

Abstract
The Saccharomyces cerevisiae protein kinase Rim15p was identified previously as a stimulator of meiotic gene expression. Here, we show that loss of Rim15p causes an additional pleiotropic phenotype in cells grown to stationary phase on rich medium; this phenotype includes defects in trehalose and glycogen accumulation, in transcriptional derepression of HSP12, HSP26, and SSA3, in induction of thermotolerance and starvation resistance, and in proper G1 arrest. These phenotypes are commonly associated with hyperactivity of the Ras/cAMP pathway. Tests of epistasis suggest that Rim15p may act in this pathway downstream of the cAMP-dependent protein kinase (cAPK). Accordingly, deletion of RIM15 suppresses the growth defect of a temperature-sensitive adenylate-cyclase mutant and, most importantly, renders cells independent of cAPK activity. Conversely, overexpression of RIM15 suppresses phenotypes associated with a mutation in the regulatory subunit of cAPK, exacerbates the growth defect of strains compromised for cAPK activity, and partially induces a starvation response in logarithmically growing wild-type cells. Biochemical analyses reveal that cAPK-mediated in vitro phosphorylation of Rim15p strongly inhibits its kinase activity. Taken together, these results place Rim15p immediately downstream and under negative control of cAPK and define a positive regulatory role of Rim15p for entry into both meiosis and stationary phase.
Keywords: Rim15p, protein kinase A, yeast, Ras, cAMP, nutrient signaling
The cAMP-dependent protein kinase (cAPK) pathway in the yeast Saccharomyces cerevisiae is required for proper regulation of growth, cell cycle progression, and development in response to nutritional conditions. Cells deficient in cAPK activity stop growth, arrest in G1, and show physiological changes normally associated with nutrient deprivation; these changes include the accumulation of trehalose and glycogen, enhanced expression of various genes (e.g., SSA3, HSP12, HSP26, CTT1, UBI4, and ADH2), and increased resistance to heat stress. In contrast, cells carrying mutations that yield elevated cAPK activity fail to arrest in G1, are defective for trehalose and glycogen accumulation, rapidly lose viability, and remain highly sensitive to heat stress upon nutrient starvation (for review, see Tatchell 1986; Thevelein 1994). On the basis of these results, it was suggested that the central role of the yeast cAPK pathway is in signaling the nutrient status, thereby participating in the cell’s decision to enter a quiescent state in G1 that is equivalent to the G0 state of higher eukaryotes. Accordingly, low levels of cAPK activity promote exit from the mitotic cell cycle and entry into G0, while high levels of cAPK activity preclude access to G0.
The activity of S. cerevisiae cAPK is regulated by a complex signaling pathway that includes two yeast homologs of mammalian ras proteins. The yeast RAS1 and RAS2 gene products (Ras) are small GTP-binding proteins that are activated by a GTP-exchange factor (GEF; encoded by CDC25) and inactivated by stimulation of the intrinsic GTPase-activity via GTPase-activating proteins (GAP; encoded by IRA1 and IRA2). Activated GTP-bound Ras stimulates adenylate cyclase (encoded by CYR1/CDC35) to yield increased levels of cAMP that can be degraded by the low- and high-affinity phosphodiesterases encoded by the PDE1 and PDE2 genes, respectively. Binding of cAMP to the regulatory subunits of cAPK (encoded by BCY1) results in their dissociation from the catalytic subunits (encoded by three functionally redundant genes, TPK1, TPK2, and TPK3) and in stimulation of cAPK activity (for review and further details on the Ras/cAMP pathway, see Tatchell 1986; Gibbs and Marshall 1989; Broach and Deschenes 1990; Thevelein 1994). While the components of the Ras/cAMP pathway required for activation of cAPK are well established, little is known about the mechanisms of activation of the pathway by biological signals (i.e., nutrients) or about the potential biochemical targets of cAPK. These include enzymes involved in carbohydrate and phospholipid metabolism and various regulators of transcription, as well as proteins involved in synthesis and degradation of cAMP. Many of these potential targets, however, have not been unequivocally shown to be directly phosphorylated by cAPK, and it is likely that cAPK impinges on some of these targets rather indirectly (for review, see Broach and Deschenes 1990).
A fairly well established example of direct regulation by cAPK-catalyzed phosphorylation, by means of genetic, physiological, and biochemical analyses, is provided by the trehalose degrading enzyme trehalase (for review, see Thevelein 1996). Accumulation of the nonreducing disaccharide trehalose is an element of the adaptive response of yeast cells to nutrient starvation (Lillie and Pringle 1980). In general, resumption and stimulation of growth upon readdition of nutrients to starved cells are associated with mobilization of trehalose by rapid cAPK-dependent activation of neutral trehalase. Interestingly, it was suggested that not only the neutral trehalase but also the second key enzyme of trehalose metabolism, namely the trehalose-6-phosphate (Tre6P) synthase, may be regulated by cAPK-catalyzed phosphorylation (Panek et al. 1987). Accordingly, trehalose levels may be finely tuned in response to the nutritional status by reciprocal regulation (i.e., activation of neutral trehalase and inactivation of Tre6P synthase) via cAPK-mediated phosphorylation of the key enzymes of trehalose metabolism. However, although control of neutral trehalase by cAPK-dependent phosphorylation seems well established, there has been controversy as to whether Tre6P synthase is also regulated by cAPK-catalyzed phosphorylation (Vandercammen et al. 1989).
Tre6P synthase itself is part of a multimeric protein complex that is composed of at least four different subunits encoded by the genes TPS1, TPS2, TPS3, and TSL1. Recent studies indicated that Tps1p and Tps2p carry the catalytic activities of Tre6P synthase and Tre6P phosphatase, respectively, whereas Tps3p and Tsl1p were suggested to have regulatory and/or structural functions (Bell et al. 1992; De Virgilio et al. 1993; Vuorio et al. 1993; Reinders et al. 1997). A particularly surprising aspect of studies of the Tre6P synthase/phosphatase complex in S. cerevisiae was the finding that tps1 mutants (including various allelic mutants) are defective not only for Tre6P synthesis but also for growth on glucose, apparently because of an uncontrolled influx of glucose into the glycolytic pathway. Therefore, it has been suggested that Tps1p may be involved in the control of glycolysis (for review, see Thevelein and Hohmann 1995; and references therein).
In view of this newly discovered role of Tps1p in regulation of the glycolytic pathway and its own possible regulation via the Ras/cAMP signaling pathway, identification of proteins that can interact with Tps1p may reveal novel regulatory mechanisms of both glucose influx and/or signal transduction via the Ras/cAMP pathway. To isolate such potential regulatory proteins, we undertook a two-hybrid screen for proteins that interact with Tps1p. Here, we describe the identification of the Rim15p protein kinase, previously identified as a stimulator of meiotic gene expression (Vidan and Mitchell 1997), as a Tps1p-interacting protein. Deletion of RIM15 results in a defective response of mutant cells to nutrient limitation, including a defect in trehalose accumulation, that is reminiscent of the effects caused by mutations that constitutively activate cAPK (e.g., bcy1 and RAS2Val19). Tests of epistasis as well as biochemical studies suggest that Rim15p acts immediately downstream of cAPK to control a broad range of physiological adaptations necessary for proper entry into stationary phase.
Results
Cloning and sequence analysis of RIM15
To identify previously uncharacterized genes whose products interact with the Tre6P synthase (Tps1p) of S. cerevisiae, we performed a two-hybrid screen (Fields and Sternglanz 1994; Zervos et al. 1993; see Materials and Methods). We rescued library plasmids from cells (EGY48; Table 1) in which both reporter genes showed galactose-dependent transcription and assigned the plasmids to three different classes by restriction mapping. Partial sequencing of plasmids of one class showed them to contain a previously unidentified gene with high homology to protein kinases. We decided to first study this gene and named it TAK1 (for Tps1p-associated protein kinase). During the course of our studies, however, the same gene was identified as RIM15 in an independent screen for mutations that cause reduced expression of IME2 (Vidan and Mitchell 1997). In addition, the TAK1/RIM15 sequence is identical to the open reading frame YFL033C identified as part of the Yeast Genome Sequencing Project (Murakami et al. 1995). We will refer to this gene and its product as RIM15 and Rim15p, respectively. In accordance with Vidan and Mitchell (1997), we found RIM15 to specify a 1770-residue polypeptide comprising a domain with high homology to serine/threonine protein kinases of the protein kinase C and protein kinase A subfamilies (see also Hunter and Plowman 1997).A notable feature of the predicted Rim15p amino acid sequence is the presence of five consensus sites for cAPK phosphorylation, Arg Arg X Ser (Edelman et al. 1987).
Table 1.
List of yeast strains
| Strain
|
Genotype
|
|
Source
|
|---|---|---|---|
| YEF473 | MATa/α | his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 | Bi and Pringle (1996) |
| AR1 | MATa/α | his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 rim15Δ::kanMX2/RIM15 | this study |
| AR1-1B | MATα | his3 leu2 lys2 trp1 ura3 rim15Δ::kanMX2 | segregant from AR1 |
| AR1-1C | MATa | his3 leu2 lys2 trp1 ura3 rim15Δ::kanMX2 | segregant from AR1 |
| AR2 | MATa/α | his3/his3 leu2/leu2 lys2/lys2 trp1/trp1 ura3/ura3 rim15Δ::kanMX2/rim15Δ::kanMX2 | AR1-1B X AR1-1C |
| SP1 | MATa | ade8 his3 leu2 trp1 ura3 | Toda et al. (1985) |
| T16-11A | MATa | his3 leu2 trp1 ura3 bcy1-1 | Toda et al. (1985) |
| PD6517 | MATα | ade8 leu2 trp1 cdc35-10 | Becher dos Passos et al. (1992) |
| NB11 | MATα | ade8 leu2 trp1 cdc35-10 rim15Δ::kanMX2 | this study |
| S7-7A × S7-5A | MATa/α | ade8/ade8 his3/his3 leu2/leu2 trp1/trp1 ura3/ura3 TPK1/tpk1::URA3 TPK2/tpk2::HIS3 TPK3/tpk3::TRP1 | Toda et al. (1987) |
| NB13 | MATa/α | ade8/ade8 his3/his3 leu2/leu2 trp1/trp1 ura3/ura3 TPK1/tpk1::URA3 TPK2/tpk2::HIS3 TPK3/tpk3::TRP1 RIM15/rim15Δ::kanMX2 | this study |
| NB13-1D | MATα | ade8 his3 leu2 trp1 ura3 tpk2::HIS3 tpk3::TRP1 rim15Δ::kanMX2 | segregant from NB13 |
| NB13-14D | MATa | ade8 his3 leu2 trp1 ura3 tpk1::URA3 tpk2::HIS3 tpk3::TRP1 rim15Δ::kanMX2 | segregant from NB13 |
| EGY48 | MATα | his3 trp1 ura3 LEU2::pLexAop6–LEU2 | Zervos et al. (1993) |
Analysis of the nine RIM15 library plasmids isolated in the two-hybrid screen revealed that they all contained the same fragment of RIM15, corresponding to amino acids 761–1051 of the predicted full-length gene product. As shown in Table 2, this Rim15p761–1051-activation domain fusion (AD) interacted with the Tps1p–DNA-binding domain fusion (DBD), but not with Tps2p–DBD, Tps3p–DBD, or Tsl1p–DBD. Because all four predicted proteins, Tps1p, Tps2p, Tps3p, and Tsl1p, share high homology over the entire length of the Tps1p sequence, these two-hybrid data indicate that the observed interaction between Rim15p761–1051–AD and Tps1p–DBD is highly specific.
Table 2.
Two-hybrid interactions between Rim15p and the subunits of the trehalose-6-phosphate synthase complex in Saccharomyces cerevisiae
| AD fusion
|
DBD fusions
|
|||
|---|---|---|---|---|
|
TPS1
|
TPS2
|
TPS3
|
TSL1
|
|
| RIM15-P | 46.8 ± 12.4 | 6.0 ± 4.0 | 1.3 ± 0.6 | 2.4 ± 1.0 |
| pJG4-5 | 2.0 ± 1.4 | 1.1 ± 0.3 | 3.1 ± 2.5 | 2.3 ± 1.4 |
Possible interactions between Rim15p and Tps1p, Tps2p, Tps3p, or Tsl1p were detected using the two-hybrid system as described in Materials and Methods. Numbers represent mean β-galactosidase activites (in Miller units) ± s.d.s from three independent transformants for each pair of plasmids. pJG4-5 indicates the AD vector without insert. Note that the Tps1p–DBD, Tps2p–DBD, Tps3p–DBD, and Tsl1p–DBD fusions were all shown previously to interact with a Tps1p–AD fusion (Reinders et al. 1997). RIM15-P codes for an internal part of Rim15p comprising a section of the kinase domain (amino acids 761–1051).
RIM15 is required for proper entry into stationary phase
To determine the consequences of the loss of Rim15p, we replaced the complete RIM15 coding region by a PCR-based gene deletion method using the kanMX2 module (Wach et al. 1994; see Materials and Methods). The heterozygous RIM15/rim15Δ diploid AR1 was sporulated and the deletion was shown to segregate 2:2 as judged by geneticin resistance of the resulting colonies. Two tetrads were checked by PCR and Southern blotting, and, in each case, the presence of the selectable marker correlated with the shift in fragment size expected for the deleted alleles (data not shown). Thus, RIM15 is not essential for growth or germination.
For further analysis of the effects of rim15Δ we constructed a homozygous diploid rim15Δ/rim15Δ strain (AR2) by mating an appropriate pair of geneticin-resistant segregants of AR1 (Table 1). The rim15Δ/rim15Δ strain had no obvious growth defect at 30°C or 37°C on all carbon sources we tested, including glucose, fructose, sucrose, raffinose, maltose, glycerol, and ethanol (data not shown). Because our two-hybrid analyses suggested that Rim15p may interact with Tps1p, we assayed the rim15Δ/rim15Δ strain for its capacity to accumulate trehalose during a mild heat shock and in stationary phase. Although the rim15Δ/rim15Δ mutant accumulated wild-type levels of trehalose during a 1-hr heat shock at 42°C (i.e., 0.345 ± 0.033 and 0.377 ± 0.009 gram/gram protein for the rim15Δ/rim15Δ mutant and the isogenic wild type, respectively), it was found to contain very low amounts of trehalose in stationary phase when compared with the isogenic wild type (Table 3). Surprisingly, this deficiency in stationary phase-induced trehalose accumulation was not reflected in any detectable changes in the activities of the two key enzymes of trehalose metabolism, namely Tre6P synthase and neutral trehalase.
Table 3.
Effects of RIM15 deletion
|
|
RIM15/RIM15a
|
rim15Δ/rim15Δa
|
||
|---|---|---|---|---|
| log
|
stat
|
log
|
stat
|
|
| Enzymes and metabolites | ||||
| Tre6P synthase (μmole/sec per gram protein) | 0.33 | 1.32 | 0.37 | 1.17 |
| trehalase (μmole/sec per gram protein) | 0.09 | 0.17 | 0.05 | 0.16 |
| invertase (μmole/sec per gram protein) | 2.00 | 12.93 | 1.93 | 12.06 |
| SSA3–lacZ inductionb (Miller units) | 4.30 | 105.20 | 2.40 | 16.30 |
| trehalose (gram/gram protein) | <0.001 | 0.199 | <0.001 | 0.009 |
| glycogen (mg/gram protein) | 1.63 | 38.53 | 0.93 | 11.36 |
| Thermotolerancec (% survival) | 0.13 | 35.60 | 0.07 | 0.03 |
| Stationary phase survivald (%) | 100.00 | 20.20 | ||
| Budded cellse (%) | 65.70 | 0.90 | 65.70 | 24.30 |
Wild-type and rim15Δ/rim15Δ strains were YEF473 and AR2, respectively. All experiments were carried out on YPD (2% glucose) medium using either log phase (log) or 4-day-old stationary phase (stat) cells, except where otherwise stated. Values represent means of at least three independent experiments; s.d.s were, in each case, <10% of the corresponding means.
β-Galactosidase activities were measured to monitor the induction of an SSA3–lacZ fusion gene (from plasmid pWB204Δ–236).
Thermotolerance was measured as the percent survival following a heat shock for 8 min at 50°C (log-phase cells) or 20 min at 53°C (stationary-phase cells).
The percentage of viable cells was determined by the colony-forming efficiency on YPD agar of 10-day-old stationary-phase cultures.
The percentage of budded cells was determined by microscopic examination of at least 200 cells.
As the rim15Δ/rim15Δ mutant was found to be defective for trehalose synthesis upon entry into stationary phase, we also determined a variety of other phenotypic traits characteristic of stationary phase cells (Werner-Washburne et al. 1993). The rim15Δ/rim15Δ mutant was found to be normal for derepression of invertase (Table 3). However, it was impaired in its ability to accumulate glycogen, to induce SSA3 transcription (as measured by induction of an SSA3–lacZ fusion gene), to acquire thermotolerance, and to arrest properly in G1 (as measured by the percentage of budded cells) upon entry into stationary phase (Table 3). As a result, the strain was also highly sensitive to prolonged starvation (e.g., 10 days in stationary phase). Finally, the rim15Δ/rim15Δ mutant also exhibited much lower sporulation efficiency (0.6% ± 0.5) than the wild type (31.1% ± 5.6). Thus, the RIM15 gene is required for sporulation and for proper entry into stationary phase.
The observed pleiotropic response to nutrient limitation and the defect in sporulation caused by deletion of RIM15 are reminiscent of the effects of two previously described mutations in the Ras/cAMP pathway, namely RAS2Val19 and bcy1, which both result in uncontrolled activation of cAPK (Kataoka et al. 1984; Cannon and Tatchell 1987; Toda et al. 1987). Thus, Rim15p might be involved in signaling the status of nutrient supply (or limitation) at some point in the Ras/cAMP pathway. Therefore, we analyzed the transcription patterns in wild-type and rim15Δ/rim15Δ cells of various genes (including SSA3, HSP12, UBI4, and ADH2) that are known to be under negative control of cAPK activity (Tanaka et al. 1988; Cherry et al. 1989; Boorstein and Craig 1990; Varela et al. 1995). A comparison of transcript levels during exponential phase, diauxic shift, post-diauxic shift, and stationary phase in wild-type and rim15Δ/rim15Δ cells is shown in Figure 1. Although the wild-type strain showed the expected pattern of transcriptional repression during exponential growth and transcriptional derepression after the diauxic shift for all five of these genes, the rim15Δ/rim15Δ strain was seriously defective for transcriptional derepression after the diauxic shift of SSA3, HSP12, and HSP26, but not for derepression of UBI4 and ADH2. In fact, derepression of ADH2, the only gene of this group whose expression is known to be controlled by cAPK-dependent inactivation of the transcriptional activator Adr1p (Cherry et al. 1989), was even found to be enhanced during the post-diauxic phase and in stationary phase. We also examined the expression pattern of the cold-inducible SSB1 gene, a member of the Hsp70p subfamily, whose transcription is repressed upon entry into stationary phase (Werner-Washburne et al. 1989). No significant difference in the SSB1 repression pattern in wild-type and rim15Δ/rim15Δ cells during and after the diauxic shift was found (Fig. 1). Taken together, these results indicate that Rim15p is required for the stationary phase-induced transcriptional derepression (or activation) of a subset of genes (SSA3, HSP12, and HSP26) known to be negatively controlled by cAPK. In accordance with such a role of Rim15p, expression of RIM15 itself was found to be very weak during exponential growth but to be highly induced during the diauxic shift and the subsequent post-diauxic and stationary phases (Fig. 1).
Figure 1.

Abundance of various mRNA species as wild-type (YEF473) and rim15Δ/rim15Δ (AR2) mutant cells grow to stationary phase in YPD medium. Total RNAs extracted from cells in exponential phase (0.5 day), diauxic shift phase (1 day), postdiauxic shift phase (2 and 3 days), and stationary phase (4–8 days) were extracted at the times indicated and equal amounts (10 μg) were probed with RIM15, SSA3, HSP12, HSP26, UBI4, SSB1, and ADH2 fragments after electrophoresis and blotting. The application and transfer of equal amounts of RNA were verified by ethidium bromide staining.
Loss of Rim15p suppresses cdc35ts and total loss of cAPK
To elucidate whether Rim15p may act in the Ras/cAMP pathway, we determined whether the loss of Rim15p could suppress the conditional growth of a temperature-sensitive adenylate cyclase mutant. To this end, the rim15::kanMX2 deletion was introduced into strain PD6517 (cdc35-10), and three geneticin-resistant colonies were tested for growth at the non-permissive temperature of 35°C. In all cases, deletion of RIM15 was found to allow growth at this temperature. Thus, cdc35-10 (PD6517) and cdc35-10 rim15Δ (NB11) cultures had an OD600 of 0.15 ± 0.06 and 4.47 ± 0.26, respectively, after inoculation (OD600 0.05) and growth for 2 days at 35°C in liquid YPD (2% glucose) medium.
To examine whether Rim15p may function immediately downstream of Cdc35p or at a later step in the Ras/cAMP pathway, we tested whether deletion of RIM15 could suppress the complete loss of cAPK. To this end, a heterozygous TPK1/tpk1 TPK2/tpk2 TPK3/tpk3 diploid strain (S7-7A × S7-5A) was transformed to geneticin-resistance with the rim15::kanMX2 deletion construct and allowed to sporulate, and the asci were dissected on YPD agar. As expected tpk1 tpk2 tpk3 RIM15 spores failed to germinate (as judged by segregation of the auxotrophic markers). However, tpk1 tpk2 tpk3 rim15Δ (tpk rim15Δ) spores were viable. In a complementary experiment, a tpk rim15Δ strain was transformed either with a plasmid that allows galactose-inducible expression of RIM15 (YCpIF2–RIM15) or with the corresponding control plasmid (YCpIF2). As expected, tpk rim15Δ cells containing the control plasmid were able to grow on both glucose- and galactose-containing medium. In contrast, tpk rim15Δ cells containing YCpIF2-RIM15 were able to grow on glucose-containing medium but failed to grow on galactose-containing medium (Fig. 2). Together, these results show that the loss of Rim15p suppresses the lethal effect of total loss of cAPK.
Figure 2.

Suppression of the tpk growth defect by rim15Δ. tpk1 tpk2 tpk3 rim15Δ cells (NB13-14D) were transformed with plasmids YCpIF2 (control) and YCpIF2-RIM15 (allowing galactose-dependent expression of RIM15), streaked on SD media containing either glucose or galactose as carbon source, and incubated for 4 days at 30°C.
Because tpk rim15Δ cells were viable, we were able to compare TPK rim15Δ and tpk rim15Δ cells and to examine whether, as expected for a downstream effector, rim15Δ was epistatic over tpk mutations with respect to various stationary-phase-associated phenotypes. SSA3, HSP12, and HSP26 were repressed in logarithmically growing wild-type cells and induced in stationary phase cells (Fig. 3, lanes 1–3). In rim15Δ mutants, these genes were found to be repressed both in logarithmically growing and in stationary phase cells independent of the presence or absence of a functional TPK gene (Fig. 3, lanes 4–9). As a control for the physiological status of the cells, SSB1 was shown to be highly expressed in log phase cells and repressed in stationary phase cells of all three strains (Fig. 3, lanes 1–9). Like TPK rim15Δ cells, tpk rim15Δ cells were also defective for the accumulation of trehalose (0.027 ± 0.005 gram/gram protein vs. < 0.01 gram/gram protein for TPK rim15Δ cells). Thus, rim15Δ is largely epistatic over tpk mutations with respect to these hallmarks of stationary phase (i.e. transcriptional activation/derepression of SSA3, HSP12, and HSP26 and trehalose accumulation). In contrast, tpk rim15Δ cells had reduced growth rates at 30°C (0.16 ± 0.01/hr versus 0.27 ± 0.01/hr for TPK rim15Δ cells), exhibited higher levels of thermotolerance (6.3 ± 1.6% vs. 0.07 ± 0.01% survival after incubation for 20 min at 53°C for TPK rim15Δ cells) and higher rates of survival in stationary phase (50.7 ± 9.8% vs. 4.6 ± 0.8% after 10 days in stationary phase for TPK rim15Δ cells), and hyperaccumulated glycogen (as measured by iodine staining) when compared with TPK rim15Δ cells. Thus, the TPK status still affected these phenotypes in rim15Δ cells. The significance of these results is discussed below.
Figure 3.

Northern blot analysis of gene expression in exponentially growing and stationary phase TPK RIM15 (SP1), TPK rim15Δ (NB13-1D), and tpk rim15Δ (NB13-14D) cells. Total RNA was extracted from log phase (L) and stationary phase (S1, 2 days; S2, 4 days after glucose depletion) cells grown in YPD medium. For further details see Fig. 1, legend.
RIM15 overexpression suppresses bcy1-1, exacerbates the growth defect of a strain partially compromised for cAPK activity, and partially mimics a nutrient limited state in wild-type cells
The data presented above suggest that Rim15p activity may be under direct or indirect negative control by cAPK and that Rim15p may be responsible for the induction of a subset of the physiological changes triggered by nutrient limitation including the synthesis of trehalose and the transcriptional derepression (or activation) of SSA3, HSP12, and HSP26. If this model were correct, one would expect that overexpression of RIM15 should, on the one hand, revert phenotypes associated with uncontrolled, constitutive activation of cAPK and, on the other hand, exacerbate phenotypes associated with attenuated cAPK activity. In accordance with these expectations, overexpression of RIM15 under the control of the ADH1 promoter (YCpADH1-RIM15) partially suppressed the defect in trehalose accumulation and completely suppressed the defects in SSA3–lacZ induction (from pWB204Δ-236) and thermotolerance acquisition of bcy1-1 cells entering stationary phase (Table 4). In addition, overexpression of RIM15 caused a significant increase of stationary-phase-induced trehalose accumulation, SSA3–lacZ induction, and thermotolerance acquisition in wild-type cells (Table 4; see also below). Furthermore, overexpression of RIM15 was found to exacerbate the growth defect of a temperature-sensitive adenylate cyclase mutant (cdc35-10): Although this particular mutant grew well at 34°C and at 27°C when harboring the control plasmid, it was defective for growth at 34°C, but not at 27°C, when overexpressing RIM15 (Fig. 4).
Table 4.
Suppression of bcy1-1 phenotypes by overexpression of RIM15
| Strain
|
Relevant genotype
|
Plasmid
|
Trehalose (gram/gram protein)
|
SSA3–lacZ induction (Miller units)
|
Thermotolerance (% survival)
|
|---|---|---|---|---|---|
| SP1 | BCY1 | YCpADH1 | 0.39 ± 0.05 | 137.8 ± 37.5 | 39.8 ± 3.7 |
| SP1 | BCY1 | YCpADH1–RIM15 | 0.56 ± 0.11 | 299.9 ± 76.0 | 73.0 ± 2.4 |
| T16-11A | bcy1-1 | YCpADH1 | 0.12 ± 0.06 | 41.4 ± 11.1 | 12.6 ± 2.9 |
| T16-11A | bcy1-1 | YCpADH1–RIM15 | 0.24 ± 0.02 | 123.9 ± 25.7 | 43.0 ± 10.9 |
Cells were grown to stationary phase (5 days) on SD medium containing 1% glucose. Values represent means ± s.d.s of three to six independent experiments. β-Galactosidase activities were measured to monitor the induction of an SSA3–lacZ fusion gene (from plasmid pWB204Δ–236). Thermotolerance was measured as the survival following a heat shock for 20 min at 50°C.
Figure 4.

Overexpression of RIM15 exacerbates the temperature-sensitive growth defect of a cdc35-10 mutant. Strain PD6517 (cdc35-10) was transformed with plasmids YCpADH1 and YCpADH1-RIM15, streaked on SD medium, and incubated for 3 days at the temperatures indicated.
To study further the effects of RIM15 overexpression in wild-type cells, strain YEF473 containing the reporter plasmid pWB204Δ-236 was transformed with either YCpIF2-RIM15 (allowing galactose-inducible expression of RIM15) or YCpIF2 (control) and analyzed for trehalose levels, SSA3–lacZ induction, and thermotolerance after growth for 16 hr (log phase) or 4 days (stationary phase) on galactose-containing medium. As shown in Table 5, overexpression of RIM15 significantly enhanced the levels of trehalose, SSA3–lacZ induction, and thermotolerance in log as well as in stationary-phase cells when compared with the corresponding control. Thus, physiological adaptations associated with stationary phase are partially induced in log-phase cells and enhanced in stationary-phase cells by overexpression of RIM15.
Table 5.
Effects of RIM15 overexpression
| Strain
|
Relevant genotype
|
Trehalose (gram/gram protein)
|
SSA3–lacZ induction (Miller units)
|
Thermotolerance (% survival)
|
|||
|---|---|---|---|---|---|---|---|
| log
|
stat
|
log
|
stat
|
log
|
stat
|
||
| YEF473 | wild-type (YCpIF2) | 0.006 ± 0.002 | 0.238 ± 0.030 | 12.6 ± 3.0 | 429.0 ± 61.9 | 0.9 ± 0.5 | 77.1 ± 8.5 |
| YEF473 | wild-type (YCpIF2-RIM15) | 0.063 ± 0.008 | 0.364 ± 0.027 | 37.0 ± 3.7 | 589.0 ± 43.7 | 4.9 ± 1.6 | 98.5 ± 18.0 |
Cells were grown to log phase (log) or stationary phase (stat; 4 days) on SD medium containing 2% galactose and 1% raffinose to induce GAL1-driven transcription of RIM15 in YCpIF2–RIM15. Values represent means ± s.d.s of three experiments. β-Galactosidase activites were measured to monitor the induction of an SSA3–lacZ fusion gene (from plasmid pWB204Δ–236). Thermotolerance was measured as the survival following a heat shock for 4 min (log-phase cells) or 20 min (stationary-phase cells) at 50°C.
Rim15p protein kinase activity is negatively regulated by cAPK-dependent phosphorylation
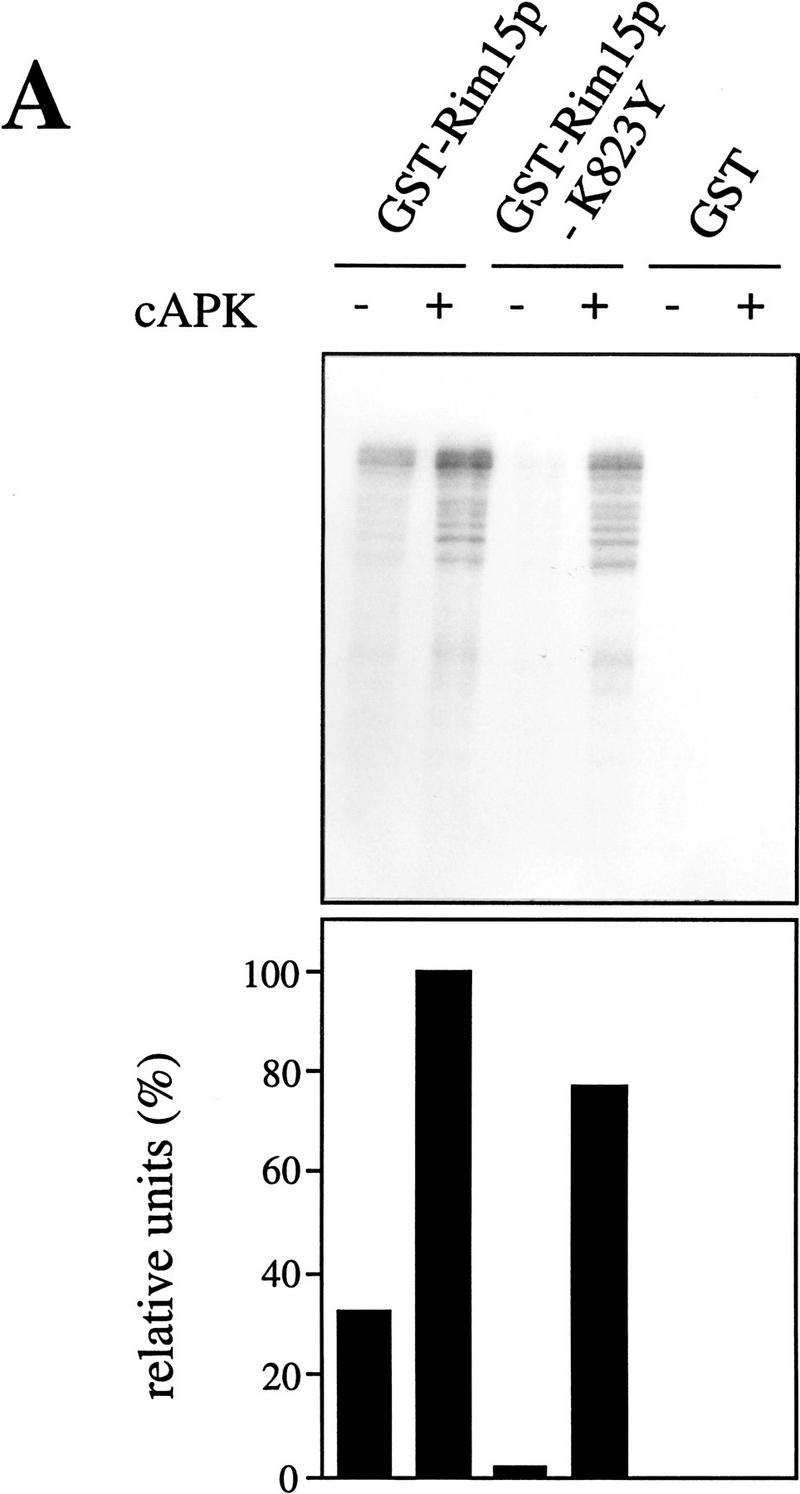
The epistasis analyses presented above suggest that Rim15p serves to inhibit cell growth and to promote stationary phase entry and that its kinase activity may be attenuated by direct cAPK-dependent phosphorylation. This would mark Rim15p as the first protein kinase known to constitute a downstream effector of cAPK in S. cerevisiae. To test this model, we first analyzed whether Rim15p has protein kinase activity and whether it could phosphorylate itself in vitro. To this end, a functional GST–Rim15p fusion protein was expressed in wild-type S. cerevisiae cells, purified by glutathione affinity chromatography, and incubated with [γ-32P]ATP. This procedure resulted in phosphorylation of a 223-kD polypeptide (expected size for GST–Rim15p) and of a variety of smaller polypeptides (Fig. 5A). Immunoblot analysis with anti-GST antibodies also revealed the presence of a 223-kD polypeptide as well as a similar array of smaller polypeptides that were not present in GST control extracts (data not shown). From these data, we infer that the 223-kD band corresponds to the GST–Rim15p and that the smaller bands represent proteolytic fragments of it. To eliminate the possibility that in vitro Rim15p phosphorylation is catalyzed by a protein kinase coprecipitating with GST–Rim15p and to assess the extent of autophosphorylation, we examined the autophosphorylation activity of an ATP-binding-deficient GST–Rim15p–K823Y fusion protein. This mutant fusion protein has lysine 823, corresponding to the conserved lysine shown to be required for ATP binding and kinase activity of known protein kinases (Saraste et al. 1990), replaced by a tyrosine. As shown in Figure 5A, autophosphorylation of GST–Rim15p-K823Y was reduced by about 90%. Thus, the major part of the Rim15p in vitro phosphorylation results from an autocatalytic reaction.
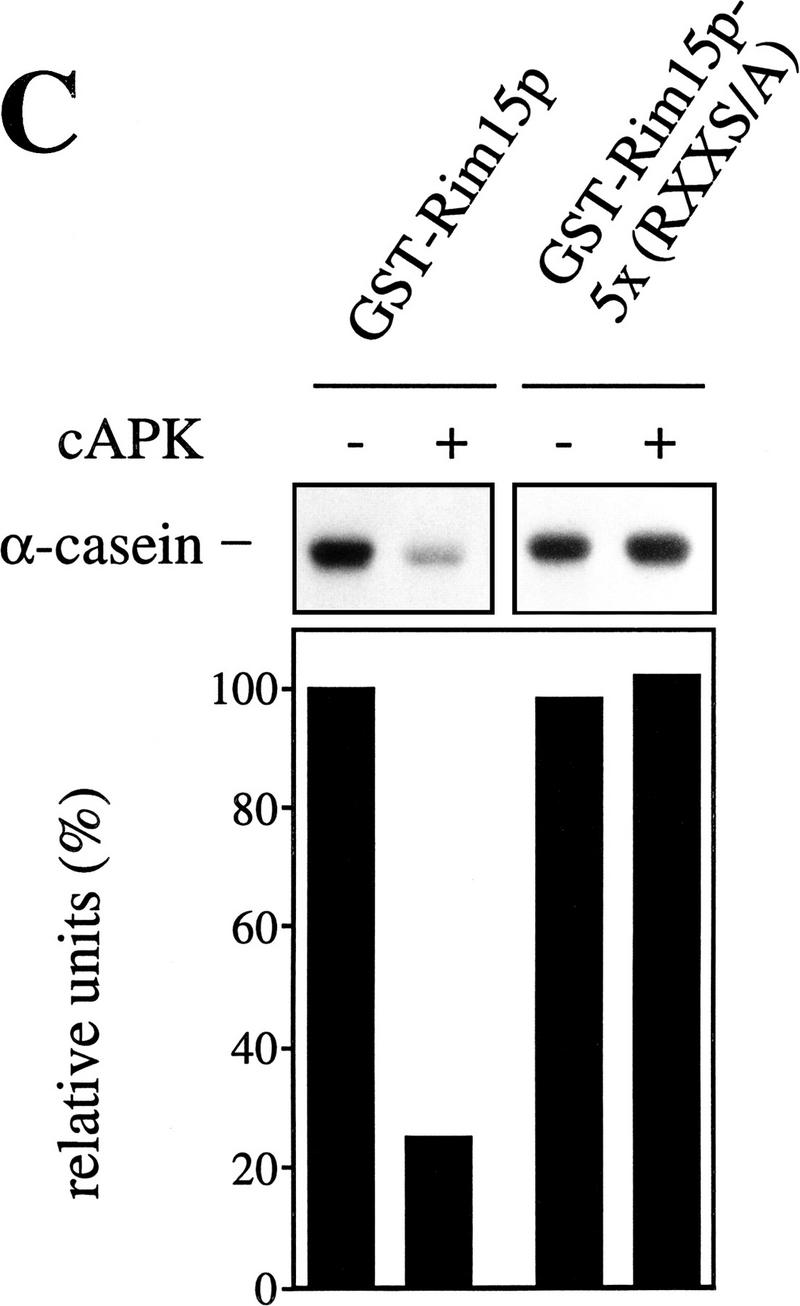
Figure 5.

cAPK-dependent phosphorylation of Rim15p inhibits its kinase activity. (A) GST–Rim15p, GST–Rim15p-K823Y, and GST proteins were purified and analyzed for autophosphorylation activity and their potential to be phosphorylated by cAPK. Accordingly, equal amounts of the fusion proteins (verified by immunoblotting with anti-GST antibodies) were incubated with [γ-32P]ATP either in the absence (−cAPK) or in the presence (+cAPK) of cAPK as described in Materials and Methods. Phosphorylation levels were quantitated by PhosphorImager analysis and expressed as percent of the GST–Rim15p-phosphorylation level (+cAPK). (B) To analyze the effect of cAPK-dependent phosphorylation of Rim15p, equal amounts of GST–Rim15p, GST–Rim15p-K823Y, and GST (verified by immunoblotting as in A) were preincubated with unlabeled ATP and either no further additions, with cAPK, with cAPK and cAPK inhibitor, or with cAPK inhibitor alone, as indicated. The samples were washed extensively and assayed for α-casein phosphorylation in the presence of cAPK inhibitor and [γ-32P]ATP. The levels of α-casein phosphorylation were quantitated by PhosphorImager analysis and expressed as percent of the control (level of α-casein phosphorylation after preincubation of GST–Rim15p in the absence of both cAPK and cAPK inhibitor). (C) Equal amounts of GST–Rim15p and GST–Rim15p-S709A/S1094A/S1416A/ S1463A/S1661A [GST–Rim15p-5x(RRXS/A)] were analyzed as in B.
Then we examined the role of cAPK in Rim15p phosphorylation. Incubation of GST–Rim15p and GST–Rim15p–K823Y with bovine cAPK and [γ-32P]ATP led to extensive phosphorylation of both fusion proteins (Fig. 5A; similar results were obtained using bacterially expressed GST–Tpk1p instead of bovine cAPK. Since we found the activity of GST–Tpk1p to be highly unstable, we used bovine cAPK in our experiments). The lower phosphorylation level after cAPK-treatment of GST–Rim15p-K823Y compared with GST–Rim15p may be explained by the absence of autophosphorylation in the former fusion protein. Because Rim15p can be phosphorylated by cAPK, we addressed the significance of this event for Rim15p activity. For this assay, α-casein, which was found to be efficiently phosphorylated by Rim15p, was used as exogenous substrate. As shown in Figure 5B, the ability of Rim15p to phosphorylate α-casein was significantly reduced (>75%) by preincubation with bovine cAPK. This cAPK-mediated reduction could be completely prevented if cAPK inhibitor was present during the preincubation. In control experiments with GST or GST–Rim15p–K823Y we found no detectable or only residual (<4%) phosphorylation of α-casein, respectively. In accordance with these results, the corresponding GST and GST–rim15K823Y genes, but not the GST–RIM15 gene, failed to complement the rim15Δ mutation (data not shown).
As mentioned above, the predicted Rim15p amino acid sequence contains five Arg Arg X Ser consensus sites for cAPK phosphorylation (Edelman et al. 1987). If Rim15p kinase activity is down-regulated by cAPK-dependent phosphorylation, replacement of Ser with Ala at these five consensus sites should yield a cAPK-unresponsive Rim15p mutant protein. As expected, we found that a GST–Rim15p-S709A/S1094A/S1416A/S1463A/S1661A quintuple mutant protein [GST–Rim15p-5x(RRXS/A)] could not be inhibited in vitro by preincubation with cAPK (Fig. 5C). Taken together, these data show that cAPK-mediated phosphorylation of Rim15p inhibits its kinase activity.
Discussion
We identified the Rim15p protein kinase in a two-hybrid screen for potential interactors with Tps1p, the small subunit of the Tre6P synthase. Loss of Rim15p was found to cause a highly pleiotropic phenotype in cells entering stationary phase, including a defect in trehalose synthesis, which suggests a role for Rim15p in nutrient signal transduction. Our results are most simply interpreted in a model in which Rim15p functions in the Ras/cAMP pathway downstream of cAPK to regulate a set of physiological adaptations necessary for proper entry into stationary phase. Several observations support such a model. First, deletion of RIM15 results in a defective response to nutrient limitation of mutant cells entering stationary phase that is reminiscent of the effects caused by mutations that constitutively activate cAPK (e.g., bcy1 and RAS2Val19). Accordingly, rim15Δ cells are seriously defective in trehalose accumulation, in transcriptional derepression of SSA3, HSP12, and HSP26, in induction of thermotolerance and starvation resistance, as well as in proper G1 arrest when entering stationary phase. Second, deletion of RIM15 suppresses the growth defect at elevated temperatures of a temperature-sensitive adenylate cyclase mutant (cdc35-10) and allows growth of cells that lack all three cAPK-encoding genes. Third, overexpression of RIM15 suppresses the defects in trehalose accumulation, transcriptional derepression of SSA3, and thermotolerance acquisition in stationary cells of a bcy1-1 mutant, exacerbates the growth defect of a strain compromised for cAPK activity (cdc35-10), and partially induces a starvation response in logarithmically growing wild-type cells. Fourth, cAPK-mediated phosphorylation of Rim15p, but not of a GST-Rim15p-5x(RRXS/A) quintuple mutant protein with Ser-to-Ala mutations at the consensus sites for cAPK phosphorylation, results in a strong reduction of the Rim15p protein kinase activity in vitro.
Despite the broad physiological overlap of the effects of cAPK hyperactivity and of loss of Rim15p, there are some notable differences. In contrast to rim15Δ, mutations that cause constitutive activation of cAPK such as bcy1 or RAS2Val19 are generally associated with constitutive activation of neutral trehalase, complete inability to accumulate glycogen, and gluconeogenic growth defects (Kataoka et al. 1984; Toda et al. 1987; Cameron et al. 1988). A model that may account for these differences is that cAPK constitutes a point of divergence in the Ras/cAMP pathway and that Rim15p acts beyond this specific point (Fig. 6). Accordingly, cAPK may on the one hand transmit the signal of the Ras/cAMP pathway to Rim15p and on the other hand directly control several other cellular mechanisms such as the activation of neutral trehalase (for review, see Thevelein 1996), the inhibition of glycogen synthesis (Hardy et al. 1994 and references therein), and the inhibition of gluconeogenic growth (Müller and Holzer 1981; Mazón et al. 1982; François et al. 1984; Cherry et al. 1989). If this model were correct, rim15Δ should be epistatic to tpk mutations with respect to some but not all phenotypes. Our results support such a conclusion. Accordingly, we found that a tpk rim15Δ strain, like a TPK rim15Δ strain, was defective for derepression of SSA3, HSP12, and HSP26 as well as for trehalose accumulation upon entry into stationary phase, suggesting that these processes are downstream of and controlled by Rim15p. In contrast, the tpk rim15Δ strain was still found to exhibit a reduced growth rate, to acquire higher levels of thermotolerance, to hyperaccumulate glycogen, and—possibly as a result of the higher glycogen content—to have significantly enhanced levels of stationary phase survival when compared to the TPK rim15Δ strain. Thus, control over these processes may be exerted to some extent at the level of cAPK and therefore be at least partially independent of Rim15p. In this context, the seemingly contradictory finding that stationary TPK rim15Δ mutants had reduced levels of glycogen may be reconciled with our model if Rim15p would be involved in mechanisms of cAPK feedback inhibition. We are currently exploring this possibility in further detail.
Figure 6.

Model for the cAMP-dependent protein kinase (cAPK)–Rim15p pathway. Glucose-repression as well as post-translational inhibition via cAPK-dependent phosphorylation, and potentially an additional yet-to-be-identified mechanism, result in low Rim15p kinase activity in log phase. During the postdiauxic shift, glucose derepression of RIM15 (illustrated by the increase in size of the Rim15p protein), down-regulation of cAPK-dependent inhibition, and potential activation by yet-to-be-identified mechanisms result in high Rim15p kinase activity. Rim15p acts as a positive regulator of stationary phase entry and, consequently, as a negative regulator of growth. (Arrow) Positive interaction; (bar) negative interaction. Bold arrows and bars denote high activity. Dashed arrows and bars refer to potential interactions. Accumulation of trehalose and glycogen is indicated with bold letters. Inactive proteins are on white and active proteins are on shaded backgrounds. Nth1p, Tps1p, and Tps2p denote neutral trehalase, Tre6P synthase, and Tre6P phosphatase, respectively. For further details, see text.
Several protein kinases whose functions may partially overlap with the ones of the Ras/cAMP pathway have been described previously. One of these protein kinases is Snf1p, which functions primarily in glucose repression by its negative control of transcriptional factors such as Mig1p (for review, see Ronne 1995). Genetic studies indicated that not all Snf1p functions are mediated by Mig1p, though, and that Snf1p controls glycogen accumulation, acquisition of thermotolerance, and proper G1 arrest upon depletion of external glucose in parallel and antagonistically to cAPK (Thompson-Jaeger et al. 1991; Woods et al. 1994; Huang et al. 1996). In this context, two other genes encoding protein kinases, namely SCH9 and YAK1, have been isolated by genetic screens for growth-related effectors of cAPK (Toda et al. 1988; Garrett and Broach 1989). However, their corresponding gene products may both define separate nutrient signaling pathways that also act parallel to the Ras/cAMP pathway (Toda et al. 1988; Denis and Audino 1991; Garrett et al. 1991; Thompson-Jaeger et al. 1991; Hartley et al. 1994). Thus, even though the roles of Snf1, Sch9, and Yak1 in the Ras/cAMP pathway are not understood, the available evidence suggests that all three protein kinases may functionally overlap with and act in parallel to the Ras/cAMP pathway. Therefore, Rim15p appears to be the first protein kinase identified as constituting a downstream effector of cAPK in S. cerevisiae.
Consistent with both the proposed role of Rim15p as an activator of stationary phase entry and the observation that rim15Δ caused no obvious defect in exponentially, glucose-grown cells, the level of RIM15 mRNA and Rim15p protein were found to be very low during growth on glucose and to increase significantly upon glucose exhaustion (Vidan and Mitchell 1997; this study). Because RIM15 is transcriptionally repressed in the presence of glucose, it is not surprising that replacement of RIM15 with the rim155x(RRXS/A) mutant gene did not cause any significant effects in exponentially growing cells (data not shown). However, we also found that the effects of rim155x(RRXS/A) overexpression did not significantly differ from the ones observed following RIM15 overexpression (i.e., both genes caused a similar, partial induction of stationary phase characteristics in exponentially growing cells; Table 5 and data not shown). This observation may be explained if overexpression of RIM15 results in Rim15p protein levels that exceed a certain threshold level beyond which Rim15p may escape cAPK-dependent down-regulation. Accordingly, maximal induction of stationary phase characteristics in log phase cells would already be achieved by overexpression of the RIM15 wild-type gene. The rather moderate level of induction of these stationary phase characteristics in log phase cells may be attributable to the presence of activated cAPK, which indirectly counteracts the effects of Rim15p (e.g., by activation of neutral trehalase), and/or to potential cAPK-independent mechanisms involved in the control of Rim15p kinase activity. The latter would raise the intriguing possibility that Rim15p—besides the observed regulation by glucose repression and cAPK-dependent inhibition—is subjected to down-regulation and/or activation by at least one other, yet unidentified cAMP-independent nutrient signaling pathway. In this context, it is worth noting that a previous report suggested the existence of cAMP-independent mechanisms for regulation of proper stationary phase entry including trehalose accumulation, glycogen synthesis, and the development of thermotolerance (Cameron et al. 1988). Therefore, future studies should address the question of whether any of the potentially separate nutrient signaling pathways may converge at and be integrated by Rim15p.
A further interesting aspect of our study is the possibility that the TPS1-encoded Tre6P synthase may be a direct target of Rim15p. This suggestion is supported by several observations. First, Rim15p shows specific two-hybrid interaction with Tps1p. Second, loss of Rim15p causes a defect in stationary phase-induced trehalose accumulation. Third, overexpression of RIM15 not only significantly induces trehalose synthesis in log-phase cells but also enhances trehalose accumulation in stationary phase cells. Even though these data do not exclude the possibility that Rim15p activates Tre6P synthase rather indirectly, they would be consistent with a model in which Tps1p is positively regulated by Rim15p-dependent phosphorylation. As intriguing as this model may be, it is not easily reconciled with the observation that the in vitro-measured Tre6P synthase of stationary phase rim15Δ/rim15Δ cells was found to be fully active, even though no trehalose was formed in vivo. This inconsistency may be explained, however, if Tre6P synthase were artificially activated by limited proteolysis during the sampling procedure as has been suggested earlier (Londesborough and Vuorio 1991; Vicente-Soler et al. 1991). Taken together, it is clear that further detailed biochemical characterization of Tps1p is necessary to elucidate whether its activity may be regulated via phosphorylation/dephosphorylation and whether Rim15p is directly or rather indirectly involved in such a process. Because recent evidence suggested that Tps1p is also involved in the control of glucose flux into glycolysis, possibly through a specific mechanism independent of trehalose synthesis (for review, see Thevelein and Hohmann 1995), such analyses may not only contribute to our understanding of the regulation of trehalose synthesis but may also help defining the potential trehalose synthesis-independent function of the yeast Tps1p protein.
Recently, Rim15p was identified independently through a mutation that caused reduced expression of IME2 (Vidan and Mitchell 1997). Current knowledge suggests that Rim15p may act to stimulate formation of the Ime1–Ume6 complex, which in turn acts as a transcriptional activator of early meiotic gene expression (Bowdish et al. 1995; Rubin-Bejerano et al. 1996; Vidan and Mitchell 1997). In view of both these recent findings on Rim15p and the long standing suggestion that entry into meiosis is at least partially regulated by starvation through the Ras/cAMP pathway (Matsumoto et al. 1983; Smith and Mitchell 1989; Matsuura et al. 1990), our present study, placing Rim15p immediately downstream of cAPK, may finally link the Ras/cAMP pathway to early meiotic gene expression.
Materials and methods
Strains, media, and microbiological and recombinant DNA methods
The S. cerevisiae strains used in this study are listed in Table 1. Escherichia coli strain JMB9 ([r−m+] trpF) was used to rescue pJG4-5-based plasmids from strain EGY48 as described earlier (De Virgilio et al. 1996). Other plasmid manipulations were performed in E. coli strain DH5α (GIBCO BRL) with standard procedures (Sambrook et al. 1989). Yeast and E. coli media, including the rich, glucose-containing medium (YPD), the defined media (SD with appropriate supplements), and sporulation medium were prepared by standard recipes (Sambrook et al. 1989; Rose et al. 1990). Standard procedures of yeast genetics and molecular biology were used (Guthrie and Fink 1991; Sambrook et al. 1989). Yeast transformations were performed with a modification of the Li+-ion method (Gietz et al. 1992). Sporulation experiments were performed essentially as described by De Virgilio et al. (1996).
DNA sequencing and sequence analyses
Sequences were obtained from double-stranded plasmid DNA, by use of the dideoxy chain termination method (Sanger et al. 1977) with ΔTth DNA polymerase (Toyobo, Japan) and [α-33P]dATP (Hartmann Analytic, Braunschweig, Germany). The University of Wisconsin Genetics Computer Group (GCG) programs were used to compile and analyze sequence data. Alignments were performed with the GAP, PILEUP, and PRETTY comparison programs.
Two-hybrid analysis
The interactions of Rim15p with the subunits of the Tre6P synthase complex were tested by two-hybrid analysis (Fields and Sternglanz 1994) with the LexA system described in detail elsewhere (Gyuris et al. 1993). The various full-length Tre6P synthase complex subunits were fused to the LexA DBD coding sequences in plasmid pEG202 (Zervos et al. 1993) as described by Reinders et al. (1997). Plasmid pJG4-5–RIM15-P, which contains a fragment of RIM15 fused to the AD moiety of pJG4-5, was isolated in a two-hybrid screen for proteins that interact with Tps1p. Strain EGY48 (Table 1) containing the LexAop–lacZ reporter plasmid pSH18-34 (Gyuris et al. 1993) was cotransformed with a pEG202-derived plasmid expressing a LexA DBD fusion protein and with pJG4-5 or pJG4-5–RIM15-P. β-Galactosidase activities (in Miller units) were then assessed in three independent clones of each strain grown for 16 hr at 27°C in minimal medium (containing 2% wt/vol galactose, 1% wt/vol raffinose, and 20 μg/ml leucine).
Cloning and deletion of RIM15
The 0.9-kb EcoRI fragment of one RIM15 two-hybrid library plasmid was labeled with [α-32P]dATP (Hartmann Analytic) by use of the Prime-It II Random Primer Labeling Kit (Stratagene) and subsequently used to screen a genomic DNA library (in pSey8; kindly provided by M. Hall, Biocenter, Basel, Switzerland) as described by Sambrook et al. (1989). Three positive clones were found to contain overlapping fragments that together covered the entire RIM15 gene. A segment of 6313 bp containing the entire RIM15 gene including flanking sequences was sequenced on both strands (submitted as TAK1, EMBL database accession no. AJ001030).
The complete RIM15 coding region was deleted by the PCR method (Wach et al. 1994) with plasmid pFA6a (kanMX2) as template and Taq DNA polymerase (Boehringer Mannheim). Oligonucleotides that contained 43 and 40 nucleotides immediately upstream and downstream, respectively, of the RIM15 coding region, and 19 and 22 nucleotides upstream and downstream, respectively, of the kanMX2 module were used to create a PCR product that contained flanking sequences of RIM15 separated by the kanMX2 module. This DNA was used to transform strains YEF473, PD6517, and S7-7A × S7-5A to construct AR1, NB11, and NB13, respectively (Table 1). Transformants that had RIM15 replaced with the kanMX2 module were identified by their geneticin-resistant growth and confirmed by PCR and/or Southern blot analysis (data not shown).
Enzyme assays and determination of metabolite levels
The activities of Tre6P synthase and neutral trehalase were measured in permeabilized cells essentially as described by De Virgilio et al. (1993, 1994). Invertase activity was determined in crude extracts according to an established protocol (Goldstein and Lampen 1975). β-Galactosidase activity was assayed using o-nitrophenyl-β-d-galactoside as substrate (Miller 1972). Trehalose and glycogen were measured by the procedures of De Virgilio et al. (1993) and Lillie and Pringle (1980), respectively. Protein concentrations were either measured by the Bio-Rad protein assay according to the manufacturer’s instructions or by a modified Lowry assay (Peterson 1977) with BSA as standard.
Preparation and Northern analysis of RNA
Extraction of total cellular RNA was performed as described previously (Piper 1994). For Northern analysis, 10 μg of total RNA was separated on 1.1% agarose gels containing 0.65 m formaldehyde, transferred to nitrocellulose membranes (BA 83; Schleicher and Schuell, Germany) in 20× SSC, and hybridized with [32P]dATP-labeled DNA fragments that were amplified by PCR from genomic DNA. The primers used for PCR were as follows, with the forward primers listed first and the fragment sizes generated given in parentheses: RIM15, 5′-CTGATTCGCCGTCACAAGTTTGTCCCACATAAGTCG-3′ and 5′-CGTATTGGTAGCTGCGATAACGTCTGAAGATAATAG-3′ (0.52 kb); SSA3, 5′-ATGTCTAGAGCAGTTGGT-3′ and 5′-ATCAACCTCTTCCACTGT-3′ (1.95 kb); HSP12, 5′-ATGTCTGACGCAGGTAGAAAAGGATTC-3′ and 5′-TTACTTCTTGGTTGGGTCTTCTTCACC-3′ (0.33 kb); HSP26, 5′-ATGTCATTTAACAGTCCATTTTTTGATTTC-3′ and 5′-TTAGTTACCCCACGATTCTTGAGAACAAAC-3′ (0.64 kb); UBI4, 5′-ATGCAGATTTTCGTCAAG-3′ and 5′-GTTACCA CCCCTCAACCT-3′ (1.14 kb); ADH2, 5′-ATGTCTATTCCAGAAACTCAAAAAGCC-3′ and 5′-TTATTTAGAAGTGTCAACAACGTATC-3′ (1.05 kb); and SSB1, 5′-ATGGCTGAAGGTGTTTTCCAAGGTGC-3′ and 5′-TTAACGAGAAGACAGGCCTTGGTGAC-3′ (1.84 kb).
Plasmid constructions
For construction of the galactose-inducible GAL1–RIM15 allele, the full-length RIM15 coding sequence was amplified by use of the Expand Long Template PCR System (Boehringer Mannheim) and genomic DNA as template. SalI and NotI restriction sites were introduced immediately upstream of the ATG start codon and 86 bp downstream of the stop codon, respectively. This PCR product was cloned at the SalI–NotI site of YCpIF2 (Foreman and Davis 1994) to yield YCpIF2-RIM15. Plasmids YCpIF2-GST and YCpIF2-GST–RIM15 were constructed by cloning of a PCR-generated SalI–SalI fragment, containing the 672 nucleotides downstream of and including the GST start codon, at the SalI site of YCpIF2 and YCpIF2-RIM15, respectively. Plasmids YCpADH1 and YCpADH1-RIM15 were constructed by replacement of the GAL1 promoter-containing ApaI–SalI fragments of YCpIF2 and YCpIF2-RIM15, respectively, with a PCR-generated ApaI–SalI fragment containing the 854 nucleotides upstream of and including the ADH1 start codon. Plasmids YCpIF2-GST–RIM15K823Y and YCpIF2-GST-RIM15S709A/S1094A/S1416A/S1463A/S1661A were constructed with the QuickChange Site-Directed Mutagenesis Kit (Stratagene) with the appropriate primers that introduced the corresponding mutations and YCpIF2–GST–RIM15 as template. All mutations introduced were confirmed by subsequent sequencing.
Protein kinase assays
GST, GST–Rim15p, GST–Rim15p-K823Y, and GST–Rim15p-S709A/S1094A/S1416A/S1463A/S1661A were expressed in wild-type S. cerevisiae cells from the GAL1 promoter. Cells were harvested in stationary phase after growth on YP medium containing 2% galactose and 1% raffinose and disrupted by vortexing in lysis buffer [50 mm Tris-HCl at pH 7.5, 0.1 m NaCl, 1 mm EDTA, 1% NP-40, and one tablet of Complete Protease Inhibitor Cocktail (Boehringer Mannheim) per 50 ml] in the presence of acid-washed glass beads (0.4-mm diameter; Merck). The extracts were clarified three times by centrifugation at 4°C in a microfuge at 17,000 rpm, 10 min each time. The various GST fusions were purified from clarified cell extracts (containing ∼30 mg protein per ml) after a 4-hr incubation at 4°C with glutathione–Sepharose beads (50 μl/ml; Pharmacia). Protein-bound beads were pelleted, washed four times with lysis buffer and three times with kinase buffer (50 mm Tris-HCl at pH 7.5, 20 mm MgCl2, 1 mm DTT, 1 mm ATP, and one tablet of Complete Protease Inhibitor Cocktail per 50 ml). Kinase assays were performed at 30°C for 30 min in reaction buffer (kinase buffer containing 50 mm NaF, 10 mm Na-orthovanadate, 15 mm p-NO2-phenylphosphate, 50 mm β-glycerophosphate, 5 mm Na-pyrophosphate, 10 μCi [γ-32P]ATP, and, where indicated, 250 μg/ml α-casein). Reactions were stopped by adding SDS-gel loading buffer and boiling for 5 min and were then subjected to SDS-PAGE. Gels were dried and exposed to X-ray film or to a PhosphorImager. Quantitation of 32P was performed with the Phosphor Analyst software (Bio-Rad).
To assay in vitro autophosphorylation and phosphorylation of Rim15p by bovine cAPK, wild-type or mutant GST–Rim15p fusion protein was purified as described above. Equal amounts of protein-bound beads (10 μl) were resuspended in reaction buffer and incubated for 30 min at 30°C in the presence or absence of 1 unit of A kinase catalytic subunit (Sigma) and/or A kinase Inhibitor (Sigma). Reactions were stopped and analyzed as described above. When the effects of cAPK-dependent phosphorylation on Rim15p activity were monitored, reactions were incubated in [γ-32P]ATP-free reaction buffer in the presence or absence of 1 unit A kinase catalytic subunit, subsequently terminated by three washes with the same buffer (without A kinase catalytic subunit), and then the mixtures were incubated with α-casein and [γ-32P]ATP as described above.
Acknowledgments
We thank J.M. Thevelein and E.A. Craig for strains and plasmids, respectively, and C. Funk and G. Vogel for assistance. This work was supported by the Swiss National Science Foundation, grants 42535.94 to A.W. and T.B. and 3100-052245.97/1 to C.D.V.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL devirgilioc@ubaclu.unibas.ch; FAX 41 61 267 2330
References
- Becher dos Passos J, Vanhalewyn M, Brandão RL, Castro IM, Nicoli JR, Thevelein JM. Glucose-induced activation of plasma membrane H+-ATPase in mutants of the yeast Saccharomyces cerevisiae affected in cAMP metabolism, cAMP-dependent protein phosphorylation and the initiation of glycolysis. Biochim Biophys Acta. 1992;1136:57–67. doi: 10.1016/0167-4889(92)90085-p. [DOI] [PubMed] [Google Scholar]
- Bell W, Klaassen P, Ohnacker M, Boller T, Herweijer M, Schoppink P, van der Zee P, Wiemken A. Characterization of the 56-kDa subunit of yeast trehalose-6-phosphate synthase and cloning of its gene reveal its identity with the product of CIF1, a regulator of carbon catabolite inactivation. Eur J Biochem. 1992;209:951–959. doi: 10.1111/j.1432-1033.1992.tb17368.x. [DOI] [PubMed] [Google Scholar]
- Bi E, Pringle JR. ZDS1 and ZDS2, genes whose products may regulate Cdc42p in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:5264–5275. doi: 10.1128/mcb.16.10.5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorstein WR, Craig EA. Regulation of a yeast HSP70 gene by a cAMP responsive transcriptional control element. EMBO J. 1990;9:2543–2553. doi: 10.1002/j.1460-2075.1990.tb07435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowdish KS, Yuan HE, Mitchell AP. Positive control of yeast meiotic genes by the negative regulator UME6. Mol Cell Biol. 1995;15:2955–2961. doi: 10.1128/mcb.15.6.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broach JR, Deschenes RJ. The function of RAS genes in Saccharomyces cerevisiae. Adv Cancer Res. 1990;54:79–139. doi: 10.1016/s0065-230x(08)60809-x. [DOI] [PubMed] [Google Scholar]
- Cameron S, Levin L, Zoller M, Wigler M. cAMP-independent control of sporulation, glycogen metabolism, and heat shock resistance in S. cerevisiae. Cell. 1988;53:555–566. doi: 10.1016/0092-8674(88)90572-7. [DOI] [PubMed] [Google Scholar]
- Cannon JF, Tatchell K. Characterization of Saccharomyces cerevisiae genes encoding subunits of cyclic AMP-dependent protein kinase. Mol Cell Biol. 1987;7:2653–2663. doi: 10.1128/mcb.7.8.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JR, Johnson TR, Dollard C, Shuster JR, Denis CL. Cyclic AMP-dependent protein kinase phosphorylates and inactivates the yeast transcriptional activator ADR1. Cell. 1989;56:409–419. doi: 10.1016/0092-8674(89)90244-4. [DOI] [PubMed] [Google Scholar]
- De Virgilio C, Bürckert N, Bell W, Jenö P, Boller T, Wiemken A. Disruption of TPS2, the gene encoding the 100-kDa subunit of the trehalose-6-phosphate synthase/phosphatase complex in Saccharomyces cerevisiae, causes accumulation of trehalose-6-phosphate and loss of trehalose-6-phosphate phosphatase activity. Eur J Biochem. 1993;212:315–323. doi: 10.1111/j.1432-1033.1993.tb17664.x. [DOI] [PubMed] [Google Scholar]
- De Virgilio C, Hottiger T, Dominguez J, Boller T, Wiemken A. The role of trehalose synthesis for the acquisition of thermotolerance in yeast. I. Genetic evidence that trehalose is a thermoprotectant. Eur J Biochem. 1994;219:179–186. doi: 10.1111/j.1432-1033.1994.tb19928.x. [DOI] [PubMed] [Google Scholar]
- De Virgilio C, DeMarini DJ, Pringle JR. SPR28, a sixth member of the septin gene family in Saccharomyces cerevisiae that is expressed specifically in sporulating cells. Microbiology. 1996;142:2897–2905. doi: 10.1099/13500872-142-10-2897. [DOI] [PubMed] [Google Scholar]
- Denis CL, Audino DC. The CCR1 (SNF1) and SCH9 protein kinases act independently of cAMP-dependent protein kinase and the transcriptional activator ADR1 in controlling yeast ADH2 expression. Mol & Gen Genet. 1991;229:395–399. doi: 10.1007/BF00267461. [DOI] [PubMed] [Google Scholar]
- Edelman AM, Blumenthal DK, Krebs EG. Protein serine/threonine kinases. Annu Rev Biochem. 1987;56:567–613. doi: 10.1146/annurev.bi.56.070187.003031. [DOI] [PubMed] [Google Scholar]
- Fields S, Sternglanz R. The two-hybrid system: an assay for protein-protein interactions. Trends Genet. 1994;10:286–292. doi: 10.1016/0168-9525(90)90012-u. [DOI] [PubMed] [Google Scholar]
- Foreman PK, Davis RW. Cloning vectors for the synthesis of epitope-tagged, truncated and chimeric proteins in Saccharomyces cerevisiae. Gene. 1994;144:63–68. doi: 10.1016/0378-1119(94)90204-6. [DOI] [PubMed] [Google Scholar]
- François J, Van Schaftingen E, Hers H-G. The mechanism by which glucose increases fructose 2,6-bisphosphate concentration in Saccharomyces cerevisiae. A cyclic-AMP-dependent activation of phosphofructokinase 2. Eur J Biochem. 1984;145:187–193. doi: 10.1111/j.1432-1033.1984.tb08539.x. [DOI] [PubMed] [Google Scholar]
- Garrett S, Broach J. Loss of Ras activity in Saccharomyces cerevisiae is suppressed by disruptions of a new kinase gene, YAK1, whose product may act downstream of the cAMP-dependent protein kinase. Genes & Dev. 1989;3:1336–1348. doi: 10.1101/gad.3.9.1336. [DOI] [PubMed] [Google Scholar]
- Garrett S, Menold MM, Broach JR. The Saccharomyces cerevisiae YAK1 gene encodes a protein kinase that is induced by arrest early in the cell cycle. Mol Cell Biol. 1991;11:4045–4052. doi: 10.1128/mcb.11.8.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JB, Marshall MS. The ras oncogene—an important regulatory element in lower eucaryotic organisms. Microbiol Rev. 1989;53:171–185. doi: 10.1128/mr.53.2.171-185.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz D, St. Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A, Lampen JO. β-d-fructofuranoside fructohydrolase from yeast. Methods Enzymol. 1975;42:504–511. doi: 10.1016/0076-6879(75)42159-0. [DOI] [PubMed] [Google Scholar]
- Guthrie, C. and G.R. Fink. 1991. Guide to yeast genetics and molecular biology. Methods Enzymol. 194. [PubMed]
- Gyuris J, Golemis E, Chertkov H, Brent R. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- Hardy TA, Huang D, Roach PJ. Interactions between cAMP-dependent and SNF1 protein kinases in the control of glycogen accumulation in Saccharomyces cerevisiae. J Biol Chem. 1994;269:27907–27913. [PubMed] [Google Scholar]
- Hartley AD, Ward MP, Garrett S. The Yak1 protein kinase of Saccharomyces cerevisiae moderates thermotolerance and inhibits growth by an Sch9 protein kinase-independent mechanism. Genetics. 1994;136:465–474. doi: 10.1093/genetics/136.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Farkas I, Roach PJ. Pho85p, a cyclin-dependent protein kinase, and the Snf1p protein kinase act antagonistically to control glycogen accumulation in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:4357–4365. doi: 10.1128/mcb.16.8.4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T, Plowman GD. The protein kinases of budding yeast: Six score and more. Trends Biochem Sci. 1997;22:18–22. doi: 10.1016/s0968-0004(96)10068-2. [DOI] [PubMed] [Google Scholar]
- Kataoka T, Powers S, McGill C, Fasano O, Strathern J, Broach J, Wigler M. Genetic analysis of yeast RAS1 and RAS2 genes. Cell. 1984;37:437–445. doi: 10.1016/0092-8674(84)90374-x. [DOI] [PubMed] [Google Scholar]
- Lillie SH, Pringle JR. Reserve carbohydrate metabolism in Saccharomyces cerevisiae: Responses to nutrient limitation. J Bacteriol. 1980;143:1384–1394. doi: 10.1128/jb.143.3.1384-1394.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londesborough J, Vuorio O. Trehalose-6-phosphate synthase/phosphatase complex from baker’s yeast: Purification of a proteolytically activated form. J Gen Microbiol. 1991;137:323–330. doi: 10.1099/00221287-137-2-323. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Uno I, Ishikawa T. Initiation of meiosis in yeast mutants defective in adenylate cyclase and cyclic AMP-dependent protein kinase. Cell. 1983;32:417–423. doi: 10.1016/0092-8674(83)90461-0. [DOI] [PubMed] [Google Scholar]
- Matsuura A, Treinin M, Mitsuzawa H, Kassir Y, Uno I, Simchen G. The adenylate cyclase/protein kinase cascade regulates entry into meiosis in Saccharomyces cerevisiase through the gene IME1. EMBO J. 1990;9:3225–3232. doi: 10.1002/j.1460-2075.1990.tb07521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazón MJ, Gancedo JM, Gancedo C. Inactivation of yeast fructose-1,6-bisphosphatase. In vivo phosphorylation of the enzyme. J Biol Chem. 1982;257:1128–1130. [PubMed] [Google Scholar]
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- Müller D, Holzer H. Regulation of fructose-1,6-bisphosphatase in yeast by phosphorylation/dephosphorylation. Biochem Biophys Res Comm. 1981;103:926–933. doi: 10.1016/0006-291x(81)90899-8. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Naitou M, Hagiwara H, Shibata T, Ozawa M, Sasanuma S, Sasanuma M, Tsuchiya Y, Soeda E, Yokoyama K, Yamazaki M, Tashiro H, Eki T. Analysis of the nucleotide sequence of chromosome VI from Saccharomyces cerevisiae. Nat Genet. 1995;10:261–268. doi: 10.1038/ng0795-261. [DOI] [PubMed] [Google Scholar]
- Panek AC, de Araujo PS, Neto VM, Panek AD. Regulation of the trehalose-6-phosphate synthase complex in Saccharomyces. Curr Genet. 1987;11:459–465. doi: 10.1007/BF00384607. [DOI] [PubMed] [Google Scholar]
- Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- Piper PW. Measurement of transcription. In: Johnston JR, editor. Molecular genetics of yeast. A practical approach. Oxford, UK: IRL Press; 1994. pp. 135–146. [Google Scholar]
- Reinders A, Bürckert N, Hohmann S, Thevelein JM, Boller T, Wiemken A, De Virgilio C. Structural analysis of the subunits of the trehalose-6-phosphate synthase/phosphatase complex in Saccharomyces cerevisiae and their function during heat shock. Mol Microbiol. 1997;24:687–695. doi: 10.1046/j.1365-2958.1997.3861749.x. [DOI] [PubMed] [Google Scholar]
- Ronne H. Glucose repression in fungi. Trends Genet. 1995;11:12–17. doi: 10.1016/s0168-9525(00)88980-5. [DOI] [PubMed] [Google Scholar]
- Rose MD, Winston F, Hieter P. Methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- Rubin-Bejerano I, Mandel S, Robzyk K, Kassir Y. Induction of meiosis in Saccharomyces cerevisiae depends on conversion of the transcriptional repressor Ume6 to a positive regulator by its regulated association with the transcriptional activator Ime1. Mol Cell Biol. 1996;16:2518–2526. doi: 10.1128/mcb.16.5.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M, Sibbald PR, Wittinghofer A. The P-loop—a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- Smith HE, Mitchell AP. A transcriptional cascade governs entry into meiosis in Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:2142–2152. doi: 10.1128/mcb.9.5.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Matsumoto K, Toh-e A. Dual regulation of the expression of the polyubiquitin gene by cyclic AMP and heat shock in yeast. EMBO J. 1988;7:495–502. doi: 10.1002/j.1460-2075.1988.tb02837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatchell K. RAS genes and growth control in Saccharomyces cerevisiae. J Bacteriol. 1986;166:364–367. doi: 10.1128/jb.166.2.364-367.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevelein JM. Signal transduction in yeast. Yeast. 1994;10:1753–1790. doi: 10.1002/yea.320101308. [DOI] [PubMed] [Google Scholar]
- ————— . Regulation of trehalose metabolism and its relevance to cell growth and function. In: Brambl R, Marzluf GA, editors. The Mycota III. Biochemistry and molecular biology. Berlin, Germany: Springer-Verlag; 1996. pp. 395–420. [Google Scholar]
- Thevelein JM, Hohmann S. Trehalose synthase: Guard to the gate of glycolysis in yeast? Trends Biochem Sci. 1995;20:3–10. doi: 10.1016/s0968-0004(00)88938-0. [DOI] [PubMed] [Google Scholar]
- Thompson-Jaeger S, François J, Gaughran JP, Tatchell K. Deletion of SNF1 affects the nutrient response of yeast and resembles mutations which activate the adenylate cyclase pathway. Genetics. 1991;129:697–706. doi: 10.1093/genetics/129.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, Uno I, Ishikawa T, Powers S, Kataoka T, Brock D, Cameron S, Broach J, Matsumoto K, Wigler M. In yeast, RAS proteins are controlling elements of adenylate cyclase. Cell. 1985;40:27–36. doi: 10.1016/0092-8674(85)90305-8. [DOI] [PubMed] [Google Scholar]
- Toda T, Cameron S, Sass P, Zoller M, Scott JD, McMullen B, Hurwitz M, Krebs EG, Wigler M. Cloning and characterization of BCY1, a locus encoding a regulatory subunit of the cyclic AMP dependent protein kinase in Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:1371–1377. doi: 10.1128/mcb.7.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, Cameron S, Sass P, Wigler M. SCH9, a gene of Saccharomyces cerevisiae that encodes a protein distinct from, but functionally and structurally related to, cAMP-dependent protein kinase catalytic subunits. Genes & Dev. 1988;2:517–527. doi: 10.1101/gad.2.5.517. [DOI] [PubMed] [Google Scholar]
- Vandercammen A, François J, Hers H-G. Characterization of trehalose-6-phosphate synthase and trehalose-6-phosphate phosphatase of Saccharomyces cerevisiae. Eur J Biochem. 1989;182:613–620. doi: 10.1111/j.1432-1033.1989.tb14870.x. [DOI] [PubMed] [Google Scholar]
- Varela JCS, Praekelt UM, Meacock PA, Planta RJ, Mager WH. The Saccharomyces cerevisiae HSP12 gene is activated by the high-osmolarity glycerol pathway and negatively regulated by protein kinase A. Mol Cell Biol. 1995;15:6232–6245. doi: 10.1128/mcb.15.11.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Soler J, Argüelles JC, Gacto M. Proteolytic activation of α,α-trehalose-6-phosphate synthase from Candida utilis. Microbiol Lett. 1991;82:157–162. doi: 10.1016/0378-1097(91)90326-6. [DOI] [PubMed] [Google Scholar]
- Vidan S, Mitchell AP. Stimulation of yeast meiotic gene expression by the glucose-repressible protein kinase Rim15p. Mol Cell Biol. 1997;17:2688–2697. doi: 10.1128/mcb.17.5.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorio OE, Kalkkinen N, Londesborough J. Cloning of two related genes encoding the 56-kDa and 123-kDa subunits of trehalose synthase from the yeast Saccharomyces cerevisiae. Eur J Biochem. 1993;216:849–861. doi: 10.1111/j.1432-1033.1993.tb18207.x. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pöhlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Werner-Washburne M, Becker J, Kosic-Smithers J, Craig EA. Yeast Hsp70 RNA levels vary in response to the physiological status of the cell. J Bacteriol. 1989;171:2680–2688. doi: 10.1128/jb.171.5.2680-2688.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner-Washburne M, Braun E, Johnston GC, Singer RA. Stationary phase in the yeast Saccharomyces cerevisiae. Microbiol Rev. 1993;57:383–401. doi: 10.1128/mr.57.2.383-401.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Munday MR, Scott J, Yang X, Carlson M, Carling D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J Biol Chem. 1994;269:19509–19515. [PubMed] [Google Scholar]
- Zervos AS, Gyuris J, Brent R. Mxi1, a protein that specifically interacts with Max to bind Myc-Max recognition sites. Cell. 1993;72:223–232. doi: 10.1016/0092-8674(93)90662-a. [DOI] [PubMed] [Google Scholar]