

Abstract
Smooth muscle cell (SMC) proliferation has been accepted as a common event in the pathophysiology of vascular diseases, including atherogenesis and intimal hyperplasia. Delivery of the nitric oxide synthase (NOS) substrate L-arginine, pharmacological nitric oxide (NO) donors, NO gas or over-expression of NOS proteins can inhibit SMC proliferation and reduce the injury responses within the blood vessel wall. Though commercial development of NO-donors that attempt to provide exogenous delivery of NO has accelerated over the last few years. None of the currently available products can provide controlled, sustained, time tunable release of NO. Nitrosamine-based NO donors, prepared in our laboratory, present a unique and innovative alternative for possible treatments for long-term NO deficiency related diseases including atherosclerosis, asthma, erectile dysfunction, cancer and neurodegenerative diseases. A family of secondary amines prepared via nucleophilic aromatic displacement reactions could be readily N-nitrosated to produce NO donors. NO release takes place in three distinct phases. During the initial phase, the release rate is extremely fast. In the second phase, the release is slower and the rate remains essentially the same during the final stage. These compounds inhibited up to 35% human aortic smooth muscle cell (HASMC) proliferation in a concentration-dependent manner.
Keywords: nitric oxide, nitric oxide donors, nitric oxide synthase, human aortic smooth muscle cells, atherosclerosis, N-nitrosamines, proliferation
INTRODUCTION
SMC proliferation has been accepted as a common event in the pathophysiology of many vascular diseases, including atherosclerosis, atherogenesis and intimal hyperplasia [1,2].The pathophysiology of atherosclerosis is complex and involves endothelial injury, which results in the accumulation of lipids and their uptake by monocytes [3]. Nitric oxide is an important anti-atherosclerotic autocoid with anti-aggregatory effects on platelets and antiproliferative and dilatory effects on vasculature [4]. Delivery of the NO synthase (NOS) substrate L-arginine, pharmacological NO donors, NO gas or overexpression of NOS proteins can inhibit SMC proliferation and reduce the injury responses within the blood vessel wall [5]. In blood, NO can inhibit the aggregation and adhesion of platelets, and attenuate leukocyte activation and leukocyte adhesion to the endothelium. This allows blood pressure to be regulated to allow for smooth blood flow [6]. Earlier findings suggest that nitric oxide plays a critical role in many cardiovascular diseases including atherosclerosis and thickening of the endothelium layer [7,8].
Nitric oxide facilitates relaxation of smooth muscle cells (SMC). Deficiency of in vivo NO production can have significant deleterious effects associated with increased SMC proliferation, which include cardiovascular disease (CVD) [9], erectile dysfunction (ED), stroke, and asthma. SMC found in different tissues vary in their cell-signaling systems and hormone receptivity [10,11]. However, commonalities exist for the NO-cyclic guanosine monophosphate (cGMP) signaling pathway for controlling SMC relaxation [12], where NO levels are implicated in SMC proliferation [13]. Thus, deficiency of NO disturbs cGMP-controlled cascades. This event is associated with disease states (e.g. ED, asthma) [13].
The need to ameliorate NO deficiency has generated demand for exogenous NO from chemical sources, termed NO-donors, for in vitro studies to gain insight into NO mediated biological processes. A vast array of NO-donors including nitrates, nitrites, N-nitroso, C-nitroso, S-nitroso, certain heterocyclics, metal/NO complexes, and diazeniumdiolates have been reported [14–16]. Underlying these activities is the relentless pursuit to find the desirable NO-donors, which can amelioratesymptoms arising from pathological NO deficiencies.
Clinically approved NO-donors such as glyceryl trinitrate (GTN), isosorbide dinitrate (ISDN), sodium nitropusside (SNP), isosorbide-5-mononitrate (IS-5N), amyl nitrite, and pentaerythrityl tetranitrate (PETN) are in use in spite of associated nitrate tolerance [17,18].Nitric oxide action is concentration dependent. For example, at low (pico-molar to nano-molar)concentrations, NO exerts beneficial effects [19]. GTN, the most notable NO-donating drug, produces NO in bursts of high concentrations (micro-molar) [20]. This NO release process is catalyzed by mitochondrial aldehyde dehydrogenase (ALDH2) [17].The major clinical side-effect of GTN use is headache, possibly caused by large quantity of NO released over a short time frame [18].The onset of nitrate tolerance occurs with long- term usage, which is characterized by reduced nitrate-induced vasodilation and a blunting of blood pressure lowering effects [20].Nitrate tolerance is believed to stem in part from the action of organic nitrates in promoting an increase in superoxide radical formation by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and uncoupled endothelial NOS (eNOS) [20]. Superoxide and vascular NO rapidly form peroxynitrite, which aggravates tolerance by promoting eNOS uncoupling and inhibition of soluble guanylyl cyclase (sGC) and prostacyclin synthase [21]. At higher concentrations, NO generates a variety of active molecules, collectively termed reactive nitrogen species (RNS), which can react with reactive oxygen species (ROS) to form RNS and peroxynitrite (ONOO−). Moreover, these highly active ionic moieties exert a wide variety of toxic effects [22]. Furthermore, NO mediates posttranslational modifications of proteins including nitrosylation and nitration, which may be highly deleterious [23,24]. Therefore, there exists a clear need for NO-donors that can release NO in a slow, sustainable and rate tunable fashion. For this reason, any drug or pro-drug which can release nitric oxide must be carefully tested to determine the release profile of nitric oxide under biological conditions.
Although commercial development of NO-donors that provide exogenous delivery of NO has accelerated over the last few years, none of the products currently available can provide controlled, sustained, time tunable formulations of NO. Nitrosamine-based NO releasing agents (NO-donors) provide this unique and innovative alternative as possible treatments for long-term NO deficiency related diseases. N-nitrosamines have the advantages of improved compatibility in intra-arterial devices and can serve as important tools in cardiovascular biomedical research [25].Furthermore it is possible to modulate the release profile of NO by changing the spacer groups of these NO donors.
We report herein the preparation of secondary amines with aliphatic groups or aromatic groups, which were subsequently nitrosated to yield N-nitrosamines. One family of these compounds was used to assess the effects of released NO on the proliferation of human aortic smooth muscle cells (HASMC).
EXPERIMENTAL
Materials
Chemicals were purchased from Aldrich and used without the further purification unless otherwise specified. The amines were purified by distillation under reduced pressure. The solid amines were recrystallized from low boiling (b.p. 35 C–50 C) ligroin and then dried in a vacuum oven at room temperature. N-methyl pyrrolidinone (NMP) and dimethyl acetamide (DMAC) were distilled over calcium hydride at reduced pressure. Toluene was used as received. Phosphate buffered saline (PBS) (Fisher Chemical) and a colorimetric non-enzymatic assay kit for nitric oxide testing (Oxford Biomedical Research, Inc.) were used as received. HASMC were purchased from Sciencell Research Laboratories. Cell culture labware was purchased from Corning. Cell viability assay kit (MTT) was purchased from American Type Culture Collection (ATCC) biological resource center, and used as received.
Measurements
Nuclear magnetic resonance (NMR) spectra (1H and 13C) were recorded on a Varian Mercury-Plus 300MHz Spectrometer using deuterated trichloromethane as the solvent. All NMR chemical shifts are reported in parts per million (δ) relative to tetramethylsilane standard (TMS δ=0). Infrared (IR) spectra (neat) were acquired with a Nicolet IR 584 Fourier Transform spectrometer (Nicolet Instrument Corporation) using thin films of the materials on sodium chloride plates. Mass spectra were obtained using a Hewlett Packard 5997 GC/MS with programmed temperature inlet or direct insertion as appropriate. The ionization potential was 70 eV. All mass spectral data are reported in Daltons. NO release absorbance measurements were conducted on a Dynatech Laboratories Minireader II plate reader with 570 nm filter, suitable for measuring the absorbance of the azo compound produced in the Greiss Reagent test for NO concentration [26]. Smooth muscle cell viability (MTT) test absorbance measurements were conducted at 570 nm on a BioTek ELx808 micro plate reader with a reference filter of 600 nm.
Procedure for synthesis of compound b
4,4'-sulfonylbis(fluorobenzene) (2.571 g, 0.0102 mol) was transferred to a 50 mL, 3-necked round-bottomed flask, fitted with a dropping funnel, a condenser, a thermometer, and a magnetic stir bar. The reaction vessel was cooled in an ice bath. A mixture of 7.5 mL sulfuric acid and 7.5 mL nitric acid in a beaker was immersed in an ice bath for 15 min. The acid mixture was transferred to the dropping funnel, and then added to the reaction vessel at the rate of about 1 drop per second. After complete addition, the reaction was allowed to continue with stirring for 3 hours. The reaction mixture was poured into 300 mL ice/water mixture to precipitate the crude product. The crude product was collected by vacuum filtration and rinsed with copious amounts of water (1000 mL). The product was then dissolved in dichloromethane and dried over anhydrous magnesium sulfate and filtered. The filtrate, which was pale yellow in color, was transferred to a 250 mL round-bottomed flask. Solvents were removed using a rotary evaporator under reduced pressure to yield a pale yellow solid crude product. The crude product was purified further by crystallization from acetic acid (yield: 75.43%, mp: 160 °C). Spectroscopic analysis for compound b: 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.68 (dd, 4H), 8.25 (m, 4H), 7.58 (dd, 4H); 13C NMR (300 MHz, CDCl3): δ (ppm) 167.50, 141.90, 139.56, 130.99, 130.51; IR (neat, ν > 1400 cm−1): 2860, 1618, 1570. 1522, 1472; MS (m/z) (% base peak): 344 (30.23%); 188 (90%); 142 (39.14%); 94 (100%).
Procedure for synthesis of compounds 1–9
A detailed reaction procedure for the synthesis of 4,4'-sulfonylbis(N-hexyl-2-nitroaniline), 1, is provided. Compounds 2–9 were prepared in a similar manner. The reaction apparatus consisted of a three-necked, 50 mL round-bottomed flask, a nitrogen inlet, a thermometer, and a Dean-Stark trap fitted with a condenser. An oil bath was used as the external heat source. The reaction vessel was charged with 0.9350 g (0.0075 mol) of anhydrous potassium carbonate. Hexan-1-amine (1.073 g, 0.01 mol) was weighed in a one-dram glass vial and dissolved in 5 mL of NMP. The solution was transferred to the reaction vessel and the vial was subsequently washed with 5mL of NMP to ensure complete transfer of hexan-1-amine. Mechanical stirring was initiated. A 1.320 g (0.005 mol) sample of 4,4'-sulfonylbis(1-fluoro-2-nitrobenzene) was weighed in a one-dram glass vial and dissolved in 5 mL of NMP. The solution was transferred to the reaction vessel. An exotherm of 10 °C was observed. The reaction mixture was bright yellow in color and slightly opaque. The reaction vessel was charged with 20 mL toluene. The progress of the reaction was monitored by thin layer chromatography. The reaction mixture was heated to 60 °C and the reaction was allowed to continue at this temperature for 30 minutes. The temperature of the reaction mixture was gradually raised to 130 C over a period of 2 hours. Water, the by-product of the reaction, was removed by azeotropic distillation with toluene through the Dean-Stark trap. The reaction vessel was cooled to room temperature after the complete removal of water. At the completion of the reaction, 25 mL dichloromethane was added into the reaction mixture, which was then filtered under reduced pressure over a celite bed. The reaction vessel was washed with approximately 30 mL of dichloromethane to ensure complete removal of products and filtered under reduced pressure over a celite bed. The filtrate, which was bright yellow in color, was transferred to a 250 mL round-bottom flask. Solvents were removed using a rotary evaporator under reduced pressure to yield dark brown viscous liquid. The liquid mixture was distilled at 80 C under vacuum to remove NMP to yield a bright yellow solid crude product. The crude product was dissolved in a minimal amount of dichloromethane and transferred to a separatory funnel. The solution was washed 3 times with 100 mL deionized water. The organic layer was dried over anhydrous magnesium sulfate, and filtered. The filtrate was evaporated using a rotary evaporator to remove dichloromethane. The resulting product, 1, was a bright yellow colored powder. The product was recrystallized from hexane. Analytical data for compounds 1–9 are supplied as supplemental material.
General procedure for the preparation of N-nitrosamines 1’–9’
A detailed reaction procedure for the synthesis of 1’ is provided. N-nitrosamines 2’–9’ were prepared in a similar manner. Compound 1 (0.057 g, 0.00025 mol) was dissolved in 4 mL of tetrahydrofuran (THF) and then added to the single-necked, 25 mL round-bottomed flask. Glacial acetic acid (6 mL) was added to adjust the pH to 2–3. The reaction mixture was bright orange in color. The reaction vessel was immersed in an ice bath for 15 min. After the temperature of the reaction mixture had reached 0 °C, sodium nitrite (0.12 g, 0.002 mol, and molar ratio 8:1 to compound 1) was added. The reaction vessel was then capped tightly and covered with aluminum foil. The reaction mixture was allowed to come to room temperature (approximately 2 hours) and the reaction was allowed to continue with stirring an additional 6 hours. At the completion of the reaction, the reaction mixture, light yellow colored slurry, was transferred to 250 mL beaker. Sodium hydroxide solution (4M, 27 mL) was added at 0 °C with ice present in the beaker to neutralize the acid and to precipitate the desired compound. The mixture was then transferred to a separatory funnel and extracted twice with 30 mL dichloromethane. The organic layer was washed three times with 100 mL deionized water followed by 10% aqueous sodium bicarbonate solution (50 mL). The organic layer was dried over anhydrous magnesium sulfate, filtered and the filtrate was evaporated under reduced pressure to yield a yellow liquid. The product was then stored in sealed vials under argon atmosphere at 4 °C.
Cell culture
Primary cultures of HASMC were maintained in Smooth Muscle Cell Medium (SMCM) consisting of 2% fetal bovine serum (FBS), 1% smooth muscle cell growth supplement and 1% penicillin/streptomycin solution. Cells were utilized between passages 5 to 10.
Cell proliferation assays
Cells were transferred to 96 well plates (Corning). Each well contained about 2,000–3,000 cells. Cells were synchronized for 24–48 h by replacing the growth medium with serum-free supplemented medium. The serum-free medium was changed to complete growth medium. A series of concentrations (0.4 μM, 0.08 μM, 0.016 μM, and 0.003 μM) of a NO donor in DMSO [25] solution were added into each well of a 96-well plate and subsequently incubated at 37 C in a 95% air-5% CO2 incubator for 48 hours. Approximately 12 wells were treated with identical concentrations of the NO-donor to address statistical analysis. The byproduct of the NO-donor, a secondary amine, was used as the control. Into each well, 10 μL MTT solution was added (viable cells convert MTT to formazan). The plate was incubated at 37 C for 4 hours. Medium was removed and 200 μL detergent (ATCC) was added into each well to dissolve the formazan formed during incubation by pipetting the solution up and down several times. The absorbances of the solution in each well were measured on the plate reader with a test wavelength of 570 nm and a reference wavelength of 600 nm to obtain sample signals. Comparisons between treatment groups were made by t-test using the Minitab software program. A value of P < 0.05 was considered statistically significant.
RESULTS AND DISCUSSION
Preparation and characteristics of compounds 1–9 and 1’–9’
In our previous communication we have reported in detail the preparation and properties of the N-nitrosamines with one electron withdrawing group at the ortho position in detail.26 The reaction of compound a (Scheme 1) with two equivalents of hexylamine did not result in the formation of the desired secondary amines, with the displacement of fluorine atoms. This was due to the fact that amines are weak nucleophiles and cannot displace the fluorine atoms by SNAr (aromatic nucleophilic substitution reactions). Therefore, it was necessary to introduce additional electron withdrawing groups into the benzene rings ortho to the fluorine atoms in order to render the fluorine atoms more labile. One of the most convenient approaches would be to introduce electron withdrawing -NO2 group. 4,4'-sulfonylbis(1-fluoro-2-nitrobenzene), b, was synthesized to achieve this objective. Secondary amines 1–9 could be prepared using b and a series of aliphatic and aromatic amines (Scheme 1). The structural features of N-nitrosamines 1’–9’ and their NO release rate provided for a better understanding of the effects these structural variations on the NO release rates.
Scheme 1.

Preparation of secondary amines and N-nitroso compounds.
The reaction of 4,4'-sulfonylbis(1-fluoro-2-nitrobenzene), b, and hexan-1-amine was conducted in NMP at 50 °C for 3hrs. The reaction temperature was critical for this reaction. At elevated reaction temperature (90 °C), in the absence to the primary amines, loss of 4,4'-sulfonylbis(1-fluoro-2-nitrobenzene) was observed with the formation of a major product in addition to other minor products. The major product was separated by column chromatography and analyzed by H-1 NMR (broad s, 12 ppm) and IR (3400 cm−1 band) spectroscopic techniques. The presence phenolic hydroxyl group, and the loss of fluorine atoms (F-19 NMR) were evident. These observations suggested that with the introduction of the nitro groups the fluorine atoms become highly labile and react with residual water present in NMP. Therefore attempts to make a high molecular weight polymer from 4,4'-sulfonylbis(1-fluoro-2-nitrobenzene) and 1,6-diaminohexane was abandoned, since it would be impossible to maintain 1:1 stoichiometry between the diamines and the activated difluoride during the reaction at a temperature above 150 °C due to the undesirable side reaction mentioned above. Identical results were obtained when DMAC instead of NMP was used as the solvent.
The reaction of 4,4'-sulfonylbis(1-fluoro-2-nitrobenzene) and hexylamine to yield 1 could be quantitatively accomplished in NMP with relative ease by lowering the reaction temperature and by increasing the molar excess of hexylamine. Thus, the competitive reaction with H2O could be prevented. The 1H-NMR of compound 1 is displayed in Fig. 1. An examination of Fig.1 indicates the vastly different chemical environments of the three aromatic protons. Furthermore, the absorbance due to the two methylene units adjacent to the secondary amine groups appear at ~3.2 ppm-significantly more downfield than the remainder of the aliphatic protons.
Figure 1.
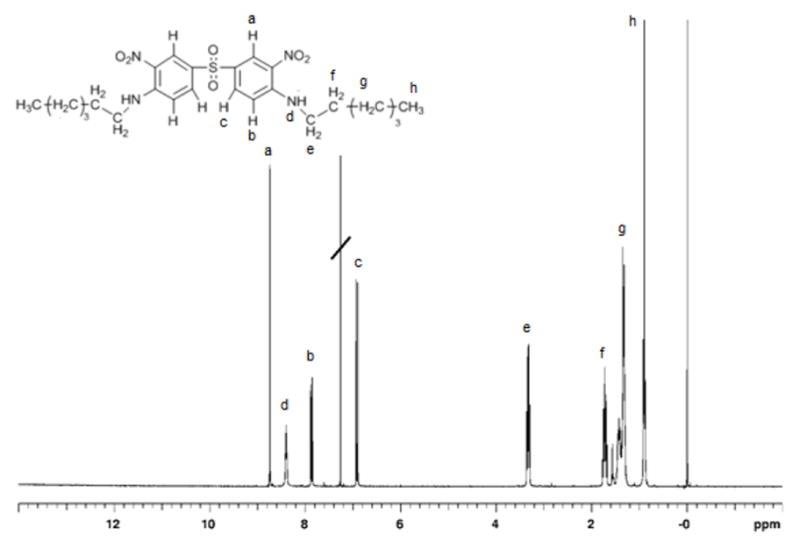
1H NMR spectrum (CDCl3) of compound 1.
Measurements of concentration NO content in 1’–9’ and released NO
N-nitrosated model compounds release NO readily in solution. This precluded the use of NMR analyses to measure the conversion of N-NH to N-NO moiety. Therefore, IR spectroscopy was used to monitor the extent of nitrosation [26]. The N-N bond of nitrosamines exhibits a strong absorbance at ~1040 cm−1. This is evident from an examination of the IR spectrum of 1’(Fig. 2). The other strong absorbance ~1450 cm−1 due to N=O group was difficult to identify due to the presence of aromatic nitro groups. The absorbance corresponding to the N-H stretch in secondary amine at 3370 cm−1, decreases in intensity after nitrosation. Thus the conversion from secondary amines into N-nitrosated materials could be easily determined by measuring the change of the ratios of the absorbances of N-H and the C=C groups (1608 cm−1, benzene ring) in the IR spectra. The percent of nitrosation of compounds 1’–9’varied between 46–86%. Details are provided as supplementary data (Tab. 1).
Figure 2.
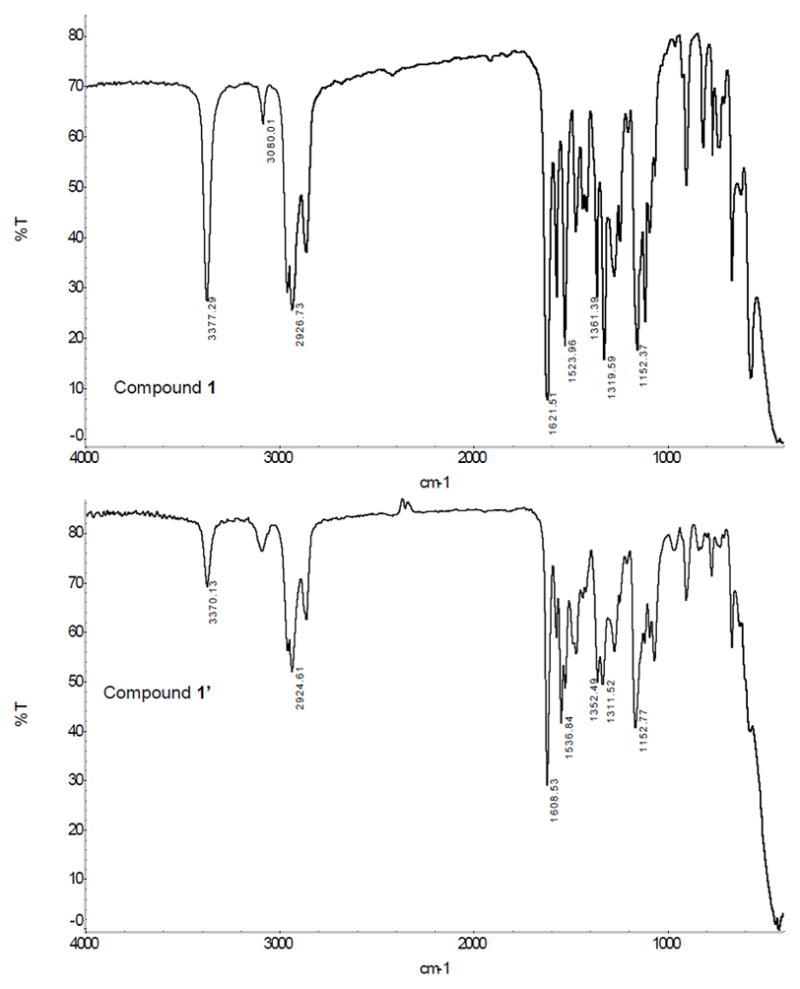
IRspectra (Neat) of compounds1 and 1’.
The intensity of the absorbance corresponding to N-H stretch of the secondary amine (compound 1) at 3370 cm−1, decreases after nitrosation. Nitrosamine (compound 1’) also exhibits a strong absorbances due to N-N bond at ~1040 cm−1and at 1608 cm−1 due to C=C bonds of the benzene rings.
NO release rates of N-nitroso derivatives (1’–9’) were monitored in PBS over a period of 30 days using Griess reagent. Data from these experiments are displayed in Fig. 3. This study was undertaken for two important reasons. First, it helped us understand the effect of variation of aliphatic chain length on the NO release rates. Second, it allowed for the measurements of their amount and rates of NO release, which are of crucial importance for possible future in vivo applications. The absorbance of the sample was used to calculate NO concentration using the calibration plot provided by the supplier. Moles of released NO, was divided by the moles the NO donor to obtain percentage of released NO. The number of moles of NO donor was calculated from the weight, degree of nitrosation and the molecular weights of the secondary amine and that of the nitrosated ones. The data obtained were then plotted versus time (Fig. 3a and 3b). An examination of Fig. 3 suggests that NO release takes place in three distinct phases. During the initial phase, the release rate is extremely fast. In the second phase, it is slower and the rate slows down further and remains essentially the same during the final stage.
Figure 3.

NO Release Profiles of N-nitrosamines 1’–9’.
Percentages of released NO obtained are plotted versus time. Aliphatic compounds 1’– 5’ are displayed in panel a; aromatic compounds 6’–9’ are displayed in panel b. NO release takes place in three distinct phases. During the initial phase, the release rate is extremely fast. In the second phase, it is slower and the rate slows down further and remains essentially the same during the final stage.
An analysis of Figure 3a indicates that the NO release rate increases systematically with the increasing the number of methylene groups. This increase may be because of the electron donating effect of the increasing number of these methylene units, which allows for the stabilization of the protonated N atom bearing the nitroso group prior to the release of the NO moiety. The same conjecture applies equally well if NO leaves as a radical. These observations point to the significance of the length of the aliphatic units attached directly to the N-nitroso groups. These observations were substantiated further by means of the NO release profiles of aromatic N-nitroso compound 1’–9’.
The NO release profiles for aromatic N-nitroso compound 6’–9’ are displayed in Figure 3b. From an examination of this figure, it is clear that the nature of the substituent, Y, plays a critical role in determining the NO-release rates. Compound 6’with a hydrogen atom at the para position with respect to the N-nitroso group exhibits the lowest NO release rate. However this rate increases significantly when the hydrogen atom is replaced with alkyl groups (e.g. isopropyl, t-butyl) with increasing electron donating inductive effects. Maximum NO release rate was achieved with the methoxy substituent (a five fold increase in the total NO release compared to 6’). The exceptional electron donating ability of a methoxy group is well documented [16].
Inhibitory effect of released NO on human aortic smooth muscle cell
In order to evaluate the inhibitory effects of the released NO on SMC proliferation, HASMC were chosen as the target. The inhibitory effect of NO on smooth muscle cell proliferation was investigated using N-nitroso compounds (1’–5’). Data obtained from cell culture studies with HASMC using 1’ at various μM concentrations are displayed in Figure 4.
Figure 4.
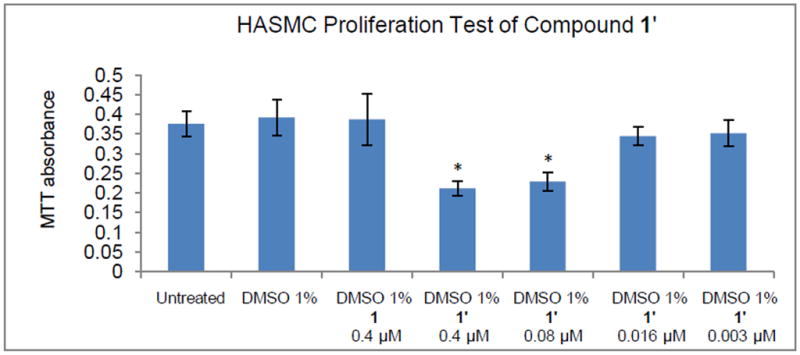
HASMC proliferation test using compound 1’. * indicates statistically significant difference (P < 0.05) from control (untreated).
An examination of the Fig. 4 clearly suggests the following. HASMC treated with 1% DMSO has almost (not significant, P > 0.01) the same absorbance as untreated HASMC, indicating that 1% DMSO does not interfere with MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] method for the measurement of cell proliferation test of HASMC. HASMC treated with 1% DMSO also has almost (not significant, P > 0.01) the same absorbance as HASMC treated with 0.4 μM compound 1 in 1% DMSO. This similarity indicates that secondary amine 1 is not cytotoxic to HASMC and does not interfere with the results of MTT cell proliferation test. A significant ~35% (P < 0.05) decrease of SMC proliferation was achieved at 0.4 μM and ~30% (P < 0.05) decrease at 0.08 μM concentration level of NO donor 1’ compared to controls (Fig. 4). This observation suggests that the NO inhibition of proliferation can be achieved using the DMSO soluble N-nitroso NO-donor 1’. With decreased concentration of NO donor 1’ (0.016 μM and 0.003 μM) the inhibition of proliferation was insignificant (~10%). Attempts at using higher concentrations of N-nitrosamines (>0.4 μM) failed due to the poor solubility of these N-nitrosamines in aqueous solution. The N-nitrosamines precipitated from the cell culture medium, which interfered with optical measurements at 570 nm.
Inhibitory effect data of five NO donors 1’–5’ are displayed in Table 1. Data are expressed as percentage of control (assigned 100%), where the control represents cells grown in the absence of added test agents. HASMC treated with 1% DMSO also had a significantly (P < 0.05) higher absorbance than HASMC treated with 0.4 μM compound 4 in 1% DMSO, indicating that secondary amine 4 exhibits cytotoxic properties or it interferes with the MTT cell proliferation test. Significant decreases of SMC proliferation at concentrations ranging from 0.4 μM to 0.003 μM (compound 4’) were not observed. Data obtained from cell culture studies with HASMC using 5’ at various μM concentrations shows similar results as seen with compound 4’. It is likely that the cytotoxicities of compounds 4’ and 5’ are due to the hydrophobicity of the longer aliphatic chain.
Table 1.
Percent Inhibition of HASMC with N-nitrosamine 1’–5’
| Concentration (μM) | Inhibition percentage (untreated HASMC assigned 100%) | ||||
|---|---|---|---|---|---|
| 1’ | 2’ | 3’ | 4’ | 5’ | |
| 0.4 | 35%* | 40%* | 40%* | 20% | 15% |
| 0.08 | 30%* | 35%* | 30%* | 15% | 15% |
| 0.016 | 10% | 15% | 15% | 10% | 15% |
| 0.003 | 9% | 15% | 15% | 5% | 20% |
signifies statistically significant difference (P < 0.05) from control (untreated)
N-nitrosamines 1’–3’ each inhibited HASMC proliferation in a concentration-dependent manner (Table 1, 2Figure 5). Compound ’, which was more potent than compound 1’ and 3’, exhibited the most significant inhibitory effect at concentrations of 0.016 μM and 0.4 μM. The by-product of N-nitrosamines (0.4 μM in DMSO) after NO release, a secondary amine, as well as 100% DMSO did not exhibit cytotoxic behaviors in our studies. More importantly, the inhibition of HASMC proliferation was achieved at a very low NO-donor concentration compared to conventional NO-donors. For example, SMC proliferation was significantly inhibited after the administration of 50 μM of spermine-based diazenuimdiolate [27] and 10 μM diethylenetriamine-NO (DETA-NO) [28].
Figure 5.

Inhibitions of HASMC proliferation by N-nitrosamines, 1’–3’. Data were expressed as percentage of control (assigned 100%), where control represents cells grown in the absence of added test agents. * signifies statistically significant difference (P < 0.05) from control (untreated).
NO reacts rapidly with oxygenated heme proteins to produce peroxynitrite [29]. Peroxynitrite can be decomposed by water soluble metalloporphyrin. Excessively high levels of NO can cause accumulation of peroxynitrite, which leads to the release of cytochrome c from mitochondria, activation of caspases, and initiation of cell apoptosis [29]. The concentration dependent effects of NO on cell proliferation (pico to nano molar) and apoptosis (from micro to milli molar) suggest that there are other pathways involved in anti-proliferation effect of NO. Indeed recent evidence suggested that NO was a potent inducer of heme oxygenase 1(HO-1) gene expression in vascular SMC, resulting in an increase in production of carbon monoxide (CO), which in turn inhibited cellular proliferation [30]. Exogenous NO donors induce SMCs apoptosis in a concentration- and time-dependent manner. The membrane-permeable cGMP analogue, dibutyryl-cGMP, did not induce SMCs apoptosis, and the highly selective inhibitor of cGMP-dependent protein kinase, KT5823, was unable to inhibit the induction of NO-induced SMCs apoptosis [31]. These findings indicate that NO inhibits SMC proliferation through cGMP-independent mechanism. NO may have anti apoptosis effects by inactive modification of key proteins, which regulate apoptosis. The original work by Jiang and colleagues [32] demonstrated that S-nitrosylation of caspase-3 is the mechanism by which fibroblasts manifest lower apoptosis. NO donor S-nitroso albumin (SNO-Alb) reported by Zhang’s group revealed a protective role in rat pulmonary artery SMC apoptosis [33]. As discussed above, additional studies are warranted to understand the role of NO involved in inhibiting cell proliferation.
In conclusion, N-nitrosamines reported herein exhibit a slow and sustained release of NO, which is of significant advantage in biological studies. At low (pico-molar to nano-molar)levels [34], NO exerts beneficial effects, including inhibition of SMC proliferation and reduces the injury responses within the blood vessel wall [35]. Since NO can pass through cell membranes freely, excess NO released in bursts of high concentrations (micro-molar) is likely to react with superoxide resulting to form copious quantities of reactive nitrogen species (RNS) which are more cytotoxic than NO [31]. In addition, NO donors, which release NO in bursts, usually exhibit relatively short half-lives. Furthermore, high levels of RNS cause depletion of reductive species (GSH) resulting in programmed cell death [36]. Repeated treatment of the cells with high concentrations of NO would exacerbate the cytotoxic effects of these NO donors [37]. These observations suggest that suppressing HASMC proliferation effects of slow and sustained release NO donors may have important therapeutic benefits for atherosclerosis, hypertension and endothelial dysfunction related diseases.
Supplementary Material
Acknowledgments
The work was supported in part by National Institute of Health (NIH) Award Number R15HL 106600 from the National Heart, Lung and Blood Institute (NHLB). The content is solely the responsibility of the authors and does not represent the official views of NHLB or NIH. Discussions with Professor David Ash during the course of this study and during manuscript preparation were extremely helpful.
References
- 1.Bian K, Doursout MF, Murad F. Vascular system: role of nitric oxide in cardiovascular diseases. J Clin Hypertens (Greenwich) 2008;10:304–10. doi: 10.1111/j.1751-7176.2008.06632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Govers R, Rabelink TJ. Cellular regulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol. 2001;280:F193–206. doi: 10.1152/ajprenal.2001.280.2.F193. [DOI] [PubMed] [Google Scholar]
- 3.Anderson TJ. Nitric oxide, atherosclerosis and the clinical relevance of endothelial dysfunction. Heart Fail Rev. 2003;8:71–86. doi: 10.1023/a:1022199021949. [DOI] [PubMed] [Google Scholar]
- 4.Cohen RA. The role of nitric oxide and other endothelium-derived vasoactive substances in vascular disease. Prog Cardiovasc Dis. 1995;38:105–28. doi: 10.1016/s0033-0620(05)80002-7. [DOI] [PubMed] [Google Scholar]
- 5.Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–76. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 6.Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol. 1987;92:639–46. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83:1774–7. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooke JP, Dzau VJ. Nitric oxide synthase: role in the genesis of vascular disease. Annu Rev Med. 1997;48:489–509. doi: 10.1146/annurev.med.48.1.489. [DOI] [PubMed] [Google Scholar]
- 9.Calabrese V, Cornelius C, Rizzarelli E, Owen JB, Dinkova-Kostova AT, Butterfield DA. Nitric oxide in cell survival: a janus molecule. Antioxid Redox Signal. 2009;11:2717–39. doi: 10.1089/ars.2009.2721. [DOI] [PubMed] [Google Scholar]
- 10.Sawa T, Zaki MH, Okamoto T, Akuta T, Tokutomi Y, Kim-Mitsuyama S, et al. Protein S-guanylation by the biological signal 8-nitroguanosine 3',5'-cyclic monophosphate. Nat Chem Biol. 2007;3:727–35. doi: 10.1038/nchembio.2007.33. [DOI] [PubMed] [Google Scholar]
- 11.Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333:664–6. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 12.Napoli C, Ignarro LJ. Nitric oxide-releasing drugs. Annu Rev Pharmacol Toxicol. 2003;43:97–123. doi: 10.1146/annurev.pharmtox.43.100901.140226. [DOI] [PubMed] [Google Scholar]
- 13.Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–28. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- 14.Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu G, del Soldato P. Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A. 2001;98:4202–8. doi: 10.1073/pnas.071054698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang PG, Xian M, Tang X, Wu X, Wen Z, Cai T, et al. Nitric oxide donors: chemical activities and biological applications. Chem Rev. 2002;102:1091–134. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 16.Wang PG, Tingwei BC, Holder AA. In: Nitric Oxide Donors For Pharmaceutical and Biological Applications. Wang PG, Cai TB, Taniguchi N, editors. Wiley-VCH, Weinheim; Germany: 2005. pp. 3–31. [Google Scholar]
- 17.Chen Z, Stamler JS. Bioactivation of nitroglycerin by the mitochondrial aldehyde dehydrogenase. Trends Cardiovasc Med. 2006;16:259–65. doi: 10.1016/j.tcm.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Daiber A, Oelze M, Wenzel P, Wickramanayake JM, Schuhmacher S, Jansen T, et al. Nitrate tolerance as a model of vascular dysfunction: roles for mitochondrial aldehyde dehydrogenase and mitochondrial oxidative stress. Pharmacol Rep. 2009;61:33–48. doi: 10.1016/s1734-1140(09)70005-2. [DOI] [PubMed] [Google Scholar]
- 19.Wimalawansa SJ. Nitric oxide: new evidence for novel therapeutic indications. Expert Opin Pharmacother. 2008;9:1935–54. doi: 10.1517/14656566.9.11.1935. [DOI] [PubMed] [Google Scholar]
- 20.Davis KL, Martin E, Turko IV, Murad F. Novel effects of nitric oxide. Annu Rev Pharmacol Toxicol. 2001;41:203–36. doi: 10.1146/annurev.pharmtox.41.1.203. [DOI] [PubMed] [Google Scholar]
- 21.Ekmekcioglu S, Tang CH, Grimm EA. NO news is not necessarily good news in cancer. Curr Cancer Drug Targets. 2005;5:103–15. doi: 10.2174/1568009053202072. [DOI] [PubMed] [Google Scholar]
- 22.Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev. 2007;27:317–52. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 23.Neeb L, Reuter U. Nitric oxide in migraine. CNS Neurol Disord Drug Targets. 2007;6:258–64. doi: 10.2174/187152707781387233. [DOI] [PubMed] [Google Scholar]
- 24.Sawa T, Arimoto H, Akaike T. Regulation of redox signaling involving chemical conjugation of protein thiols by nitric oxide and electrophiles. Bioconjug Chem. 2010;21:1121–9. doi: 10.1021/bc900396u. [DOI] [PubMed] [Google Scholar]
- 25.Asmis L, Tanner FC, Sudano I, Luscher TF, Camici GG. DMSO inhibits human platelet activation through cyclooxygenase-1 inhibition. A novel agent for drug eluting stents? Biochem Biophys Res Commun. 2010;391:1629–33. doi: 10.1016/j.bbrc.2009.12.102. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Teng Y, Yu H, Oh-Lee J, Mohanty DK. Preparation and Properties of Polyamines: Part II-Controlled and sustained Release of Nitric Oxide (NO) from Nitrosated Polymers. Polymer Journal. 2009;41(9):715–725. [Google Scholar]
- 27.Wedgwood S, Black SM. Molecular mechanisms of nitric oxide-induced growth arrest and apoptosis in fetal pulmonary arterial smooth muscle cells. Nitric Oxide. 2003;9:201–10. doi: 10.1016/j.niox.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 28.Jobgen WS, Fried SK, Fu WJ, Meininger CJ, Wu G. Regulatory role for the arginine-nitric oxide pathway in metabolism of energy substrates. J Nutr Biochem. 2006;17:571–88. doi: 10.1016/j.jnutbio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Su J, Groves JT. Mechanisms of peroxynitrite interactions with heme proteins. Inorg Chem. 2010;49:6317–6329. doi: 10.1021/ic902157z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang YF, Tian H, Tang CS, Jin HF, Du JB. Nitric oxide modulates hypoxic pulmonary smooth muscle cell proliferation and apoptosis by regulating carbon monoxide pathway. Acta Pharmacol Sin. 2007;28:28–35. doi: 10.1111/j.1745-7254.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- 31.Nishio E, Fukushima K, Shiozaki M, Watanabe Y. Nitric oxide donor SNAP induces apoptosis in smooth muscle cells through cGMP-independent mechanism. Biochem Biophys Res Commun. 1996;221:163–168. doi: 10.1006/bbrc.1996.0563. [DOI] [PubMed] [Google Scholar]
- 32.Jiang ZL, Fletcher NM, Diamond MP, Abu-Soud HM, Saed GM. S-nitrosylation of caspase-3 is the mechanism by which adhesion fibroblasts manifest lower apoptosis. Wound Repair Regen. 2009;17:224–229. doi: 10.1111/j.1524-475X.2009.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li HH, Xu J, Wasserloos KJ, Li J, Tyurina YY, Kagan VE, Wang X, Chen AF, Liu ZQ, Stoyanovsky D, et al. Cytoprotective effects of albumin, nitrosated or reduced, in cultured rat pulmonary vascular cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L526–533. doi: 10.1152/ajplung.00282.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouley R, Breton S, Sun T, McLaughlin M, Nsumu NN, Lin HY, et al. Nitric oxide and atrial natriuretic factor stimulate cGMP-dependent membrane insertion of aquaporin 2 in renal epithelial cells. J Clin Invest. 2000;106:1115–26. doi: 10.1172/JCI9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–9. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 36.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–42. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 37.Bauer PM, Fukuto JM, Buga GM, Pegg AE, Ignarro LJ. Nitric oxide inhibits ornithine decarboxylase by S-nitrosylation. Biochem Biophys Res Commun. 1999;262:355–8. doi: 10.1006/bbrc.1999.1210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.