

Abstract
CB1 receptor antagonists that are peripherally restricted were targeted. Compounds with permanent charge as well as compounds that have increased polar surface area were made and tested against CB1 for binding and activity. Sulfonamide and sulfamide with high polar surface area and good activity at CB1 were rationally designed and pharmacologically tested. Further optimization of these compounds and testing could lead to the development of a new class of therapeutics to treat disorders where the CB1 receptor system has been implicated.
Keywords: CB1, peripheral, antagonist, cannabinoid, topological polar surface area
Cannabinoid receptors (CBRs) belong to the endocannabinoid (EC) system, which consists of receptors, transporters, endocannabinoids, and the enzymes involved in synthesis and degradation of endocannabinoids.1 To date, two different cannabinoid receptors have been identified CB1 and CB2. Both CB1 and CB2 are G protein–coupled receptors (GPCRs) primarily activating inhibitory G proteins (Gi/o).2, 3 The EC system regulates many important physiological processes and several components of the EC system are under evaluation as targets to treat a diverse array of indications including obesity, liver disease, diabetes, pain and inflammation.4
The CB1 receptor is prominently expressed in the central nervous system (CNS) and also in peripheral tissues. It has emerged as an important target to treat metabolic disorders including obesity and diabetes. The first CB1 receptor selective drug developed for medical use was rimonabant 1 (SR141716A), an inverse agonist/antagonist. Rimonabant was designed to treat obesity and other related disorders that have both CNS and peripheral components. However, rimonabant was withdrawn from European markets and denied approval in the United States due to CNS-related side effects including anxiety, depression and suicidal ideation.5 Subsequently, development of other CB1 receptor antagonists taranabant, otenabant, and ibipinabant were halted due to regulatory concerns.6
A strategy to minimize CNS-related side effects noted with CB1 antagonists while maintaining beneficial effects of blocking the receptor in peripheral tissues is to develop compounds that do not cross the blood-brain barrier (BBB). This approach is being pursued by several groups and some compounds have been reported (Fig 1).7–11 However, most of these reported compounds need to be fully characterized and their clinical efficacies remain unproven. Herein we describe our efforts in developing CB1 antagonists selective for the periphery using two strategies. First, since charged compounds do not normally cross the BBB unless transported by specific transporters,12 permanently charged compounds, such as alkyl pyridinium salts and N-oxides, were targeted. The second strategy was driven by topological polar surface area (TPSA). The total surface area of polar atoms in a molecule has been shown to correspond with passive transport through membranes, where higher TPSA corresponds to lower penetration into the CNS.13 Therefore, compounds with relatively high TPSAs, such as sulfonamide and sulfamide, were targeted. In addition to the significantly increased TPSA, these neutral sulfonamides and sulfamides also have hydrogen available for hydrogen bonding, offering the opportunity for further interaction with the receptor and thus possibly leading to improved potency.
Figure 1.

Structure of CB1 antagonists
Charged compounds were synthesized as described in Scheme 1. Acid A is readily available using a procedure developed in our laboratories.14, 15 The acid was then coupled by first making an acid chloride using oxalyl chloride and a catalytic amount of DMF followed by amide formation with the appropriate aminopyridine and triethylamine; or by the use of standard BOP coupling conditions.16 Alkyl pyridinium salts (6, 9, 12) were made by reacting B with methyl iodide in dichloromethane or methanol.17 Pyridine N-oxides (7, 10, 13) were readily obtained by reacting B with m-CPBA in dichloromethane.18
Scheme 1.

Reagents and conditions: (i) (a) oxalyl chloride, CH2Cl2, DMF cat., rt, 2 h; (b) aminopyridine, CH2Cl2, Et3N, rt; (ii) aminopyridine, BOP, i-Pr2EtN, DMF, rt, 16h; (iii) methyl iodide, CH2Cl2 or MeOH, 2–7 d; (iv) m-CPBA, CH2Cl2, 16 h.
Sulfonamide and sulfamide compounds were synthesized by the route described in Scheme 2. Acid A was coupled to diamines via a method previously described by our group.16 The diastereomeric ratio of compound 19 could be enriched by careful column chromatography. These amines were converted to the corresponding sulfonamide compound (14, 17, 20, 22, 24, 27) with methanesulfonyl chloride and triethylamine in THF. The desired sulfamides (15, 18, 21, 23, 26, 28) were synthesized by reacting the appropriate amine with sulfamide at an elevated temperature, as has been previously described.19
Scheme 2.

Reagents and conditions: (i) diamine, BOP, THF, rt; (ii) methanesulfonyl chloride, Et3N, THF, rt, 16h; (iii) sulfamide, dioxane, 85° C, 16 h. X is used to designate a spacer of any kind.
All compounds were purified on a Teledyne ISCO CombiFlash Companion system using RediSepRf prepacked columns and characterized by 1H NMR (300 MHz), TLC, and mass spectroscopy. All compounds were evaluated for antagonism using a calcium mobilization assay. Each compound was pharmacologically characterized for antagonism using a functional fluorescent CB1 activated Gαq16-coupled intracellular calcium mobilization assay in CHO-K1 cells as has been described in our previous publication.16 Briefly, the ability of each compound to antagonize the concentration-response of CP55940 at CB1 was measured.20 Antagonism was noted by a right-ward shift of the CP55940 concentration-response curve upon pre-incubation with the test agent. The EC50 values in the presence and absence of an antagonist was used to determine its apparent antagonist dissociation equilibrium constant (Ke).21 Further characterization of select compounds was performed using radioligand displacement of [3H]1 and equilibrium dissociation constant (Ki) values were determined.16 Selectivity of these compounds at CB1 versus CB2 was also determined by obtaining Ki values at either receptor using displacement of [3H]CP55940 in membranes of CHO-K1cells over-expressing either receptor. Data reported are average values from 3–6 measurements.16
The pyridinium compounds were charged analogs of a reported methylpyridine amide similar to 11.22 To date, only limited activity has been observed with alkyl pyridinium salts and pyridine N-oxides (Table 1). The parent pyridines (5, 8, 11) were more potent than their alkyl pyridinium salt or N-oxide analogues in all cases. All pyridinium salt analogues made to date have Ke values of greater than 8 µM against CB1. The pyridine N-oxides were modestly activewith compounds 7 and 10 both having Ke values less than 2 µM, making them interesting for future interrogation.
Table 1.
Alkyl pyridinium salts and N-oxides derivatives 1–9 via Scheme 1
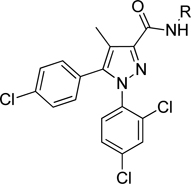 | |||
|---|---|---|---|
| Compound | R | Ke CB1 (µM) | Yield %a |
| 5 | 0.117 | 90% | |
| 6 | 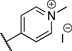 |
>10 | 55% |
| 7 | 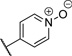 |
1.39 | 70% |
| 8 | 0.384 | 91% | |
| 9 | 8.41 | 61% | |
| 10 | 1.20 | 99% | |
| 11 | 0.980 | 19% | |
| 12 | 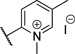 |
8.59 | 16% |
| 13 | 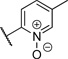 |
4.58 | 20% |
Yield of the final step only.
The initial CB1 antagonists with high TPSAs, sulfonamide 14 and sulfamide 15, were both active at CB1 receptor (Table 2) and have significantly higher TPSAs than rimonabant 1 (rimonabant’s TPSA is 50 and the TPSAs for 14 and 15 are 101 and 127 respectively). With compounds 14 and 15 in hand, attempts were made to improve potency for these CB1 receptor antagonists while maintaining high TPSAs. Analogs that contain a rigid element in the amide functionality were targeted in hopes of improving potency. The initial partially constrained analogues 16–18 had little or no activity. However, our laboratories had previously discovered that more polar functionality could be tolerated if linkers of sufficient length are used.16 Therefore, compounds 19–21, which have longer spacers (X, Scheme 2), were targeted. Compounds 20 and 21, as a 1:1 cis/trans mixture, were functionally potent (Ke~100 nM) and bound CB1 with high affinity (Ki ~ 10 nM). Compounds 22 and 23, enriched in the cis- isomer (cis:trans ~ 7:1) compared to 20 and 21 and were demonstrated to be even more potent.23 Compound 24 suggested that basic spacer groups with sulfonamides were not tolerated. Basic groups at the terminus of the linker were not tolerated (16, 19) either. Compounds 25–28 were prepared to study the effect of in the nature of the spacer on activity. Unfortunately, aromatic groups used as spacers were deemed detrimental for CB1 activity. Further exploration of constrained analogues is under way.
Table 2.
Sulfonamide and sulfamide derivatives 14–28 via Scheme 2
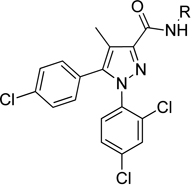 | |||
|---|---|---|---|
| Compound | R | Ke CB1 (µM) | tPSA |
| 14 | 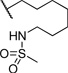 |
0.207 | 101 |
| 15 | 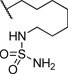 |
0.304 | 127 |
| 16 | >10 | 73 | |
| 17 | 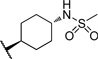 |
>10 | 101 |
| 18 | 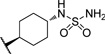 |
9.43 | 127 |
| 19a | 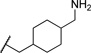 |
3.76 | 73 |
| 20a | 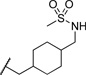 |
0.113 | 101 |
| 21a | 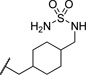 |
0.106 | 127 |
| 22b | 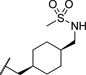 |
0.030 | 101 |
| 23b | 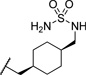 |
0.093 | 127 |
| 24 | 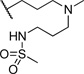 |
5.34 | 105 |
| 25 | 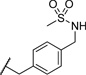 |
2.93 | 101 |
| 26 | 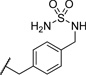 |
0.376 | 127 |
| 27 | 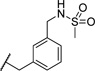 |
2.83 | 101 |
| 28 | 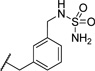 |
4.20 | 127 |
Compounds isolated are approximately 1:1 mixture of cis and trans isomers.
Compounds are 7:1 mixture of cis/trans isomers. A 7:1 mixture of 15 was isolated after careful column chromatography. Assignment of relative stereochemistry was tentatively made by H1NMR. Further efforts to isolate pure cis- and trans- isomers are ongoing.
Select compounds (Table 3) were chosen for further study in radioligand displacement assays using radiolabeled SR141716 ([3H]1). Several of these compounds demonstrated excellent Ki values in the low nM range, with 22 having a Ki of 8 nM. Selectivity against the CB2 receptor was determined by comparing the compound displacement of radiolabeled CP55940 at CB1 and CB2 receptors. In general, most compounds were selective for CB1 over CB2.
Table 3.
Radioligand displacement data for select compounds
| No | Ki(µM) CB1 SR141716 |
Ki(µM) CB1 CP55940 |
Ki(µM) CB2 CP55940 |
CB2:CB1 CP55940 |
|---|---|---|---|---|
| 5 | 0.013 | 0.056 | 1.74 | 31 |
| 7 | 0.061 | 0.294 | 4.52 | 15 |
| 8 | 0.026 | 0.102 | 4.01 | 39 |
| 9 | 0.786 | 1.698 | 2.27 | 1.3 |
| 14 | 0.060 | 0.158 | 0.78 | 4.9 |
| 15 | 0.020 | 0.049 | 1.01 | 21 |
| 20 | 0.011 | 0.055 | 0.90 | 16 |
| 21 | 0.021 | 0.135 | 2.31 | 17 |
| 22 | 0.008 | 0.036 | 0.96 | 27 |
| 23 | 0.015 | 0.107 | 1.79 | 17 |
Charged compounds generally showed poor activity in the calcium flux assay. However, an interesting piece of data was the good affinity of compound 7 for CB1 versus 3H-SR141716 (Ki of 61 nM) in contrast to its Ke = 1.39 M for calcium flux. An explanation for this disparity could be that binding of 7 induced a conformational change to the CB1 receptor, which in turn selectively abrogated signaling through the Gαq16 pathway coupled to calcium mobilization. This interesting result needs further investigation in the future.
Two compounds were selected to advance into an in vitro model of brain penetration. The MDCK-mdr1 (Madin-Darby Canine Kidney (MDCK) stably expressing mdr1 (multi-drug resistance gene 1)) cell line has been found to be useful in identifying compounds that passively distribute into the CNS.24, 25 When compounds 22 and 23 were tested in the MDCK-mdr1 transport assay they were found to have less than 1% transport from apical to basal side of the membrane.26 This would be consistent with little are no brain penetration if no active transport was taking place. By contrast, rimonabant and otenabant 6 were used as control compounds and these compounds were transported ~15% and ~90% across MDCK-mdr1 cells respectively.
In conclusion, two classes of CB1 antagonists were rationally targeted for exclusion from the CNS. First, compounds with permanent charge were synthesized and tested. However, these compounds showed poor activity in the calcium flux assay. Charge at other positions on the pyrazole ring is being explored. Compounds with TPSAs greater than rimonabant have been synthesized. Polar compounds with sufficient potency and selectivity have been identified. These compounds have poor in vitro permeability and appear to be more promising for further development and refinement. Select compounds are being advanced into in vivo experiments.
Acknowledgments
The authors would like to thank David Perrey for graciously providing all of acid A that was used in this work. We would also like to thank Anne Gilliam for her assistance in performing binding assays. Patricia Reggio helped in our understanding of possible membrane penetration issues faced by our charged compounds. We express our gratitude to the NIDA drug supply program for providing radiolabeled probes. This research was funded by research grants 1R21AA019740-01 and 1R03AA017514-01 to R.M. from NIAAA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Pacher P, Batkai S, Kunos G. Pharmacol Rev. 2006;58:389. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouaboula M, Bianchini L, McKenzie FR, Pouyssegur J, Casellas P. FEBS Lett. 1999;449:61. doi: 10.1016/s0014-5793(99)00395-6. [DOI] [PubMed] [Google Scholar]
- 3.Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, Barth F, Calandra B, Pecceu F, Lupker J, Maffrand JP, Le Fur G, Casellas P. J Biol Chem. 1997;272:22330. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- 4.Janero DR, Makriyannis A. Expert Opin Emerg Drugs. 2009;14:43. doi: 10.1517/14728210902736568. [DOI] [PubMed] [Google Scholar]
- 5.Di Marzo V. Drug Discov Today. 2008;13:1026. doi: 10.1016/j.drudis.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Lee HK, Choi EB, Pak CS. Curr Top Med Chem. 2009;9:482. doi: 10.2174/156802609788897844. [DOI] [PubMed] [Google Scholar]
- 7.Tam J, Vemuri VK, Liu J, Batkai S, Mukhopadhyay B, Godlewski G, Osei-Hyiaman D, Ohnuma S, Ambudkar SV, Pickel J, Makriyannis A, Kunos G. The Journal of clinical investigation. 2010;120:2953. doi: 10.1172/JCI42551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LoVerme J, Duranti A, Tontini A, Spadoni G, Mor M, Rivara S, Stella N, Xu C, Tarzia G, Piomelli D. Bioorg Med Chem Lett. 2009;19:639. doi: 10.1016/j.bmcl.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hortala L, Rinaldi-Carmona M, Congy C, Boulu L, Sadoun F, Fabre G, Finance O, Barth F. Bioorg Med Chem Lett. 2010;20:4573. doi: 10.1016/j.bmcl.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 10.Receveur JM, Murray A, Linget JM, Norregaard PK, Cooper M, Bjurling E, Nielsen PA, Hogberg T. Bioorg Med Chem Lett. 2010;20:453. doi: 10.1016/j.bmcl.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Sasmal PK, Reddy DS, Talwar R, Venkatesham B, Balasubrahmanyam D, Kannan M, Srinivas P, Kumar KS, Devi BN, Jadhav VP, Khan SK, Mohan P, Chaudhury H, Bhuniya D, Iqbal J, Chakrabarti R. Bioorg Med Chem Lett. 2010;21:562. doi: 10.1016/j.bmcl.2010.10.055. [DOI] [PubMed] [Google Scholar]
- 12.Chen RZ, Frassetto A, Lao JZ, Huang RR, Xiao JC, Clements MJ, Walsh TF, Hale JJ, Wang J, Tong X, Fong TM. Eur J Pharmacol. 2008;584:338. doi: 10.1016/j.ejphar.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 13.Clark DE, Pickett SD. Drug Discov Today. 2000;5:49. doi: 10.1016/s1359-6446(99)01451-8. [DOI] [PubMed] [Google Scholar]
- 14.Seltzman HH, Carroll FI, Burgess JP, Wyrick CD, Burch DF. Journal of Labelled Compounds & Radiopharmaceuticals. 2002;45:59. [Google Scholar]
- 15.Zhang Y, Burgess JP, Brackeen M, Gilliam A, Mascarella SW, Page K, Seltzman HH, Thomas BF. Journal of medicinal chemistry. 2008;51:3526. doi: 10.1021/jm8000778. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Gilliam A, Maitra R, Damaj MI, Tajuba JM, Seltzman HH, Thomas BF. J Med Chem. 2010;53:7048. doi: 10.1021/jm1006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang S, Wong JC, Leung AK, Chan YM, Wong L, Fernendez MR, Miller AK, Wu W. Tetrahedron letters. 2009;50:5018. doi: 10.1016/j.tetlet.2009.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang YQ, Yuen J, Lautens M. The Journal of organic chemistry. 2007;72:5152. doi: 10.1021/jo070460b. [DOI] [PubMed] [Google Scholar]
- 19.Jones C, Pass M, Rudge D. World Intellectual Property Organization Patent. WO 2007138277. Patent Appl. PCT Int. Appl. 2007
- 20.Each compound was pharmacologically characterized using a functional fluorescent CB1 activated Gαq16-coupled intracellular calcium mobilization assay in CHO-K1 cells as has been described in our previous publication and apparent affinity (Ke) values were determined.16 Further characterization of select compounds was performed using radioligand displacement of [3H]1 and equilibrium dissociation constant (Ki) values were determined.16 Selectivity of these compounds at CB1 versus CB2 was also determined by obtaining Ki values at either receptor using displacement of [3H]CP55940 in membranes of CHO-K1cells over-expressing either receptor. Data reported are average values from 3–6 measurements.
- 21.Kosterlitz HW, Lees GM, Wallis DI, Watt AJ. Br J Pharmacol. 1968;34:691P. [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng L, Jonforsen M, Schell P. WO 2007010217 Patent Appl. PCT Int. Appl. 2007
- 23.Efforts to isolate diastereomerically pure compounds are ongoing.
- 24.Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, Hidalgo IJ. Int J Pharm. 2005;288:349. doi: 10.1016/j.ijpharm.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 25.Garberg P, Ball M, Borg N, Cecchelli R, Fenart L, Hurst RD, Lindmark T, Mabondzo A, Nilsson JE, Raub TJ, Stanimirovic D, Terasaki T, Oberg JO, Osterberg T. Toxicol In Vitro. 2005;19:299. doi: 10.1016/j.tiv.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 26.MDCK-mdr1 cells were grown on Transwell type filters (Corning) for 4 days to confluence. Compounds were added to the apical side at a concentration of 3.16 µM and incubated for 1 hr at 37°C. Compounds selected for MDCK-mdr1 cell assays were infused on an Applied Biosystems API-4000 mass spectrometer to optimize for analysis using multiple reaction monitoring (MRM). Flow injection analysis was also conducted to optimize for mass spectrometer parameters. Samples from the apical and basolateral side of the MDCK cell assay were dried under nitrogen on a Turbovap LV. The chromatography was conducted with an Agilent 1100 binary pump with a flow rate of 0.5 mL/min. Mobile phase solvents were A, 0.1% formic acid in water, and B, 0.1% formic acid in methanol. The initial solvent conditions were 10% B for 1 minute, then a gradient was used by increasing to 95% B over 5 minutes, then returning to initial conditions.