

Abstract
The spontaneously hypertensive rat (SHR) is widely used as a model of attention-deficit/hyperactivity disorder (ADHD). Deficits in central nicotinic receptors (nAChRs) have previously been observed in SHRs, which is interesting since epidemiological studies have identified an association between smoking and ADHD symptoms in humans. Here we examine whether nAChR deficits in SHRs compared to Wistar Kyoto rat (WKY) controls are nAChR subtype-specific and whether these deficits correlate with changes at the level of mRNA transcription in specific brain regions. Levels of binding sites (Bmax) and dissociation constants (Kd) for nAChRs were determined from saturation curves of high-affinity [3H]epibatidine- and [3H]MLA binding to membranes from cortex, striatum, hippocampus and cerebellum. In additional brain regions, nAChRs were examined by autoradiography with [125I]A-85380 and [125I]α-bungarotoxin. Levels of mRNA encoding nAChR subunits were measured using quantitative real-time PCR (qPCR). We show that the number of α4β2 nAChR binding sites is lower globally in the SHR brain compared to WKY in the absence of significant differences in mRNA levels, with the exception of lower α4 mRNA in cerebellum of SHR compared to WKY. Further, nAChR deficits were subtype- specific because no strain difference was found in α7 nAChR binding or α7 mRNA levels. Our results suggest that the lower α4β2 nAChR number in SHR compared to WKY may be a consequence of dysfunctional post-transcriptional regulation of nAChRs.
Keywords: Attention-deficit/hyperactivity disorder, mRNA, nicotinic receptor, post translational, spontaneous hypertensive rat, Wistar Kyoto rat
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a frequent diagnosis in children with a prevalence of 8 to 12% worldwide (Faraone et al. 2003). Typical behavioral symptoms include hyperactivity, impulsivity and difficulty in concentration (Biederman 2005) with negative outcomes during adulthood if symptoms are left untreated (Spencer et al. 2007). Although the exact etiology of ADHD is unclear, twin studies have suggested that ADHD is highly heritable (Biederman 2005), and both animal and human studies have suggested that dysregulation of frontal-subcortical-cerebellar circuits contributes to the pathophysiology of ADHD (Biederman & Faraone 2005). In addition, as with many psychiatric and developmental disorders, environmental risk factors are correlated with symptoms of ADHD (Thapar et al. 2007). In particular, maternal smoking has been found to correlate with increased ADHD symptoms (Linnet et al. 2003, Milberger et al. 1996). In addition, adults with ADHD are more likely to smoke and fail in smoking cessation when compared to control populations (Humfleet et al. 2005, Pomerleau et al. 1995), suggesting that ADHD could be related to cholinergic perturbations. Given that symptoms of ADHD generally precede the onset of smoking, and that nicotine treatment has been shown to attenuate dysfunction in ADHD (Levin et al. 2001), it has been suggested that the increased smoking in ADHD may result from self-medication to improve symptoms of ADHD (Gehricke et al. 2007).
The primary targets for nicotine in the brain are nicotinic acetylcholine receptors (nAChRs), which are pentameric ligand-gated ion channels composed of a number of subunits (α2–α7 and β2–β4) (Millar & Gotti 2009) expressed widely in the central nervous system. A clinical trial of an agonist of the high affinity α4β2 nAChR subtype (ABT-418) resulted in improvement of ADHD symptoms (Wilens et al. 1999), suggesting that a deficit in nAChR signaling may contribute to attentional dysfunction. The involvement of nAChRs in ADHD has been supported by evidence from one of the most commonly used animal models of ADHD (Davids et al. 2003), the spontaneous hypertensive rat (SHR), where previous studies found that both α4β2 and α7 nAChR binding sites were lower in several brain regions of SHRs compared to Wistar Kyoto (WKY) controls (Gattu et al. 1997a, Hohnadel et al. 2005, Terry et al. 2000). As a model of ADHD, the SHR shows good face validity and exhibits a constellation of behavioral characteristics reminiscent of ADHD including deficits in sustained attention (Sagvolden 2000), motor impulsivity (Berger & Sagvolden 1998) and hyperactivity in response to novel situations (Sagvolden et al. 1992a). These behavioral abnormalities in SHR are responsive to stimulants such as methylphenidate and amphetamine (Sagvolden et al. 1992b, Myers et al. 1982), which adds predictive validity to this animal model of ADHD. Moreover, juvenile SHRs (4–6-week-old) exhibit behavioral symptoms of ADHD (Qian et al. 2010, Ueno et al. 2002, Whitehorn et al. 1983) prior to the development of hypertension which occurs at around age 7-weeks (Cruzado et al. 2005). The SHR line was derived from the WKY rat strain (Okamoto & Aoki 1963) suggesting a genetic and construct validity of this model when compared to control animals from the WKY strain (Sagvolden et al. 2009).
Although previous reports indicate changes in both the α4β2- and α7 nAChR subtype in SHRs (Gattu et al. 1997a, Gattu et al. 1997b, Hohnadel et al. 2005, Terry et al. 2000), most of these studies have employed adult SHRs, with potential confounding effects caused by the hypertension in these animals. In addition, while nAChR impairments may contribute to ADHD-like symptoms in SHRs, little is known about the mechanism underlying these receptor changes. Here, using highly specific in vitro binding techniques, we investigated distinct nAChR subtypes in selected brain regions of juvenile pre-hypertensive SHRs compared to WKY rats. Moreover, we examined whether lower numbers of nAChRs in SHRs compared to WKY controls were coupled with concomitant changes at the level of mRNA transcription. Our data provide an in-depth determination of specific nAChRs and nAChR subunit expression in the brain of this animal model of ADHD.
Material and methods
Materials
[3H]epibatidine (30–70 Ci/mmol), [125I]α-bungarotoxin (10–20 µCi/µg) and [125I]A-85380 (2200 Ci/mmol) were purchased from Perkin-Elmer (Waltham, MA, USA). [3H]Methyllycaconitine (MLA) (60–100 Ci/mmol) was purchased from American Radiolabeled Chemicals Inc (St. Louis, MO, USA). Additional reagents were purchased from standard commercial sources.
Animals
Male SHRs were obtained from Charles River (Sulzfeld, Germany) while male WKY rats were obtained from Harlan UK (Blacktorn, UK). Animals were kept under conditions of constant temperature (22 ± 2°C) and humidity (55 ± 5%), a 12 h alternating light/dark cycle and with free access to food and water. Animals were treated according to the Norwegian Animal Welfare Act and the European Communities Council Directive of 24 November 1986 (86/609/EEC) and efforts were made to minimize animal suffering and to reduce the number of animals used.
Preparation of membranes
4-week-old SHR and WKY rats were anesthetized with CO2, decapitated and the cortex, striatum, hippocampus and cerebellum were removed. The brain tissue was rapidly frozen and stored at −80°C. Subsequent preparation steps included thawing of the frozen brain tissue at 23°C, homogenization in 15 volumes of 50 mM Tris-HCl (pH 7.4) (450 rpm, 12 strokes, glass-teflon homogenizer) before centrifugation at 100 000 g for 30 min (Ti80 rotor, Beckman Optima™ LE-80K Ultracentrifuge, 4°C). The supernatant was discarded and the resulting pellet was resuspended in 15 volumes of 50 mM Tris buffer. The suspension was incubated for 30 min at 23°C and then centrifuged at 100 000 g for 30 min. The supernatant was discarded before the resulting pellet was resuspended in 4 volumes of either [3H]epibatidine- or [3H]MLA incubation buffer.The membrane preparations were stored at −80 °C prior to experiments and protein quantification by the BCA Protein Assay Kit (Pierce, Rockford, IL, USA).
High-affinity [3H]epibatidine binding to membranes
α4β2 nAChR sites account for approximately 90% of high-affinity [3H]-epibatidine binding in whole-brain preparations (Marks et al. 2006), and was measured as described (Marks et al. 2009) with minor adjustments. In brief, saturation curves for high-affinity binding were determined by incubating membranes (0.1 mg protein) with [3H]epibatidine (0.005–0.4 nM) for 3.0 hours at 23°C in glass tubes containing incubation buffer (NaCl, 144 mM; KCl, 1.5 mM, CaCl2, 2 mM; MgSO4, 1 mM; HEPES, 20 mM; pH 7.4) in a total volume of 3 mL. Non-specific binding was determined in the presence of 100 µM nicotine. After incubation, the mixture was diluted with 3 mL ice-cold incubation buffer and vacuum filtered through a GF/B filter (Whatman), which had been presoaked for 60 min in 0.5% polyethylenimine. Filters were immediately washed twice with 3 mL ice-cold incubation buffer, dissolved in Filter-Count and counted for retained radioactivity in a Liquid Scintillation Analyzer (Tri-Carb 3100TR).
[3H]MLA binding to membranes
[3H]MLA binding was measured as described (Davies et al. 1999) with minor adjustments. In brief, saturation curves were determined by incubating membranes (0.6 mg protein) with [3H]MLA (0.25–10 nM) for 1.5 hours at 23°C in glass tubes containing incubation buffer (Na2HPO4, 20 mM; KH2PO4, 5 mM; NaCl, 150 mM; BSA, 0.1% (w/v); pH 7.4) in a total volume of 0.25 mL. Non-specific binding was determined in the presence of 1 mM nicotine. After incubation, the mixture was diluted with 2 mL ice-cold incubation buffer and vacuum filtered through a GF/B filter (Whatman) which had been presoaked for 60 min in 0.5% polyethylenimine. Filters were immediately washed twice with 2 mL ice-cold incubation buffer, dissolved in Filter-Count and counted for retained radioactivity in a Liquid Scintillation Analyzer (Tri-Carb 3100TR).
Preparation of sections
4-week-old SHR and WKY rats were anesthetized with CO2, decapitated and brains were frozen and stored at −80°C. Parallel series of coronal sections (20-µm thick) from the two rat strains were obtained by sectioning in a cryostat at −20°C, thaw-mounted onto SuperFrost Plus slides (Fisher Scientific, Pittsburgh, PA, USA) and stored at −70°C until use.
[125I]A-85380 binding in brain sections
[125I]A-85380 binding was measured as described previously (Mineur et al. 2009). In brief, frozen sections were thawed to room temperature and incubated with 200 pM [125I]A-85380 for 45 min in 50 mM Tris-HCl, pH 7.4, washed three times in the same buffer and washed once in distilled water. Slides were air-dried overnight and exposed to film (Kodak MR) for 24 hours at room temperature. Quantitation of [125I]A-85380 binding was performed using Photoshop CS4 11.0 (Adobe Systems, Mountainview, CA, USA) by measuring mean grey value in each brain region. Background density was subtracted for each measurement.
[125I]α-bungarotoxin binding in brain sections
[125I]α-bungarotoxin binding was measured as described previously (Whiteaker et al. 2000). In brief, frozen sections were thawed to room temperature and incubated in binding buffer (NaCl, 144 mM; KCl, 1.5 mM; CaCl2, 2 mM; MgSO4, 1 mM; HEPES, 20 mM; BSA, 0.1% (w/v); pH = 7.5) at 23°C for 10 min. The sections were then incubated with 2 nM [125I]α-bungarotoxin in binding buffer for 4 h at 23°C, and washed as follows (all washes at 4°C): 10 min in binding buffer (twice), 5 s in 0.1X binding buffer (twice), and 2 s in 5 mM HEPES (pH = 7.5), twice. Non-specific binding was determined in the presence of 1 mM nicotine. Slides were air-dried overnight and exposed to film (Kodak MR) for 8 days at room temperature. Quantitation of [125I]α-bungarotoxin binding was performed using Photoshop CS4 11.0 by measuring mean grey value in each brain region.
Quantitative real-time PCR (qPCR) and mRNA quantitation
4 week old SHR and WKY rats were stunned and decapitated, and the cortex, striatum, hippocampus and cerebellum were removed quickly and frozen at −80°C. The frozen brain tissues were soaked overnight at −20°C in RNAlater-ICE Solution (Applied Biosystems-Ambion, Austin, TX, USA) before total RNA was extracted using the RNeasy Lipid tissue kit (Qiagen, Valencia, CA, USA). The concentration and purity of the total RNA samples were measured using the NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). 500 ng of total RNA was reverse transcribed into cDNA in a volume of 60 µL H2O and residual genomic DNA was removed using the Quantitect reverse transcription kit (Qiagen Inc., Chatsworth, CA, USA). Primers for qPCR were designed using Primer3 (http://frodo.wi.mit.edu/), and settings in this program were chosen to avoid hairpin secondary structures and self- and cross-dimers. Each pair of primers was designed to amplify short (90–150 bp) products. To ensure specificity, primer sequences were queried using Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) against all known rat mRNA sequences. Dilution series of cDNA were used to measure PCR efficiency of primers and only primers with efficiencies between 90 and 110% were retained. Given the very low level of expression of the β4 mRNA in most brain areas, we did not investigate the expression of this nAChR subunit. All primers were synthesized by Integrated DNA Technologies (Coralville, IA, USA) and primer sequences are detailed in Table 1.
Table 1.
Details of Primer Sequences used for quantitative real-time PCR
| Primer | Direction | Sequence | Reference |
|---|---|---|---|
| Chrna3 | Forward Reverse |
5'-AGTCTGTCGACGCTGTGTTG-3 5'-TGGCAACGTACTTCCAATCA-3' |
Novel |
| Chrna4 | Forward Reverse |
5'-AGCATCGACGTCACCTTCTT-3' 5'-CGGCTATGCATGCTCACTAA-3' |
Novel |
| Chrna5 | Forward Reverse |
5'-ACTCGATTGCATTCGCTACA-3' 5'-CCCAATGATTGACACCAGAA-3' |
Novel |
| Chrna7 | Forward Reverse |
5'-ATCGTGGGCCTCTCTGTAGT-3' 5'-GAAACCATGCACACCAGTTC-3' |
Novel |
| Chrnb2 | Forward Reverse |
5'-TGCGAAGTGAGGATGATGAC-3' 5'-ACGGTCCCAAAGACACAGAC-3' |
Chen et al., 2005 |
| Chrnb3 | Forward Reverse |
5'-GCCTCCGAGTCCATCAGATA-3' 5'-AAGATGCGGTCCAGAACTTG-3' |
Chen et al., 2005 |
| Hprt1 | Forward Reverse |
5'-TTGTTGGATATGCCCTTGACT-3' 5'-CCGCTGTCTTTTAGGCTTTG-3' |
Van Wijngaarden et al., 2007 |
Each cDNA sample was amplified in duplicate wells containing 1 µL from the 60 µL cDNA solution (described above), 0.6 µM of each primer and 8 µL Power SYBR Green PCR Master Mix (Applied Biosystems Inc.) in a final volume of 16 µL. qPCR was performed for 40 cycles as follows: 95°C for 10 min, followed by 40 cycles with denaturation at 95°C for 15 s and annealing and extension at 62°C for 1 min using the 7500 Real-Time PCR System (Applied Biosystems Inc, Foster City, CA, USA). Relative gene expression was determined by the -ΔΔCT method (Livak & Schmittgen 2001) with HPRT used as housekeeping gene for normalization. Negative controls (samples without cDNA) were included in all experiments. The specificity of each PCR reaction was verified by melt-curve analysis and by checking the PCR products on a 1.5% agarose gel.
Statistical analysis
Results shown are the mean ± standard error of the mean (SEM). Fitting of saturation curves to binding data, and determination of receptor density and dissociation constant were performed with GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA) using the one site-specific binding equation. For comparisons of two groups, Student's t-test for unpaired groups was used. For comparison of binding to SHR- and WKY brain sections, two-way ANOVA (with strain and brain region as factors) was carried out. Values of p < 0.05 were considered to be significant.
Results
To examine whether SHRs exhibit altered numbers of nAChRs, we estimated saturation isotherms for high-affinity [3H]epibatidine binding using membrane preparations derived from cortex, striatum, hippocampus and cerebellum of 4-week-old SHRs and WKY rats. As illustrated by the linear scatchard plots shown in Fig. 1, over a concentration range of 0.005–0.4 nM, [3H]epibatidine appeared to bind a single class of high-affinity receptors, with a regional distribution corresponding to the α4β2 nAChR subtype (Marks et al. 2009). The dissociation constant (Kd) for [3H]epibatidine binding obtained in these experiments was between 11 and 22 pM. Analysis of saturation data showed that maximum [3H]epibatidine binding sites (Bmax) were lower in SHRs compared to WKY rats in all brain regions examined. Of the four brain regions, cerebellum and hippocampus showed largest strain differences with Bmax values of 10.8 ± 0.4 and 17.4 ± 1.4 fmol/mg protein (t(6) = 6.53, p < 0.0001) in cerebellum of SHRs and WKY respectively, while in hippocampus, Bmax values were 12.8 ± 0.8 and 17.0 ± 0.3 fmol/mg protein (t(4) = 4.59, p < 0.05) in SHRs and WKY respectively. Moderate differences in Bmax values of [3H]epibatidine were also observed in cortex and striatum. In cortex, Bmax values were 19.8 ± 1.0 and 24.1 ± 0.5 fmol/mg protein (t(5) = 3.39, p < 0.05) for SHRs and WKY respectively, while in striatum, Bmax values were 22.1 ± 0.3 and 26.4 ± 1.0 fmol/mg protein (t(6) = 3.97, p < 0.01) for SHRs and WKY respectively. On the other hand, no significant difference in the dissociation constant (Kd values) of [3H]epibatidine binding was found between SHRs and WKY rats in any of the examined brain regions. Given that these [3H]epibatidine membrane binding data suggested that the number of α4β2 nAChRs were globally lower in SHR brains compared to WKY, we next measured [125IA-85380 binding in brain sections from SHRs and WKY, and included additional brain regions to the analysis. Consistent with previous a report (Mukhin et al. 2000), the regional distribution of [125I]A-85380 binding in coronal rat brain sections (Fig. 2) strongly matched that of the α4β2 nAChR subtype with high levels of binding in thalamic nuclei, intermediate levels in striatum and cortex, and low levels in hypothalamus and cerebellum. Two-way ANOVA revealed a significant effect of rat strain (F(1,96) = 51.11, p < 0.0001), with approximately 30% lower [125I]A-85380 binding overall in SHR compared to WKY. In addition, there was a significant effect of brain region F(11,96) = 41.93, p < 0.0001), with no interaction between strain and brain region F(11,96) = 0.62, p = 0.80), thereby supporting the conclusion that α4β2 nAChR binding sites appeared lower globally in SHR brains compared to WKY controls.
Figure 1.

Specific [3H]epibatidine binding to SHR and WKY membranes from cortex (A), striatum (B), hippocampus (C) and cerebellum (D). Corresponding Scatchard plots are shown in the inset. Maximum binding sites (Bmax) were lower in SHRs compared to WKY rats in all four brain regions, with no significant difference in dissociation constants (Kd values). In cortex, Bmax values were 19.8 ± 1.0 and 24.1 ± 0.5 fmol/mg protein (p < 0.05), with Kd = 11.6 ± 0.9 and 11.8 ± 0.5 pM for SHR and WKY respectively (p > 0.05). In striatum, Bmax values were 22.1 ± 0.3 and 26.4 ± 1.0 fmol/mg protein (p < 0.01), with Kd = 15.7 ± 1.3 and 15.9 ± 1.9 pM for SHR and WKY respectively (p > 0.05). In hippocampus, Bmax values were 12.8 ± 0.8 and 17.0 ± 0.3 fmol/mg protein (p < 0.05), with Kd = 16.6 ± 2.1 and 21.9 ± 2.7 pM for SHR and WKY respectively (p > 0.05). In cerebellum, Bmax values were 10.8 ± 0.4 and 17.4 ± 1.4 fmol/mg protein (p < 0.0001), with Kd = 17.7 ± 2.1 and 21.7 ± 2.6 pM for SHR and WKY respectively (p > 0.05). Membranes (~0.1 mg protein) and [3H]epibatidine were incubated for 3.0 hours at 23°C. Non-specific binding was determined in the presence of 100 µM nicotine. Each data point represents mean ± SEM of 3 to 4 rats.
Figure 2.

Distribution of receptors labeled by [125I]A-85380 autoradiography in brain sections from SHR and WKY (A). Quantitative analysis of [125I]A-85380 binding in brain regions from SHR and WKY (B). Two-way ANOVA revealed a significant effect of rat strain (p < 0.0001), with approximately 30% lower [125I]A-85380 binding overall in SHR compared to WKY. Sections were incubated with 200 pM [125I]A-85380 for 45 min. Each value represents mean ± SEM of 5 to 6 rats. Cx, cerebral cortex; Cg, cingulate cortex; Cpu, caudate putamen; Acb, accumbens nucleus; Ml, molecular layer of dentate gyrus; Sl-m, stratum-lacunosum molecular; Mhb, medial habenular nucleus; Th, thalamus; Amy, amygdala; Hyp, hypothalamus; Snc, substantia nigra compact part; Cb, cerebellum. Two-way ANOVA revealed a
We next examined whether SHRs exhibit altered numbers of α7 nAChRs compared to WKY controls. For this purpose, we estimated saturation isotherms for [3H]MLA binding using membrane preparations derived from cortex, striatum, hippocampus and cerebellum of 4-week-old SHR and WKY rats. As illustrated in Fig. 3, [3H]MLA appeared to bind a single class of receptors, with a regional distribution corresponding to the α7 nAChR subtype, as reported previously (Davies et al. 1999). Analysis of saturation data found no significant change in Bmax between SHRs and WKY in any of the examined brain regions. Similarly, no significant change in Kd was found between the strains. To confirm this lack of α7 nAChR alterations in SHRs, we also measured [125I]α-bungarotoxin binding in brain sections from SHRs and WKY. Consistent with a previous report (Chen & Patrick 1997), the regional distribution of [125I]α-bungarotoxin binding matched that of the α7 nAChR subtype with high levels of binding in hippocampus, intermediate levels in cortex and low levels in striatum and cerebellum (Fig. 4 and S1). Two-way ANOVA showed a significant effect of brain region (F(3,32) = 12.03, p < 0.0001), but no significant effect of strain (F(1,32) = 0.32, p = 0.65). In addition, no interaction between strain and brain region was found (p = 0.99). Thus, in examined brain regions, α7 nAChRs appeared unchanged in SHRs compared to WKY.
Figure 3.

Specific [3H]MLA binding to SHR and WKY membranes from cortex (A), striatum (B), hippocampus (C) and cerebellum (D). Corresponding Scatchard plots are shown in the inset. Both maximum binding sites (Bmax) and dissociation constants (Kd values) were unchanged in SHRs compared to WKY rats in all four brain regions. In cortex, Bmax values were 21.0 ± 1.9 and 20.9 ± 1.5 fmol/mg protein (p > 0.05), with Kd = 1.8 ± 0.5 and 1.9 ± 0.4 nM for SHR and WKY respectively (p > 0.05). In striatum, Bmax values were 9.8 ± 2.6 and 7.6 ± 1.4 fmol/mg protein (p > 0.05), with Kd = 1.4 ± 0.7 and 0.8 ± 0.4 nM for SHR and WKY respectively (p > 0.05). In hippocampus, Bmax values were 44.0 ± 1.7 and 45.8 ± 1.7 fmol/mg protein (p > 0.05), with Kd = 1.7 ± 0.2 and 1.7 ± 0.2 pM for SHR and WKY respectively (p > 0.05). In cerebellum, Bmax values were 4.9 ± 2.5 and 6.2 ± 5.2 fmol/mg protein (p > 0.05), with Kd = 1.4 ± 1.4 and 1.9 ± 2.8 nM for SHR and WKY respectively (p > 0.05). Membranes (~0.6 mg protein) and [3H]MLA were incubated for 1.5 hours at 23°C. Non-specific binding was determined in the presence of 1 mM nicotine. Each data point represents mean ± SEM of 3 to 4 rats.
Figure 4.

Quantitative analysis of [125I]α-bungarotoxin binding in brain regions from SHR and WKY. Sections were incubated with 2 nM [125I]α-bungarotoxin for 45 min. Each value represents mean ± SEM of 5 to 6 rats.
We next measured the abundance of nAChR subunit encoding mRNAs in cortex, striatum, hippocampus and cerebellum of SHRs compared to WKY, using qPCR. In cerebellum, which showed the largest decrease in the number of α4β2 nAChR binding sites in SHR, we found a parallel decrease in α4 mRNA levels (t(13) = 2.32, p < 0.05) (Fig. 5). No significant difference in β2 mRNA levels could be detected between the strains in cerebellum, although there was a trend towards a decrease in SHRs. In contrast to the decreased number of α4β2 nAChR binding sites in cortex, striatum and hippocampus, no significant strain differences in α4 or β2 mRNA levels could be detected in these brain regions. In agreement with the unchanged α7 nAChR binding in SHRs compared to WKY, no significant difference in the levels of α7 mRNA could be detected in any brain region. Interestingly, α3 mRNA levels were significantly higher in SHRs compared to WKY rats in several brain regions, such as cortex (t(13) = 2.22, p < 0.05), hippocampus (t(14) = 2.36, p < 0.05) and cerebellum (t(14) = 2.95, p < 0.01). In contrast, we could not detect any significant strain differences in mRNA levels of α5 or β3 nAChR subunits in any of the brain regions.
Figure 5.

Relative levels of mRNAs encoding nAChR subunits in SHR and WKY in cortex (A), striatum (B), hippocampus (C) and cerebellum (D). Quantitative real-time PCR and mRNA quantitation was performed as described in the Material and Methods. Reliable expression of the α5 nAChR subunit in cerebellum or the β3 nAChR subunit in striatum and hippocampus could not be measured due to a very low level of expression. Each value represents mean ± SEM of 6 to 8 rats. *, p < 0.05.
Discussion
We investigated binding properties and transcriptional regulation of specific nAChR subtypes and nAChR subunits in selected brain regions of the juvenile SHR rat model of ADHD. Our main finding emphasizes that the number of α4β2 nAChR binding sites is globally lower in the SHR brain compared to WKY controls without changes in mRNA expression, with the exception of a decrease of both α4β2 nAChR binding and α4 mRNA in the cerebellum. These nAChR decreases appear to be subtype-specific because no strain differences were found in α7 nAChR binding or α7 mRNA levels. Our data suggest that behavioral abnormalities in SHRs such as attention- and memory deficits could in fact be related to a low number of α4β2 nAChRs in the brain.
Using pre-hypertensive SHRs, a previous study reported decreased binding of the α4β2 nAChR partial agonist [3H]cytisine in several brain regions (Gattu et al. 1997b). In our study, using binding measurements with high subtype specificity ([125I]A-85380) (Mukhin et al. 2000, Sullivan et al. 1996), we first confirmed both that these changes represent the α4β2 nAChR subtype, but also that these changes are present in juvenile rats prior to development of hypertension. Previous studies using adult rats have also reported lower nAChR binding in brain regions of SHRs compared to WKY (Gattu et al., 1997a; Hohnadel et al., 2005; Terry et al., 2000), and these observations could have been difficult to interpret given the possible confounding effects of hypertension in these adult SHRs. However, in agreement with our findings, Gattu et al (1997b) showed that although antihypertensive drug treatment prevents development of hypertension in adult SHR, it does not forestall differences in [3H]cytisine binding in SHR brain compared to WKY. Both our findings and those by Gattu et al (1997b) consequently show that α4β2 nAChR deficiencies are probably not related to the hypertensive state in SHRs, which add validity to the SHR as a useful model to investigate α4β2 nAChRs in ADHD.
Although two studies have reported lower α7 nAChR binding in brain regions such as cortex in adult SHRs compared to WKY (Gattu et al. 1997a, Terry et al. 2000), here we show that in the juvenile SHR brain, prior to the development of hypertension, α7 nAChRs are unchanged both at the level of mRNA expression and binding properties. Given that behavioral symptoms of ADHD are present in SHRs prior to the development of hypertension (Qian et al. 2010, Ueno et al. 2002, Whitehorn et al. 1983), our results therefore suggest that α7 nAChR changes might have limited relevance in the SHR as a model of ADHD. Moreover, the discrepancy between previously observed α7 nAChR changes in hypertensive SHRs and the lack of α7 nAChRs differences in pre-hypertensive SHRs indicate that α7 nAChR changes might even be a consequence of the hypertension present in adult SHRs. Additional support to this conclusion was found in a study of a sub-strain of SHRs showing that antihypertensive drug treatment can in fact counteract α7 nAChR binding decreases in cortex of these rats (Ferrari et al. 1999).
Examining the mechanism underlying nAChR changes in SHRs, we generally found that α4β2 binding was lower in SHR compared to WKY without accompanying changes in α4- or β2 subunit mRNA levels, with the exception of lower α4 mRNA in cerebellum of SHR compared to WKY. This lack of correlation between nAChR binding and subunit mRNA levels raises the possibility of a post-transcriptional regulatory mechanism. Interestingly, in a previous study it was shown that following repeated nicotine exposure, SHRs do not show the upregulation of high-affinity nAChR binding routinely observed in control rats after such nicotine treatment (Hohnadel et al. 2005). Since this hallmark nAChR upregulation is believed to be mediated through post-transcriptional events such as nAChR assembly in the endoplasmic reticulum, transport to the cell surface and decreased receptor turnover (Corringer et al. 2006, Peng et al. 1994, Sallette et al. 2005), these findings support the notion that post-transcriptional nAChR regulation might be altered in this animal model of ADHD.
The dopaminergic hypothesis of ADHD, which states that this disorder is caused by a deficit in dopamine signaling in the brain (Swanson et al. 2007), implies that modulation of dopamine signaling is highly relevant both for ADHD- pathology and therapeutics. Activation of α4β2 nAChRs can stimulate dopamine release and blockade of β2 containing nAChRs suppresses tonic dopamine signaling in rodent striatum (Zhang et al. 2009). One should therefore expect SHRs with low numbers of β2 nAChRs to display suppressed dopamine signaling. Our findings indicate that in critical SHR brain regions such as cortex, striatum and hippocampus, lower α4β2 nAChR numbers could give rise to impaired dopamine signaling and in turn influence ADHD-like symptoms. Supporting the involvement of α4β2 nAChRs, the β2 nAChR subunit knockout mouse has previously been proposed as a model of ADHD (Granon & Changeux 2006), and other studies have identified trends towards increased locomotor activity in β2 nAChR subunit knockout mice (King et al. 2004, Mineur et al. 2009). Several polymorphisms in the α4 nAChR subunit have also been associated with ADHD in humans (Lee et al. 2008, Guan et al. 2009, Brookes et al. 2006, Comings et al. 2000, Todd et al. 2003), although other studies were conflicting (Bobb et al. 2005, Kent et al. 2001), possibly reflecting different phenotypes used for subject inclusion. Importantly, evidence from Alzheimer’s disease supports a critical role for nAChRs in cognitive functions with significant nAChR binding reductions in the brains of these patients (Kellar et al. 1987, Nordberg et al. 1992, Nordberg & Winblad 1986, Perry et al. 1995, Perry et al. 1987), and later studies have confirmed that such nAChR deficits correlate well with cognitive impairments (Kadir et al. 2006, Nordberg et al. 1995).
Similar to the ADHD medications methylphenidate and amphetamine, α4β2 nAChR activation can potentiate dopamine release, making this nAChR subtype a potential therapeutic target in ADHD. In fact, it has been reported that in a sub-strain of the SHR, nicotine treatment improves spontaneous alternation behavior through a mechanism involving activation of α4β2, but not α7 nAChRs (Ueno et al. 2002). Other studies in rats also showed that nicotine can improve attention in a five-choice serial reaction time task through α4β2 nAChR activation (Blondel et al. 2000, Grottick & Higgins 2000). Importantly, human studies support α4β2 nAChRs as targets for ADHD therapeutics, since a trial of the full α4β2 agonist ABT-418 improved symptoms in ADHD patients (Wilens et al. 1999). Taken together with our findings, these data strongly suggest that the efficacy of nAChR agonists in ADHD could rely on the ability of these agents to counteract deficits in α4β2 transmission that may underlie this disorder.
The connection between ADHD and nicotinic transmission is also illustrated by the relationship between smoking and ADHD. For instance, it has been shown that ADHD patients are more likely to smoke and fail in smoking cessation when compared to the general population (Humfleet et al. 2005, Pomerleau et al. 1995). Nicotine, the principal psychoactive element in smoked tobacco, can both activate and desensitize nAChRs, and these two seemingly opposing effects are responsible for the behavioral effect of nicotine (Picciotto et al. 2008). Our findings might indicate that ADHD is related to low numbers of α4β2 nAChRs, and therefore that increased smoking represents self-medication of this deficit, or alternatively that low α4β2 nAChRs leads to a blunted response to smoking, giving a compensating shift towards increased smoking behavior.
We also found dramatically increased α3 mRNA levels in SHRs compared to WKY in several brain regions such as cortex, hippocampus and cerebellum. In the nervous system, α3 containing nAChRs are highly expressed in the periphery with a more restricted expression in CNS (Gotti & Clementi 2004), and functional α3 containing nAChRs are formed through co-expression of α3 and β4 subunits (McGehee & Role 1995, Nelson & Lindstrom 1999). Previously, a lower density of α3 containing nAChRs was reported in the superior colliculus of SHRs compared to WKY (Hernandez et al. 2003), supporting a possible role for this subunit in SHR pathology. Our study showing a significant mismatch between the increased mRNA levels for the α3 nAChR subunit and the simultaneous decreased number of nAChRs, suggests that α3* nAChRs may be implicated in the etiology of ADHD-like behaviors in SHRs, and that feedback mechanisms to compensate for nAChR changes are not effective.
Although some have questioned the SHR as a useful model of ADHD (Alsop 2007) and the WKY as an adequate control (van den Bergh et al. 2006), a recent review concludes that the SHR obtained from Charles River (Sulzfeld, Germany) constitutes the best validated animal model of ADHD, and that WKY obtained from Harlan (Blacktorn, UK) is the most appropriate control (Sagvolden et al. 2009). In fact, this study found that the use of outbred Wistar, Sprague Dawley or other rat strains as controls could produce spurious neurobiological differences compared to SHR. As a result, in the present study, we employ WKY as the control strain for the SHR. Finally, as previous studies in SHRs have found abnormalities in other neurotransmitter systems besides the cholinergic, such as differences in glutamate (Jensen et al. 2009) and dopamine (Roessner et al. 2010) systems in SHRs compared to WKY, it should also be noted that the cholinergic differences in SHRs at this point should be considered correlative rather than necessarily responsible for behavioral abnormalities in SHRs. However, given that human studies support α4β2 nAChRs as targets for ADHD therapeutics and the strong relationship between smoking and ADHD, our results suggest that nAChRs could play a role in ADHD-like behavior displayed by SHRs.
In summary, our study shows that SHRs exhibit a global decrease in α4β2 nAChRs in the brain. We suggest that this decrease in α4β2 nAChR numbers is likely to be a consequence of dysfunctional post-transcriptional regulation of these receptors. This nAChR subtype has a potential role both in the pathology of and therapeutics for ADHD. Our results therefore support a role for central α4β2 deficiencies in ADHD, and in the elevated smoking behavior displayed by ADHD patients.
Supplementary Material
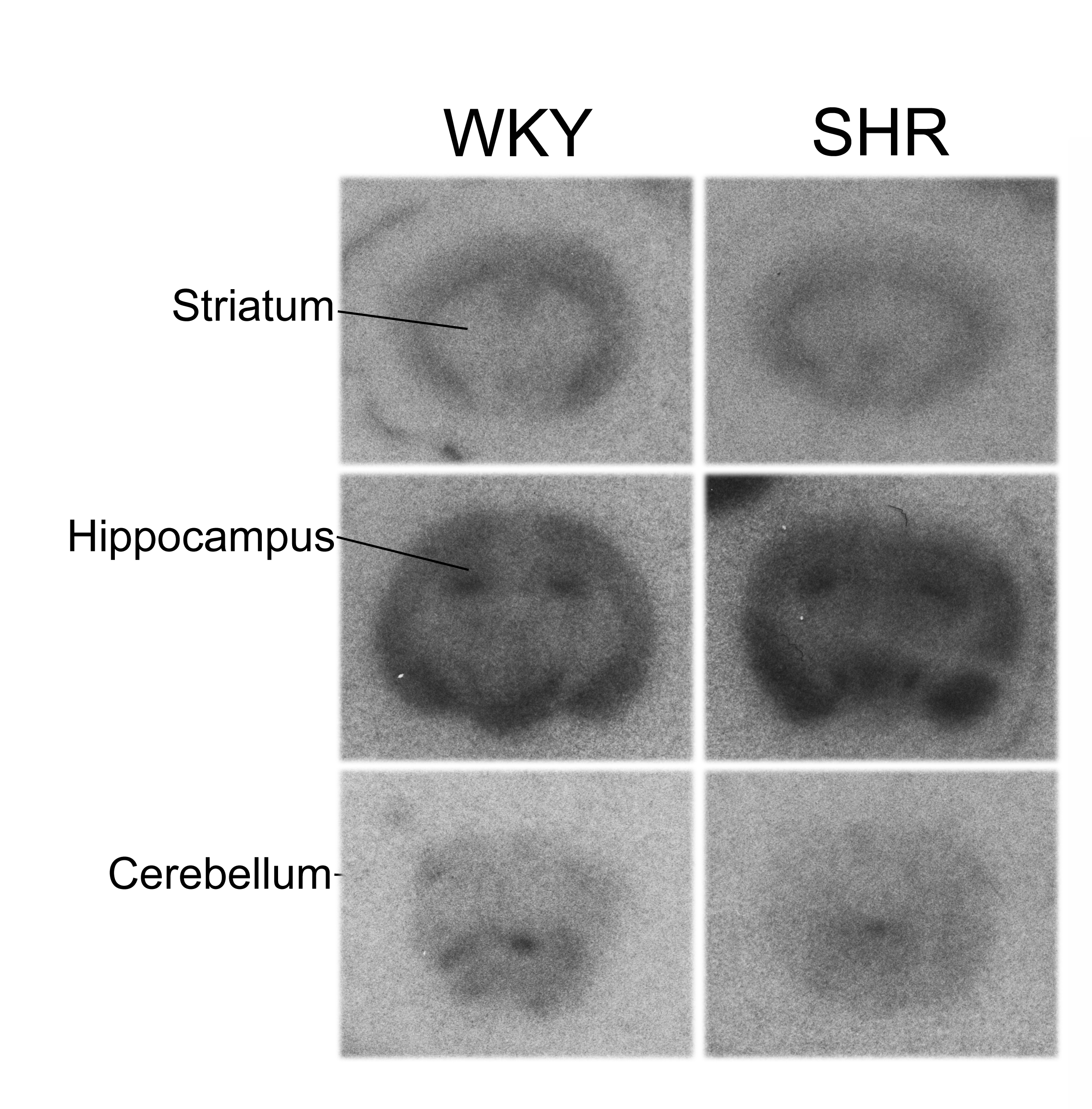
Distribution of receptors labeled by [125I]α-bungarotoxin autoradiography in brain sections from SHR and WKY. Sections were incubated with 2 nM [125I]α-bungarotoxin for 45 min.
{kind=link}
Acknowledgements
This research was supported by the Jahre foundation for Medical Research, Norway. MBW was supported by Ringstiftelsen, Norway. YSM, CJH and MRP were supported by grant DA10455 from the National Institutes of Health, USA. The authors would like to thank Kine Dervola and Ole Rostad for assistance with binding experiments.
Abbreviations used
- ADHD
Attention-deficit/hyperactivity disorder
- Bmax
maximum binding sites
- Kd
dissociation constant
- MLA
methyllycaconitine
- nAChR
nicotinic acetylcholine receptor
- SHR
spontaneous hypertensive rat
- WKY
Wistar Kyoto rat
Footnotes
The authors declare no conflict of interest.
REFERENCES
- Alsop B. Problems with spontaneously hypertensive rats (SHR) as a model of attention-deficit/hyperactivity disorder (AD/HD) J Neurosci Methods. 2007;162:42–48. doi: 10.1016/j.jneumeth.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Berger DF, Sagvolden T. Sex differences in operant discrimination behaviour in an animal model of attention-deficit hyperactivity disorder. Behav Brain Res. 1998;94:73–82. doi: 10.1016/s0166-4328(97)00171-x. [DOI] [PubMed] [Google Scholar]
- Biederman J. Attention-deficit/hyperactivity disorder: a selective overview. Biol Psychiatry. 2005;57:1215–1220. doi: 10.1016/j.biopsych.2004.10.020. [DOI] [PubMed] [Google Scholar]
- Biederman J, Faraone SV. Attention-deficit hyperactivity disorder. Lancet. 2005;366:237–248. doi: 10.1016/S0140-6736(05)66915-2. [DOI] [PubMed] [Google Scholar]
- Blondel A, Sanger DJ, Moser PC. Characterisation of the effects of nicotine in the five-choice serial reaction time task in rats: antagonist studies. Psychopharmacology (Berl) 2000;149:293–305. doi: 10.1007/s002130000378. [DOI] [PubMed] [Google Scholar]
- Bobb AJ, Addington AM, Sidransky E, et al. Support for association between ADHD and two candidate genes: NET1 and DRD1. Am J Med Genet B Neuropsychiatr Genet. 2005;134B:67–72. doi: 10.1002/ajmg.b.30142. [DOI] [PubMed] [Google Scholar]
- Brookes K, Xu X, Chen W, et al. The analysis of 51 genes in DSM-IV combined type attention deficit hyperactivity disorder: association signals in DRD4, DAT1 and 16 other genes. Mol Psychiatry. 2006;11:934–953. doi: 10.1038/sj.mp.4001869. [DOI] [PubMed] [Google Scholar]
- Chen D, Patrick JW. The alpha-bungarotoxin-binding nicotinic acetylcholine receptor from rat brain contains only the alpha7 subunit. J Biol Chem. 1997;272:24024–24029. doi: 10.1074/jbc.272.38.24024. [DOI] [PubMed] [Google Scholar]
- Comings DE, Gade-Andavolu R, Gonzalez N, et al. Multivariate analysis of associations of 42 genes in ADHD, ODD and conduct disorder. Clin Genet. 2000;58:31–40. doi: 10.1034/j.1399-0004.2000.580106.x. [DOI] [PubMed] [Google Scholar]
- Corringer PJ, Sallette J, Changeux JP. Nicotine enhances intracellular nicotinic receptor maturation: a novel mechanism of neural plasticity? J Physiol Paris. 2006;99:162–171. doi: 10.1016/j.jphysparis.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Cruzado MC, Risler NR, Miatello RM, Yao G, Schiffrin EL, Touyz RM. Vascular smooth muscle cell NAD(P)H oxidase activity during the development of hypertension: Effect of angiotensin II and role of insulinlike growth factor-1 receptor transactivation. Am J Hypertens. 2005;18:81–87. doi: 10.1016/j.amjhyper.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Davids E, Zhang K, Tarazi FI, Baldessarini RJ. Animal models of attention-deficit hyperactivity disorder. Brain Res Brain Res Rev. 2003;42:1–21. doi: 10.1016/s0165-0173(02)00274-6. [DOI] [PubMed] [Google Scholar]
- Davies AR, Hardick DJ, Blagbrough IS, Potter BV, Wolstenholme AJ, Wonnacott S. Characterisation of the binding of [3H]methyllycaconitine: a new radioligand for labelling alpha 7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology. 1999;38:679–690. doi: 10.1016/s0028-3908(98)00221-4. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Sergeant J, Gillberg C, Biederman J. The worldwide prevalence of ADHD: is it an American condition? World Psychiatry. 2003;2:104–113. [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Frasoldati A, Leo G, Torri C, Zini I, Agnati LF, Zoli M. Changes in nicotinic acetylcholine receptor subunit mRNAs and nicotinic binding in spontaneously hypertensive stroke prone rats. Neurosci Lett. 1999;277:169–172. doi: 10.1016/s0304-3940(99)00879-4. [DOI] [PubMed] [Google Scholar]
- Gattu M, Pauly JR, Boss KL, Summers JB, Buccafusco JJ. Cognitive impairment in spontaneously hypertensive rats: role of central nicotinic receptors. I. Brain Res. 1997a;771:89–103. doi: 10.1016/s0006-8993(97)00793-2. [DOI] [PubMed] [Google Scholar]
- Gattu M, Terry AV, Jr.,, Pauly JR, Buccafusco JJ. Cognitive impairment in spontaneously hypertensive rats: role of central nicotinic receptors. Part II. Brain Res. 1997b;771:104–114. doi: 10.1016/s0006-8993(97)00794-4. [DOI] [PubMed] [Google Scholar]
- Gehricke JG, Loughlin SE, Whalen CK, Potkin SG, Fallon JH, Jamner LD, Belluzzi JD, Leslie FM. Smoking to self-medicate attentional and emotional dysfunctions. Nicotine Tob Res. 2007;9 Suppl 4:S523–S536. doi: 10.1080/14622200701685039. [DOI] [PubMed] [Google Scholar]
- Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Granon S, Changeux JP. Attention-deficit/hyperactivity disorder: a plausible mouse model? Acta Paediatr. 2006;95:645–649. doi: 10.1080/08035250600719747. [DOI] [PubMed] [Google Scholar]
- Grottick AJ, Higgins GA. Effect of subtype selective nicotinic compounds on attention as assessed by the five-choice serial reaction time task. Behav Brain Res. 2000;117:197–208. doi: 10.1016/s0166-4328(00)00305-3. [DOI] [PubMed] [Google Scholar]
- Guan L, Wang B, Chen Y, Yang L, Li J, Qian Q, Wang Z, Faraone SV, Wang Y. A high-density single-nucleotide polymorphism screen of 23 candidate genes in attention deficit hyperactivity disorder: suggesting multiple susceptibility genes among Chinese Han population. Mol Psychiatry. 2009;14:546–554. doi: 10.1038/sj.mp.4002139. [DOI] [PubMed] [Google Scholar]
- Hernandez CM, Hoifodt H, Terry AV., Jr Spontaneously hypertensive rats: further evaluation of age-related memory performance and cholinergic marker expression. J Psychiatry Neurosci. 2003;28:197–209. [PMC free article] [PubMed] [Google Scholar]
- Hohnadel EJ, Hernandez CM, Gearhart DA, Terry AV., Jr Effect of repeated nicotine exposure on high-affinity nicotinic acetylcholine receptor density in spontaneously hypertensive rats. Neurosci Lett. 2005;382:158–163. doi: 10.1016/j.neulet.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Humfleet GL, Prochaska JJ, Mengis M, Cullen J, Munoz R, Reus V, Hall SM. Preliminary evidence of the association between the history of childhood attention-deficit/hyperactivity disorder and smoking treatment failure. Nicotine Tob Res. 2005;7:453–460. doi: 10.1080/14622200500125310. [DOI] [PubMed] [Google Scholar]
- Jensen V, Rinholm JE, Johansen TJ, Medin T, Storm-Mathisen J, Sagvolden T, Hvalby O, Bergersen LH. N-methyl-D-aspartate receptor subunit dysfunction at hippocampal glutamatergic synapses in an animal model of attention-deficit/hyperactivity disorder. Neuroscience. 2009;158:353–364. doi: 10.1016/j.neuroscience.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Kadir A, Almkvist O, Wall A, Langstrom B, Nordberg A. PET imaging of cortical 11C-nicotine binding correlates with the cognitive function of attention in Alzheimer's disease. Psychopharmacology (Berl) 2006;188:509–520. doi: 10.1007/s00213-006-0447-7. [DOI] [PubMed] [Google Scholar]
- Kellar KJ, Whitehouse PJ, Martino-Barrows AM, Marcus K, Price DL. Muscarinic and nicotinic cholinergic binding sites in Alzheimer's disease cerebral cortex. Brain Res. 1987;436:62–68. doi: 10.1016/0006-8993(87)91556-3. [DOI] [PubMed] [Google Scholar]
- Kent L, Middle F, Hawi Z, Fitzgerald M, Gill M, Feehan C, Craddock N. Nicotinic acetylcholine receptor alpha4 subunit gene polymorphism and attention deficit hyperactivity disorder. Psychiatr Genet. 2001;11:37–40. doi: 10.1097/00041444-200103000-00007. [DOI] [PubMed] [Google Scholar]
- King SL, Caldarone BJ, Picciotto MR. Beta2-subunit-containing nicotinic acetylcholine receptors are critical for dopamine-dependent locomotor activation following repeated nicotine administration. Neuropharmacology. 2004;47 Suppl 1:132–139. doi: 10.1016/j.neuropharm.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Lee J, Laurin N, Crosbie J, et al. Association study of the nicotinic acetylcholine receptor alpha4 subunit gene, CHRNA4, in attention-deficit hyperactivity disorder. Genes Brain Behav. 2008;7:53–60. doi: 10.1111/j.1601-183X.2007.00325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Conners CK, Silva D, Canu W, March J. Effects of chronic nicotine and methylphenidate in adults with attention deficit/hyperactivity disorder. Exp Clin Psychopharmacol. 2001;9:83–90. doi: 10.1037/1064-1297.9.1.83. [DOI] [PubMed] [Google Scholar]
- Linnet KM, Dalsgaard S, Obel C, et al. Maternal lifestyle factors in pregnancy risk of attention deficit hyperactivity disorder and associated behaviors: review of the current evidence. Am J Psychiatry. 2003;160:1028–1040. doi: 10.1176/appi.ajp.160.6.1028. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Laverty DS, Whiteaker P, Salminen O, Grady SR, McIntosh JM, Collins AC. John Daly's compound, epibatidine, facilitates identification of nicotinic receptor subtypes. J Mol Neurosci. 2009;40:96–104. doi: 10.1007/s12031-009-9264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Whiteaker P, Collins AC. Deletion of the alpha7, beta2, or beta4 nicotinic receptor subunit genes identifies highly expressed subtypes with relatively low affinity for [3H]epibatidine. Mol Pharmacol. 2006;70:947–959. doi: 10.1124/mol.106.025338. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Role LW. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol. 1995;57:521–546. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- Milberger S, Biederman J, Faraone SV, Chen L, Jones J. Is maternal smoking during pregnancy a risk factor for attention deficit hyperactivity disorder in children? Am J Psychiatry. 1996;153:1138–1142. doi: 10.1176/ajp.153.9.1138. [DOI] [PubMed] [Google Scholar]
- Millar NS, Gotti C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology. 2009;56:237–246. doi: 10.1016/j.neuropharm.2008.07.041. [DOI] [PubMed] [Google Scholar]
- Mineur YS, Brunzell DH, Grady SR, Lindstrom JM, McIntosh JM, Marks MJ, King SL, Picciotto MR. Localized low-level re-expression of high-affinity mesolimbic nicotinic acetylcholine receptors restores nicotine-induced locomotion but not place conditioning. Genes Brain Behav. 2009;8:257–266. doi: 10.1111/j.1601-183X.2008.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhin AG, Gundisch D, Horti AG, et al. 5-Iodo-A-85380, an alpha4beta2 subtype-selective ligand for nicotinic acetylcholine receptors. Mol Pharmacol. 2000;57:642–649. doi: 10.1124/mol.57.3.642. [DOI] [PubMed] [Google Scholar]
- Myers MM, Musty RE, Hendley ED. Attenuation of hyperactivity in the spontaneously hypertensive rat by amphetamine. Behav Neural Biol. 1982;34:42–54. doi: 10.1016/s0163-1047(82)91397-8. [DOI] [PubMed] [Google Scholar]
- Nelson ME, Lindstrom J. Single channel properties of human alpha3 AChRs: impact of beta2, beta4 and alpha5 subunits. J Physiol. 1999;516(Pt 3):657–678. doi: 10.1111/j.1469-7793.1999.0657u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg A, Alafuzoff I, Winblad B. Nicotinic and muscarinic subtypes in the human brain: changes with aging and dementia. J Neurosci Res. 1992;31:103–111. doi: 10.1002/jnr.490310115. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Lundqvist H, Hartvig P, Lilja A, Langstrom B. Kinetic analysis of regional (S)(-)11C-nicotine binding in normal and Alzheimer brains--in vivo assessment using positron emission tomography. Alzheimer Dis Assoc Disord. 1995;9:21–27. doi: 10.1097/00002093-199505000-00006. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Winblad B. Reduced number of [3H]nicotine and [3H]acetylcholine binding sites in the frontal cortex of Alzheimer brains. Neurosci Lett. 1986;72:115–119. doi: 10.1016/0304-3940(86)90629-4. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Aoki K. Development of a strain of spontaneously hypertensive rats. Jpn Circ J. 1963;27:282–293. doi: 10.1253/jcj.27.282. [DOI] [PubMed] [Google Scholar]
- Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol. 1994;46:523–530. [PubMed] [Google Scholar]
- Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, Irving D, Brown A, Perry RH. Alteration in nicotine binding sites in Parkinson's disease, Lewy body dementia and Alzheimer's disease: possible index of early neuropathology. Neuroscience. 1995;64:385–395. doi: 10.1016/0306-4522(94)00410-7. [DOI] [PubMed] [Google Scholar]
- Perry EK, Perry RH, Smith CJ, Dick DJ, Candy JM, Edwardson JA, Fairbairn A, Blessed G. Nicotinic receptor abnormalities in Alzheimer's and Parkinson's diseases. J Neurol Neurosurg Psychiatry. 1987;50:806–809. doi: 10.1136/jnnp.50.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not "either/or": activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol. 2008;84:329–342. doi: 10.1016/j.pneurobio.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerleau OF, Downey KK, Stelson FW, Pomerleau CS. Cigarette smoking in adult patients diagnosed with attention deficit hyperactivity disorder. J Subst Abuse. 1995;7:373–378. doi: 10.1016/0899-3289(95)90030-6. [DOI] [PubMed] [Google Scholar]
- Qian Y, Lei G, Castellanos FX, Forssberg H, Heijtz RD. Deficits in fine motor skills in a genetic animal model of ADHD. Behav Brain Funct. 2010;6:51. doi: 10.1186/1744-9081-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessner V, Sagvolden T, Dasbanerjee T, Middleton FA, Faraone SV, Walaas SI, Becker A, Rothenberger A, Bock N. Methylphenidate normalizes elevated dopamine transporter densities in an animal model of the attention-deficit/hyperactivity disorder combined type, but not to the same extent in one of the attention-deficit/hyperactivity disorder inattentive type. Neuroscience. 2010;167:1183–1191. doi: 10.1016/j.neuroscience.2010.02.073. [DOI] [PubMed] [Google Scholar]
- Sagvolden T. Behavioral validation of the spontaneously hypertensive rat (SHR) as an animal model of attention-deficit/hyperactivity disorder (AD/HD) Neurosci Biobehav Rev. 2000;24:31–39. doi: 10.1016/s0149-7634(99)00058-5. [DOI] [PubMed] [Google Scholar]
- Sagvolden T, Hendley ED, Knardahl S. Behavior of hypertensive and hyperactive rat strains: hyperactivity is not unitarily determined. Physiol Behav. 1992a;52:49–57. doi: 10.1016/0031-9384(92)90432-2. [DOI] [PubMed] [Google Scholar]
- Sagvolden T, Johansen EB, Woien G, et al. The spontaneously hypertensive rat model of ADHD--the importance of selecting the appropriate reference strain. Neuropharmacology. 2009;57:619–626. doi: 10.1016/j.neuropharm.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagvolden T, Metzger MA, Schiorbeck HK, Rugland AL, Spinnangr I, Sagvolden G. The spontaneously hypertensive rat (SHR) as an animal model of childhood hyperactivity (ADHD): changed reactivity to reinforcers and to psychomotor stimulants. Behav Neural Biol. 1992b;58:103–112. doi: 10.1016/0163-1047(92)90315-u. [DOI] [PubMed] [Google Scholar]
- Sallette J, Pons S, Devillers-Thiery A, Soudant M, Prado de Carvalho L, Changeux JP, Corringer PJ. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46:595–607. doi: 10.1016/j.neuron.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Spencer TJ, Biederman J, Mick E. Attention-deficit/hyperactivity disorder: diagnosis, lifespan, comorbidities, and neurobiology. J Pediatr Psychol. 2007;32:631–642. doi: 10.1093/jpepsy/jsm005. [DOI] [PubMed] [Google Scholar]
- Sullivan JP, Donnelly-Roberts D, Briggs CA, et al. A-85380 [3-(2(S)-azetidinylmethoxy) pyridine]: in vitro pharmacological properties of a novel, high affinity alpha 4 beta 2 nicotinic acetylcholine receptor ligand. Neuropharmacology. 1996;35:725–734. doi: 10.1016/0028-3908(96)84644-2. [DOI] [PubMed] [Google Scholar]
- Swanson JM, Kinsbourne M, Nigg J, et al. Etiologic subtypes of attention-deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychol Rev. 2007;17:39–59. doi: 10.1007/s11065-007-9019-9. [DOI] [PubMed] [Google Scholar]
- Terry AV, Jr, Hernandez CM, Buccafusco JJ, Gattu M. Deficits in spatial learning and nicotinic-acetylcholine receptors in older, spontaneously hypertensive rats. Neuroscience. 2000;101:357–368. doi: 10.1016/s0306-4522(00)00377-8. [DOI] [PubMed] [Google Scholar]
- Thapar A, Langley K, Asherson P, Gill M. Gene-environment interplay in attention-deficit hyperactivity disorder and the importance of a developmental perspective. Br J Psychiatry. 2007;190:1–3. doi: 10.1192/bjp.bp.106.027003. [DOI] [PubMed] [Google Scholar]
- Todd RD, Lobos EA, Sun LW, Neuman RJ. Mutational analysis of the nicotinic acetylcholine receptor alpha 4 subunit gene in attention deficit/hyperactivity disorder: evidence for association of an intronic polymorphism with attention problems. Mol Psychiatry. 2003;8:103–108. doi: 10.1038/sj.mp.4001257. [DOI] [PubMed] [Google Scholar]
- Ueno K, Togashi H, Matsumoto M, Ohashi S, Saito H, Yoshioka M. Alpha4beta2 nicotinic acetylcholine receptor activation ameliorates impairment of spontaneous alternation behavior in stroke-prone spontaneously hypertensive rats, an animal model of attention deficit hyperactivity disorder. J Pharmacol Exp Ther. 2002;302:95–100. doi: 10.1124/jpet.302.1.95. [DOI] [PubMed] [Google Scholar]
- van den Bergh FS, Bloemarts E, Chan JS, Groenink L, Olivier B, Oosting RS. Spontaneously hypertensive rats do not predict symptoms of attention-deficit hyperactivity disorder. Pharmacol Biochem Behav. 2006;83:380–390. doi: 10.1016/j.pbb.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Marks MJ, Grady SR, Lu Y, Picciotto MR, Changeux JP, Collins AC. Pharmacological and null mutation approaches reveal nicotinic receptor diversity. Eur J Pharmacol. 2000;393:123–135. doi: 10.1016/s0014-2999(00)00052-2. [DOI] [PubMed] [Google Scholar]
- Whitehorn D, Atwater DG, Low WC, Gellis JE, Hendley ED. Independence of blood pressure and locomotor hyperactivity in normotensive and genetically hypertensive rat. Behav Neural Biol. 1983;37:357–361. doi: 10.1016/s0163-1047(83)91501-7. [DOI] [PubMed] [Google Scholar]
- Wilens TE, Biederman J, Spencer TJ, et al. A pilot controlled clinical trial of ABT-418, a cholinergic agonist, in the treatment of adults with attention deficit hyperactivity disorder. Am J Psychiatry. 1999;156:1931–1937. doi: 10.1176/ajp.156.12.1931. [DOI] [PubMed] [Google Scholar]
- Zhang L, Doyon WM, Clark JJ, Phillips PE, Dani JA. Controls of tonic and phasic dopamine transmission in the dorsal and ventral striatum. Mol Pharmacol. 2009;76:396–404. doi: 10.1124/mol.109.056317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of receptors labeled by [125I]α-bungarotoxin autoradiography in brain sections from SHR and WKY. Sections were incubated with 2 nM [125I]α-bungarotoxin for 45 min.