

Abstract
FK506 and rapamycin are immunosuppressive drugs with a unique mode of action. Prior to binding to their protein targets, these drugs form a complex with an endogenous chaperone FK506-binding protein 12 (FKBP12). The resulting composite FK506-FKBP and rapamycin-FKBP binding surfaces recognize the relatively flat target surfaces of calcineurin and mTOR, respectively, with high affinity and specificity. To test whether this mode of action may be generalized to inhibit other protein targets, especially those that are challenging to inhibit by conventional small molecules, we have developed a parallel synthesis method to generate a 200-member library of bifunctional cyclic peptides as FK506 and rapamycin analogues, which were referred to as “rapalogs”. Each rapalog consists of a common FKBP-binding moiety and a variable effector domain. The rapalogs were tested for binding to FKBP12 by a fluorescence polarization competition assay. Our results show that FKBP12 binds to most of the rapalogs with high affinity (KI values in the nanomolar to low micromolar range), creating a large repertoire of composite surfaces for potential recognition of macromolecular targets such as proteins.
Keywords: Cyclic peptides, FKBP, FK506, rapamycin, structure-activity relationship
INTRODUCTION
FK506 and rapamycin are natural products that are produced by soil microorganisms. Both molecules are effective immunosuppressive drugs that are used for organ transplantation1,2. In addition, rapamycin has shown activity as an anti-cancer agent3. The molecular target of FK506 is the protein phosphatase calcineurin whereas that of rapamycin is a protein kinase, mammalian target of rapamycin (mTOR). Unlike most small-molecules drugs, FK506 and rapamycin by themselves show only very modest affinity for their respective targets. To be efficacious, FK506 and rapamycin must first form a binary complex with the endogenous FK506-binding protein 12 (FKBP12), a peptidyl-prolyl isomerase that is widely expressed in human cells. The resulting FK506-FKBP12 and rapamycin-FKBP12 complexes bind with high affinity and specificity and inhibit the enzymatic activities of calcineurin and mTOR, respectively4–7. Compared to the drugs alone, the drug-FKBP12 binary complexes show increased binding affinities because the FKBP12 protein surface also makes favorable molecular interactions with the target protein surface8. Thus, by binding to FKBP12, FK506 and rapamycin create composite binding surfaces that allow both drug-target and FKBP12-target interactions.
FK506 and rapamycin are bifunctional molecules (Figure 1). One part of the molecules binds to FKBP12, while the second part (the effector domain) interacts with the target protein and provides target specificity. The co-crystal structures of FK506-FKBP12-calcineurin and rapamycin-FKBP12-mTOR reveal that the composite FKBP12-drug binding surfaces are large and bind to relatively flat surfaces on their target proteins9–11. The two-dimensional binding character of the composite FKBP-drug binding surface is in stark contrast to that of most small-molecule drugs, which typically bind in deep binding pockets that provide three-dimensional binding spaces. A deep binding pocket in a target protein allows for the establishment of many molecular interactions between a small molecule and the protein so that high affinity binding can occur. Proteins with distinct binding pockets are usually enzymes, ion channels and receptors. These classes of proteins make up the majority of all drug targets that have already been exploited12. On the other hand, many potential drug targets exert their biological activities through protein-protein interactions. Protein-protein interactions often take place via large and flat surfaces that are considered “undruggable” because small molecules cannot establish sufficient molecular interactions with these flat target surfaces in order to bind with high affinity and selectivity.
Figure 1.

Structures of FK506, rapamycin, meridamycin, and rapalogs SLF, 1 and 2.
Through the formation of composite drug-FKBP binding surfaces, Nature has found a solution to successfully target flat protein surfaces with small molecules and has repeatedly exploited this approach. In addition to FK506 and rapamycin, meridamycin, a macrolide produced by the soil bacterium Streptomyces hygroscopicus, is also a high-affinity FKBP ligand that antagonizes the immunosuppressive activity of FK50613. The molecular target of the meridamycin-FKBP complex is currently unknown. Likewise, antascomicins A-E bind to FKBP12 with about the same nanomolar affinity as FK506 and rapamycin and the antascomycin-FKBP complexes inhibits the function of a yet unknown cellular target(s)14. Nature has also used other proteins to form composite binding surfaces. One such example is the peptidyl-prolyl isomerase cyclophilin A (CypA). The natural products that bind to cyclophilin and form drug-chaperon complexes include cyclosporin A (CsA) and sanglifehrin A. CsA is a cyclic undecapeptide and the CsA-CypA complex creates a composite surface that binds and inhibits calcineurin15. Both CsA and CypA make molecular contacts with calcineurin and contribute to the overall affinity and specificity16. Sanglifehrin A has shown exceptionally high affinity for CypA in a cell free assay (IC50 = ~7 nM)17. The Sanglifehrin-CypA complex has immunosuppressive activity, although the target of the complex remains to be identified.
Despite the different effector domains of FK506, rapamycin, meridamycin and antascomycins, which allow them to bind to different target proteins, the four compounds possess a common FKBP-binding moiety, which consists of a triketo pipecolyl core (Figure 1). We hypothesize that it may be possible to generalize the mechanism-of-action of FK506 and rapamycin to target otherwise intractable protein sites. To test this hypothesis, we have developed a synthetic route that allows the rapid synthesis of large libraries of cyclic bifunctional molecules. These synthetic molecules (or rapalogs) consist of a common FKBP12-binding moiety but different effector domains which replace the calcineurin/mTOR-binding motifs of FK506/rapamycin (compound 2, Figure 1). When bound to FKBP12, these rapalogs should create diverse composite binding surfaces on FKBP12, which may be screened for binding to otherwise intractable protein targets.
RESULTS AND DISCUSSION
Our objective is to synthesize a large library of rapalogs that retain the ability to bind to FKBP12 but each have a different effector domain in order to create a diverse array of composite surfaces for screening against a target protein. To achieve this goal, we must first devise a minimal structural unit that is capable of binding to FKBP12 with high affinity and specificity. Next, we need to replace the effector domain of rapamycin with structurally diverse moieties. In the present work, we chose peptides as the effector domains, because peptides of diverse structures can be generated from a small set of amino-acid building blocks and are synthetically accessible, although other types of structures may also be used.
Identification of a Minimal FKBP12-Binding Motif
Many FK506 and rapamycin analogues have previously been synthesized and tested, resulting in ample SAR data with respect to FKBP12 binding. For example, the work of Holt and coworkers18 established that a macrocycle containing a 3,3-dimethyl-2-ketobutyryl-L-pipecolinate (Dkb-Pip) core (compound 1a, Figure 1) retains much of the FKBP-binding activity of rapamycin (apparent Ki of 30 nM). Addition of an (R)-1-(phenylethyl)benzyl group (1b) to the core further increases the binding affinity by ~30-fold. To facilitate later library synthesis, we elected to replace the (R)-1-(phenylethyl)benzyl moiety with a simpler, more readily available building block such as an amino acid (R1 in 2, Figure 1). In addition, we felt that the residue immediately N-terminal to the Dkb-Pip core (R2 in 2, Figure 1) might also affect FKBP12 binding. To identify a competent, minimal FKBP12-binding motif, we designed 15 cyclic peptides that featured 10 different R1 residues including L-alanine, D-alanine, (S)-2-aminobutyric acid (D-Abu), (R)-3-aminoadipic acid (D-β-Glu), (R)-3-amino-5-phenylpentanoic acid (D-β-homoPhe), (S)-3-amino-5-phenylpentanoic acid (L-β-homoPhe), D-homophenylalanine (D-homoPhe), L-β-isoleucine (L-β-Ile), L-isoleucine, and D-isoleucine and three R2 residues (D-Ala, L-Thr, and L-Phe) (Table 1, compounds 2a–n). To facilitate solid-phase synthesis, an invariant glutamine was added to the C-terminal side of the R1 residue, for the purpose of backbone peptide cyclization and providing an anchor for attachment to the solid support. For this set of compounds, the dipeptide Ala-Ala was used as the effector domain (Figure 1).
Table 1.
SAR of Rapalogs 2a-ya
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | Building Blocks
|
IC50 (μM) | |||||
| R1 | R2 | R3 | R4 | R5 | R6 | ||
| 2a | L-Ala | D-Ala | L-Ala | L-Ala | 240 ± 7 | ||
| 2b | D-Ala | D-Ala | L-Ala | L-Ala | 298 ± 80 | ||
| 2c | D-Abu | D-Ala | L-Ala | L-Ala | 303 ± 27 | ||
| 2d | D-β-Glu | D-Ala | L-Ala | L-Ala | 310 ± 2 | ||
| 2e | D-β-homoPhe | D-Ala | L-Ala | L-Ala | 13 ± 4 | ||
| 2f | D-homoPhe | D-Ala | L-Ala | L-Ala | 147 ± 23 | ||
| 2g | L-β-Ile | D-Ala | L-Ala | L-Ala | 165 ± 42 | ||
| 2h | L-Ile | D-Ala | L-Ala | L-Ala | 316 ± 46 | ||
| 2i | D-Ile | D-Ala | L-Ala | L-Ala | 266 ± 46 | ||
| 2j | L-β-homoPhe | L-Phe | L-Ala | L-Ala | 26 ± 3 | ||
| 2k | D-β-homoPhe | L-Phe | L-Ala | L-Ala | 18 ± 2 | ||
| 2l | L-β-Ile | L-Phe | L-Ala | L-Ala | 250 ± 57 | ||
| 2m | D-β-Glu | L-Phe | L-Ala | L-Ala | 386 ± 43 | ||
| 2n | D-β-homoPhe | L-Thr | L-Ala | L-Ala | 4 ± 1 | ||
| 2o | D-β-homoPhe | D-Ala | L-Ala | D-Ala | L-Ala | 20 ± 6 | |
| 2p | D-β-homoPhe | D-Ala | L-Ala | L-Ala | L-Ala | 6 ± 2 | |
| 2q | D-β-homoPhe | D-Ala | D-Ala | D-Ala | D-Ala | 13 ± 2 | |
| 2r | D-β-homoPhe | L-Thr | L-Ala | L-Ala | L-Ala | 9 ± 3 | |
| 2s | D-β-homoPhe | L-Phe | L-Ala | D-Ala | L-Ala | 31 ± 6 | |
| 2t | D-β-homoPhe | L-Phe | L-Ala | L-Ala | L-Ala | 10 ± 1 | |
| 2u | D-β-homoPhe | L-Thr | 37 ± 10 | ||||
| 2v | D-β-homoPhe | D-Ala | 18 ± 6 | ||||
| 2w | D-β-homoPhe | D-Ala | L-Ala | D-Ala | L-Ala | L-Ala | 13 ± 2 |
| 2x | D-β-homoPhe | D-Ala | L-Ala | L-Ala | L-Ala | L-Ala | 5 ± 1 |
| 2y | D-β-homoPhe | L-Phe | L-Ala | D-Ala | L-Ala | L-Ala | 8 ± 2 |
| FK506 | 0.22 ± 0.08 | ||||||
| Rapamycin | 0.057 ± 0.02 | ||||||
| SLF | 2.6 ± 1.5 | ||||||
The IC50 values reported were mean ± SD from multiple independent titration experiments (n) (rapalogs, n = 2–4; FK506, n = 5; rapamycin, n = 3; SLF, n = 18).
Synthesis of cyclic peptides 2a–n began with the preparation of a key building block 3 (Scheme 1). Starting from the commercially available N-Fmoc-L-pipecolinic acid, its carboxyl group was protected as an allyl ester by treatment with allyl bromide under basic conditions. Removal of the Fmoc group with piperidine gave amine 4, which was acylated with dihydro-4,4-dimethyl-2,3-furandione to give alcohol 518. Coupling of alcohol 5 with three different N-Fmoc amino acids followed by allyl removal with Pd(PPh3)4 afforded the building block 3a-c in good yields (~45% overall). Next, the 15 cyclic peptides were synthesized in parallel on Rink amide resin (Scheme 2). N-Fmoc-Glu-α-allyl ester was coupled to the amino group of Rink resin using O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) as the coupling agent. After removal of the Fmoc group, the N-terminal amine was acylated with the 10 different N-Fmoc amino acids (R1) described above. Subsequent addition of building blocks 3a-c and L-Ala-L-Ala were carried out using standard peptide chemistry. It should be noted that following the coupling of R1 and R3 residues, removal of the Fmoc groups with piperidine should be carried out quickly and the resulting amines should be immediately acylated with the next amino acid to avoid undesired cyclization at the ester moieties. Prior to peptide cyclization, the C-terminal allyl group was removed by treatment with a catalytic amount of Pd(PPh3)4 in the presence of N-methylaniline and the N-terminal Fmoc group was removed by piperidine. Peptide cyclization was achieved by using benzotriazole-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBop) as the coupling reagent. Finally, treatment with 50% trifluoroacetic acid (TFA) in dichloromethane released the peptides from the resin and deprotected the amino acid side chains. The resulting crude peptides were quickly passed through a silica gel column to remove the salts and used directly in activity assays.
Scheme 1.

The synthetic route for key building blocks of rapalogs library
Reagents and conditions: a) 5 equiv allyl bromide, 2 equiv Cs2CO3, acetone, 2 h; b) 20% piperidine in DCM, 20 min; c) 1.5 equiv dihydro-4,4-dimethyl-2,3-furandione, 10% DMAP, Ar gas, toluene, reflux, overnight; d) DIC, Fmoc- amino acids (R2), 5% DMAP,1 h; e) 5% Pd(Ph3P)4, 3 equiv N-methylaniline, THF.
Scheme 2.

Solid-Phase Synthesis of Rapalogs 2a-n.
Reagents and conditions: a) N-Fmoc-Glu-α-allyl ester, HOBT, HBTU, NMM, 2 h; b) 20% piperidine in DMF; c) N-Fmoc-amino acids (R1), HOBt, HBTU, NMM, 2 h; d) building block 3, HOBt, HBTU, NMM, 2 h; e) standard Fmoc/HBTU chemistry; f) 20% Pd(Ph3P)4, 9 eq N-methylaniline, THF; g) PyBop, HOBT, NMM, overnight; h) 50% TFA in DCM.
Peptides 2a-n were tested for binding to FKBP12 by a fluorescence polarization competition assay19,20. A previously reported synthetic ligand of FKBP1221 (SLF in Figure 1), was labeled with the fluorescent dye fluorescein20. Binding of the labeled ligand to FKBP12 increases its fluorescence anisotropy (FA) value. Addition of an unlabeled rapalog to the reaction inhibits the binding of SLF to FKBP12 and decreases the FA value (Figure 2). By using this competition assay in the presence of increasing concentrations of the rapalogs 2a-n, we determined the IC50 values (concentrations of rapalogs at which the FA value of SLF is reduced by 50%) for the peptides (Table 1). The results show that a D-β-homoPhe at the R1 position resulted in the best binding affinity for FKBP12 (IC50 = 4–18 μM) and L-β-homoPhe was slightly less effective (IC50 = 26 μM), whereas the other eight building blocks were much less effective (IC50 ≥ 147 μM) (Table 1). This is not surprising, since D-β-homoPhe is structurally similar to the 2-cyclohexylethyl-β-hydroxyketone moiety of rapamycin and the (R)-1-(phenylethyl)benzyl moiety in SLF and compound 1b (Figure 1). Among the three R2 residues examined, the smaller residues (L-Thr and D-Ala) were somewhat more effective than L-Phe (Table 1 compare compounds 2e, 2k, and 2n). Therefore, we chose the tetrapeptide L-Thr (or D-Ala)-Dkb-Pip-D-β-homoPhe as the minimal structural unit for binding to FKBP12.
Figure 2.

Representative titration curves showing the competition of FK506 or rapalogs 2o–2y (1–200 μM) with SLF-fluorescein (100 nM) for binding to FKBP12 (200 nM) as monitored by fluorescence anisotropy (FA). The percentage of FA was plotted against the rapalog concentration (in logarithmic scale).
Effect of Ring Size and Building Blocks in the Effector Domain
For our approach to be successful, FKBP12 must be able to tolerate structurally diverse effector domains of different ring sizes and building blocks. As an initial test of the feasibility, we synthesized compounds 2o-y, which all contain L-Thr (or D-Ala, L-Phe)-Dkb-Pip-D-β-homoPhe as the FKBP12-binding domain but have zero to four L- or D-Ala residues as the effector domain (Table 1). All of the compounds bound to FKBP12 with high affinity with IC50 values in the range of 5–37 μM. In comparison, rapamycin, FK506, and SLF had IC50 values of 0.057, 0.22, and 2.6 μM, respectively, under the same conditions. These results suggest that FKBP12 can tolerate a wide variety of effector domains including different ring sizes and both L- and D-amino acids as building blocks.
Synthesis and Evaluation of a Rapalog Library
To gain further insight into the SAR with respect to the influence of the effector domain structure on FKBP12-binding activity, we synthesized a 200-member rapalog library on Rink amide resin in parallel by following a procedure similar to that described in Scheme 2. All of the compounds contained the tetrapeptide D-Ala-Dkb-Pip-D-β-homoPhe as the FKBP12-binding domain. For half of the library members (compound 11), a dipeptide of random sequence (R3 and R4) was employed as the effector domain, while a tripeptide (R3-R4-L-Ala) was used as the effector domain for the other half (compound 12) (Table 2). At each random position (R3 and R4), 10 different amino acids (D-Thr, Gly, Ala, D-Val, Pro, Lys, Trp, Asp, D-Phe, and Nle) were used. After synthesis, side-chain deprotection, and cleavage from the resin, the rapalogs were analyzed by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) and each showed a single species with the expected m/z value (Figures S3 and S4 in Supporting Information).
Table 2.
Binding Affinities (IC50, μM) of Rapalog Library Members to FKBP12a
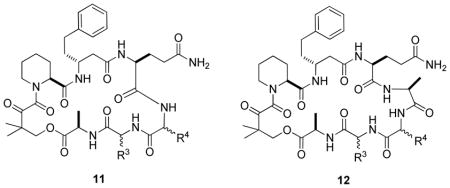 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R3 | R4 | D-Thr | Gly | Ala | D-Val | Pro | Lys | Trp | Asp | D-Phe | Nle |
| 11 | 17 ± 1 | 27 ± 2 | 15 ± 1 | 18 ± 5 | 19 ± 1 | > 100 | 37 ± 10 | 12 ± 1 | 65 ± 4 | >100 | |
| D-Thr | 12 | 5 ± 1 | 14 ± 4 | 4 ± 1 | ND | >100 | 80 ± 35 | 8 ± 3 | 25 ± 5 | 28 ± 2 | 57 ± 18 |
| 11 | 13 ± 5 | 25 ± 7 | ND | 25 ± 6 | 17 ± 1 | 30 ± 7 | 18 ± 4 | 24 ± 9 | 21 ± 6 | 32 ± 6 | |
| Gly | 12 | 16 ± 2 | 26 ± 7 | 5 ± 2 | 70 ± 16 | 49 ± 12 | 5 ± 2 | 9 ± 1 | 10 ± 1 | 95 ± 25 | 38 ± 11 |
| 11 | 23 ± 4 | 28 ± 13 | ND | 14 ± 4 | >100 | 63 ± 11 | 9 ± 3 | 17 ± 6 | ND | 28 ± 9 | |
| Ala | 12 | ND | 14 ± 5 | 6 ± 1 | 41 ± 4 | 57 ± 6 | >100 | ND | 9 ± 3 | 8 ± 4 | 71 ± 17 |
| 11 | 55 ± 5 | 5 ± 1 | 62 ± 2 | >100 | 12 ± 5 | 12 ± 4 | 9 ± 3 | 10 ± 1 | >100 | 48 ± 8 | |
| D-Val | 12 | 89 ± 16 | 2 ± 1 | 35 ± 6 | >100 | >100 | 52 ± 25 | 8 ± 2 | 13 ± 7 | >100 | 36 ± 17 |
| 11 | 41 ± 8 | ND | 33 ± 10 | 64 ± 10 | 27 ± 6 | 36 ± 8 | 21 ± 6 | 21 ± 2 | 46 ± 2 | 71 ± 15 | |
| Pro | 12 | 38 ± 11 | 6 ± 3 | 21 ± 3 | 43 ± 1 | ND | 29 ± 5 | ND | 3 ± 1 | 87 ± 5 | 26 ± 7 |
| 11 | 62 ± 8 | 93 ± 31 | >100 | 80 ± 28 | 58 ± 10 | >100 | 12 ± 1 | 22 ± 4 | 47 ± 10 | 67 ± 10 | |
| Lys | 12 | >100 | 22 ± 4 | >100 | ND | 15 ± 5 | 37 ± 6 | ND | 4 ± 1 | 45 ± 12 | >100 |
| 11 | 75 ± 16 | 12 ± 6 | 16 ± 2 | 34 ± 10 | 41 ± 9 | 53 ± 15 | 21 ± 4 | 8 ± 2 | ND | 13 ± 2 | |
| Trp | 12 | 89 ± 12 | 14 ± 1 | 6 ± 1 | 15 ± 3 | 66 ± 2 | ND | 42 ± 10 | 15 ± 4 | 22 ± 1 | 44 ± 6 |
| 11 | 13 ± 2 | 7 ± 2 | 13 ± 4 | 11 ± 2 | 13 ± 1 | 10 ± 3 | 4 ± 1 | 19 ± 4 | 15 ± 1 | 5 ± 1 | |
| Asp | 12 | 18 ± 5 | 12 ± 2 | 5 ± 2 | 50 ± 17 | 60 ± 4 | 7 ± 1 | 4 ± 2 | 2 ± 1 | 21 ± 6 | 10 ± 2 |
| 11 | 50 ± 16 | 40 ± 15 | 84 ± 24 | 38 ± 14 | 71 ± 23 | >100 | 69 ± 6 | 4 ± 1 | 34 ± 5 | ND | |
| D-Phe | 12 | 81 ± 16 | 38 ± 6 | ND | 50 ± 11 | >100 | >100 | 6 ± 3 | 4 ± 1 | 35 ± 1 | ND |
| 11 | 72 ± 19 | 24 ± 5 | 31 ± 9 | 68 ± 12 | 13 ± 1 | >100 | 14 ± 1 | ND | 33 ± 5 | 28 ± 6 | |
| Nle | 12 | >100 | 24 ± 6 | 35 ± 11 | 23 ± 13 | 79 ± 2 | 40 ± 11 | 11 ± 3 | 8 ± 1 | ND | 17 ± 2 |
The IC50 values reported were mean ± SD from 2–4 independent titration experiments.
The 200 compounds were individually assayed for their binding affinities to FKBP12 by the FA competition assay and their IC50 values are listed in Table 2. Evaluation of the 200 compounds revealed several important trends. First, out of the 182 compounds whose IC50 values were reliably determined, the great majority of them (163 compounds) bound to FKBP12 with excellent to respectable affinities (IC50 of 2–93 μM). Some of the compounds were more potent than SLF and only ~10-fold less potent than FK506 (IC50 = 0.22 μM). Second, although for a particular peptide sequence the two different ring sizes may result in as much as 10-fold difference in the IC50 values, in general, the compounds from the two sub-libraries of different ring sizes have similar potencies, consistent with our earlier observations. Third, at the R3 position, an Asp resulted in the most potent ligands against FKBP12 (IC50 = 2.0–25 μM), whereas Lys generally gave poorer activities. Finally, the R4 residue had minimal effect on the binding affinity and all 10 amino acids were well tolerated. The only exception was Asp, which gave high activities for most compounds, even when the R3 residue was not optimal (e.g. when R3 was Lys).
To confirm the library screening results, which were carried out with crude samples, we chose four of the rapalogs that had relatively high activities to FKBP12 and purified them to homogeneity by HPLC [compounds 11a, 12a (or 2p), 12b, and 12c in Table 3). The pure samples were assayed for binding to FKBP12 under the same conditions to give IC50 values of 6.3, 4.9, 7.0, and 6.3 μM, respectively (Table 3). These values were ≤2-fold different from those derived from the crude samples; these differences were well within the margin of error for the competition assay method.
Table 3.
Comparison of the Binding Affinities of Crude vs Purified Rapalogs to FKBP12.
| Rapalog (R3, R4) | Crude sample | Purified sample | ||
|---|---|---|---|---|
| IC50 (μM) | KI (μM) | IC50 (μM) | KI (μM) | |
| 11a (Asp, D-Phe) | 15 ± 1 | 0.48 ± 0.04 | 6.3 ± 1 | 0.20 ± 0.01 |
| 12a (2p) (Ala, Ala) | 6.0 ± 2.0 | 0.19 ± 0.03 | 4.9 ± 1.4 | 0.15 ± 0.04 |
| 12b (Asp, Ala) | 5.0 ± 2.0 | 0.16 ± 0.06 | 7.0 ± 2.1 | 0.22 ± 0.10 |
| 12c (Gly, Ala) | 4.7 ± 2.1 | 0.16 ± 0.06 | 6.3 ± 2.8 | 0.20 ± 0.09 |
The IC50 values reported were mean ± SD from 2–4 independent titration experiments.
Molecular Modeling of the FKBP-Rapalog Complexes
To gain some structural insight into the above observed SAR, molecular modeling was carried out for the binding of FKBP12 to rapalogs 11b (R3 = Ala, R4 = Asp) and 11c (R3 = Asp, R4 = Ala) using a two stage protocol as described under Experimental Section. In the model, the D-Ala-Dkb-Pip-D-β-homoPhe FKBP-binding motif is mostly buried in the active site, whereas the effector domain is largely exposed to the solvent (Figure 3a and b). A key FKBP-rapalog binding interaction occurs in the form of CH-π interactions between the side chain of Pip and the hydrophobic pocket formed by the side chains of Tyr-26, Phe-46, Trp-59, and Phe-99 of FKBP12 (Figure 3c). The carbonyl groups of Dkb and Pip also make two key hydrogen bonds with the side chain of Tyr-82 and the mainchain –NH of Ile-56, respectively. The pro-(S) methyl group of Dkb makes hydrophobic contact with Phe-36 side chain, while its pro-(R) methyl group interacts with the side chain of His-87. In addition, the phenyl ring of D-β-homoPhe contacts the side chains of Tyr-82 and His-87. The side chain of the D-Ala (R2) points away from the protein, consistent with the tolerance of different side chains at this position (e.g., D-Ala, Thr, and Phe). Some of the effector domain residues also contribute to the overall binding affinity. The side chain of Asp at position R3 is physically proximal to and engages in charge-charge interaction with the guanidinium group of FKBP Arg-42 (Figure 3c). A similar electrostatic interaction also occurs when an Asp or Glu is placed at the R4 position (Figure 3a and b). This is in excellent agreement with our experimental observations (Table 2). Finally, the side chain amide of the anchoring Gln is 3.82 Å from the side chain of Glu-54, suggesting that they may be involved in electrostatic interactions as well. Overall, the modeled binding mode explains why FKBP12 is capable of binding to the wide variety of rapalogs in this work.
Figure 3.

Top (a) and side view (b) of the overall FKBP12-rapalog 11b complex. FKBP12 is rendered as a surface with a probe radius of 1.4 Å and rapalog is in a licorice representation with hydrogen atoms omitted for clarity. C, O and N atoms are colored in cyan, red, and blue, respectively. (c) Stereo view of the complex between FKBP12 (shown in silver color except for the O and N atoms involved in binding) and rapalog 11c [color scheme: C (cyan), O (red), and N (blue)]. Dash lines indicate intermolecular hydrogen bonding, electrostatic, and CH-π interactions between the rapalog and the active-site residues of FKBP12.
Comparison with Previous Approaches
The unique mode-of-action of FK506, rapamycin, and other natural products has inspired other investigators as well as us to develop several approaches to modulate the biological activity of small molecules. For example, we demonstrated that the affinity of a linear peptide for the Fyn SH2 domain can be enhanced when the peptide is coupled to an FKBP ligand and bound to FKBP.22 This affinity enhancement is likely due to the establishment of favorable FKBP-SH2 interactions. In another application it was shown that conjugation of an FKBP ligand to Congo Red, a dye molecule that binds to beta-amyloid peptide, improved the ability of the dye molecule to disrupt amyloid plaque formation.23 Presumably, binding of FKBP to the molecule creates a steric block FKBP that hinders the beta-amyloid peptide aggregation. In a third experiment, it was shown that linking a HIV protease inhibitor to an FKBP ligand increased the half-life of the drug in mice.24 It was thought that the association of the bifunctional molecule with FKBP in mammalian cells may have slowed the molecule’s metabolism and excretion. These previous studies required a pre-existing ligand with high affinity and specificity to the target of interest; however, for many high-value targets such as the flat surfaces involved in protein-protein interactions, no such ligand is available (or possible). To recognize these flat surfaces, which are generally considered as “undruggable” by the conventional small-molecule approaches, we are exploring an alternative approach to recapitulate the mode of action of rapamycin and other natural products. Our strategy is to generate a large library of bifunctional cyclic peptides, which contain a common FKBP-binding motif on one side and diverse effector domains on the other side. These cyclic peptides should bind to FKBP12 via their shared FKBP-binding motif to form a library of FKBP-cyclic peptide complexes, each of which displays a unique and relatively flat surface formed by both the FKBP protein and the variable face of the cyclic peptide. Screening of the library of composite surfaces against a target surface may identify complexes that can inhibit protein-protein interactions. In the current study, we have shown that the tetrapeptide D-Ala-Dkb-Pip-D-β-homoPhe acts as an effective minimal structural motif that binds to FKBP12 with high affinity and specificity. When the motif was incorporated into ~200 cyclic peptides of different ring sizes and amino acid building blocks, the vast majority of them (177 out of the 196 peptides tested) were capable of binding to FKBP12 with IC50 values of 2–95 μM as determined by the FKBP competition assay, corresponding to KI values of 60–3000 nM (Table 1 and 2). By using the same assay, a KI value of 7.0 nM was obtained for FK506, in reasonable agreement with the KI value of 1–2 nM previously reported for inhibition of FKBP rotamase activity by FK506.18, 25 Thus, the binding affinities of our rapalogs for FKBP12 are generally one to three orders of magnitude lower than that of FK506. Most importantly, our results as well as the previously reported examples18, 26, 27 demonstrate that essentially any cyclic peptide or other types of macrocycles should be able to bind to FKBP12, as long as they contain a FKBP-binding motif such as the tetrapeptide identified in this work in a proper conformation. The solid-phase synthesis method we have developed may be easily adapted for the combinatorial synthesis of one-bead-one-compound (OBOC) libraries. In fact, the combinatorial synthesis and screening of large OBOC libraries of rapalogs are currently underway in our laboratories and will be reported in due course.
Potential Applications
We envision at least two important applications for the above rapalog libraries. First, the rapalog-FKBP composite surfaces may be screened for inhibition of protein-protein interactions. Protein-protein interaction is ubiquitous in biology and provides an exciting class of largely unexploited drug targets.28 Unlike the conventional small-molecule drug targets, which typically contain deep pockets or clefts to make three-dimensional interactions with the small-molecule drugs, protein-protein interaction often involves large, flat surfaces, which are challenging targets for traditional small molecules. As has been repeatedly demonstrated in nature, the drug-protein composite surfaces are flat and sufficiently large in areas and should be able to bind to the flat surfaces involved in protein-protein interactions. Second, the rapalog-FKBP composite surfaces may be utilized for isoform-specific inhibition of proteins and enzymes that belong to large families of structurally similar proteins such as protein kinases and protein tyrosine phosphatases (PTPs). The human genome encodes 500 protein kinases and ~100 PTPs.29, 30 Each enzyme family has a highly conserved active site, making it difficult to develop specific inhibitors against a particular kinase or PTP. Indeed, most of the kinase inhibitors so far designed target the ATP-binding site. But because the ATP-binding site is conserved, few of these inhibitors are truly selective for the intended kinase target. For the same reason, few selective PTP inhibitors are currently available. On the other hand, for both kinases and PTPs, the protein surfaces outside the active site are highly divergent. It is conceivable to design or screen for a rapalog-FKBP composite surface that recognizes a unique surface area outside (but near) the kinase/PTP active site. While binding to such a site by a conventional small molecule may not have significant effect on the biological activity of the enzymes, association with a rapalog-FKBP complex would create a steric block to the active site, preventing the access of kinase/PTP substrates, which are large protein molecules. This is precisely the mechanism by which FK506 and cyclosporin A inhibit the serine/threonine phosphatase calcineurin.9
EXPERIMENTAL SECTION
Materials
N-Fmoc amino acids were purchased from Advanced ChemTech (Louisville, KY), Peptides International (Louisville, KY), or NovaBiochem (La Jolla, CA). HBTU and 1-hydroxybenzotriazole hydrate (HOBt) were from Peptides International. TFA was purchased from Sigma-Aldrich. Dihydro-4,4-dimethyl-2,3-furandione, tetrakis(triphenylphosphine)palladium, allyl bromide, cesium carbonate and solvents were purchased from Aldrich, Fisher Scientific (Pittsburgh, PA), or VWR (West Chester, PA). Rink resin (0.20 mmol/g, 100–200 μm) was purchased from Advanced ChemTech. 1H and 13C NMR spectra were recorded on a 400 MHz spectrometer (operated at 400 and 100 MHz, respectively). Chemical shifts are reported as δ values (ppm). NMR data were collected by using DMSO-d6 or CDCl3 as solvent. Reaction progress in solution phase was monitored by thin-layer chromatography (TLC), using 0.25 mm silica gel plates with visualization by irradiation with a UV lamp. Reaction progress in solid phase was monitored by ninhydrin test, whenever possible. HRMS data were collected with electrospray ionization mass spectrometry or direct probe ionization. MALDI-TOF mass analysis was performed on a Bruker III MALDI-TOF instrument in an automated manner at Campus Chemical Instrument Center of The Ohio State University. The data obtained were analyzed by either Moverz software (Proteometrics LLC, Winnipeg, Canada) or Bruker Daltonics flexAnalysis 2.4 (Bruker Daltonic GmbH, Germany). Flash column chromatography was carried out on silica gel 40.
Allyl Nα-Fluorenyloxycarbonyl-L-Pipecolate
To a solution of 1.0 equiv of Fmoc-L-pipecolinic acid (0.5 mmol, 176 mg), dissolved in 10 mL of acetone (saturated by K2CO3), 2 equiv of Cs2CO3 (1 mmol, 326 mg) and 5 equiv allyl bromide (2.5 mmol, 302 mg) were added. The solution was stirred for 4 h at room temperature. The crude product was purified by using flash column chromatography on a silica gel column with hexane/ethyl acetate (2:1) as eluent to give a clear oil (yield 99%): 1H NMR (400 MHz CDCl3) δ1.30–1.74 (m, 6H), 2.30 (t, 1H), 3.10 (dt, 1H), 4.12 (t, 1H), 4.33 (m, 3H), 4.60 (dd, 2H), 5.30 (dd, 2H), 5.95 (m, 1H), 7.28–7.81(m, 8H). 13C NMR (100 MHz, CDCl3) δ 171.3, 156.5, 144.1, 141.3, 131.8, 127.7, 127.1, 125.1, 120.0, 118.6, 67.7, 65.7, 54.5, 47.3, 41.8, 26.9, 24.7, 20.7. HRMS (ESI): calcd for C24H25NO4Na (M + Na+) 414.1682, found 414.1697.
Allyl-L-pipecolinate (4)
Allyl Nα-fluorenyloxycarbonyl-L-pipecolate was added to 20% piperidine in DCM solution. The solution is stirred for 20–25 min at room temperature and monitored by TLC. After evaporation of solvent, the crude product was purified by flash chromatography on a silica gel column eluted with hexane/ethyl acetate/EtOH/diisopropylethylamine (40:40:19:1) (80% yield). 1H NMR (400 MHz CDCl3) δ 1.42–2.01 (m, 6H), 2.64 (t, 1H), 3.05 (d, 1H), 3.45 (dd, 1H), 4.59 (dd, 2H), 5.25 (dd, 2H), 5.89 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 171.2, 132.0, 118.4, 65.2, 58.6, 45.7, 29.2, 25.6, 24.1. HRMS: (ESI): calcd for C9H15NO2 (M + H+) 170.1171, found 170.1181,
Allyl Nα-(3,3-dimethyl-4-hydroxy-2-ketobutyryl)-L-pipecolinate (5)
To a solution of amine 4 (0.33 mmol, 55 mg) dissolved in 1.5 mL of toluene was added dihydro-4,4-dimethyl-2,3-furandione (0.49 mmol, 62 mg) and 4-dimethylaminopyridine (DMAP) (0.033 mmol, 4 mg). The solution was stirred for 17–20 h at reflux temperature under argon atmosphere. After removal of solvent, the crude product was purified by flash chromatography on a silica gel column with hexane/ethyl acetate (2:1) as eluent (81% yield). 1H NMR (400 MHz CDCl3) δ 1.50 (s, 6H), 1.23–1.80 (m, 6H), 3.22 (dt. 1H), 3.50 (d, 1H), 3.56 (q, 1H), 4.44 (s, 2H), 4.66 (dd, 2H), 5.30 (dd, 2H), 5.90 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 205.9, 170.1, 168.1, 131.4, 119.3, 69.3, 66.2, 51.6, 49.5, 44.2, 26.3, 24.8, 21.3, 20.9. HRMS (ESI) calcd for C15H23NO5Na (M + Na+) 320.1474 found 320.1476.
Building Block 3
To a solution of allyl ester 5 (1.0 mmol) in freshly distilled DCM (3 mL) was added the proper Fmoc-amino acid (1.05 mmol), N,N′-diisopropyl carbodiimide (2.0 mmol), and DMAP (0.05 mmol). The resulting mixture was stirred for 1 h at room temperature. The crude allyl ester products were purified by silica gel column chromatography using hexane/ethyl acetate (3:1) as eluent (90–95% yield). Next, the allyl ester (0.9 mmol) was dissolved in 4 mL of distilled THF, and Pd(Ph3P)4 (0.045 mmol) and N-methylaniline (2.7 mmol) were added. The solution was stirred for 40 min at room temperature under argon atmosphere and the color of the solution changed from light yellow to brown. The crude products were purified by flash silica gel column chromatography using hexane/ethyl acetate/AcOH (66:33:1) as eluent (70–80% yields).
3a
1H NMR (400 MHz CDCl3) δ 1.27 (s, 6H), 1.32 (d, 3H), 1.24–1.59 (m, 6H), 2.97 (m, 1H), 3.26 (m, 1H), 3.37 (m, 1H), 4.22–4.39 (m, 6H), 7.28–7.65 (m, 8H). 13C NMR (100 MHz, CDCl3) δ 206.1, 203.5, 178.3, 172.5, 170.0, 163.3 157.0, 143.8, 141.3, 135.1, 132.2, 128.6, 127.8, 127.4, 125.1, 120.0, 70.0, 67.8, 67.1, 47.1, 46.6, 42.4, 41.6, 38.9, 30.9, 19.2. HRMS (ESI) calcd for C30H34N2O8Na (M + Na+) 573.2213, found 573.2216.
3b
1H NMR (400 MHz CDCl3) δ 1.16–1.65 (m, 6H), 1.34 (s, 6H), 2.90 (s, 2H), 3.00 (m, 1H), 3.30 (m, 1H), 3.36 (m, 1H), 4.16–446 (m, 6H), 7.10–7.78 (m, 13H). 13C NMR (100 MHz, CDCl3) δ 205.0, 204.0, 174.1, 172.0, 170.4, 166.3, 158.0, 156.8, 143.7, 141.3, 137.8, 128.6, 127.8, 127.2, 125.1, 120.0, 118.3, 74.1, 70.0, 67.8, 67.1, 47.1, 46.6, 42.4, 40.6, 38.9, 30.9, 19.3. HRMS (ESI) calcd for C36H38N2O8Na (M + Na+) 649.2526, found 649.2529.
3c
1H NMR (400 MHz CDCl3) δ 1.14 (s, 9H), 1.17 (s, 3H), 1.47 (s, 6H), 1.23–1.79 (m, 6H), 3.10 (m, 1H), 3.30 (m, 1H), 3.51 (m,1H), 4.01–4.59 (m, 7H), 5.30 (bs, 1H), 7.19–7.80 (m, 8H). 13C NMR (100 MHz, CDCl3) δ 205.2, 204.4, 174.0, 172.4, 170.4, 166.3, 158.0, 156.8, 143.7, 141.3, 137.8, 128.6, 127.8, 127.2, 125.1, 120.0, 118.3, 74.1, 70.0, 67.8, 59.0, 56.2, 47.1, 43.3, 33.3, 28.3, 27.3, 24.0, 20.0. HRMS (ESI) calcd for C35H44N2O9Na (M + Na+) 659.2944 found 659.2941.
Synthesis of Rapalog Library
The library was synthesized on 0.50 g of Rink resin (0.20 mmol/g, 100–200 μm). Standard Fmoc/HBTU peptide chemistry was employed for all of the synthesis steps unless otherwise noted. The coupling reactions typically employed 2 equiv of Fmoc-amino acids, 2 equiv HBTU, 2 equiv HOBt and 4 equiv NMM for 2 h and were monitored by Ninhydrin tests. The Fmoc group was removed by treatment with 20% piperidine in DMF for 5–25 min. After each step, the beads were exhaustively washed with DMF and DCM. Starting from the Fmoc-protected rink resin, the Fmoc group was removed with piperidine and the exposed amine was acylated with N-Fmoc-Glu-α-allyl ester (2 equiv), followed by the coupling of Fmoc-D-β-homoPhe (2 equiv). The resin was treated with 20% piperidine for 5 min, exhaustively washed with DMF, and immediately coupled to building block 3a (1.5 equiv). The resulting resin was split into 10 equal aliquots and each aliquot was coupled to a different Fmoc-amino acid (D-Thr, Gly, Ala, D-Val, Pro, Lys, Trp, Asp, D-Phe, and Nle). After the coupling reaction was complete, each aliquot was further split into ten equal portions to give 100 individual samples. Each portion (5 mg) was individually treated with 20% piperidine (5 min) to remove the Fmoc group, washed exhaustively, and immediately coupled to one of the ten Fmoc-amino acids described above. At this point, each of the 100 samples was split into two equal portions. The first half was washed and stored for later cyclization to produce the sublibrary 11, while the second half was subjected to another round of coupling reaction (to Fmoc-L-Ala) to expand the ring size of the cyclic peptides (sublibrary 12). Prior to cyclization, the C-terminal allyl group was removed by treatment with Pd(PPh3)4 (0.2 equiv) and N-methylaniline (9.0 equiv) in anhydrous THF (45 min). The resin was washed sequentially with THF, DMF and DCM, and treated with 20% piperidine to remove the N-terminal Fmoc group. For peptide cyclization, the resin was suspended in a solution of PyBOP/HOBt/NMM (5, 5, and 10 equiv, respectively) in DMF and the mixture was incubated on a carousel shaker for 17–20 h. The cyclization reaction was terminated when Ninhydrin tests showed negative results. The resulting resin was washed with DMF and DCM, dried under vacuum, and stored at 4 °C. Cleavage of the peptides from the resin and side-chain deprotection was achieved by treating the resin with 50% trifluoroacetic acid in DCM for 1.5 h. The solvents were evaporated under vacuum and the crude peptides were dissolved in DCM containing 10% diethylpropylamine. The solution was quickly passed through a silica gel column to remove the salts, evaporated to dryness, and stored at 4 °C until use. Compounds 2a–y were prepared in a similar manner.
Evaluation of the Rapalog Library
To check the quality of library synthesis, each of the compounds from above (2a–y, sublibrary 11, and sublibrary 12) was dissolved in 0.1% TFA (in water) and analyzed by ESI or MALDI-TOF MS. Most of the compounds showed single species of the expected m/z ratios (Figure S2–S4 in Supporting Information). Four of the compounds were selected for further purification by HPLC on a semi-preparative C-18 column, which was eluted by a linear gradient of 0–100% CH3CN in water (containing 0.05% trifluoroacetic acid) over 20 min. The pure samples were assayed against FKBP12 and compared to the results obtained with the crude samples (Table 3). Because of the small sample quantities and lack of suitable optical activity, the sample concentrations were determined as follows. The amount of crude compound (0.25 mg for a compound of the molecular mass of 1000) was estimated on the basis of the resin loading capacity (0.20 mmol/g) and a 50% overall synthesis (isolated) yield. For the purified compounds, it was assumed that HPLC purification had 70% sample recovery. Sample partition was carried out by dissolving the crude compound in 1 mL of DCM and withdrawing appropriate volumes of the stock solution for different experiments (e.g., activity assay and HPLC purification). For activity assay, the samples were evaporated to dryness and redissolved in appropriate volumes of DMSO.
FKBP Binding Assay
The fluorescence polarization based competition assay was performed in a 384-well plate. Each reaction (20 μL total volume) contained 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.76 mM KH2PO4, pH 7.4, 200 nM recombinant FKBP12, 100 nM fluorescent probe FLU-SLF (kindly provided by I. Graef of Stanford University), and varying concentrations of rapalogs (0–5000 μM). After the addition of the rapalogs as the last component, the binding reactions were incubated for 1 h at room temperature to reach binding equilibrium. Anisotropy values were measured on a FlexStation 3 plate reader (Molecular Devices, Sunnyvale, CA). KI values were calculated from the corresponding IC50 values using the method by Cheng-Prusoff31 [KI = IC50/(1 + D/KDF)], where D is the concentration of FLU-SLF (100 nM) and KDF is the binding affinity of FLU-SLF for FKBP (3.3 nM).20
Molecular modeling
At the first stage, three dimensional structures were generated for rapaplogs 11a and 11b by geometry optimization with the SPARTAN 02 program. Since the rapalogs have similar FKBP-binding motif to compound 13 of Holt et al.,18 the coordinates for atoms in the binding motif were modeled using the coordinates of compound 13 in a 2.2-Å resolution x-ray crystal structure of its complex with FKBP12. The binding motif was then fused with the cyclic peptide sequences of rapalogs 11a and 11b at both ends to generate an initial geometry. The latter was optimized at the HF/6–31+G* level with the geometry of the binding motif fixed. The optimized structure appears to be quite rigid, due to the formation of multiple hydrogen bonds between backbone amide -NH and carbonyl oxygens, as in antiparallel β-strands. During the second stage, the optimized rapalog structure was docked with FKBP12 by means of quantum mechanics/molecular mechanics (QM/MM) optimization to form the FKBP12-rapalog complex. Initially, the rapalog was placed into the binding site of FKBP12 utilizing the crystal structure of a previously reported FKBP12-rapalog complex18 (PDB access number 1FKI) as a template. The QM layer contained the rapalog and the protein residues that directly interact with the rapalog, and was treated at the PM3 level. The MM layer included the rest of the FKBP12 protein and was described by the AMBER force field. The QM/MM optimization was performed using the Gaussian 03 package.32 The QM/MM two-layer partitioning was performed as previously described.33
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM062820 and CA132855), the FORE Cancer Research Foundation and the Jack Roth Fund for Lung Cancer.
ABBREVIATIONS
- CypA
peptidyl-prolyl isomerase cyclophilin A
- CsA
cyclosporin A
- SAR
structure-activity relationship
- D-Abu
(S)-2-aminobutyric acid
- D-β-homoPhe
(R)-3-amino-5-phenylpentanoic acid
- L-β-homoPhe
(S)-3-amino-5-phenylpentanoic acid
- D-homoPhe
D-homophenylalanine
- L-β-Ile
L-β-isoleucine
- HBTU
O-benzotriazole-N,N,N,N-tetramethyluronium hexafluorophosphate
- NMM
N-methylmorpholine
- PyBop
benzotriazole-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
- DMAP
4-(dimethylamino) pyridine
- FA
fluorescence anisotropy
- OBOC
one-bead-one-compound
- HOBt
1-hydroxybenzotriazole hydrate
Footnotes
SUPPORTING INFORMATION. HPLC, NMR, MS, data of target peptides. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiotics. 1975;28:721–726. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 2.Tanaka H, Kuroda A, Marusawa H, Hatanaka H, Kino T, et al. Structure of FK506, a novel immunosuppressant isolated from Streptomyces. J Am Chem Soc. 1987;109:5031–5033. [Google Scholar]
- 3.Yuan R, Kay A, Berg WJ, Lebwohl D. Targeting tumorigenesis: development and use of mTOR inhibitors in cancer therapy. J Hematol Oncology. 2009;2:45. doi: 10.1186/1756-8722-2-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, et al. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 5.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 6.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 7.Chiu MI, Katz H, Berlin V. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc Natl Acad Sci U S A. 1994;91:12574–12578. doi: 10.1073/pnas.91.26.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Futer O, DeCenzo MT, Aldape RA, Livingston DJ. FK506 binding protein mutational analysis. Defining the surface residue contributions to stability of the calcineurin co-complex. J Biol Chem. 1995;270:18935–18940. doi: 10.1074/jbc.270.32.18935. [DOI] [PubMed] [Google Scholar]
- 9.Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, et al. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell. 1995;82:507–522. doi: 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- 10.Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, et al. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature. 1995;378:641–644. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 11.Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;273:239–242. doi: 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- 12.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 13.Salituro GM, Zink DL, Dahl A, Nielsen J, Wu E, Huang L, Kastner C, Dumont FJ. Meridamycin: A novel nonimmunosuppressive FKBP12 ligand from Streptomyces Hygroscopicus. Tetrahedron Lett. 1995;36:997–1000. [Google Scholar]
- 14.Fehr TS, J-J, Schuler W, Gschwind L, Ponelle M, Schilling W, Wioland C. Antascomicins A, B, C, D, and E. Novel FKBP12 binding compounds from a Micromonospora strain. J Antibiotics. 1996;49:230–233. doi: 10.7164/antibiotics.49.230. [DOI] [PubMed] [Google Scholar]
- 15.Jin L, Harrison SC. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc Natl Acad Sci U S A. 2002;99:13522–13526. doi: 10.1073/pnas.212504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Etzkorn FAC, ZY, Stolz LA, Walsh CT. Cyclophilin residues that affect noncompetitive inhibition of the protein serine phosphatase activity of calcineurin by the cyclophilin-cyclosporin A complex. Biochemistry. 1994;33:2380–2388. doi: 10.1021/bi00175a005. [DOI] [PubMed] [Google Scholar]
- 17.Zenke G, Strittmatter U, Fuchs S, Quesniaux VF, Brinkmann V, et al. Sanglifehrin A, a novel cyclophilin-binding compound showing immunosuppressive activity with a new mechanism of action. J Immunol. 2001;166:7165–7171. doi: 10.4049/jimmunol.166.12.7165. [DOI] [PubMed] [Google Scholar]
- 18.Holt DA, Luengo JL, Yamashita D, Oh H-J, Konilian AL, Yen H-K, Rozamus LW, Brandt M, Bossard MJ, Levy MA, Eggleston D, Liang J, Schults LW, Stout T, Clardy J. Design, Synthesis and Kinetic Evaluation of High-Affinity FKBP Ligands and the X-ray Crystal Structures of Their Complexes with FKBP12. J Am Chem Soc. 1993;115:9925–9938. [Google Scholar]
- 19.Dubowchik GM, Ditta JL, Herbst JJ, Bollini S, Vinitsky A. Fluoresceinated FKBP12 ligands for a high-throughput fluorescence polarization assay. Bioorg Med Chem Lett. 2000;10:559–562. doi: 10.1016/s0960-894x(00)00045-7. [DOI] [PubMed] [Google Scholar]
- 20.Braun PD, Wandless TJ. Quantitative analyses of bifunctional molecules. Biochemistry. 2004;43:5406–5413. doi: 10.1021/bi035839g. [DOI] [PubMed] [Google Scholar]
- 21.Maynard-Smith LA, Chen LC, Banaszynski LA, Ooi AG, Wandless TJ. A directed approach for engineering conditional protein stability using biologically silent small molecules. J Biol Chem. 2007;282:24866–24872. doi: 10.1074/jbc.M703902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Briesewitz R, Ray GT, Wandless TJ, Crabtree GR. Affinity modulation of small-molecule ligands by borrowing endogenous protein surfaces. Proc Natl Acad Sci U S A. 1999;96:1953–1958. doi: 10.1073/pnas.96.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gestwicki JE, Crabtree GR, Graef IA. Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science. 2004;306:865–869. doi: 10.1126/science.1101262. [DOI] [PubMed] [Google Scholar]
- 24.Marinec PS, Chen L, Barr KJ, Mutz MW, Crabtree GR, et al. FK506-binding protein (FKBP) partitions a modified HIV protease inhibitor into blood cells and prolongs its lifetime in vivo. Proc Natl Acad Sci U S A. 2009;106:1336–1341. doi: 10.1073/pnas.0805375106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison RK, Stein RL. Substrate specificities of the peptidyl prolyl cis-trans isomerase activities of cyclophilin and FK-506 binding protein: evidence for the existence of a family of distinct enzymes. Biochemistry. 1990;29:3813–3816. doi: 10.1021/bi00468a001. [DOI] [PubMed] [Google Scholar]
- 26.Somers PK, Wandless TJ, Schreiber SL. Synthesis and analysis of 506BD, a high-affinity ligand for the immunophilin FKBP. J Am Chem Soc. 1991;113:8045–8056. [Google Scholar]
- 27.Kawai M, Lane BC, Hsieh GC, Mollison KW, Carter GW, Luly JR. Structure-activity profiles of macrolactam immunosuppresant FK506 analogues. FEBS Lett. 1993;316:107–113. doi: 10.1016/0014-5793(93)81196-7. [DOI] [PubMed] [Google Scholar]
- 28.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 29.Johnson SA, Hunter T. Kinomics: methods for deciphering the kinome. Nat Methods. 2005;2:17–25. doi: 10.1038/nmeth731. [DOI] [PubMed] [Google Scholar]
- 30.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustellin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 31.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmaco. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 32.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C03. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 33.Liu YM, Hu XH. Molecular determinants for binding of ammonium ion in the ammonia transporter AmtB - A quantum chemical analysis. J Phys Chem A. 2006;110:1375–1381. doi: 10.1021/jp054261c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.