

Abstract
As part of our continuing search for new amino acid inhibitors of metalloenzymes, we now report the synthesis and biological evaluation of the trifluoromethylketone analogue of L-arginine, (S)-2-amino-8,8,8-trifluoro-7-oxo-octanoic acid (10). While this novel amino acid was initially designed as a potential inhibitor of human arginase I, it exhibits no measurable inhibitory activity against this enzyme. Surprisingly, however, 10 is a potent inhibitor of human histone deacetylase 8, with IC50 = 1.5 ± 0.2 μM. Additionally, 10 weakly inhibits the related bacterial enzyme, acetylpolyamine amidohydrolase, with IC50 = 110 ± 30 μM. The lack of inhibitory activity against human arginase I may result from unfavorable interactions of the bulky trifluoromethyl group of 10 in the constricted active site. Since the active site of histone deacetylase 8 is less constricted, we hypothesize that it accommodates 10 as the gem-diol, which mimics the tetrahedral intermediate and its flanking transition states in catalysis. Therefore, we suggest that 10 represents a new lead in the design of an amino acid or peptide-based inhibitor of histone deacetylases with simpler structure than previously studied trifluoromethylketones.
Arginase is a binuclear manganese metalloenzyme that catalyzes the hydrolysis of L-arginine to form L-ornithine and urea through a metal-activated hydroxide mechanism.1–3 Two isozymes are present in humans;4, 5 arginase I is predominantly found in the liver, where it catalyzes the final cytosolic step of the urea cycle,6, 7 but it is also found in extrahepatic tissues where it functions to regulate cellular concentrations of L-arginine and L-ornithine for subsequent biosynthetic transformations, e.g., nitric oxide biosynthesis or polyamine biosynthesis.8–10 The highest concentrations of arginase II are found in the kidney, but this isozyme, too, functions in L-arginine homeostasis in various tissues and cell types.11, 12 Both arginases are increasingly prominent as drug targets due to the fact that arginase inhibitors enhance L-arginine bioavailability to nitric oxide (NO) synthase, and thereby amplify endogenous NO levels and NO-dependent processes such as vasodilation in penile erection13–17 or bronchodilation in the asthmatic airway.18–20 Thus, the exploration of new L-arginine analogues for evaluation as potentially therapeutically useful arginase inhibitors continues to challenge the field of medicinal chemistry.
Interestingly, the arginases and the metal-dependent histone deacetylases (HDACs) share a common α/β protein fold despite exhibiting insignificant amino acid sequence identity.21–23 The conservation of a metal binding site between these two enzyme families despite substantial evolutionary drift further indicates that they divergently evolved from a common metalloprotein ancestor.3 HDACs catalyze the hydrolysis of acetyl-L-lysine residues to yield acetate and L-lysine in histone and non-histone protein substrates. HDAC8 is the most studied isozyme in terms of structure-function relationships,24–28 although numerous structures of other isozymes29–31 and HDAC-related deacetylases22, 32–34 such as acetylpolyamine amidohydrolase (APAH)33 are available to guide inhibitor design efforts. Given that HDAC activity facilitates cancer cell proliferation, HDAC inhibitors are rapidly emerging as chemopreventive and chemotherapeutic drugs for cancer treatment. To date, two HDAC inhibitors are approved for cancer chemotherapy and several more are in clinical trials, so there is a great deal of current effort focused on the discovery of new inhibitor designs.35–37
In order to assess structure-function relationships between the arginases and the HDACs, we now report the synthesis of a new trifluoromethylketone analogue of L-arginine, (S)-2-amino-8,8,8-trifluoro-7-oxo-octanoic acid (10) (Scheme 1). As initially outlined by Gelb and colleagues,38 the introduction of fluorine atoms adjacent to a ketone carbonyl group of an enzyme inhibitor enhances the tendency of the ketone to add a nucleophile such as a water molecule, which explains why difluoromethyl and trifluoromethyl ketones exist in aqueous solution predominantly as the gem-diol hydrates. As such, appropriately designed trifluoromethyl ketones make effective inhibitors of metallohydrolases because the gem-diol hydrate form mimics the tetrahedral intermediate and its flanking transition states in a hydrolytic reaction involving direct attack of a solvent nucleophile at the scissile amide linkage of the substrate. This was first demonstrated in the binding of a trifluoromethyl ketone inhibitor to the prototypical zinc enzyme carboxypeptidase A,39 leading to the design of trifluoromethylketone inhibitors of other metal-dependent hydrolases such as the HDACs.29, 40, 41 However, to date no trifluoromethylketone analogue of L-arginine has been prepared and evaluated against arginase and arginase-related deacetylases, thereby motivating the current study.
Scheme 1.

Synthesis of (S)-2-amino-8,8,8-trifluoro-7-oxo-octanoic acid (10).
The synthesis of 10 is summarized in Scheme 1. The commercially available L-glutamic acid derivative, (S)-2-tert-butoxycarbonylamino-pentandioic acid 1-tert-butyl ester (1) was converted into the corresponding methyl ester 2 using methyl chloroformate in methylene chloride. The relatively acidic carbamate proton was replaced by a second tert-butoxycarbonyl (Boc) group to allow for the selective reduction of the less sterically hindered methyl ester (intermediate 3) with diisobutylaluminum hydride (DIBAH), thus yielding the corresponding aldehyde 4.20, 42 Wittig reaction of 4 with methyl(triphenylphosphoranylidene)acetate, followed by Pd/C-catalyzed hydrogenation of the double bond in intermediate 5 generated ester 6, which was selectively reduced with DIBAH to form aldehyde 7.43, 44 Nucleophilic trifluoromethylation of 7 with trifluoromethyltrimethylsilane (TMSCF3) and tetra-n-butylammonium fluoride (TBAF) as initiator, using a combination of modified classic procedures,45–47 afforded (S)-tert-butyl-2-bis(tert-butoxycarbonyl)amino-8,8,8-trifluoro-7-hydroxyoctanoate (8) in 76% yield.48 Dess-Martin periodinane (DMP) alcohol oxidation49, 50 yielded the corresponding ketone derivative 9 as a mixture of the ketone and hydrate forms in a 2:1 molar ratio as determined by 19F-NMR,51 as similarly observed for other trifluoromethyl ketones solubilized in polar solvents.52 Deprotection with trifluoroacetic acid (TFA) in methylene chloride generated 10 in 93% yield (ketone:hydrate 15:1 molar ratio by 19F-NMR).53
The in vitro inhibitory potency of 10 against recombinant human arginase I was assessed using the fixed point assay with [14C-guanidino]-L-arginine.54 Briefly, recombinant human arginase I was prepared as previously described.55 The final working concentrations in the reaction tubes were 1 mM for unlabeled L-arginine and 0.07 μg/μL for human arginase I; the concentration of trifluoromethylketone 10 ranged 0 – 800 mM. Data analysis was based on the kinetic replot of v0/vi as a function of inhibitor concentration, where v0 and vi were the observed velocities in the absence and presence of inhibitor, respectively. Since v0/vi ~ 1 at all inhibitor concentrations tested, no inhibition of human arginase I by 10 was detected.
The inhibitory potency of 10 against HDAC8 and the HDAC-related deacetylase APAH was measured using the commercially available Fluor-de-Lys deacetylase substrates N-acetyl-L-Arg-L-His-L-Lys(ε-acetyl)-L-Lys(ε-acetyl)-coumarin or L-Lys(ε-acetyl)-coumarin, respectively (Enzo Life Sciences).56 The deacetylation of this assay substrate allows for proteolytic cleavage of the C-terminal fluorophore to result in a fluorescence shift. For inhibition of either HDAC8 or APAH, assays were run at 25 °C and each 50 μL reaction sample contained 0.5 – 1 μM enzyme, 50 – 150 μM substrate, 25 mM Tris (pH 8.2), 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2. For HDAC8 assays, concentrations of trifluoromethylketone inhibitor 10 were 0.1 μM, 0.5 μM, 1.0 μM, 8.0 μM, 10.0 μM, 30.0 μM, 50.0 μM, 75.0 μM, and 500 μM; for APAH assays, inhibitor concentrations were 1 μM, 10 μM, 100 μM, 1 mM and 10 mM. The inhibitor was incubated with the enzyme for 15 minutes prior to starting the reaction by the addition of substrate. After 20 min (HDAC8) or 30 min (APAH), reactions were stopped by the addition of 100 μM M344 (Sigma) and the appropriate Fluor-de-Lys developer. Fluorescence was measured using a Fluoroskan II plate reader (ex = 355 nm, em = 460 nm). The IC50 value was calculated using Prism 5 and plots of inhibitory activity are shown in Figure 1 (GraphPad Software). All measurements were made in triplicate. For HDAC8, these experiments yielded IC50 = 1.5 ± 0.2 μM; for APAH, IC50 = 110 ± 30 μM.
Figure 1. Inhibition of HDAC8 and APAH by 10.

Inhibition of HDAC8 (A) and APAH (B) deacetylase activity by the trifluoromethyketone inhibitor 10 was measured using the fluorescence-based Fluor-de-Lys assay56 as described in the text. For HDAC8 assays, a deacetylated standard was used to convert fluorescence readings into product concentrations; for APAH, only raw fluorescence readings were recorded. All measurements were made in triplicate and IC50 values were calculated using Prism 5 (GraphPad).
It is surprising that 10 does not inhibit human arginase I, especially since the corresponding aldehyde, (S)-2-amino-7-oxoheptanoic acid, binds as the gem-diol hydrate with Kd = 60 ± 8 μM.44 Possibly, the α-trifluoro moiety of 10 is too bulky to fit in the constricted active site; additionally, the partial negative charge on the fluorine atoms may make an unfavorable electrostatic interaction with the negatively charged side chain of E277 at the base of the active site. This residue accepts a hydrogen bond from the η1-NH2 group of the substrate L-arginine, which is isosteric with the η-CF3 group of 10. We were unsuccessful in cocrystallizing the human arginase I-10 complex for structural analysis, presumably due to the relatively weak affinity of inhibitor binding.
It is further surprising that while 10 was designed, but ultimately failed, as an inhibitor of arginase, it exhibits unusually potent, low micromolar activity against HDAC8. It is 73-fold less potent against the related bacterial deacetylase APAH, possibly due to unfavorable interactions in the more sterically constrained active site cleft of APAH.33 In comparison with other trifluoromethylketone inhibitors of HDACs, we believe that 10 is the smallest inhibitor known that exhibits a low micromolar IC50 value (Table 1).29, 40, 41 We hypothesize that the relatively high affinity of 10 arises from its propensity to form the gem-diol hydrate in aqueous solution. The gem-diol form of a trifluoromethylketone mimics the tetrahedral intermediate and flanking transition states for an enzyme-catalyzed hydrolytic reaction, as first demonstrated in the binding of a trifluoromethylketone gem-diol to the zinc metalloenzyme carboxypeptidase A.57 More recently, a trifluoromethylketone inhibitor has been shown to bind as a gem-diol in the active site of HDAC4 with IC50 = 0.367 μM,29 so it is reasonable to expect a comparable binding mode for 10 in the active site of HDAC8. Interestingly, that 10 is an amino acid avoids the poor solubility issues encountered with more lipophilic trifluoromethylketones, such as those illustrated in Table 1.40 Moreover, a single amino acid comprises a more efficient inhibitor scaffold in comparison with bigger and bulkier cyclic tetrapeptide trifluoromethylketones (Table 1); “downsizing” the peptide inhibitor incurs only a 4-fold loss of affinity against HDAC8.41 Accordingly, we suggest that 10 represents a simplified new lead for the design of trifluoromethylketone inhibitors of HDAC8 and perhaps other HDAC enzymes.
Table 1.
Trifluoromethylketone HDAC inhibitors
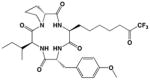
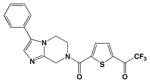
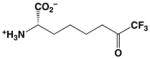
In order to visualize possible binding interactions in the enzyme-inhibitor complex, we constructed a model of the gem-diol form of 10 and manually docked it in the active site of HDAC8 using the structure of an HDAC4-trifluoromethylketone complex as a template (Figure 2A). The molecular structures of HDAC8 and 10 were superimposed on the structures of HDAC4 and its cognate trifluoromethylketone inhibitor, respectively. The conformation of 10 was subsequently adjusted to more closely resemble the conformation of acetyl-L-lysine observed in the Y306F HDAC8-tetrapeptide complex.26 Based on this model, we hypothesize that the gem-diol moiety of 10 is stabilized by Zn2+ coordination and hydrogen bond interactions with H142, H143, and Y306 (Figures 2A, 2B). As such, the unusually high affinity of 10 likely results from the resemblance of the gem-diol form (Figure 2B) to the tetrahedral intermediate (and its flanking transition states) in catalysis (Figure 2C).
Figure 2. Trifluoromethylketone gem-diol binding to HDAC8 mimics the tetrahedral intermediate in catalysis.

(A) Stereoview of the gem-diol form of 10 modeled into the HDAC8 active site shows possible metal coordination and hydrogen bond interactions (black and orange dashed lines, respectively). Atoms are color-coded as follows: C = green (inhibitor) or gray (protein), N = blue, O = red, F = cyan; the catalytic Zn2+ ion is shown as a lavender sphere. (B) The Zn2+ coordination geometry observed in the HDAC4-trifluoromethylketone gem-diol complex29 and potentially formed by the HDAC8-10 complex mimics that expected for the tetrahedral intermediate in catalysis by HDAC8 (C).
In summary, while the new trifluoromethylketone amino acid isostere of L-arginine 10 is a poor inhibitor of arginase, it is a surprisingly good inhibitor of HDAC8. Importantly, HDACs are validated targets for cancer chemotherapy, so structure-activity relationships now established for 10 can serve as a foundation for the design of a new class of amino acid inhibitors of the HDACs. Such inhibitors might prove to be pharmaceutically useful, especially if their relatively compact size does not enable interactions with other biological targets. Future work in this area will be reported in due course.
Acknowledgments
We thank the NIH for grant GM49758 in support of this research and Dr. Rakesh Kohli for recording high resolution mass spectra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Ash DE, Cox JD, Christianson DW. Met Ions Biol Syst. 2000;37:407. [PubMed] [Google Scholar]
- 2.Ash DE. J Nutr. 2004;134:2760S. doi: 10.1093/jn/134.10.2760S. [DOI] [PubMed] [Google Scholar]
- 3.Christianson DW. Acc Chem Res. 2005;101:191. doi: 10.1021/ar040183k. [DOI] [PubMed] [Google Scholar]
- 4.Morris SM., Jr Annu Rev Nutr. 2002;22:87. doi: 10.1146/annurev.nutr.22.110801.140547. [DOI] [PubMed] [Google Scholar]
- 5.Morris SM., Jr Br J Pharmacol. 2009;157:922. doi: 10.1111/j.1476-5381.2009.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krebs HA, Henseleit K. Z Physiol Chem. 1932;210:33. [Google Scholar]
- 7.Herzfeld A, Raper SM. Biochem J. 1976;153:469. doi: 10.1042/bj1530469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris SM., Jr . In: Nitric Oxide Biology and Pathobiology. Ignarro LJ, editor. San Diego: Academic Press; 2000. pp. 187–197. [Google Scholar]
- 9.Mori M, Gotoh T. In: Nitric Oxide Biology and Pathobiology. Ignarro LJ, editor. San Diego: Academic Press; 2000. pp. 199–208. [Google Scholar]
- 10.Tabor CW, Tabor H. Annu Rev Biochem. 1984;53:749. doi: 10.1146/annurev.bi.53.070184.003533. [DOI] [PubMed] [Google Scholar]
- 11.Morris SM, Jr, Bhamidipati D, Kepka-Lenhart D. Gene. 1997;193:157. doi: 10.1016/s0378-1119(97)00099-1. [DOI] [PubMed] [Google Scholar]
- 12.Gotoh T, Araki M, Mori M. Biochem Biophys Res Commun. 1997;233:487. doi: 10.1006/bbrc.1997.6473. [DOI] [PubMed] [Google Scholar]
- 13.Cox JD, Kim NN, Traish AM, Christianson DW. Nat Struct Biol. 1999;6:1043. doi: 10.1038/14929. [DOI] [PubMed] [Google Scholar]
- 14.Kim NN, Cox JD, Baggio RF, Emig FA, Mistry SK, Harper SL, Speicher DW, Morris SM, Jr, Ash DE, Traish A, Christianson DW. Biochemistry. 2001;40:2678. doi: 10.1021/bi002317h. [DOI] [PubMed] [Google Scholar]
- 15.Bivalacqua TJ, Hellstrom WJG, Kadowitz PJ, Champion HC. Biochem Biophys Res Commun. 2001;283:923. doi: 10.1006/bbrc.2001.4874. [DOI] [PubMed] [Google Scholar]
- 16.Cama E, Colleluori DM, Emig FA, Shin H, Kim SW, Kim NN, Traish A, Ash DE, Christianson DW. Biochemistry. 2003;42:8445. doi: 10.1021/bi034340j. [DOI] [PubMed] [Google Scholar]
- 17.Bivalacqua TJ, Burnett AL, Hellstrom WJG, Champion HC. Am J Physiol: Heart Circ Physiol. 2007;292:H1340. doi: 10.1152/ajpheart.00121.2005. [DOI] [PubMed] [Google Scholar]
- 18.Maarsingh H, Zuidhof AB, Bos IST, van Duin M, Boucher JL, Zaagsma J, Meurs H. Am J Respir Crit Care Med. 2008;178:565. doi: 10.1164/rccm.200710-1588OC. [DOI] [PubMed] [Google Scholar]
- 19.Munder M. Br J Pharmacol. 2009;158:638. doi: 10.1111/j.1476-5381.2009.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilies M, Di Costanzo L, North ML, Scott JA, Christianson DW. J Med Chem. 2010;53:4266. doi: 10.1021/jm100306a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Nature. 1996;383:554. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- 22.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Nature. 1999;401:188. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 23.Dowling DP, Di Costanzo L, Gennadios HA, Christianson DW. Cell Mol Life Sci. 2008;65:2039. doi: 10.1007/s00018-008-7554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, Di Marco S. Proc Natl Acad Sci USA. 2004;101:15064. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structure. 2004;12:1325. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 26.Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A, De Francesco R, Steinkühler C, Di Marco S. EMBO Rep. 2007;8:879. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Biochemistry. 2008;47:13554. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dowling DP, Gattis SG, Fierke CA, Christianson DW. Biochemistry. 2010;49:5048. doi: 10.1021/bi1005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Neddermann P, De Francesco R, Steinkühler C, Gallinari P, Carfi A. J Biol Chem. 2008;283:26694. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuetz A, Min J, Allali-Hassani A, Schapira M, Shuen M, Loppnau P, Mazitschek R, Kwiatkowski NP, Lewis TA, Maglathin RL, McLean TH, Bochkarev A, Plotnikov AN, Vedadi M, Arrowsmith CH. J Biol Chem. 2008;283:11355. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, De Jong R, O’Connell S, Grimshaw CE, Navre M, Gangloff AR. Bioorg Med Chem Lett. 2010;20:3142. doi: 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen TK, Hildmann C, Dickmanns A, Schwienhorst A, Ficner R. J Mol Biol. 2005;354:107. doi: 10.1016/j.jmb.2005.09.065. [DOI] [PubMed] [Google Scholar]
- 33.Lombardi PM, Angell HD, Whittington DA, Flynn EF, Rajashankar KR, Christianson DW. Biochemistry. 2011;50:1808. doi: 10.1021/bi101859k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abendroth J, Gardberg AS, Robinson JI, Christensen JS, Staker BL, Myler PJ, Stewart LJ, Edwards TE. J Struct Funct Genomics. 2011 doi: 10.1007/s10969-011-9101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolden JE, Peart MJ, Johnstone RW. Nature Rev Drug Discov. 2006;5:769. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 36.Mwakwari SC, Patil V, Guerrant W, Oyelere AK. Curr Topics Med Chem. 2010;10:1423. doi: 10.2174/156802610792232079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guan P, Fang H. Drug Discov Ther. 2010;4:388. [PubMed] [Google Scholar]
- 38.Gelb MH, Svaren JP, Abeles RH. Biochemistry. 1985;24:1813. doi: 10.1021/bi00329a001. [DOI] [PubMed] [Google Scholar]
- 39.Christianson DW, Lipscomb WN. J Am Chem Soc. 1986;108:4998. doi: 10.1021/ja00263a052. [DOI] [PubMed] [Google Scholar]
- 40.Frey RR, Wada CK, Garland RB, Curtin ML, Michaelides MR, Li J, Pease LJ, Glaser KB, Marcotte PA, Bouska JJ, Murphy SS, Davidsen SK. Bioorg Med Chem Lett. 2002;12:3443. doi: 10.1016/s0960-894x(02)00754-0. [DOI] [PubMed] [Google Scholar]
- 41.Jose B, Oniki Y, Kato T, Nishino N, Sumida Y, Yoshida M. Bioorg Med Chem Lett. 2004;14:5343. doi: 10.1016/j.bmcl.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 42.Adamczyk M, Johnson DD, Reddy RE. Tetrahedron: Asymmetry. 1999;10:775. [Google Scholar]
- 43.Padrón JM, Kokotos G, Martín T, Markidis T, Gibbons WA, Martín VS. Tetrahedron: Asymmetry. 1998;9:3381. [Google Scholar]
- 44.Shin H, Cama E, Christianson DW. J Am Chem Soc. 2004;126:10278. doi: 10.1021/ja047788w. [DOI] [PubMed] [Google Scholar]
- 45.Prakash SGK, Krishnamurti R, Olah GA. J Am Chem Soc. 1989;111:393. [Google Scholar]
- 46.Singh RP, Shreeve JM. Tetrahedron. 2000;56:7613. [Google Scholar]
- 47.Enders D, Herriger C. Eur J Org Chem. 2007;7:1085. [Google Scholar]
- 48.Synthesis of (S)-tert-butyl-2-bis(tert-butoxycarbonyl)amino-8,8,8-trifluoro-7-hydroxyoctanoate (8). Aldehyde 7 (0.5 g, 1.2 mmol) was reacted with neat TMSCF3 and TBAF (molar ratio 7:TMSCF3:TBAF 1:1.5:1) at 0 °C (45 min), and then stirred at room temperature until 1H NMR monitoring showed the disappearance of signals for the aldehyde group (3 hrs). The reaction mixture was quenched with sat. aq. NaHCO3 and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine (10 mL), dried (Na2SO4), and rotoevaporated to yield 8 as a light-yellow oil (0.44 g, 76%) that was further used without additional purification. 1H NMR (360 MHz, CDCl3): (ppm) 4.69 (dd, J = 5.1, 9.6 Hz, 1H, CHN), 3.97 (d, J = 9.4 Hz, 1H, CHCF3), 3.00 (dd, J = 5.5, 31.3 Hz, 1H, HA, CHAHBCHCF3), 2.13-1.95 (m, 2H, HB, CHAHBCHCF3 + HA, NCHC HAHB), 1.94-1.76 (m, 1H, HB, NCHCHAHB), 1.74-1.56 (m, 2H, HA, NCHCH2CHAHB + HA, CHAHBCH2CHCF3), 1.53-1.34 (m, 2H, HB, NCHCH2CHAHB + HB, CHAHBCH2CHCF3), 1.49 (s, 18H, 2Boc), 1.43 (s, 9H, NCHCOOC(CH3)3). F NMR (282.4 MHz, CDCl3):19 (ppm) −77.3 (s). HRMS m/z 508.2480 (calcd for M+Na, 508.2497).
- 49.Dess DB, Martin JC. J Org Chem. 1983;48:4156. [Google Scholar]
- 50.Linderman RJ, Graves DM. J Org Chem. 1989;54:661. [Google Scholar]
- 51.Synthesis of (S)-tert-butyl-2-bis(tert-butoxycarbonyl)amino-8,8,8-trifluoro-7-oxooctanoate (9). A 10% CH2Cl2 solution of 8 (0.4 g, 0.82 mmol) was added to a 15% CH2Cl2 solution of DMP (molar ratio 8:DMP 1:4) at room temperature, with stirring under N2. The reaction mixture was stirred at room temperature for 4 hrs (TLC monitoring, hexane/ethyl acetate 3/1), then diluted with ether (50 mL) and poured into a 0.26 M solution of sodium thiosulfate in sat. aq. NaHCO3 (molar ratio DM:Na2S2O3 1:7). The resulting immiscible layers were separated and the aqueous layer was extracted with Et2O (2 × 15 mL). The combined organic extracts were washed with sat. aq. NaHCO3 (15 mL) and brine (10 mL), dried (Na2SO4), and rotoevaporated. Purification by flash column chromatography (hexane/methyl-tert-butyl ether gradients) afforded 9 as a light-yellow oil (0.35 g, 88%). 1H NMR (360 MHz, CDCl3): (ppm) (ketone:hydrate 2:1) 4.72-4.63 (m, 1H, CHN), 3.73 (bs, OH from hydrate), 2.71 (t, J = 7.2 Hz, 2H, CH2COCF3), 2.13-1.96 (m, 1H, HA, NCHCHAHB), 1.94-1.60 (m, 3H, HB, NCHCHAHB + CH2CH2COCF3), 1.54-1.30 (m, 2H, NCHCH2CH2), 1.49 (s, 18H, 2Boc), 1.43 (s, 9H, NCHCOOC(CH3)3). 13C NMR (90.6 MHz, CDCl3): (ppm) 191.6 (q, J = 35 Hz, COCF3), 170.0 (NCHCOOtBu), 152.9 (2C, 2NCOOtBu), 115.6 (q, JCF = 292 Hz, CF3), 83.2 (2C, 2NCOOCMe3), 81.6 (NCHCOOCMe3), 58.9 (CHN), 36.5 (CH2COCF3), 29.2 (CH2CHN), 28.3 (9C, 3COOC(CH3)3), 25.9 (CH2CH2COCF3), 22.4 (CH2CH2CH2COCF3). 19F NMR (282.4 MHz, CDCl3): (ppm) −79.5 (s, COCF3), −86.1 (s, C(OH)2CF3). HRMS m/z 484.2530 (calcd for M+H, 484.2521).
- 52.Walter MW, Adlington RM, Baldwin JE, Schofield CJ. J Org Chem. 1998;63:5179. [Google Scholar]
- 53.Synthesis of (S)-2-amino-8,8,8-trifluoro-7-oxo-octanoic acid (10). The Boc-protected derivative 9 (0.35 g, 0.72 mmol) was dissolved in CH2Cl2 (4 mL), cooled at 0 °C, and treated dropwise with TFA (4 mL) with constant stirring. After reaching room temperature, the reaction mixture was stirred until TLC monitoring (hexane/ethyl acetate 3/1) revealed complete deprotection (3 hrs). Rotovaporation, washing with CH2Cl2, and purification by flash column chromatography (CHCl3/MeOH/NH3 gradients) afforded 10 as a light-yellow oil (0.15 g, 93%).1H NMR (360 MHz, D2O): (ppm) (ketone:hydrate 15:1) 3.76 (t, J = 5.8 Hz, 1H, CHN), 2.12-1.80 (m, 4H, NCHCH2 + CH2COCF3), 1.65-1.33 (m, 4H, NCHCH2CH2 + CH2CH2COCF3). 13 C NMR (90.6 MHz, D2O): (ppm) 177.9 (CO, COOH), 162.9 (q, JCF = 35.3 Hz, CO, COCF3), 116.3 (q, JCF = 291.8 Hz, CF3), 55.1 (CHN), 31.7, 29.2, 24.6, 20.7. 19F NMR (282.4 MHz, D2O): (ppm) −75.6 (s, COCF3), −85.3 (s, C(OH)2CF3). HRMS m/z 228.0851 (calcd for M+H, 228.0847).
- 54.Rüegg UT, Russell AS. Anal Biochem. 1980;102:206. doi: 10.1016/0003-2697(80)90340-1. [DOI] [PubMed] [Google Scholar]
- 55.Di Costanzo L, Sabio G, Mora A, Rodriguez PC, Ochoa AC, Centeno F, Christianson DW. Proc Natl Acad Sci USA. 2005;102:13058. doi: 10.1073/pnas.0504027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoffmann K, Brosch G, Loidl P, Jung M. Nucl Acids Res. 1999;27:2057. doi: 10.1093/nar/27.9.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Christianson DW, Lipscomb WN. J Am Chem Soc. 1986;108:4998. doi: 10.1021/ja00263a052. [DOI] [PubMed] [Google Scholar]