

Abstract
LuxS (S-ribosylhomocysteinase) catalyzes the cleavage of the thioether linkage of S- ribosylhomocysteine (SRH) to produce homocysteine and 4,5-dihydroxy-2,3-pentanedione (DPD), the precursor to a small signaling molecule that mediates interspecies bacterial communication called autoinducer 2 (AI-2). Inhibitors of LuxS should interfere with bacterial interspecies communication and potentially provide a novel class of antibacterial agents. In this work, SRH analogues containing substitution of a nitrogen atom for the endocyclic oxygen as well as various deoxyriboses were synthesized and evaluated for LuxS inhibition. Two of the [4-aza]SRH analogues showed modest competitive inhibition (KI ~40 μM), while most of the others were inactive. One compound that contains a hemiaminal moiety exhibited time-dependent inhibition, consistent with enzyme-catalyzed ring opening and conversion into a more potent species (KI* = 3.5 μM). The structure-activity relationship of the designed inhibitors highlights the importance of both the homocysteine and ribose moieties for high-affinity binding to LuxS active site.
Keywords: Azahemiacetals, Azasugars, homocysteine, LuxS, S-ribosylhomocysteinase
Introduction
Quorum sensing (QS) is a type of bacterial cell-to-cell communication mediated through the production, release and detection of the small signaling molecules called autoinducers (AIs).1-3 Such communication allows bacterial control of crucial functions in united communities for enhancement of symbiosis, virulence, antibiotic production, biofilm formation, and many other processes.4-5 Hence, there have been great interests in the synthesis of small molecules that can modulate QS pathways.6-8 S-Ribosylhomocysteinase (LuxS) is a key enzyme in the biosynthetic pathway of type II autoinducer, which mediates the interspecies quorum sensing among both Gram-positive and Gram-negative bacteria.
The biosynthesis of AI-2 starts with the dual substrate-specific microbial enzyme 5′-methylthioadenosine/AdoHcy nucleosidase (MTAN), which catalyzes the depurination of S-adenosyl-L-homocysteine (SAH), a byproduct of many S-adenosyl-L-methionine-dependent methyltransferases reactions, to form S-ribosyl-L-homocysteine (SRH, Figure 1).9-10 SRH is subsequently converted to L-homocysteine and 4,5-dihydroxy-2,3-pentadione (DPD) by the LuxS enzyme.11 DPD undergoes spontaneous cyclization to 1e and complexation with borate to form a furanosyl borate diester, which acts as the AI-2 in some bacteria.11-12 Chemical synthesis of the unstable DPD has been accomplished recently by the groups of Janda13 and Semmelhack,14 which allowed the vital complexation properties of DPD with borate15 to be studied and provided DPD as a reliable standard for investigation of AI-2 regulated QS processes.
Figure 1.

Biosynthetic pathway to AI-2. Enzymatic conversion of SRH to DPD by LuxS .
LuxS is a small metalloenzyme (157 amino acids in the Bacillus subtilis enzyme) containing Fe2+ coordinated by His-54, His-58, Cys-126, and a water molecule. The native enzyme is unstable under aerobic conditions, but substitution of Co2+ for Fe2+ gives a highly stable variant with essentially wild-type catalytic activity.16-18 In the proposed catalytic mechanism, LuxS catalyzes consecutive aldose-ketose (1a → 1b) and ketose-ketose (1b → 1c) isomerization steps and then β-elimination of Hcy from a 3-keto intermediate (1c → 1d) to form DPD.11,19 LuxS-catalyzed cleavage of the C5–S thioether bond in SRH is analogous to that of SAH hydrolase, which effects cleavage of an equivalent thioether bond in SAH by first oxidizing the C3′ secondary alcohol into a ketone with an NAD+ cofactor.20-21
Zhou et al. designed and synthesized two LuxS substrate analogues, the S-(anhydroribosyl)-L-homocysteine (2) and S-(homoribosyl)-L-cysteine compounds, which blocked initial and final mechanistic steps, respectively (Figure 2).22 Pei et al. have prepared a series of stable analogues of the putative enediolate intermediate, some of which showed submicromolar inhibition of the enzyme (e.g., KI = 0.72 μM for isostere 3).23 Zhang et al. found that the brominated furanones 4 covalently modify and inactivate LuxS.24 Recognizing structural similarities between substrates of mammalian AdoHcy hydrolase and bacterial S-ribosylhomocysteine (SRH) hydrolase (LuxS enzyme), we designed and synthesized SRH analogues with 6-(fluoro)vinyl moiety in place of the C5 and sulfur atoms which acted as weak/moderate inhibitors of LuxS enzyme.25 The SRH analogues 5 lacking enolizable hydroxyl group at C3 were found to be competitive substrate of LuxS.26-27 The time dependence inhibition with C3 halogenated substrates was caused by enzyme-catalyzed elimination of halide ions.27
Figure 2.

LuxS inhibitors
Here, we report synthesis of [4-aza]SRH mimics in which the furanose ring oxygen has been replaced by a nitrogen atom. The resulting hemiaminals should have different stabilities28 relative to the O,O-hemiacetals present in SRH and as a result different rates of metabolic alteration. The higher basicity of the aza analogues is expected to have different binding strengths and rates for productions of the open chain aldehyde form–necessary for the first isomerization to occur. Also, the aminosugars can be protonated at physiological pH and the corresponding positive charge may have an effect on binding to the enzymatic active site. Azasugars29 have been found to be potent inhibitors of glycosidases and glycosyltransferases30-31 and have been targeted as transition-state models.32-33 The 4′-azanucleosides34 function as transition-state inhibitors of MTAN at the femtomolar level.35-36
Results and Discussion
Chemistry
Our first target was 1,4-dideoxy-[4-aza]SRH 12 lacking the hydroxyl group at C1 (Scheme 1). Compound 12 cannot undergo ring opening (which will preclude the initial step of the LuxS-catalyzed reaction) and may act as a competitive inhibitor of LuxS. Synthesis of 12 started with the protected 1-amino-1,4-anhydro-1-deoxy-D-ribitol 6, which was readily prepared from the commercially available D-gulonic acid γ-lactone.37 However, attempted mesylation of the N-benzyl protected 6 resulted in the formation of piperidine derivative 13 as a mixture of two diastereomers (~3:1). Presumably, the mesylated pyrrolidine underwent a rearrangement reaction into the piperidines through an aziridine intermediates.38-39 We found that replacement of the benzyl protecting group at ring nitrogen of 6 with a Boc group suppressed the nucleophilicity of the nitrogen and prevented ring expansion, allowing the formation of stable 5-O-mesyl derivatives. Thus, silylation of 6 with TBDMSCl and subsequent hydrogenation (5% Pd/C) in the presence of (Boc)2O39-40 yielded 8 (97% from 5). Desilylation of 8 (70%) followed by mesylation gave 10 as a stable compound (96%). Displacement of the mesylate group with a thiolate, generated by reduction of properly protected L-homocystine19 with water soluble tris(2-carboxyethyl)phosphine hydrochloride,26 gave thioether 11 (86%). Treatment of 11 with TFA effected simultaneous removal of the N-Boc, acetonide and t-butyl ester protection groups to give the desired [4-aza]SRH analogue 12 in good yields (66%).
Scheme 1.

Reagents and conditions: (a) TBDMSCl/imidazole/DMAP/CH2Cl2/rt; (b) H2/Pd-C/(Boc)2O/Et3N/EtOH/rt; (c) MsCl/Et3N/CH2Cl2/rt; (d) TBAF/THF/rt; (e) BocNHCH(CH2CH2SH)CO2t-Bu/LDA/DMF; (f) (i) TFA, (ii) TFA/H2O.
The second target was γ-lactam 21, which contains an amide carbonyl at C1 and nitrogen as a replacement of the ring oxygen (Scheme 2). It is noteworthy that, as opposed to the [4-aza]SRH analogue 12 (or 23), the lactam nitrogen cannot be protonated at physiological pH. Selective oxidation of the 5-O-TBDMS-azasugar 7 at C1 with RuO2/NaIO4 under EtOAc/H2O biphasic conditions41 produced N-benzyl lactam 14a (65%) and a small amount (18%) of the corresponding N-benzoylpyrrolidinone byproduct, resulted from oxidation of the benzylic carbon of the N-protecting group. Desilylation of 14a with TBAF, followed by mesylation and displacement of the mesylate group with protected Hcy gave thioether 18. Treatment of 18 with TFA removed all of the acid-labile protection groups to yield N-benzyl protected [4-aza]SRH lactam 20 (48%). However, all attempts to remove the N-benzyl group from 18 or 20 (to yield 21) were unsuccessful [e.g., H2/Pd-C or Pd(OH)2-C, Na/NH3(liq.), BCl3]. Our attempt to mesylate the N-Boc protected 15b (prepared by RuO2-catalyzed oxidation of 8 and desilylation of the resulting 15a) failed to produce 17, yielding only the starting material 15b. Fortunately, oxidation of the 5-O-mesyl and N-Boc protected pyrrolidine 10 with RuO2/NaIO4 afforded 17 efficiently (95%). Coupling of 17 with homocysteinate afforded thioether 19a with concomitant loss of Boc group at ring nitrogen. Subsequent deprotection with TFA followed by TFA/H2O gave [4-aza]SRH lactam 21 (58%).
Scheme 2.

Reagents and conditions: (a) NaIO4/RuO2 x H2O/EtOAc/H2O/rt; (b) TBAF/THF/rt; (c) MsCl/Et3N/CH2Cl2/rt; (d) BocNHCH(CH2CH2SH)CO2t-Bu/LDA/DMF; (e) (i) TFA, (ii) TFA/H2O; (f) (Boc)2O/Et3N/CH2Cl2/rt; (g) LiEt3BH/THF/−78 °C.
Our next target was hemiaminal 23. Since only the open aldehyde form of SRH is catalytically active, we were interested in the effect of the nitrogen substitution on the ring opening. The existence of azahemiacetals in equilibrium with dehydrated form (imine) as well as with open aldehyde and dimeric forms was reported for 4-azapentofuranoses.42-43 It is noteworthy that sugar N,O-acetals were found to be stable enough to undergo coupling with nucleoside bases,41 or transformation to proline.44 Although direct reduction of lactam 21 (or 19a) with LiBEt3H failed to yield hemiaminal 23 (or 22), the protection of the ring nitrogen with a Boc group facilitated the reduction reaction.44 Thus, treatment of 19a with (Boc)2O/DMAP gave N-Boc protected lactam 19b (93%) which upon treatment with LiBEt3H produced hemiaminal 22 (92%) as a mixture of two anomers. Deprotection of 22 with TFA followed by TFA/H2O gave desired [4-aza]SRH (N,O-acetal) analogue 23 (72%) as a mixture of α/β anomers. Interestingly, no free aldehyde or imine proton peaks were visible on 1H NMR spectra. Compound 23 is stable when kept at 4 °C but decomposes slowly in solution at ambient temperature especially at basic pH.
To determine whether the cyclic [4-aza]SRH exists in equilibrium with the open chain aldehyde form, we carried out a limited model study. Thus, N-Boc protected lactam 15a44 was reduced with LiBEt3H to afford protected hemiaminal 24a (Scheme 3). Desilylation with TBAF yielded 24b, which was treated with TFA to give deprotected hemiaminal 25a as a mixture of anomers susceptible to dehydration at pH higher than 7 to form imine 25c.43 Subsequent treatment of 25a with O-benzylhydroxylamine gave expected oxime 26 as the only product. The formation of oxime 26 indicates that azasugar 25a exists in equilibrium with the open aldehyde form (25b) and that the equilibrium could be shifted by subsequent transformations.
Scheme 3.

Reagents and conditions: (a) LiEt3BH/THF/−78 °C; (b) TBAF/THF/rt; (c) (i) TFA/0 °C, (ii) TFA/H2O/0 °C; (d) BnONH2/pyr/rt.
2,3,4-Trideoxy-[4-aza]SRH 38 lacking the enolizable hydroxyl groups at C2 and C3 was next prepared to examine the importance of C2 and C3-OH groups for LuxS binding and catalysis. The key starting material (S)-5-(bromomethyl)-2-pyrrolidone (27) was conveniently prepared from L-pyroglutamic acid45 (Scheme 4). Displacement of the bromide in 27 with the L-homocysteinate afforded thioether 33 (79%), which was deprotected with TFA quantitatively to give 2,3-dideoxy-4-azaSRH analogue 34 as a trifluoroacetate. Displacement with the unprotected D/L-homocysteine produced racemic 36 (75%) as a sodium salt, which upon treatment with TFA was also converted to its trifluoroacetate salt. As expected, 1H NMR spectrum of 34 showed only one set of peaks which are present in the spectrum of racemic 36. Treatment of 33 with (Boc)2O/DMAP gave the N-Boc protected lactam 35, which was reduced with LiBEt3H to give hemiaminal 37. Subsequent deprotection with TFA produced 38.
Scheme 4.

Reagents and conditions: (a) RSH/NaH/DMF; (b) (Boc)2O/DMAP/CH2Cl2; (c) LiEt3BH/THF/CH2Cl2/−78 °C; (d) TFA; (e) BocNHCH(CH2CH2SH)CO2t-Bu/LDA/DMF; (f) D/L-Hcy/NaH/DMF; (g) BnONH2/pyr
The 5-S-alkyl-2,3-dideoxy-[4-aza]SRH (e.g., 28/29) and the 5-S-alkyl-[4-aza]SRH analogues with different length of the alkylthio chain were also prepared.46 These cyclic azahemiacetals and their ancestor lactams were found to modulate Pseudomonas aeruginosa QS.46 The alkylthiomethyl azahemiacetal 28/29 existed in solution as an equilibrium mixture of anomers along with the open chain aldehydes [5-25%, 1 H NMR (δ 8.90), 13C NMR (δ 180.8)] and the corresponding imines 30/31 [3-30%; 1 H NMR (δ 7.63), 13C NMR (δ 167.0)].46 Treatment of 28 with O-benzylhydroxylamine also produced the expected oxime 32,46 as observed for 25a.
To explore the possibility of the LuxS-mediated addition of a water across carbon-nitrogen double bond, we synthesized an imine-type analogue 43 (Scheme 5). The precursor 1-methyliminocyclitol 39 was prepared by the Moriarty rearrangement47 of the exo-imino to endo-iminocyclitol, which involves inversion at C4 of the L-lyxo sugar to give the D-ribo azasugar. The imine 3947 was mesylated at the primary alcohol to give 40 (85%), which was coupled with protected L-Hcy to give 41 (Scheme 5, 85%). Treatment of 41 with TFA for a short time gave only isopropylidene protected 42. We found that the protons at the C1-methyl group are exchangeable with deuterium within few hours when compound 42 is dissolved in D2O. Treatment of 42 with aqueous TFA (9:1) yielded fully deprotected 43 in quantitative yield. Protons at C1-methyl group of 43 were also exchangeable with deuterium. These exchange indicate that 1,4-ketimine-SRH analogue 43 might be expected to undergo enzyme-catalyzed hydrolysis to generate a [4-aza]SRH analogue with a methyl ketone rather than an aldehyde at C1. This change might affect the regioselectivity and rate of the first isomerization step in the LuxS-catalyzed reaction. We also proved that the methyl group protons in 39 are not susceptible to exchange even if 39 was dissolved in D2O for several hours. Additionally, we noticed that observed low rate of exchange in 39 (relatively to 42 and 43) can be enhanced exclusively in the presence of acid or amino acid (TFA and glycine were used, respectively). Attempted, one-step deprotection of 41 with BCl3 led to a partial loss of chirality at C9 giving 43 as a mixture of diastereomers (2:3).
Scheme 5.

Reagents and conditions: (a) MsCl/Et3N/CH2Cl2; (b) BocNHCH(CH2CH2SH)CO2t-Bu/LDA/DMF; (c) TFA/rt; (d) TFA/H2O.
Our attempt to prepare the imine derivative of [4-aza]SRH was unsuccessful. Thus, debenzylation of 7 and treatment of the aminoribitol 44 with N-chlorosuccinimide (NCS) followed by dehydrochlorination of the resulting N-chloroamine with lithium tetramethylpiperidine gave unstable aldoimine of type 39 (H instead of CH3 ), as reported.48 However, couplings of such aldoimine with Hcy to give the imine SRH analogue failed. Acid-catalyzed hydrolysis of such imine analogue could serve as an alternative route to 4-azaSRH 23. Also, enzyme-mediated protonation of the imine nitrogen atom and the addition of water might generate 23 and/or new species with an “amino group” within the enzyme active site.
A nitrone analogue of SRH 49 was also targeted. Since nitrones are more electrophilic than imines such analogue might act as irreversible inhibitors by forming a covalent adduct(s) with enzyme. It is noteworthy that nitrones are overall neutral and cannot be protonated at physiological pH. Thus, treatment of the aminoribitol 44 with SeO2/H2O2 gave nitrone 4549 (74%; Scheme 6). Desilylation and subsequent mesylation gave 47 (56%). Coupling of 47 with protected Hcy afforded a nitrone-SRH derivative 48 (43%). Deprotection of 48 with TFA produced unstable nitrone derivative 49 (40%).
Scheme 6.

Reagents and conditions: (a) H2/Pd-C/EtOH/rt; (b) SeO2/H2O2/Me2CO/−4°C; (c) TBAF/THF; (d) MsCl/Et3N/CH2Cl2/−4 °C; (e) BocNHCH(CH2CH2SH)CO2t-Bu/LDA/DMF/−20 °C; (f) (i) TFA/−4°C, (ii) TFA/H2O/−4 °C.
Inhibition of LuxS
Compounds 12, 20, 21, 23, 28, 36, 38, and 43 were evaluated as potential inhibitors of Co(II)-substituted B. subtilis LuxS. Compound 12 inhibited LuxS in a concentration-dependent manner that is consistent with competitive inhibition (Figure 3a), with a KI value of 48 μM (Table 1). Similarly, lactam 21 also behaved as a competitive inhibitor with KI value of 37 μM. As expected, the lactam 20, which contains a bulky benzyl group at the ring nitrogen, was found to be inactive, likely due to steric reasons. Compounds 36 and 38 were both inactive toward LuxS, highlighting the importance of the ribose hydroxyl groups for enzyme binding. The proposed mechanism predicts that the C2 and C3 hydroxyl groups directly coordinate with the catalytic metal ion during different catalytic steps (Figure 1). The lack of activity of compound 43, which contains a methyl group instead of a hydroxyl group at the C1 position, may be caused by both loss of favorable interactions with the OH group and the bulky size of the methyl group. Collectively, these results suggest that proper interactions between the ribose ring and the enzyme active site critically contribute to the formation of a productive E-S complex and subsequent catalysis.
Figure 3.

Inhibition of LuxS by compounds 12 and 23. (A) Reaction progress curves in the presence of increasing concentrations of inhibitor 12 (0, 200, 400, 800, 1600, and 3200 μM). The last two curves were control reactions in the absence of LuxS. Inset, plot of remaining LuxS activity as a function of inhibitor 12 concentration. (B) Reaction progress curves of LuxS in the presence of increasing concentrations of inhibitor 23 (0, 20, 40, and 50 μM) (without preincubation). Inset, plot of remaining LuxS activity as a function of inhibitor 12 concentration (after 30 min preincubation).
Table 1.
Inhibition constants of [4-aza]SRH analogous against B. subtilis LuxS
| Compound | KI or KI* (μM) |
|---|---|
| 12 | 48 |
| 21 | 37 |
| 23 | 3.5 |
Unlike the other analogues described above, inhibition of LuxS by the hemiaminal-containing analogue 23 was time dependent (Figure 3b). Its inhibition kinetics can be described by the slow-binding equation
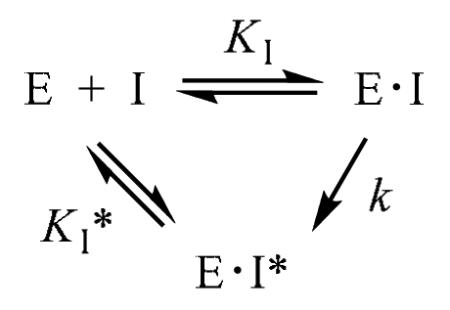 |
where KI is the equilibrium constant for the formation of the initial E•I complex, k is the rate constant for the conversion of the E•I complex to the tighter E•I* complex, and KI* represents the dissociation constant of the E•I* complex. To assess its potency, different concentrations of compound 23 were preincubated with LuxS for 30 min at 4 °C and the residual enzymatic activity was measured. Plot of the residual activity against the inhibitor concentration resulted in an IC50 value of 60 μM, from which a KI* value of 3.5 μM was estimated (Table 1). Unfortunately, the complex inhibition kinetics precluded an accurate determination of the KI value. While further work is clearly necessary to determine the exact mechanism of inhibition by 23, we propose a working hypothesis to explain the observed time dependence (Figure 4). Since compounds 12 and 21, which are structurally similar to 23, did not exhibit time-dependent inhibition and our model study shows that the hemiaminal 25a exists in equilibrium with the free aldehyde form (25b), we propose that hemiaminal 23 may undergo ring opening to form aldehyde 23a. Due to its structural similarity to catalytic intermediate 1a (Figure 1), 23a may undergo the aldose-ketose isomerization reaction to form 2-ketone 23b, which presumably binds to the LuxS active site with higher affinity than the ribose analogue 23. This behavior is very similar to that of a class of halogenated SRH analogues (e.g., [3-F]SRH and [3-Br]SRH), which have been shown to undergo LuxS-catalyzed ring opening to form open-chain species that are more potent LuxS inhibitors than the initial ribose analogues.27
Figure 4.

Proposed mechanism for the time-dependent inhibition of LuxS by [4-aza]SRH hemiaminal 23.
The remaining compound 28 and its ancestor lactam showed no significant inhibition of LuxS.
Conclusions
We have synthesized [4-aza] S-ribosylhomocysteine analogues in which the furanose ring oxygen has been substituted by a nitrogen atom having also the additional modifications at anomeric carbon. Coupling of the protected 4-amino-5-O-methanesulfonyl-4-deoxy-D-ribono-1,4-lactam with homocysteinate and subsequent deprotection with TFA gave [4-aza]SRH with an amide carbonyl at anomeric carbon. Reduction of the N-Boc protected lactam with LiBEt3H and acid catalyzed deprotection produced [4-aza]SRH hemiaminal analogue. The [4-aza]SRH analogue lacking the hydroxyl group at C1 and the corresponding lactam derivative showed modest competitive inhibition (KI ~40-50 μM) of LuxS. The hemiaminal analogue exhibited time-dependent inhibition (KI* = 3.5 μM), consistent with the enzyme-catalyzed ring opening and generation of 2- and/or 3-ketone intermediates, which presumably bind to the LuxS active site with higher affinity than the ribose natural substrate.
Experimental Procedure
The 1H (400 or 600 MHz) and 13C (100 MHz) NMR spectra were determined with solutions in CDCl3 unless otherwise noted. Mass spectra (MS) were obtained with atmospheric pressure chemical ionization (APCI) technique and HRMS in AP-ESI or TOF-ESI mode. TLC was performed with Merck kieselgel 60-F254 sheets products were detected with 254 nm light or by visualization with Ce(SO4)2/(NH4)6Mo7O24•4H2O/H2SO4/H2O reagent. Merck kieselgel 60 (230-400 mesh) was used for column chromatography. HPLC purifications were performed using XTerra® preparative RP18 OBD™ column (5μm 19 × 150 mm) with gradient program using CH3CN/H2O as a mobile phase. Reagent grade chemicals were used, and solvents were dried by reflux over and distillation from CaH2 (except for THF/potassium) under argon.
1-Amino-1,4-anhydro-N-benzyl-5-O-tert-butyldimethylsilyl-1-deoxy-2,3-O-isopropylidene-D-ribitol (7)
To a stirred solution of 637 (150 mg, 0.57 mmol) in anhydrous CH2Cl2 (5 mL) at rt under Ar atmosphere were added DMAP (7 mg, 0.05 mmol) and imidazole (93 mg, 1.36 mmol) followed by TBDMSCl (103 mg, 0.68 mmol). The mixture was then stirred for 10 h and partitioned (CH2Cl2// NaHCO3/H2O). The organic layer was washed (brine), dried (MgSO4) and evaporated. The residue was purified by column chromatography (15% EtOAc/hexane) to give 750 (204 mg, 98%) as a colorless oil. 1H NMR δ −0.13 (s, 3, CH3), 0.00 (s, 3, CH3), 0.83 (s, 9, t-Bu), 1.26 (s, 3, CH3), 1.49 (s, 3, CH3), 2.65 (dd, J = 2.7, 10.3 Hz, 1, H1), 2.94 (‘q’, J =, 2.2 Hz, 1, H4), 3.04 (dd, J = 5.5, 10.3 Hz, 1, H1′), 3.57 (dd, J = 4.1, 10.6 Hz, 1, H5), 3.64 (d, J = 13.4 Hz, 1, Bn), 3.71 (dd, J = 4.3, 10.6 Hz, 1, H5′), 3.94 (d, J = 13.4 Hz, 1, Bn), 4.49 (dd, J = 2.0, 6.5 Hz, 1, H3), 4.58 (‘dt’, J = 2.7, 6.2 Hz, 1, H2), 7.13-7.23 (m, 5, Bn); 13C NMR δ −5.5, (CH3), −5.4, (CH3), 18.2 (t-Bu), 25.2 (CMe2), 25.7 (CH3), 25.9 (CH3), 27.2 (CMe2), 56.9 (Bn), 59.3 (C1), 63.2 (C5), 68.9 (C4), 79.4 (C2), 83.3 (C3), 111.9 (CMe2), 128.2, 128.5, 126.8, 139.4 (Bn); MS (APCI) m/z 378 (100, MH+).
1-Amino-1,4-anhydro-N-tert-butoxycarbonyl-5-O-tert-butyldimethylsilyl-1-deoxy-2,3-O-isopropylidene-D-ribitol (8)
A solution of 7 (145 mg, 0.38 mmol), triethylamine (0.105 mL, 0.76 mmol), di-tert-butyldicarbonate (126 mg, 0.57 mmol) and Pd/C (5%, 300 mg) in ethanol (6 mL) was stirred under an atmosphere of hydrogen at room temperature for 6 h. The reaction mixture was filtered through Celite to remove the catalyst. The Celite was washed with ethanol (5 mL) and washings and the filtrate were combined and evaporated. The residue was partitioned (EtOAc// NaHCO3/H2O). The organic layer was washed (brine), dried (MgSO4) and evaporated. The residue was column chromatographed (20 → 30% EtOAc/hexane) to give 8 (147 mg, 99%) with spectral properties as reported.44
1-Amino-1,4-anhydro-N-tert-butoxycarbonyl-1-deoxy-2,3-O-isopropylidene-D-ribitol (9)
TBAF (1 M/THF; 0.25 mL, 0.25 mmol) was added to a stirred solution of 8 (66 mg, 0.17 mmol) in THF (5 mL) at ambient temperature. After stirring for 30 min, the reaction mixture was partitioned (EtOAc// NaHCO3/H2O). The organic layer was washed (brine), dried (MgSO4) and evaporated. The residue was column chromatographed (50 → 60% EtOAc/hexane) to give 9 (32 mg, 70%) with spectral properties as reported.51
1-Amino-1,4-anhydro-N-tert-butoxycarbonyl-1-deoxy-2,3-O-isopropylidene-5-O-methanesulfonyl-D-ribitol (10). Procedure A
Triethylamine (99 μL, 0.71 mmol) and MsCl (25 μL, 0.33 mmol) were added dropwise to stirred solution of 9 (60 mg, 0.22 mmole) in anhydrous CH2Cl2 (6 mL) at 0 °C (ice-bath). After 5 min, ice-bath was removed and the reaction mixture was allowed to stir at ambient temperature for 30 min. The reaction mixture was quenched with saturated NaHCO3/H2O and was extracted with CH2Cl2. The organic layer was washed (brine), dried (MgSO4) and evaporated to give 10 (73 mg, 96%) as a mixture (~3:2) of two rotamers of sufficient purity to be directly used in next step: 1H NMR δ 1.28 (s, 3, CH3), 1.42 (s, 12H, t-Bu, CH3), 2.96 (s, 1.2, Ms), 2.98 (s, 1.8, Ms), 3.39 (dd, J = 4.8, 12.5 Hz, 0.4, H1), 3.46 (dd, J = 4.8, 12.5 Hz, 0.6, H1), 3.69 (d, J = 12.5 Hz, 0.6, H1′), 3.82 (d, J = 12.5 Hz, 0.4, H1′), 4.10-4.14 (m, 0.4, H4), 4.22-4.30 (m, 0.6, H4), 4.22-4.29 (m, 1.4, H5,5′), 4.45 (dd, J = 4.1, 10.1 Hz, 0.6, H5), 4.65 (‘d’, J = 5.9 Hz, 1, H3); 4.72 (‘t’, J = 5.3 Hz, 1, H2); 13C NMR (major rotamer) δ 24.9 (CMe2), 26.9 (CMe2), 29.6 (t-Bu), 37.1 (Ms), 52.5 (C1), 62.4 (C4), 68.9 (C5), 79.2 (C2), 80.4 (t-Bu), 81.7 (C3), 112.1 (CMe2),154.2 (NHCO); 13C NMR (minor rotamer) δ 24.9 (CMe2), 26.9 (CMe2), 29.6 (t-Bu), 37.5 (Ms), 53.1 (C1), 62.6 (C4), 68.6 (C5), 78.5 (C2), 80.6 (t-Bu), 82.5 (C3), 112.1 (CMe2), 153.6 (NHCO); MS (APCI) m/z 352 (10, MH+), 252 (100, [MH2-Boc]+).
S-(1-Amino-1,4-anhydro-N-tert-butoxycarbonyl-1,5-dideoxy-2,3-O-isopropylidene-D-ribitol-5-yl)-N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester (11). Procedure B
Step a. H2O (0.4 mL) and tris(2-carboxyethyl)phosphine hydrochloride (140 mg, 0.5 mmol) were added to a stirred solution of N,N’-di(tert-butoxycarbonyl)-L-homocystine di(tert-butyl) ester19 (250 mg, 0.4 mmol) in anhydrous DMF (4 mL) at ambient temperature under Ar atmosphere. After 24 h, the reaction mixture [TLC (EtOAc/hexane, 2:8) showed conversion of disulfide (Rf 0.55) into thiol (Rf 0.65)] was partitioned between EtOAc and saturated NaHCO3/H2O. Aqueous layer was extracted with EtOAc, and the combined organic layer was washed with brine, dried (MgSO4) and concentrated to give N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester19 (240 mg, 99%) as colorless oil of sufficient purity to be directly used in next step. Step b. LDA (85 μL, 2.0 M/THF and heptane, 0.17 mmol) was added dropwise (10 min) to a stirred solution of freshly prepared thiol from step a (200 mg, 0.6 mmol) in anhydrous DMF (5 mL) under a vigorous stream of argon at 0 °C (ice-bath). After an additional 10 min, 10 (100 mg, 0.2 mmol) in anhydrous DMF (5 mL) was added via a syringe. After 15 min ice-bath was removed and the reaction mixture was stirred for 24 h at ambient temperature. Ice-cold saturated NH4Cl/H2O was added and the resulting suspension was diluted with EtOAc. The organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic layer was washed (brine), dried (MgSO4) and was evaporated. The residue was column chromatographed (40 → 50% EtOAc/hexane) to give 11 (130 mg, 86%) as a mixture of rotamers (~1:1): 1H NMR δ 1.29 (s, 3, CH3), 1.41 (s, 12H, t-Bu, CH3), 1.44 (s, 18H, t-Bu), 1.79-1.92 (m, 2, H8,8′), 2.39-2.80 (m, 4, H5,5′,7,7′ ), 3.37 (dd, J = 4.2, 11.7 Hz, 0.5, H1 ), 3.43 (dd, J = 4.5, 11.7 Hz, 0.5, H1 ), 3.70 (d, J = 12.6 Hz, 0.5, H1′), 3.84 (d, J = 12.8 Hz, 0.5, H1′), 3.99-4.05 (m, 0.5, H4), 4.11-4.17 (m, 0.5, H4), 4.18-4.29 (m, 1, H9), 4.56 (dd, J = 5.6, 10.4 Hz, 0.5, H3), 4.60 (dd, J = 5.6, 10.4 Hz, 0.5, H3), 4.69 (d, J = 4.8 Hz, 0.5, H2), 4.71 (d, J = 4.8 Hz, 0.5, H2), 5.06 (br. d, J = 7.3 Hz, 0.5, NH), 5.29 (br. d, J = 6.1 Hz, 0.5, NH); 13C NMR δ 25.0 (CMe2), 26.9 (CMe2), 27.9 (C7), 28.0 (t-Bu), 28.3 (t-Bu), 28.4 (t-Bu), 32.2 (C5), 32.6 (C8), 32.9 (C8), 33.2 (C5), 51.7 (C1), 52.4 (C1), 53.5 (C9), 62.8 (C4), 63.2 (C4), 78.5 (C2), 78.5 (t-Bu), 79.2 (C2), 79.2 (t-Bu), 80.0 (t-Bu), 80.1 (t-Bu), 83.5 (C3), 84.1 (C3), 111.9 (CMe2), 154.1 (CO), 154.9 (CO), 155.4 (CO), 171.2 (C10), 171.5 (C10); MS (APCI) m/z 547 (100, MH+); HRMS (AP-ESI) m/z calcd for C26H47N2O8S [MH]+ 547.3048; found 547.3042.
S-(1-Amino-1,4-anhydro-1,5-dideoxy-D-ribitol-5-yl)-L-homocysteine (12). Procedure C
Step a. Compound 11 (39 mg, 0.07 mmol) dissolved in TFA (4.0 mL) was stirred at 0 °C for 3 h. Volatiles were coevaporated with toluene to give an oily residue, which was used directly in next step. Step b. Product from step a was treated with TFA/H2O (9:1, 4.0 mL) for 1 h at 0 °C. Volatiles were evaporated and the crude product was purified on RP-HPLC (5% CH3CN/H2O at 2.5 mL/min; tR = 12 min) to give 12 (12 mg, 66%) as a colorless oil: 1 H NMR (D2O) δ 2.10-2.25 (m, 2, H8,8′), 2.71-2.81 (m, 2, H7,7′), 2.87 (dd, J = 10.4, 14.5 Hz, 1, H5), 3.15 (dd, J = 4.3, 14.5 Hz, 1, H5′), 3.34 (dd, J = 1.9, 13.0 Hz, 1, H1), 3.53 (dd, J = 4.0, 13.0 Hz, 1, H1′), 3.69 (ddd, J = 4.3, 8.7, 10.4 Hz, 1, H4), 3.86 (t, J = 6.2 Hz, 1, H9), 4.14 (dd, J = 4.1, 8.7 Hz, 1, H3), 4.39 (ddd, J = 1.9, 4.0, 4.1 Hz, 1, H2); 13C NMR (D2O) δ 26.8 (C8), 30.1 (C7), 30.6 (C5), 49.2 (C1), 53.6 (C9), 59.5 (C4), 69.5 (C2), 74.3 (C3), 174.0 (C10); MS m/z 251 (100, MH+); HRMS (TOF MSESI) m/z calcd for C9H19N2O4S [M+H]+ 251.1060; found 251.1063
1-Benzyl-5-chloro-3,4-dihydroxy-3,4-O-isopropylidenepiperidine [13(3S,4S,5R/S)]
Treatment of 6 (50 mg, 0.19 mmol) with MsCl (21.9 μL, 0.28 mmol) by Procedure A [column chromatography (20 → 30% EtOAc/hexane)] gave 13 (25 mg, 46%) as a 3:1 mixture of diastereomers: The major isomer had: 1H NMR δ 1.30 (s, 3, CH3), 1.50 (s, 3, CH3), 2.13-2.19 (m, 1, H2), 2.49 (dd, J = 3.7, 13.3 Hz, 1, H6), 2.87 (dq, J = 1.7, 12.0 Hz, 1, H2′), 3.05 (‘dt’, J = 2.1, 13.3 Hz, 1, H6′), 3.50 (d, J = 13.4 Hz, 1, Bn), 3.61 (d, J = 13.4 Hz, 1, Bn), 3.93-3.99 (m, 2, H3,5), 4.19 (dd, J = 3.7, 7.5 Hz, 1, H4), 7.20-7.25 (m, 5, Bn); 13C NMR δ 26.2 (CMe2), 28.4 (CMe2), 53.5 (C6), 56.5 (C2), 58.5 (C5), 61.5 (Bn), 73.8 (C4), 79.5 (C3), 112.8 (CMe2), 127.3, 128.4, 128.9, 137.2 (Bn); MS m/z 282 (100, MH+[35Cl]), 284 (40, MH+[37Cl]); HRMS (TOF MS-ESI) m/z calcd for C15H20 35ClNO2 [M+H]+ 282.1255; found 282.1259.
4-Amino-N-benzyl-5-O-tert-butyldimethylsilyl-4-deoxy-2,3-O-isopropylidene-D-ribono-1,4-lactam (14a). Procedure D
RuO2·xH2O (4.3 mg, 0.032 mmol) was added to a stirred solution of NaIO4 (83 mg, 0.39 mmol) in H2O (1 mL) at ambient temperature. After 5 min, a solution of 7 (50 mg, 0.13 mmol) in EtOAc (1 mL) was added dropwise and the reaction mixture was continued to stir for 12 h. H2O (10 mL) and EtOAc (10 mL) were added and the separated aqueous layer was furthermore extracted with EtOAc (2 × 10 mL). The combined organic layers were washed (brine), dried (MgSO4) and evaporated. The residue was column chromatographed (50 → 60% EtOAc/hexane) to give 14a (33 mg, 65%) and N-benzoylated byproduct (10 mg, 18%). Compound 14a had: 1H NMR δ 0.01 (s, 6, 2 × CH3), 0.84 (s, 9, t-Bu), 1.35 (s, 3, CH3), 1.42 (s, 3, CH3), 3.51 (t, J = 2.1 Hz, 1, H4), 3.62 (dd, J = 2.0, 10.9 Hz, 1, H5), 3.69 (dd, J = 2.3, 10.9 Hz, 1, H5′), 3.93 (d, J = 15.2 Hz, 1, Bn), 4.50 (d, J = 5.6 Hz, 1, H3), 4.69 (d, J = 5.6 Hz, 1, H2), 5.00 (d, J = 15.2 Hz, 1, Bn), 7.22-7.32 (m, 5, Bn); 13C NMR δ −5.7, (CH3), −5.6, (CH3), 18.1 (t-Bu), 25.8 (t-Bu), 25.8 (CMe2), 27.3 (CMe2), 44.2 (Bn), 60.2 (C5), 62.0 (C4), 76.7 (C3), 78.0 (C2), 111.7 (CMe2), 127.7, 128.2, 128.7, 135.6 (Bn), 171.9 (C1); MS (APCI) m/z 392 (100, MH+). The 4-amino-N-benzoyl-5-O-tert-butyldimethylsilyl-4-deoxy-2,3-O-isopropylidene-D-ribono-1,4-lactam byproduct had: 1H NMR δ 0.01 (s, 3, CH3), 0.05 (s, 3, CH3), 0.87 (s, 9, t-Bu), 1.40 (s, 3, CH3), 1.51 (s, 3, CH3), 3.83 (dd, J = 1.5, 10.7 Hz, 1, H5), 4.19 (dd, J = 2.2, 10.7 Hz, 1, H5′), 4.58 (‘t’, J = 1.8 Hz, 1, H4), 4.65 (d, J = 5.5 Hz, 1, H3), 4.76 ( d, J = 5.5 Hz, 1, H2), 7.38-7.43 (m, 2, Bn), 7.51 (‘dt’, J = 1.3, 6.7 Hz, 2, Bn), 7.54-7.58 (m, 1, Bn); 13C NMR δ −5.7, (CH3), −5.6, (CH3), 18.2 (t-Bu), 25.3 (CMe2), 25.8 (t-Bu), 27.2 (CMe2), 61.7 (C4), 62.1 (C5), 76.3 (C3), 78.7 (C2), 112.1 (CMe2), 127.9, 128.7, 132.2, 134.1 (Bn) 170.6 (CO), 171.7 (C1); MS (APCI) m/z 406 (100, MH+).
4-Amino-N-benzyl-4-deoxy-2,3-O-isopropylidene-D-ribono-1,4-lactam (14b)
TBAF (1 M/THF; 0.18 mL, 0.18 mmol) was added dropwise to a stirred solution of 14a (49 mg, 0.12 mmol) in THF (10 mL) at 0 °C. After 5 min, the ice-bath was removed and reaction mixture was allowed to stir at ambient temperature for 2 h. The reaction mixture was quenched with water and volatiles were evaporated. The residue was partitioned (EtOAc// NaHCO3/H2O) and the organic layer was washed (brine), dried (MgSO4) and evaporated. The residue was column chromatographed (80 → 90% EtOAc/hexane) to give 14b52as a white solid (33 mg, 97%): 1H NMR δ 1.37 (s, 3, CH3), 1.47 (s, 3, CH3), 3.35 (s, 1, OH), 3.54 (‘t’, J = 1.8 Hz, 1, H4), 3.64 (d of m, J = 11.8 Hz, 1, H5), 3.85 (br. d, J = 12.0 Hz, 1, H5′), 4.08 (d, J = 15.2 Hz, 1, Bn), 4.64 (d, J = 5.6, Hz, 1, H3), 4.77 (d, J = 5.2 Hz, 1, H2), 5.04 (d, J = 15.2 Hz, 1, Bn), 7.29-7.37 (m, 5, Bn); MS (APCI) m/z 278 (100, MH+);
4-Amino-N-tert-butoxycarbonyl-5-O-tert-butyldimethylsilyl-4-deoxy-2,3-O-isopropylidene-D-ribono-1,4-lactam (15a)
Oxidation of 8 (90 mg, 0.23 mmol) with NaIO4 (126 mg, 0.7 mmol) and RuO2·xH2O (8 mg, 0.05 mmol) by Procedure D [column chromatography (50 → 60% EtOAc/hexane)] gave 15a (56 mg, 60%) as a colorless oil with data as reported.44
4-Amino-N-tert-butoxycarbonyl-4-deoxy-2,3-O-isopropylidene-D-ribono-1,4-lactam (15b)
Desilylation of 15a (50 mg, 0.12 mmol) with TBAF (1M/THF, 0.14 mL, 0.14 mmol), as described for 14b, [column chromatography (70 → 80% EtOAc/hexane)] gave 15b (27 mg, 77%) as a colorless oil: 1H NMR δ 1.36 (s, 3, CH3), 1.46 (s, 12, t-Bu, CH3), 3.88 (‘dt’, J = 3.9, 5.3 Hz, 1, H4), 4.03 (dd, J = 4.1, 11.4 Hz, 1, H5), 4.19 (dd, J = 3.8, 11.4 Hz, 1, H5′), 4.57 (dd, J = 5.3, 5.7 Hz, 1, H3), 4.63 (d, J = 5.7 Hz, 1, H2); 13C NMR δ 25.7 (CMe2), 27.0 (CMe2), 27.6 (t-Bu), 56.7 (C4), 66.6 (C5), 76.7 (C2), 77.0 (C3), 83.3 (t-Bu), 112.4 (CMe2), 153.0 (CO), 173.7 (C1); MS (APCI) m/z 288 (100, MH+); HRMS (AP-ESI) m/z calcd for C13H22NO6 [MH]+ 288.1442; found 288.1437.
4-Amino-N-benzyl-4-deoxy-2,3-O-isopropylidene-5-O-methanesulfonyl-D-ribono-1,4-lactam (16)
Mesylation of 14b (67 mg, 0.24 mmol) with MsCl (27 μL, 0.36 mmol) by Procedure A gave 16 (83 mg, 97%) as a colorless oil of sufficient purity to be used directly in next step: 1H NMR δ 1.30 (s, 3, CH3), 1.36 (s, 3, CH3), 3.07 (s, 3, Ms), 3.65 (t, J = 2.7 Hz, 1, H4), 4.06 (d, J = 15.2 Hz, 1, Bn), 4.16 (dd, J = 2.2, 11.0 Hz, 1, H5), 4.22 (dd, J = 3.0, 11.0 Hz, 1, H5′), 4.51 (d, J = 5.6 Hz, 1, H3), 4.69 (d, J = 5.6 Hz, 1, H2), 4.92 (d, J = 15.2 Hz, 1, Bn), 7.18-7.30 (m, 5, Bn); MS (APCI) m/z 388 (100, [MH+MeOH]+), 356 (40, MH+).
4-Amino-N-tert-butoxycarbonyl-5-O-methanesulfonyl-4-deoxy-2,3-O-isopropylidene-D-ribono-1,4-lactam (17)
Oxidation of 10 (80 mg, 0.32 mmol) with NaIO4 (172 mg, 0.96mmol) and RuO2·xH2O (8.5 mg, 0.064 mmol) by procedure D [column chromatography (EtOAc)] gave 17 (78 mg, 95%) as a colorless oil: 1H NMR δ 1.37 (s, 3, CH3), 1.44 (s, 3, CH3), 1.54 (s, 9H, t-Bu) 3.01 (s, 3, Ms), 4.39-4.43 (m, 2, H4,5), 4.58 (d, J = 5.5 Hz, 1, H3), 4.64 (dd, J = 3.1, 11.2 Hz, 1, H5′), 4.70 (d, J = 5.5 Hz, 1, H2); 13C NMR δ 25.6 (CMe2), 27.0 (CMe2), 28.0 (t-Bu), 37.7 (Ms), 59.2 (C4), 67.0 (C5), 74.5 (C3), 77.5 (C2), 84.7 (t-Bu), 112.8 (CMe2), 149.7 (CO), 170.2 (C1); MS (APCI) m/z 366 (5, MH+), 297 (100, [MH2-Boc+MeOH]+).
S-(4-Amino-N-benzyl-4,5-dideoxy-2,3-O-isopropylidene-D-ribono-1,4-lactam-5-yl)-N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester (18)
Treatment of 16 (85 mg, 0.24 mmol) with lithium homocysteinate (104 mg, 0.36 mmol) by Procedure B [column chromatography (60 → 70% EtOAc/hexane)] gave 18 (94 mg, 70%) as a colorless oil. 1H NMR δ 1.28 (s, 3, CH3), 1.37 (s, 12, t-Bu, CH3), 1.39 (s, 9, t-Bu), 1.74-1.86 (m, 1, H8), 1.95-2.17 (m, 1, H8′), 2.46-2.57 (m, 2, H7,7′), 2.69-2.73 (m, 2, H5,5′), 3.59-3.71 (m, 1, H4), 3.86 (d, J = 15.0 Hz, 1, Bn), 4.17-4.18 (m, 1, H9), 4.43 (d, J = 5.7 Hz, 1, H3), 4.79 (d, J = 5.5 Hz, 1, H2), 4.97 (d, J = 15.0 Hz, 1, Bn), 5.05 (br. d, J = 7.1 Hz, 1, NH), 7.17-7.27 (m, 5, Bn); 13C NMR δ 25.6 (CMe2), 27.0 (CMe2), 28.0 (t-Bu), 28.3 (t-Bu ), 29.0 (C7), 33.1 (C5), 33.2 (C8), 44.4 (Bn), 53.1 (C9), 60.3 (C4), 77.2 (C3), 77.5 (C2), 79.9 (t-Bu) 82.4 (t-Bu), 112.1 (CMe2), 127.9, 128.2, 128.8, 135.2 (Bn), 153.0 (CO), 171.1 (C10), 171.4 (C1); MS m/z 551 (100, MH+). HRMS (AP-ESI) m/z calcd for C28H43N2O7S [MH]+ 551.2785; found 551.2792.
S-(4-Amino-4,5-dideoxy-2,3-O-isopropylidene-D-ribono-1,4-lactam-5-yl)-N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester (19a)
Treatment of 17 (70 mg, 0.19 mmol) with lithium homocysteinate (83 mg, 0.28 mmol) by Procedure B [column chromatography (70%→ 80% EtOAc/hexane)] gave 19a (40 mg, 45%) as a light yellow oil. 1H NMR δ 1.36 (s, 3, CH3), 1.43 (s, 9, t-Bu), 1.45 (s, 12, t-Bu, CH3), 1.82-1.90 (m, 1, H8), 1.97-2.06 (m, 1, H8′), 2.58 (‘t’, J = 7.4 Hz, 2, H7,7′), 2.64-2.72 (m, 2, H5,5′), 3.83 (‘t’, J = 5.8 Hz 1, H4), 4.23-4.24 (m, 1, H9), 4.48 (d, J = 4.4 Hz, 1, H3), 4.69 (d, J = 4.6 Hz, 1, H2), 5.21 (br. d, J = 7.9 Hz, 1, NH); 13C NMR δ 25.6 (CMe2), 27.0 (CMe2), 28.0 (t-Bu), 28.4 (t-Bu ), 29.2 (C7), 32.9 (C8), 33.8 ( C5), 53.2 (C9), 60.4 (C4), 75.9 (C3), 77.5 (C2), 80.0 (t-Bu) 82.4 (t-Bu), 112.4 (CMe2), 155.4 (CO), 170.4 (C1), 171.2 (C10); MS (APCI) m/z 461 (100, MH+).
S-(4-Amino-N-tert-butoxycarbonyl-4,5-dideoxy-2,3-O-isopropylidene-D-ribono-1,4-lactam-5-yl)-N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester (19b). Procedure E
DMAP (18.8 mg, 0.15 mmol) and (Boc)2O (46.4 mg, 0.21 mmol) were added to a stirred solution of compound 19a (27 mg, 0.06 mmol) in CH2Cl2 (2 mL) at ambient temperature under Ar atmosphere. After 48 h, the reaction mixture was quenched with H2O (5 mL) and partitioned between CH2Cl2//NaHCO3/H2O. The organic layer was washed (brine), dried (MgSO4) and evaporated. The residue was column chromatographed (30 → 40% EtOAc/hexane) to give 19b (30 mg, 93%) as a colorless oil: 1H NMR δ 1.36 (s, 3, CH3), 1.43 (s, 9, t-Bu), 1.45 (s, 3, CH3), 1.46 (s, 9, t-Bu), 1.54 (s, 9, t-Bu), 1.82-1.87 (m, 1, H8), 2.02-2.04 (m, 1, H8′), 2.49-2.55 (m, 1, H7), 2.58-2.62 (m, 1, H7′), 2.70 (dd, J = 7.1, 14.2 Hz, 1, H5), 2.95 (dd, J = 2.5, 14.2 Hz, 1, H5′), 4.22-4.24 (m, 1, H9), 4.23 (dd, J = 2.5, 7.1 Hz, 1, H4), 4.46 (d, J = 5.5 Hz, 1, H3), 4.80 (d, J = 5.5 Hz, 1, H2), 5.08 (br. d, J = 7.9 Hz, 1, NH); 13C NMR δ 25.6 (CMe2), 26.9 (CMe2), 28.0 (t-Bu), 28.3 (t-Bu), 28.4 (t-Bu), 28.9 (C7), 33.2 (C8), 36.6 (C5), 53.2 (C9), 58.0 (C4), 76.7 (C3), 79.1 (C2), 82.3 (2 × t-Bu), 84.1 (t-Bu), 112.7 (CMe2), 149.9 (CO), 155.5 (CO), 171.2 (C10), 174.0 (C1); MS (ESI) m/z 583 (100, [M+Na]+).
S-(4-Amino-N-benzyl-4,5-dideoxy-D-ribono-1,4-lactam-5-yl)-L-homocysteine (20)
Treatment of 18 (40 mg, 0.07 mmol) with TFA by Procedure C (step a, 3 h) gave crude 20 as colorless oil. RP-HPLC purification (5% CH3CN/H2O for 30 min followed by gradient 5 → 90% CH3CN/H2O for 30 min at 2.5 mL/min; tR = 45 min) gave 20 (12 mg, 48%): 1H NMR (D2O) δ 1.98-2.11 (m, 2, H8,8′), 2.57-2.61 (m, 2, H7,7′), 2.71 (dd, J = 8.4, 14.0 Hz, 1, H5), 2.82 (dd, J = 3.9, 14.0 Hz, 1, H5′), 3.57 (dd, J = 3.6, 8.3 Hz, 1, H4), 3.78 (‘t’, J = 5.8 Hz, 1, H9), 4.36 (d, J = 5.3 Hz, 1, H3), 4.37 (d, J = 14.7 Hz, 1, Bn), 4.72 (d, J = 5.2 Hz, 1, H2), 4.78 (d, J = 14.7 Hz, 1, Bn), 7.33-7.46 (m, 5, Bn); 13C NMR (D2O) δ 27.7 (C7), 30.3 (C8), 30.8 (C5), 45.2 (Bn), 53.6 (C9), 63.9 (C4), 70.0 (C3), 70.1 (C2), 128.0, 128.1, 128.0, 135.1 (Bn), 173.9 (C1), 174.7 (C10); MS (APCI) m/z 355 (100, MH+); HRMS (AP-ESI) m/z calcd for C16H22N2NaO5S [M+Na]+ 377.1142; found 377.1156.
S-(4-Amino-4,5-dideoxy-D-ribono-1,4-lactam-5-yl)-L-homocysteine (21)
Treatment of 19a (30 mg, 0.065 mmol) with TFA by Procedure C (step a, 3h) gave crude 21 as colorless oil. RP-HPLC purification (5% CH3CN/H2O at 2.5 mL/min; tR = 16 min) gave 21 (10 mg, 58%): 1H NMR (D2O) δ 2.10-2.28 (m, 2, H8,8′), 2.75-2.77 (m, 3, H5′,7,7′), 2.83-2.84 (m, 1, H5), 3.73 (‘t’, J = 6.5 Hz, 1, H4), 3.95 (‘t’, J = 5.8 Hz, 1, H9), 4.29 (‘d’, J = 5.2 Hz, 1, H3), 4.57 (d, J = 5.2 Hz, 1, H2); 13C NMR (D2O) δ 26.2 (C7), 28.5 (C8), 32.7 (C5), 58.8 (C4), 51.1 (C9), 69.7 (C2), 70.5 (C3), 170.9 (C1), 176.1 (C10); MS (ESI) m/z 264 (100, M+); HRMS (AP-ESI) m/z calcd for C9H17N2O5S [MH]+ 265.0853; found 265.0859.
S-(4-Amino-N-tert-butoxycarbonyl-4,5-dideoxy-2,3-isopropylidene-α/β-D-ribofuranos-5-yl)-N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester (22). Procedure F
LiEt3BH (1 M/THF; 125 μL, 0.125 mmol) was added to a stirred solution of 19b (28 mg, 0.05 mmol) in anhydrous THF (1 mL) at −78 °C under N2 atmosphere. After 30 min, the solution was quenched with water and volatiles were evaporated. The residue was partitioned (EtOAc// NaHCO3/H2O), washed (brine) and dried (MgSO4). The resulting oil was chromatographed (40% EtOAc/hexane) to give 22 [26 mg, 92%; mixture of anomers (3:2) which appear as a set of rotamers as colorless oil: 1H NMR δ 1.30 (s, 3, CH3), 1.43 (s, 12, t-Bu, CH3), 1.46 (s, 9, t-Bu), 1.48 (s, 9, t-Bu), 1.83-1.94 (m, 1, H8), 2.03-2.10 (m, 1, H8′), 2.51-2.62 (m, 3, H7,7′,5), 2.83 (dd, J = 3.7, 13.5 Hz, 0.6, H5′), 2.92 (dd, J = 3.5, 13.7 Hz, 0.4, H5′), 3.45 (dd, J = 3.5, 10.6 Hz, 0.3, H4), 3.99-4.28 (m, 1.7, H4,9), 4.57 (d, J =5.8 Hz, 0.6, H3), 4.59 (d, J = 5.9 Hz, 0.4, H3), 4.66 (d, J = 6.7 Hz, 0.4, H2), 4.72 (d, J = 5.8 Hz, 0.6, H2), 5.09 (br. d, J = 7.2 Hz, 0.6, NH), 5.32 (br. d, J = 7.8 Hz, 0.4, NH), 5.39 (s, 0.4, H1), 5.50 (s, 0.6, H1); 13C NMR δ (major isomer) 24.8 (CMe2), 26.7 (CMe2), 28.0 (t-Bu), 28.3 (t-Bu), 28.4 (t-Bu), 29.7 (C7), 32.9 (C8), 34.7 (C5), 53.2 (C9), 63.9 (C4), 81.2 (2 × t-Bu), 82.3 (t-Bu), 82.8 (C2), 84.4 (C3), 87.1 (C1), 112.7 (CMe2), 154.3 (CO), 155.4 (CO), 171.3 (C10); MS (ESI) m/z 585 (100, [M+Na]+).
S-(4-Amino-4,5-dideoxy-α/β-D-ribofuranos-5-yl)-L-homocysteine (23)
Treatment of 22 (24 mg, 0.04 mmol) with TFA by Procedure C (step a, 1 h; step b, 10 h at 0°C) gave crude 23. RP-HPLC (5% CH3CN/H2O at 2.5 mL/min; tR = 14 min) yielded 23 (8 mg, 72%) as a light yellow oil of a mixture of anomers (3:2): 1H NMR (D2O) δ 2.15-2.24 (m, 1, H8), 2.26-2.37 (m, 1, H8′), 2.75-2.82 (m, 2, H7,7′), 2.84-2.98 (m, 1, H5,5′), 3.09-3.17 (m, 1, H5,5′), 3.55-3.69 (m, 0.6, H4), 3.76-3.83 (m, 0.4, H4), 4.15-4.21 (m, 2, H2,3,9), 4.23-4.25 (m, 0.4, H2), 4.34 (dd, J = 4.8, 6.3 Hz, 0.6, H3), 5.27 (d, J = 2.6 Hz, 0.6, H1), 5.41 (d, J = 2.2 Hz, 0.4, H1); 13C NMR δ (major isomer) 25.8 (C7), 28.7 (C8), 30.7 (C5), 51.3 (C9), 58.8 (C4), 72.0 (C3), 73.0 (C2), 86.5 (C1), 171.2 (C10); MS (ESI) m/z 267 (50, MH+), 249 (100, [M-17]+).
4-Amino-N-tert-butoxycarbonyl-5-O-tert-butyldimethylsilyl-4-deoxy-2,3-O-isopropylidene-α/β-D-ribofuranose (24a)
Reduction of 15a44 (68 mg, 0.17 mmol) with LiEt3BH (1 M/THF; 0.43 mL, 0.43 mmol) in anhydrous THF (2 mL) at −78 °C by the procedure F gave 24a44 (68 mg, 100%) as a colorless oil with data as reported.
4-Amino-N-tert-butoxycarbonyl-4-deoxy-2,3-O-isopropylidene-α/β-D-ribofuranose (24b)
Desilylation of 24a (55 mg, 0.13 mmol) with TBAF (1M/THF, 0.19 mL, 0.19 mmol), as described for 14b, [column chromatography (50 → 60% EtOAc/hexane)] gave 24b (39 mg, 96%) as a colorless oil of a mixture of anomers (3:2): 1H NMR δ 1.30 (s, 3, CH3), 1.40 (s, 3, CH3), 1.46 (s, 5.4, t-Bu ), 1.49 (s, 3.6, t-Bu ), 2.87 (br. s, 0.4, OH), 3.15 (br. s, 0.6, OH), 3.60-3.77 (m, 1.4, H5,5′), 3.84-3.90 (m, 0.8, H5,OH), 4.10 (br. s, 0.6, H4), 4.27 (br. s, 0.4, H4), 4.30 (br. s, 0.6, OH), 4.55 (d, J = 5.9 Hz, 1, H3), 4.73 (d, J = 5.9 Hz, 0.4, H2), 4.77 (d, J = 5.9 Hz, 0.6, H2), 5.36 (d, J = 5.7 Hz, 0.4, H1), 5.51 (s, 0.6, H1); 13C NMR (major anomer) δ 24.6 (CMe2), 26.6 (CMe2), 28.3 (t-Bu), 62.6 (C5), 65.8 (C4), 81.9 (C2), 81.3 (t-Bu), 85.3 (C3), 86.4 (C1), 111.4 (CMe2), 154.3 (CO); 13C NMR (minor anomer) δ 24.7 (CMe2), 26.7 (CMe2), 28.3 (t-Bu), 62.6 (C5), 65.8 (C4), 81.2 (C2), 81.3 (t-Bu), 86.4 (C3), 86.5 (C1), 111.4 (CMe2), 153.5 (CO); MS (APCI) m/z 272 (50, [M-OH+]), 213 [100, [MH-Boc-OH+CH3CN]+).
4-Amino-4-deoxy-α/β-D-ribofuranose (25a)
Treatment of 24b (27 mg, 0.09 mmol) with TFA by Procedure C (step a, 5 h; step b, 6 h at 0°C) gave crude 25a43 (13 mg, 92%) as a light yellow oil of a mixture of anomers (α/β, 0.65:0.35): 1H NMR (D2O; pD = 5-6 ) 3.52-3.53 (m, 0.35, H4), 3.56-3.59 (m, 0.65, H4), 3.76-3.78 (m, 0.65, H3), 3.85 (dd, J = 6.0, 13.1 Hz, 0.35, H5), 3.86 (dd, J = 2.1, 12.8 Hz, 0.65, H5), 3.97-4.00 (m, 0.35, H3), 4.06 (dd, J = 3.0, 4.0 Hz, 0.35, H2), 4.10 (dd, J = 2.8, 13.5 Hz, 0.35, H5′), 4.16 (dd, J = 3.0, 12.8 Hz, 0.65, H5′), 4.20 (‘t’, J = 3.4 Hz, 0.65, H2), 4.84 (d, J = 1.3 Hz, 0.35, H1), 5.12 (d, J = 4.0 Hz, 0.65, H1); 13C NMR (major anomer) δ 50.1 (C4), 58.9 (C5), 65.6 (C2), 69.8 (C3), 94.1 (C1); 13C NMR (minor anomers) δ 49.9 (C4), 61.5 (C5), 63.6 (C2), 69.8 (C3), 94.1 (C1); MS (APCI) m/z 150 (100, MH+).
The 1H NMR (D2O) at pD = 11 showed a singlet at 7.76 ppm which suggested formation of imine 25c.
4-Amino-4-deoxy-α/β-D-ribose O-Benzyloxime (26)
A solution of the crude 25a (13 mg. 0.09 mmol) and O-benzylhydroxylamine hydrochloride (43 mg, 0.27 mmol) in anhydrous pyridine (4 mL) was stirred under an atmosphere of nitrogen at room temperature for 12 h. Pyridine was evaporated to afford 26 of sufficient purity (~95%) for spectroscopic characterization together with the excess of BnONH2 used: 1H NMR (MeOH-d4) δ 3.52 (‘dt’, J = 4.1, 8.4 Hz, 1, H4), 3.79 (dd, J = 3.8, 11.5 Hz, 1, H5), 3.85 (dd, J = 4.4, 8.7 Hz, 1, H3), 3.94 (dd, J = 8.4, 11.5 Hz, 1, H5′), 4.13 (dd, J = 6.8, 8.7 Hz, 1, H2), 4.92-5.16 (2H, Bn; signals for benzylic protons were within the envelope of the solvent peak but cross peaks between them were observed in COSY), 7.41 (1, H1; signal for H1 was within the envelope of protons from benzyl group but cross peaks of H1 to H2 were observed in COSY), 7.35-7.47 (m, 5H, Bn);13C NMR δ 56.2 (C4), 58.8 (C5), 71.2 (C3), 71.6 (C2), 77.0 (Bn, confirmed by HETCOR), 128.9, 129.3, 129.5, 139.1 (Bn), 151.9 (C1); MS (ESI) m/z 255 (100, MH+).
S-(4-Amino-2,3,4,5-tetradeoxy-D-glycero-pentono-1,4-lactam-5-yl)-N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester (33)
Treatment of 2745 (5S; 18 mg, 0.1 mmol) with protected L-Hcy (35 mg, 0.12 mmol) by Procedure B (step a and b, 48h) gave 50.5 mg of the yellow oil. This material was column chromatographed (EtOAc/MeOH, 19:1) to give 33 (31 mg, 79%) as a colorless oil: 1H NMR δ 1.45 (s, 9), 1.48 (s, 9), 1.77-1.92 (m, 2, H3,8), 2.02-2.14 (m, 1, H8′), 2.27-2.47 (m, 3, H2,2′,3′), 2.51-2.57 (dd, J = 8.1, 13.3 Hz, 1, H5), 2.57-2.65 (m, 2, H7,7′), 2.72 (dd, J = 5.1, 13.2 Hz, 1, H5′), 3.80 (‘quin’, J = 6.4 Hz, 1, H4), 4.28 (br. ‘d’, J = 3.7 Hz, H9), 5.20 (br. d, J = 7.1 Hz, 1, NHBoc), 6.45 (br. s, 1, CONH); 13C NMR δ 26.7 (C3), 28.0 (t-Bu), 28.3 (t-Bu), 28.3 (C7), 30.1 (C2), 33.2 (C8), 38.7 (C5), 53.3 (C9), 53.7 (C4), 79.9 (t-Bu), 82.3 (t-Bu), 155.4 (CO), 171.2 (C10), 177.7 (C1); HRMS (AP-ESI) m/z calcd for C18H33N2O5S [MH]+ 389.2105; found 389.2110.
S-(4-Amino-2,3,4,5-tetradeoxy-D-glycero-pentono-1,4-lactam-5-yl)-L-homocysteine (34)
Compound 33 (9 mg, 0.02 mmol) was dissolved in TFA (0.7 mL), and the resulting mixture was stirred at ambient temperature for 60 min. The reaction mixture was evaporated, and coevaporated with toluene to give a trifluoroacetate of 34 (7.5 mg, 95%) as a colorless oil: 1H NMR (D2O) δ 1.84-1.94 (m, 1, H3), 2.12-2.23 (m, 1, H8), 2.23-2.31 (m, 1, H8′), 2.31-2.37 (m, 1, H3′,), 2.37-2.52 (m, 2, H2,2′), 2.73 (dd, J = 6.5, 13.6 Hz, 1, H5), 2.76 (t, J = 7.6 Hz, 2, H7,7′), 2.82 (dd, J = 5.4, 13.6 Hz, 1, H5′), 3.99 (‘quin’, J = 6.3 Hz, 1, H4), 4.17 (t, J = 6.4 Hz, 1, H9); 13C NMR (D2O) δ 25.3 (C3), 27.1 (C7), 29.7 (C8), 29.9 (C2), 36.8 (C5), 51.9 (C9), 54.3 (C4), 172.0 (C10), 181.5 (C1); HRMS (AP-ESI) m/z calcd for C9H17N2O3S [MH]+ 233.0954; found 233.0957.
S-(4-Amino-N-tert-butoxycarbonyl-2,3,4,5-tetradeoxy-D-glycero-pentono-1,4-lactam-5-yl)-N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester (35)
Treatment of 33 (40 mg, 0.1 mmol) in CH2Cl2 (2 mL) with DMAP (20 mg, 0.16 mmol), and (Boc)2O (63 mg, 0.29 mmol) by procedure E [column chromatography (30 → 35% EtOAc/hexane)] gave 35 (42 mg, 83%) as a colorless oil: 1H NMR (isomers ratio ~3:2) δ 1.45 (s, 9H), 1.47 (s, 9H), 1.55 (s, 9H), 1.82-1.94 (m, 1, H8), 1.99-2.22 (m, 3, H3,3′,8′), 2.44 & 2.45 (2 × ddd, J = 2.5, 9.6, 17.9 Hz, 1, H2), 2.54-2.70 (m, 4, H2′,5,7,7′), 2.94 (‘dt’, J = 3.5, 13.4 Hz, 1, H5′), 4.22-4.32 (m, 2, H4,9), 5.11 & 5.16 (2 × br. d, J = 7.9 Hz, 1, NHBoc); 13C NMR δ 22.0 (C3), 28.0 (t-Bu), 28.1 (t-Bu), 28.3 (t-Bu), 28.8 (C7), 31.1 & 31.2 (C2), 33.0 & 33.2 (C8), 35.1 & 35.5 (C5), 53.2 & 53.3 (C9), 57.2 & 57.5 (C4), 79.8 (t-Bu), 82.1 & 82.2 (t-Bu), 83.2 & 83.3 (t-Bu), 149.9 & 150.0 (CO), 155.5 (CO), 171.3 (C10), 173.7 & 173.8 (C1); MS (APCI) m/z 489 (30, MH+), 389 (100, [MH2-Boc]+).
S-(4-Amino-2,3,4,5-tetradeoxy-D-glycero-pentono-1,4-lactam-5-yl)-D/L-homocysteine (36)
Treatment of 2745 (62 mg, 0.35 mmol) with D/L-Hcy (52 mg, 0.385 mmol)/NaH (44 mg, 1.1 mmol; 60%/ mineral oil) by procedure G gave crude 36. RP-HPLC purification (5% CH3CN/H2O at 2.5 mL/min; tR = 14 min) gave 36 (60.5 mg, 75%) as a Na salt: 1H NMR (D2O) δ 1.83-1.95 (m, 2, H3,8), 1.95-2.04 (m, 1, H8′), 2.29-2.38 (m, 1, H3′), 2.38-2.53 (m, 2, H2,2′), 2.65 (t, J = 7.7 Hz, 2, H7,7′), 2.72 (dd, J = 6.5, 13.6 Hz, 1, H5), 2.81 (dd, J = 5.6, 13.6 Hz, 1, H5′), 3.47 (br. s, 1, H9), 3.99 (‘quin’, J = 6.3 Hz, 1, H4); 13C NMR (D2O) δ 25.4 (C3), 27.9 & 28.0 (C7), 29.9 & 29.9 (C2), 33.5 & 33.6 (C8), 36.8 (C5), 54.4 (C4), 54.8 (br., C9), 180.4 (br., C10), 180.5 (C1); MS (APCI) m/z 233 (100, MH+); (ESI) m/z 233 (100, MH+).
Stirring sodium salt of 36 (10 mg) in TFA (1 mL) for 1 h at ambient temperature and evaporation of volatiles gave trifluoroacetate of 36 as a mixture of diastereomers (9R/S, ~1:1): 1H NMR (D2O) δ 1.79-1.89 (m, 1, H3), 2.09-2.19 (m, 1, H8), 2.21-2.28 (m, 1, H8′), 2.28-2.33 (m, 1, H3′,), 2.34-2.48 (m, 2, H2,2′), 2.678 (dd, J = 6.5, 13.6 Hz, 0.5, H5, 9R), 2.683 (dd, J = 6.5, 13.6 Hz, 0.5, H5, 9S), 2.72 (t, J = 7.6 Hz, 2, H7,7′), 2.776 (dd, J = 5.4, 13.6 Hz, 0.5, H5′, 9R), 2.781 (dd, J = 5.4, 13.6 Hz, 0.5, H5′, 9S), 3.94 (‘quin’, J = 6.3 Hz, 1, H4), 4.181 (t, J = 6.4 Hz, 1, H9R), 4.186 (t, J = 6.4 Hz, 1, H9S). Chemical shifts observed for TFA salt of 36(9R/S) were different from its sodium salt but parallel the signals for the trifluoroacetate of 34(9S) derived from Hcy.
S-(4-Amino-N-tert-butoxycarbonyl-2,3,4,5-tetradeoxy-α/β-D-glycero-pentofuranos-5-yl)-N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester (37)
Treatment of 35 (42 mg, 0.086 mmol) in CH2Cl2 (2 mL) with LiEt3BH (1M/THF, 0.22 mL, 0.22 mmol), by procedure F [quenched with MeOH (3 mL) at low temp., column chromatography (30 → 40% EtOAc/hexane)] gave 37 (31 mg, 73%; colorless oil) as a complex mixture of isomers (1H, and 13C NMR): MS (APCI) m/z 473 (100, [M-17]+), 373 (55, [M-Boc-18]+); (ESI) m/z 473 (50, [M-17]+), 373 (100, [M-Boc-18]+).
S-(4-Amino-2,3,4,5-tetradeoxy-α/β-D-glycero-pentofuranos-5-yl)-L-homocysteine (38)
Treatment of 37 (20 mg, 0.04 mmol) with an excess of TFA (1 mL) by Procedure C (step a, 2 h at ambient temperature) gave trifluoroacetate of 38 (13 mg, 95%; light yellow oil) as a complex mixture of isomers accompanied (~10%) by the open aldehyde form [1H NMR δ 8.89 (s)]: MS (APCI) m/z 217 (100, [M-17]+).
1-Amino-1,4-anhydro-1,N-didehydro-2,3-O-isopropylidene-5-O-methanesulfonyl-1-methyl-D-ribitol (40)
Treatment of 3947 (48.5 mg, 0.26 mmol) with MsCl (0.031 mL, 45 mg, 0.39 mmol) in the presence of Et3N (0.11 mL, 80 mg, 0.79 mmol) by Procedure A [3h; column chromatography (EtOAc → 10% MeOH/EtOAc)] gave 40 (59 mg, 85%) as a colorless oil: 1H NMR δ 1.38 (s, 3), 1.39 (s, 3), 2.16 (d, J = 1.0 Hz, 3, N=CCH3), 3.00 (s, 3, Ms), 4.38 (s, 1, H4), 4.39 (dd, 1, J = 3.6, 11.2 Hz, 1, H5), 4.53 (dd, J = 4.2, 11.3 Hz, 1, H5′), 4.64 (d, J = 5.8 Hz, 1, H3), 4.95 (‘quin’, J = 5.6 Hz, 1, H2); 13C NMR δ 17.1 (N=CMe), 25.7 (CMe2), 26.8 (CMe2), 37.4 (Ms), 69.7 (C5), 75.0 (C4), 79.8 (C3), 87.4 (C2), 112.4 (CMe2), 177.0 (C=N); MS (APCI) m/z 264 (100, MH+).
S-(1-Amino-1,4-anhydro-5-deoxy-1,N-Didehydro-2,3-O-isopropylidene-1-methyl-D-ribitol-5-yl)-N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester (41)
Treatment of 40 (49.5 mg, 0.19 mmol) with protected L-Hcy (82 mg, 0.284 mmo l) by Procedure B (step a and b, 36h) gave 124 mg of yellow oil. Crude product was chromatographed (50 → 60% EtOAc/hexane) to give 41 as an colorless oil (73 mg, 85%): 1H NMR δ 1.35 (s, 3), 1.35 (s, 3),1.43 (s, 9), 1.46 (s, 9), 1.84 (‘sx’, J = 7.3 Hz, 1, C8), 1.98-2.07 (m, 1, H8′), 2.11 (d, J = 1.1 Hz, 3, N=CMe), 2.54 (‘t’, J = 7.6 Hz, 2, H7,7′), 2.65 (dd, J = 6.4, 13.5 Hz, 1, H5), 2.87 (dd, J = 4.4, 13.5 Hz, 1, H5′), 4.17-4.27 (m, 1, H9), 4.33 (br. ‘t’, J = 4.6 Hz, 1, H4), 4.48 (d, J = 5.6 Hz, 1, H2) 4.96 (d, J = 5.6 Hz, 1, H3) 5.13 (br. d, J = 7.7 Hz, 1, NH); 13C NMR δ 17.0 (N=CMe), 25.7 (CMe2), 26.8 (CMe2), 28.0 (t-Bu), 28.3 (t-Bu), 29.2 (C7), 33.1 (C8), 35.3 (C5), 53.3 (C9), 76.5 (C4), 79.8 (t-Bu), 81.8 (C2), 82.1 (t-Bu), 87.3 (C3), 111.9 (CMe2), 155.3 (CO), 171.2 (C10), 174.7 (N=C); MS (APCI) m/z 459 (100, MH+); HRMS (TOF MS-ESI) m/z calcd for C22H39N2O6S [MH]+ 459.2523; found 459.2523.
S-(1-Amino-1,4-anhydro-5-deoxy-1,N-didehydro-2,3-O-isopropylidene-1-methyl-D-ribitol-5-yl)-L-homocysteine (42)
Treatment of 41 (62 mg, 0.136 mmol) with TFA by Procedure C (step a, ambient temperature) gave 42 (41 mg, 99%) as a colorless oil: 1H NMR (D2O) δ 1.38 (s, 3), 1.41 (s, 3), 2.10-2.20 (m, 1, H8), 2.22-2.31 (m, 1, H8′), 2.57 (br. s, 3 → 0, exch. with deuterium within few hrs, N=CMe), 2.75 (t, J = 7.3 Hz, 2, H7,7′), 2.99 (d, J = 6.2 Hz, 2, H5,5′), 4.17 (t, J = 6.4 Hz, 1, H9), 4.73 (‘t’, J = 6.3 Hz, 1, H4), 4.91 (d, J = 5.4, 1, H2), 5.64 (d, J = 5.3, 1, H3); 1H NMR (DMSO) δ 1.28 (s, 3), 1.30 (s, 3), 1.95-2.07 (m, 2, H8,8′), 2.08 (s, 3, N=CMe), 2.60 (dd, J = 7.4, 13.6 Hz, 1, H5) 2.65 (‘t’, J = 7.9 Hz, 2, H7,7′), 2.82 (dd, J = 5.2, 13.7 Hz, 1, H5′), 4.01 (br. s, 1, H9), 4.21 (‘t’, J = 5.8 Hz, 1, H4), 4.51 (d, J = 5.5, 1, H2), 5.10 (d, J = 5.5, 1, H3), 7.28 (br. s, 3, +NH3); 13C NMR (D2O) δ 15.5 (‘quin’, J = 20 Hz, N=CCD3), 24.0 (CMe2), 25.4 (CMe2) 27.4 (C7), 29.5 (C8), 31.6 (C5), 51.4 (C9), 71.2 (C4), 79.0 (C2), 84.0 (C3), 114.2 (CMe2), 171.5 (C10), 191.7 (N=CCD3); MS (APCI) m/z 303 (100, MH+).
S-(1-Amino-1,4-anhydro-5-deoxy-1,N-didehydro-1-methyl-D-ribitol-5-yl)-L-homocysteine (43)
Treatment of 42 (41 mg, 0.136 mmol) with TFA/H2O by Procedure C (step b, TFA/H2O 4:1, ambient temperature, 12 h) gave homogenous 43 (36 mg, 98%) as a colorless oil: 1H NMR (D2O) δ 2.05-2.16 (m, 1, H8), 2.17-2.27 (m, 1, H8′), 2.45 (br. s, 3 → 0, exch. with deuterium within few hrs, N=CMe), 2.73 (dt, J = 3.3, 7.3 Hz, 2, H7,7′), 2.78 (dd, J = 8.3, 14.4 Hz, 1, H5), 2.92 (dd, J = 6.0, 14.3 Hz, 1, H5′) 4.14 (t, J = 6.4 Hz, 1, H9), 4.35 (‘t’, J = 7.1 Hz, 1, H4), 4.37 (d, J = 5.3, 1, H2), 5.12 (d, J = 5.5, 1, H3); 13C NMR (D2O) δ 15.4 (‘quin’, J = 21 Hz, N=CCD3), 27.0 (C7), 29.3 (C8), 30.7 (C5), 51.3 (C9), 71.6 (C2), 71.9 (C4), 76.8 (C3), 171.4 (C10), 195.8 (N=CCD3); MS (APCI) m/z 263 (100, MH+); HRMS (AP-ESI) m/z calcd for C10H19N2O4S [MH]+ 263.1060; found 263.1065.
1-Amino-1,4-anhydro-5-O-tert-butyldimethylsilyl-1-deoxy-2,3-O-isopropylidene-D-ribitol (44)
To a solution of 7 (109 mg, 0.28 mmol) in EtOH (6 mL) was added 5% Pd/C (300 mg) and stirred under an atmosphere of H2 at room temperature for 6 h. The mixture was filtered through Celite to remove the catalyst. The Celite bed was washed with ethanol (5 mL) and the filtrate and washings were combined and evaporated. The residue was column chromatographed (30% EtOAc/hexane) to give 4450 as a colorless oil (70 mg, 80%). 1H NMR δ 0.04 (s, 3, CH3), 0.04 (s, 3, CH3), 0.87 (s, 9, t-Bu), 1.32 (s, 3, CH3), 1.46 (s, 3, CH3), 2.33 (s, 1, NH), 2.98 (‘d’, J = 2.6 Hz, 2, H1,1′), 3.20 (‘dt’, J = 0.6, 5.8 Hz, 1, H4), 3.52 (dd, J = 5.9, 10.3 Hz, 1, H5), 3.62 (dd, J = 5.1, 10.3 Hz, 1, H5′), 4.63 (dd, J = 0.8, 5.8 Hz, 1, H3), 4.68 (‘dt’, J = 2.6, 5.8 Hz, 1, H2); MS (APCI) m/z 288 (100, MH+).
1-Amino-1,4-anhydro-5-O-tert-butyldimethylsilyl-1,N-didehydro-2,3-O-isopropylidene-D-ribitol N–oxide (45)
A stirred solution of 44 (70 mg, 0.24 mmol) and SeO2 (0.01 mmol, 1.1 mg) in acetone (3 mL) was cooled to −4°C under N2 atmosphere and H2O2 (25%) was added slowly (3-4 h) until the reaction was completed (as judged by TLC). Volatiles are evaporated and the residue was partitioned (EtOAc//NaHCO3/H2O).The organic layer was collected, washed (brine) and dried (MgSO4). The resulting solid was chromatographed (50% EtOAc/hexane) to give 45 (54 mg, 73%) as a white solid with data as reported.49
1-Amino-1,4-anhydro-1,N-didehydro-2,3-O-isopropylidene-D-ribitol N-oxide (46)
Desilylation of 45 (155 mg, 0.51 mmol) with TBAF (1M/THF, 0.77 mL, 0.77 mmol) at −4 °C as described for 14b [column chromatography (10% → 20% MeOH/CHCl3)] gave 46 (40 mg, 42%) as a white solid: 1H NMR δ 1.34 (s, 3, CH3), 1.41 (s, 3, CH3), 3.87 (dd, J = 2.6, 11.9 Hz, 1, H5), 4.02-4.03 (m, 1, H4), 4.14 (dd, J = 2.3, 11.9 Hz, 1, H5′), 4.96 (d, J = 6.2 Hz, 1, H3), 5.21 (‘dt’, J = 1.4, 6.2 Hz, 1, H2), 6.90 (s, 1, H1); 13C NMR δ 25.7 (CMe2), 27.2 (CMe2), 58.9 (C5), 77.2 (C3), 78.9 (C4), 80.8 (C2), 111.5 (CMe2 ), 134.6 (C1); MS (ESI) m/z 186 (100, M+).
1-Amino-1,4-anhydro-1,N-didehydro-2,3-O-isopropylidene-5-O-methanesulfonyl-D-ribitol N–oxide (47)
Treatment of 46 (40 mg, 0.21 mmol) at −4°C with MsCl (26 μL, 0.32 mmol) by Procedure A [column chromatography (80% → 90% EtOAc/hexane)] gave 47 (24 mg, 56%) as a colorless oil: 1H NMR δ 1.37 (s, 3, CH3), 1.46 (s, 3, CH3), 3.04 (s, 3, Ms), 4.23- 4.24 (m, 1, H4), 4.55 (dd, J = 1.8, 11.1 Hz, 1, H5), 4.83 (dd, J = 2.4, 11.1 Hz, 1, H5′), 4.91 (dd, J = 1.1, 6.3 Hz, 1, H3), 5.25 (‘dt’, J = 1.5, 6.4 Hz, 1, H2), 6.99 (s, 1, H1); 13C NMR δ 25.6 (CMe2), 27.1 (CMe2), 37.4 (Ms), 65.5 (C5), 75.8 (C3), 77.8 (C4), 78.4 (C2), 112.6 (CMe2), 134.1 (C1); MS (APSI) m/z 266 (100, MH+); HRMS (AP-ESI) m/z calcd for C9H16NO6S [MH]+ 266.0693; found 266.0682.
S-(1-Amino-1,4-anhydro-5-deoxy-1,N-didehydro-2,3-O-isopropylidene-D-ribitol-5-yl N–oxide)-N-tert-butoxycarbonyl-L-homocysteine tert-butyl ester (48)
Treatment of 47 (24 mg, 0.09 mmol) with L-homocysteine (38 mg, 0.13 mmol) by Procedure B (step a and b, 8h at −20°C) and purification by column chromatography (80% → 90% EtOAc/hexane) gave 48 as a colorless oil (24 mg, 43%): 1H NMR δ 1.37 (s, 3, CH3), 1.44 (s, 9, t-Bu ), 1.45 (s, 3, CH3), 1.46 (s, 9, t-Bu), 1.81-1.90 (m, 1, H8), 1.96-2.09 (m, 1, H8′), 2.52-2.70 (m, 2, H7,H7′), 3.07 (dd, J = 3.5, 14.4 Hz, 1, H5), 3.14 (dd, J = 5.2, 14.4 Hz, 1, H5′), 4.20-4.31 (m, 2, H4,9), 4.71 (d, J = 6.2 Hz, 1, H3), 5.06 (m, 1, NH), 5.31 (‘dt’, J = 1.4, 6.4 Hz, 1, H2), 6.97 (s, 1, H1); 13C NMR δ 25.6 (CMe2), 26.1 (CMe2), 28.0 (t-Bu), 28.3 (t-Bu), 29.1 (C7), 32.0 (C5), 32.8 (C8), 53.1 (C9), 77.9 (C3), 79.0 (C2), 79.1 (C4), 79.8 (t-Bu), 82.2 (t-Bu), 112.0 (CMe2), 133.4 (C1), 155.4 (CO), 171.1 (C10); MS (APCI) m/z 461 (100, MH+).
S-(1-Amino-1,4-anhydro-5-deoxy-1,N-didehydro-D-ribitol-5-yl N-Oxide)-L-homocysteine (49)
Treatment of 48 (72 mg, 0.15 mmol) with TFA by Procedure C (step a, 5 h; step b, 6 h at 0°C) gave crude 49. Purification on HPLC (5% CH3CN/H2O at 2.5 mL/min; tR = 10-14 min) afforded 49 (16 mg, 40 %) as a white solid: 1H NMR (D2O) δ 2.01-2.17 (m, 2, H8,8′), 2.83-2.89 (m, 2, H7,7′), 2.97 (dd, J = 6.3, 14.4 Hz, 1,H5), 3.06 (dd, J = 3.8, 14.4 Hz, 1, H5′), 3.73-3.77 (m, 1, H9), 4.08-4.22 (m, 1, H4), 4.40 (dd, J = 3.2, 6.0 Hz, 1, H3), 4.89-4.96 (m, 1, H2), 7.25 (s, 1, H1); 13C NMR δ 27.6 (C7), 29.9 (C5), 30.5 (C8), 53.7 (C9), 70.4 (C3), 78.3 (C4), 80.7 (C2), 141.8 (C1), 173.9 (C10); MS (APCI) m/z 265 (100, MH+); HRMS (TOF MSESI) m/z calcd for C9H16N2O5SNa [M+Na]+ 287.0672; found 287.0664.
LuxS Inhibition Assay
SRH was prepared by incubating SAH (typically 10 mM) with nucleosidase Pfs (2 μM) overnight at 4 °C and the completion of the reaction was monitored spectrophotometrically by the absorption difference between SAH and adenine (Δε276 = −1.4 mM−1 cm−1). A typical LuxS reaction (total volume = 1.0 mL) contained 50 mM HEPES (pH 7.0), 150 mM NaCl, 17.8 μM SRH, and 150 μM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB). The reaction was initiated by the addition of LuxS (final concentration 0.8 μM) and monitored continuously at 412 nm (ε = 14000 M−1 cm−1) in a Perkin-Elmer λ20 UV-vis spectrophotometer at room temperature. For compounds that showed time dependent inhibition, the inhibitor and LuxS (1.6 μM) were preincubated for 30 min at 4 °C and the reaction was then initiated by addition of SRH.
Acknowledgment
We thank NIH (SC1CA138176, AI62901, and DE019667) and FIU’s Doctoral Evidence Acquisition Fellowship (V.L.A.M) for their support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Waters CM, Bassler BL. Annu. Rev. Cell Dev. Biol. 2005;21:319. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- (2).Bassler BL, Losick R. Cell. 2006;125:237. doi: 10.1016/j.cell.2006.04.001. [DOI] [PubMed] [Google Scholar]
- (3).Winans SC. Nature. 2005;437:330. doi: 10.1038/437330a. [DOI] [PubMed] [Google Scholar]
- (4).Camilli A, Bassler BL. Science. 2006;311:1113. doi: 10.1126/science.1121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lebeer S, De Keersmaecker SCJ, Verhoeven TLA, Fadda AA, Marchal K, Vanderleyden J. J. Bacteriol. 2007;189:860. doi: 10.1128/JB.01394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ni N, Li M, Wang J, Wang B. Med. Res. Rev. 2009;29:65. doi: 10.1002/med.20145. [DOI] [PubMed] [Google Scholar]
- (7).Mattmann ME, Blackwell HE. J. Org. Chem. 2010;75:6737. doi: 10.1021/jo101237e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Galloway WRJD, Hodgkinson JT, Bowden SD, Welch M, Spring DR. Chem. Rev. 2011;111:28. doi: 10.1021/cr100109t. [DOI] [PubMed] [Google Scholar]
- (9).Lee JE, Cornell KA, Riscoe MK, Howell PL. Structure. 2001;9:941. doi: 10.1016/s0969-2126(01)00656-6. [DOI] [PubMed] [Google Scholar]
- (10).Lee JE, Cornell KA, Riscoe MK, Howell PL. J. Biol. Chem. 2003;278:8761. doi: 10.1074/jbc.M210836200. [DOI] [PubMed] [Google Scholar]
- (11).Pei D, Zhu J. Curr. Opin. Chem. Biol. 2004;8:492. doi: 10.1016/j.cbpa.2004.08.003. [DOI] [PubMed] [Google Scholar]
- (12).Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM. Nature. 2002;415:545. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- (13).Meijler MM, Hom LG, Kaufmann GF, McKenzie KM, Sun C, Moss JA, Matsushita M, Janda KD. Angew. Chem. Int. Ed. 2004;43:2106. doi: 10.1002/anie.200353150. [DOI] [PubMed] [Google Scholar]
- (14).Semmelhack MF, Campagna SR, Federle MJ, Bassler BL. Org. Lett. 2005;7:569. doi: 10.1021/ol047695j. [DOI] [PubMed] [Google Scholar]
- (15).Semmelhack MF, Campagna SR, Hwa C, Federle MJ, Bassler BL. Org. Lett. 2004;6:2635. doi: 10.1021/ol048976u. [DOI] [PubMed] [Google Scholar]
- (16).Zhu J, Dizin E, Hu X, Wavreille A-S, Park J, Pei D. Biochemistry. 2003;42:4717. doi: 10.1021/bi034289j. [DOI] [PubMed] [Google Scholar]
- (17).Zhu J, Patel R, Pei D. Biochemistry. 2004;43:10166. doi: 10.1021/bi0491088. [DOI] [PubMed] [Google Scholar]
- (18).Rajan R, Zhu J, Hu X, Pei D, Bell CE. Biochemistry. 2005;44:3745. doi: 10.1021/bi0477384. [DOI] [PubMed] [Google Scholar]
- (19).Zhu J, Hu X, Dizin E, Pei D. J. Am. Chem. Soc. 2003;125:13379. doi: 10.1021/ja0369663. [DOI] [PubMed] [Google Scholar]
- (20).Turner MA, Yang X, Yin D, Kuczera K, Borchardt RT, Howell PL. Cell Biochem. Biophys. 2000;33:101. doi: 10.1385/CBB:33:2:101. [DOI] [PubMed] [Google Scholar]
- (21).Yuan C-S, Liu S, Wnuk SF, Robins MJ, Borchardt RT. Biochemistry. 1994;33:3758. doi: 10.1021/bi00178a036. [DOI] [PubMed] [Google Scholar]
- (22).Alfaro JF, Zhang T, Wynn DP, Karschner EL, Zhou ZS. Org. Lett. 2004;6:3043. doi: 10.1021/ol049182i. [DOI] [PubMed] [Google Scholar]
- (23).Shen G, Rajan R, Zhu J, Bell CE, Pei D. J. Med. Chem. 2006;49:3003. doi: 10.1021/jm060047g. [DOI] [PubMed] [Google Scholar]
- (24).Zang T, Lee BWK, Cannon LM, Ritter KA, Dai S, Ren D, Wood TK, Zhou ZS. Bioorg. Med. Chem. Lett. 2009;19:6200. doi: 10.1016/j.bmcl.2009.08.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wnuk SF, Lalama J, Garmendia CA, Robert J, Zhu J, Pei D. Bioorg. Med. Chem. 2008;16:5090. doi: 10.1016/j.bmc.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wnuk SF, Robert J, Sobczak AJ, Meyers BP, Malladi VLA, Zhu J, Gopishetty B, Pei D. Bioorg. Med. Chem. 2009;17:6699. doi: 10.1016/j.bmc.2009.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gopishetty B, Zhu J, Rajan R, Sobczak AJ, Wnuk SF, Bell CE, Pei D. J. Am. Chem. Soc. 2009;131:1243. doi: 10.1021/ja808206w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Jencks WP. Chem. Rev. 1972;72:705. [Google Scholar]
- (29).Pearson MSM, Mathé-Allainmat M, Fargeas V, Lebreton J. Eur. J. Org. Chem. 2005:2159. [Google Scholar]
- (30).Kajimoto T, Liu KKC, Pederson RL, Zhong Z, Ichikawa Y, Porco JA, Wong CH. J. Am. Chem. Soc. 1991;113:6187. [Google Scholar]
- (31).Wong C-H, Provencher L, Porco JA, Jung S-H, Wang Y-F, Chen L, Wang R, Steensma DH. J. Org. Chem. 1995;60:1492. [Google Scholar]
- (32).Schramm VL. Acc. Chem. Res. 2003;36:588. doi: 10.1021/ar0200495. [DOI] [PubMed] [Google Scholar]
- (33).Schramm VL. Arch. Biochem. Biophys. 2005;433:13. doi: 10.1016/j.abb.2004.08.035. [DOI] [PubMed] [Google Scholar]
- (34).Yokoyama M, Momotake A. Synthesis. 1999:1541. [Google Scholar]
- (35).Lee JE, Singh V, Evans GB, Tyler PC, Furneaux RH, Cornell KA, Riscoe MK, Schramm VL, Howell PL. J. Biol. Chem. 2005;280:18274. doi: 10.1074/jbc.M414471200. [DOI] [PubMed] [Google Scholar]
- (36).Singh V, Evans GB, Lenz DH, Mason JM, Clinch K, Mee S, Painter GF, Tyler PC, Furneaux RH, Lee JE, Howell PL, Schramm VL. J. Biol. Chem. 2005;280:18265. doi: 10.1074/jbc.M414472200. [DOI] [PubMed] [Google Scholar]
- (37).Fleet GWJ, Son JC. Tetrahedron. 1988;44:2637. [Google Scholar]
- (38).Calvez O, Chiaroni A, Langlois N. Tetrahedron Lett. 1998;39:9447. [Google Scholar]
- (39).Lee J, Hoang T, Lewis S, Weissman SA, Askin D, Volante RP, Reider PJ. Tetrahedron Lett. 2001;42:6223. [Google Scholar]
- (40).Haidle AM, Myers AG. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12048. doi: 10.1073/pnas.0402111101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Qiu XL, Qing FL. J. Org. Chem. 2005;70:3826. doi: 10.1021/jo050057+. [DOI] [PubMed] [Google Scholar]
- (42).Kim YJ, Kitahara T. Tetrahedron Lett. 1997;38:3423. [Google Scholar]
- (43).Witte JF, McClard RW. Tetrahedron Lett. 1991;32:3927. [Google Scholar]
- (44).Zanardi F, Battistini L, Nespi M, Rassu G, Spanu P, Cornia M, Casiraghi G. Tetrahedron: Asymmetry. 1996;7:1167. [Google Scholar]
- (45).Otsuka M, Masuda T, Haupt A, Ohno M, Shiraki T, Sugiura Y, Maeda K. J. Am. Chem. Soc. 1990;112:838. [Google Scholar]
- (46).Malladi VLA, Sobczak AJ, Maricic N, Murugapiran SK, Schneper L, Makemson J, Mathee K, Wnuk SF. Bioorg. Med. Chem. doi: 10.1016/j.bmc.2011.07.044. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Moriarty RM, Mitan CI, Branza-Nichita N, Phares KR, Parrish D. Org. Lett. 2006;8:3465. doi: 10.1021/ol061071r. [DOI] [PubMed] [Google Scholar]
- (48).Evans GB, Furneaux RH, Gainsford GJ, Schramm VL, Tyler PC. Tetrahedron. 2000;56:3053. [Google Scholar]
- (49).Evans GB, Furneaux RH, Hausler H, Larsen JS, Tyler PC. J. Org. Chem. 2004;69:2217. doi: 10.1021/jo035744k. [DOI] [PubMed] [Google Scholar]
- (50).Horenstein BA, Zabinski RF, Schramm VL. Tetrahedron Lett. 1993;34:7213. [Google Scholar]
- (51).Murruzzu C, Riera A. Tetrahedron: Asymmetry. 2007;18:149. [Google Scholar]
- (52).Moreaux V, Warren H, Williams JM. Tetrahedron Lett. 1997;38:4655. [Google Scholar]