.1006/jmbi.1999.2782. [DOI] [PubMed] [Google Scholar]
- Osawa T, Yura T. Effects of reduced amount of RNA polymerase sigma factor on gene expression and growth of Escherichia coli: Studies of the rpoD40 (Amber) mutation. Mol & Gen Genet. 1981;184:166–173. doi: 10.1007/BF00272900. [DOI] [PubMed] [Google Scholar]
- Polyakov A, Severinova E, Darst SA. Three-dimensional structure of E. coli core RNA polymerase: Promoter binding and elongation conformations of the enzyme. Cell. 1995;83:365–373. doi: 10.1016/0092-8674(95)90114-0. [DOI] [PubMed] [Google Scholar]
- Ring BZ, Roberts JW. Function of a nontranscribed DNA strand site in transcription elongation. Cell. 1994;78:317–324. doi: 10.1016/0092-8674(94)90300-x. [DOI] [PubMed] [Google Scholar]
- Ring BZ, Yarnell WS, Roberts JW. Function of E. coli RNA polymerase σ factor σ70 in promoter-proximal pausing. Cell. 1996;86:485–493. doi: 10.1016/s0092-8674(00)80121-x. [DOI] [PubMed] [Google Scholar]
- Roberts CW, Roberts JW. Base-specific recognition of the nontemplate strand of promoter DNA by E. coli RNA polymerase. Cell. 1996;86:495–501. doi: 10.1016/s0092-8674(00)80122-1. [DOI] [PubMed] [Google Scholar]
- Severinov K, Soushko M, Goldfarb A, Nikiforov N. Rifampicin region revisited. New rifampicin-resistant and streptolydigin-resistant mutants in the beta subunit of Escherichia coli RNA polymerase. J Biol Chem. 1993;268:14820–14825. [PubMed] [Google Scholar]
- Severinov K, Fenyo D, Severinova E, Mustaev A, Chait BT, Goldfarb A, Darst SA. The σ subunit conserved region 3 is part of “5′-face” of active center of Escherichia coli RNA polymerase. J Biol Chem. 1994;269:20826–20828. [PubMed] [Google Scholar]
- Shuler MF, Tatti KM, Wade KH, Moran CP., Jr A single amino acid substitution in σE affects its ability to bind core RNA polymerase. J Bacteriol. 1995;177:3687–3694. doi: 10.1128/jb.177.13.3687-3694.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegele D, Hu J, Walter W, Gross C. Altered promoter recognition by mutant forms of the σ70 subunit of Escherichia coli RNA polymerase. J Mol Biol. 1989;206:591–603. doi: 10.1016/0022-2836(89)90568-8. [DOI] [PubMed] [Google Scholar]
- Tang H, Severinov K, Goldfarb A, Ebright R. Rapid RNA polymerase genetics: One-day, no-column preparation of reconstituted recombinant Escherichia coli RNA polymerase. Proc Natl Acad Sci. 1995;92:4902–4906. doi: 10.1073/pnas.92.11.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintut Y, Gralla JD. PCR mutagenesis identifies a polymerase-binding sequence of sigma 54 that includes a sigma 70 homology region. J Bacteriol. 1995;177:5818–5825. doi: 10.1128/jb.177.20.5818-5825.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, Messing J. New pUC-derived cloning vectors with different selectable markers and DNA replication origins. Gene. 1991;100:189–194. doi: 10.1016/0378-1119(91)90365-i. [DOI] [PubMed] [Google Scholar]
- Wilson C, Dombroski AJ. Region 1 of σ70 is required for efficient isomerization and initiation of transcription by Escherichia coli RNA polymerase. J Mol Biol. 1997;267:60–74. doi: 10.1006/jmbi.1997.0875. [DOI] [PubMed] [Google Scholar]
- Wosten MMSM. Eubacterial σ-factors. FEMS Microbiol Rev. 1998;22:127–150. doi: 10.1111/j.1574-6976.1998.tb00364.x. [DOI] [PubMed] [Google Scholar]
- Zalenskaya K, Lee J, Gujuluva C, Shin Y, Slutsky M, Goldfarb A. Recombinant RNA polymerase: inducible overexpression, purification, and assembly of Escherichia coli rpo gene products. Gene. 1990;89:7–12. doi: 10.1016/0378-1119(90)90199-2. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gross C. How a mutation in the gene encoding σ70 suppressed the defective heat shock response caused by a mutation in the gene encoding σ32. J Bacteriol. 1992;174:7128–7137. doi: 10.1128/jb.174.22.7128-7137.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]