

Abstract
The mitogen-activated protein kinase (MAPK) Kss1 has a dual role in regulating filamentous (invasive) growth of the yeast Saccharomyces cerevisiae. The stimulatory function of Kss1 requires both its catalytic activity and its activation by the MAPK/ERK kinase (MEK) Ste7; in contrast, the inhibitory function of Kss1 requires neither. This study examines the mechanism by which Kss1 inhibits invasive growth, and how Ste7 action overcomes this inhibition. We found that unphosphorylated Kss1 binds directly to the transcription factor Ste12, that this binding is necessary for Kss1-mediated repression of Ste12, and that Ste7-mediated phosphorylation of Kss1 weakens Kss1–Ste12 interaction and relieves Kss1-mediated repression. Relative to Kss1, the MAPK Fus3 binds less strongly to Ste12 and is correspondingly a weaker inhibitor of invasive growth. Analysis of Kss1 mutants indicated that the activation loop of Kss1 controls binding to Ste12. Potent repression of a transcription factor by its physical interaction with the unactivated isoform of a protein kinase, and relief of this repression by activation of the kinase, is a novel mechanism for signal-dependent regulation of gene expression.
Keywords: Protein–protein interaction, signal transduction, gene regulation, developmental control, invasive growth
Commitment of a cell to a developmental fate entails a change in the pattern of gene expression. In many instances, this gene regulation is controlled by signal-transduction pathways that affect the phosphorylation of transcription factors (or their associated proteins) by protein kinases (for review, see Karin 1994; Hill and Treisman 1995; Treisman 1996). Phosphorylation can control the localization or sequestration of many transcription factors, as shown for yeast Swi5, Drosophila dorsal and yan, SV40 large T antigen, and vertebrate NF-κB, NFAT, STATs, and SMADs (for review, see Hill and Treisman 1995; Vandromme et al. 1996; Heldin et al. 1997). Phosphorylation by protein kinases can also influence the DNA-binding activity of transcription factors. For example, DNA-binding of Elk-1 is stimulated by the Erk and Jnk mitogen-activated protein kinases (MAPKs), whereas DNA-binding of c-Jun is inhibited by CKII (for review, see Treisman 1996). The transactivation potential of transcription factors can also be regulated independently of DNA binding. For example, protein kinase A (PKA)-mediated phosphorylation of CREB, as well as Jnk-mediated phosphorylation of c-Jun, stimulates the binding of the coactivators CREB-binding protein (CBP) and P300 (for review, see Goldman et al. 1997), and Cdk-mediated phosphorylation of Rb leads to dissociation of this repressor from E2F (for review, see DePinho 1998). Finally, protein kinases can regulate transcription factor stability (Clevers and van de Wetering 1997). In all of these cases, the catalytic (phosphotransferase) activity of the protein kinase involved is an indispensable component of its regulatory action.
Recent studies of invasive growth in the yeast Saccharomyces cerevisiae have revealed an unexpected and previously unknown mode of protein kinase-mediated regulation. In this developmental response, the principal regulatory role of the MAPK Kss1 is as an inhibitor, and this function does not require its catalytic activity (Cook et al. 1997; Madhani et al. 1997). Kss1 was first characterized as a component of the signaling cascade required for response to mating pheromone (for review, see Bardwell et al. 1994b; Leberer et al. 1997). In this pathway, pheromone-receptor binding leads to activation of a MAPK module. Ste11 (a MAPK kinase kinase or MEKK) phosphorylates and activates Ste7 (a MAPK kinase or MEK); Ste7, in turn, phosphorylates and activates two MAPKs, Kss1 and Fus3 (Errede et al. 1993; Neiman and Herskowitz 1994; Bardwell et al. 1996). Kss1 and Fus3 then stimulate, directly or indirectly, the transcription factor, Ste12, permitting expression of a battery of genes involved in the mating process (Erdman et al. 1998). Deletion (or mutation that destroys the catalytic activity) of any tier of this MAPK cascade—STE11, STE7, or both KSS1 and FUS3—abolishes pheromone-induced transcription and results in sterility (Rhodes et al. 1990; Elion et al. 1991; Cairns et al. 1992; Ma et al. 1995).
Invasive (filamentous) growth is a distinct developmental option available to both haploids and diploids. Cells undergoing this process become elongated; alter their cell cycle, budding pattern, and adhesiveness; and acquire the capacity to penetrate beneath the surface of an agar plate (Gimeno et al. 1992; Kron et al. 1994; Roberts and Fink 1994). Invasive growth is regulated by a signaling network containing at least two parallel branches (Cook et al. 1997; Lo et al. 1997; Chandarlapaty and Errede 1998). It has been shown (Liu et al. 1993; Roberts and Fink 1994) that one branch utilizes proteins that are also components of the pheromone response pathway, including the protein kinases, Ste20 [a p21-activated kinase (PAK) homolog], Ste11, Ste7, and the Ste12 transcription factor. Ste12 cooperates with the Tec1 transcription factor to regulate genes specific for invasive growth (Laloux et al. 1994; Gavrias et al. 1996; Baur et al. 1997; Madhani and Fink 1997). Upstream components of the invasive growth signaling network, including Mep2 (an ammonium permease; Lorenz and Heitman 1998), Gpa2 (a Gα subunit; Kübler et al. 1997), Bmh1 and Bmh2 (14-3-3 proteins; Roberts et al. 1997), and Ras2 and Cdc42 (two small GTPases; Gimeno et al. 1992; Mösch et al. 1996), are coupled to downstream branches by mechanisms that are not completely understood.
Until recently, the MAPK target of Ste7 in invasive growth was unknown. Because deletion of either KSS1 or FUS3 (or both) only moderately affected invasive growth, whereas deletion of STE7 caused a substantial reduction in invasiveness (Liu et al. 1993; Roberts and Fink 1994), it was proposed that Ste7 has substrates other than Kss1 and Fus3, perhaps even non-MAPK targets (for review, see Hunter and Plowman 1997). This conundrum was resolved by experiments showing that invasive growth could be restored to a strain lacking STE7 by also deleting KSS1 (Cook et al. 1997; Madhani et al. 1997). This result established that Kss1 acts to inhibit invasive growth, and suggested that Ste7 functions, in response to appropriate upstream signals, to alleviate Kss1-mediated inhibition. Fus3 also inhibits invasive growth, but is less potent, and only functions in haploid cells (Cook et al. 1997). Kss1 also has a stimulatory role in invasive growth that (in contrast to its inhibitory role) requires its protein kinase activity (Cook et al. 1997; Madhani et al. 1997).
Here, we demonstrate that Kss1-mediated inhibition of invasive growth operates at the level of transcription, characterize the mechanism of this transcriptional repression, and examine its regulation by upstream signals.
Results
Ste7-mediated phosphorylation is necessary and sufficient to relieve Kss1-mediated inhibition of invasive growth
Deletion of STE7 substantially reduces invasive growth (Liu et al. 1993; Roberts and Fink 1994). We found that neither an unactivatable ste7 allele (T363V; Neiman and Herskowitz 1994) nor a catalytically inactive allele (K220R; Cairns et al. 1992) could restore invasive growth to ste7Δ cells (data not shown). Thus, both phosphorylation and activation of Ste7 (presumably by Ste11) and the subsequent phosphorylation of downstream substrates by Ste7 are required for invasive growth. Ste7 is required for invasive growth only if Kss1 is also present, implicating Kss1 as the principal target of Ste7 in this process (Cook et al. 1997; Madhani et al. 1997). To examine how Kss1-mediated inhibition of invasive growth is relieved by Ste7 function, a series of kss1 alleles (Fig. 1A, a) was introduced, on low-copy (CEN) vectors, into a derivative of Σ1278b lacking both Kss1 and Fus3, strain JCY130 (MATa STE7+ kss1Δ fus3Δ). The biochemical and genetic properties of these Kss1 mutants with respect to pheromone response have been extensively characterized (Ma et al. 1995; Bardwell et al. 1996). Two of these mutants, Kss1Y24F and Kss1K42R Q45P, contain substitutions in conserved subdomains involved in ATP binding and are catalytically inactive, but are phosphorylated by Ste7 to the same extent as wild-type Kss1 (Ma et al. 1995; Bardwell et al. 1996; see also Fig. 1C). The other three mutants are altered in the target site (183-Thr-Glu-Tyr-185) for Ste7-mediated phosphorylation: Kss1T183A, which can be phosphorylated only on Y185; Kss1Y185F, which can be phosphorylated only on T183; and Kss1AEF (containing both substitutions), which cannot be phosphorylated by Ste7 (Ma et al. 1995). These three mutants are unactivatable, and consequently lack detectable protein kinase activity (Ma et al. 1995; Bardwell et al. 1996). Nevertheless, both classes of inactive mutants repress invasive growth when Ste7 is absent (Cook et al. 1997).
Figure 1.
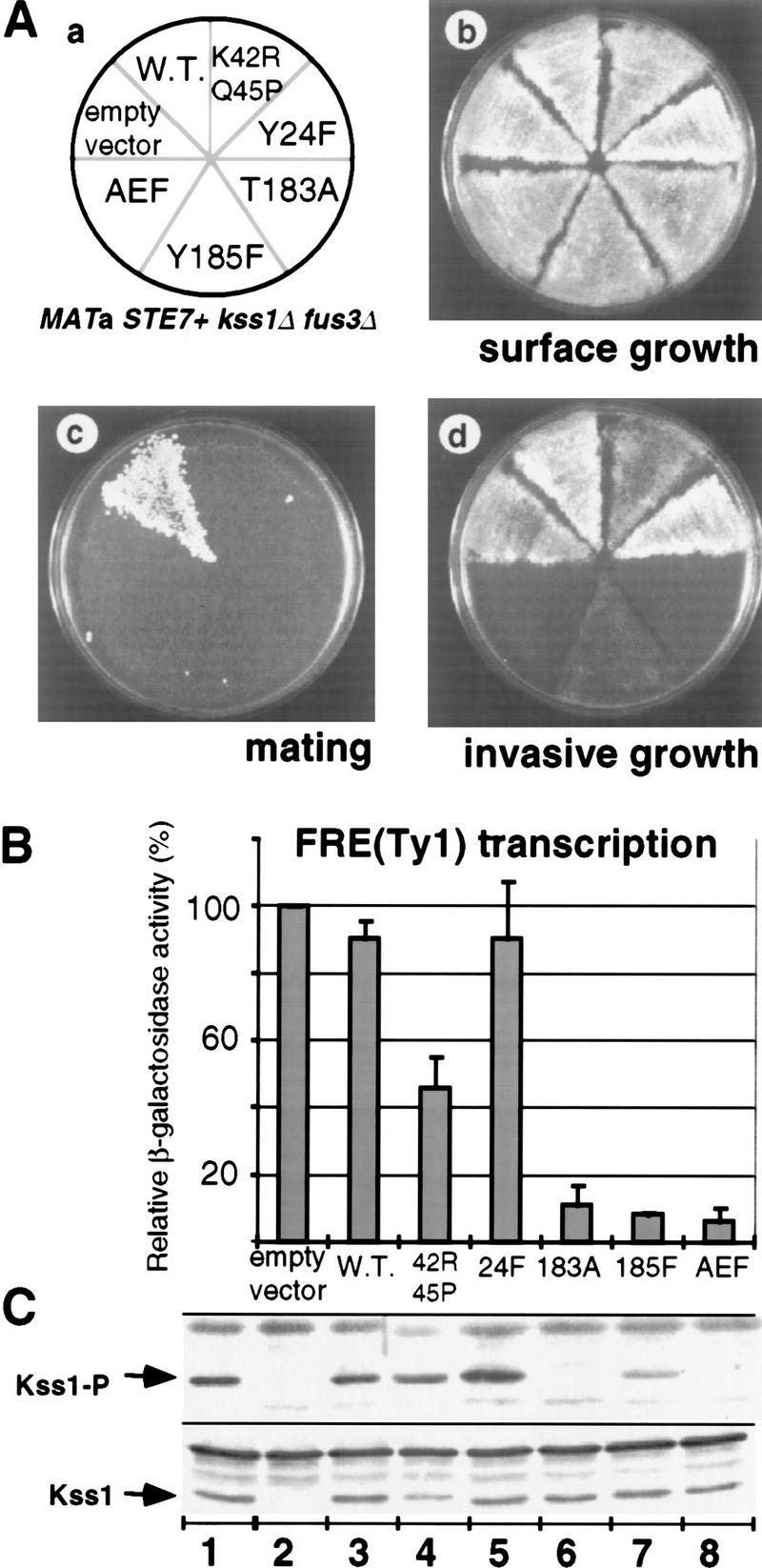
Phosphorylation of Kss1 is necessary and sufficient to permit haploid invasive growth. (A) Strain JCY130 (MATa STE7+ kss1Δ fus3Δ) was transformed with centromeric (low-copy) plasmids YCpU (empty vector), YCpU–KSS1 (W.T.), YCpU–kss1(K42R Q45P), YCpU–kss1(Y24F), YCpU–kss1(T183A), YCpU–kss1(Y185F), or YCpU–kss1(AEF) (a). The resulting transformants were streaked onto a plate selective for plasmid maintenance. After 2 days at 30°C, plates were replica-plated onto rich medium (YPD) plates and scored for surface growth (b), mating to strain DC17 (MATα his1) (c), or invasive growth after 30 hr at 30°C (d). (B) Expression of the FRETy1–lacZ reporter gene. The strains described in A were transformed with plasmid YEpL–FTyZ, grown to mid-log phase in liquid medium, and β-galactosidase-specific activity was measured. Values are normalized to that observed for JCY130 carrying YCpU (3936 nmoles/min per milligram of protein) and represent the average of measurements, made in duplicate, on protein extracts prepared from at least three independent transformants of each strain (error bars indicate standard deviation). (C) Portions (20 μg) of the protein extracts used in B, plus an additional extract (lane 1) prepared from strain JCY100 (MATa KSS1+), were resolved on a 10% SDS–polyacrylamide gel and analyzed by immunoblotting with either anti-Kss1 (bottom) or anti-phospho–MAPK (top) antiserum.
Strain JCY130 expressing each mutant, normal Kss1, or no Kss1 at all, grew equally vigorously (Fig. 1A, b). Only wild-type Kss1 supported detectable mating (Fig. 1A, c), as observed previously in kss1Δ fus3Δ strains of the S288C and W303 lineages (Elion et al. 1991; Ma et al. 1995). In the standard bioassay for invasive growth (Fig. 1A, d), neither the absence of Kss1 nor the presence of wild-type Kss1 prevented invasive growth. In contrast, none of the three unactivatable mutants (Kss1T183A, Kss1Y185F, and Kss1AEF) permitted normal invasive growth (Fig. 1A, d). This reduced level of invasiveness was quite comparable with that of an otherwise isogenic ste7Δ KSS1+ strain (data not shown). Thus, phosphorylation of Kss1 on both T183 and Y185 is required to relieve Kss1-mediated inhibition of invasive growth. Revealingly, both of the catalytically inactive (yet phosphorylatable) mutants (Kss1Y24F and Kss1K42R Q45P) permitted haploid invasive growth (Fig. 1A, d). Hence, phosphorylation of Kss1 by Ste7 suffices to relieve inhibition, even in the absence of the resultant activity of this MAPK. When Fus3 is present, however, there is a stronger requirement for the protein kinase activity of Kss1 (Cook et al. 1997).
Transcription from filamentation response elements (FREs), which contain binding sites for Ste12 and Tec1 (Baur et al. 1997; Madhani and Fink 1997), correlates with MAPK cascade signaling in the invasive growth response (Mösch et al. 1996; Madhani and Fink 1997) and reflects the degree of MAPK-mediated inhibition of invasive growth (Cook et al. 1997). Quantitative measurement of the expression of an FRE–lacZ reporter gene confirmed the results obtained by use of the bioassay. The three unactivatable alleles permitted only minimal FRE-dependent transcription, whereas the two catalytically inactive alleles allowed near wild-type levels (Fig. 1B). Immunoblot analysis of protein extracts from these strains (Fig. 1C, bottom) revealed that each Kss1 mutant (Fig. 1C, lanes 3–8) was expressed comparably with endogenous Kss1 (Fig. 1C, lane 1). Staining of these same extracts with an antibody that preferentially recognizes the MEK-phosphorylated isoforms of MAPKs of the ERK (extracellular signal-regulated kinase) family, including Kss1 (see Materials and Methods), confirmed phosphorylation of wild-type Kss1 and the catalytically inactive derivatives, and the lack (or reduction) of phosphorylation on the unactivatable Kss1 mutants (Fig. 1C, top).
Kss1 binds directly to Ste12
Because inhibition of invasive growth by unphosphorylated Kss1 is accompanied by repression of FRE-mediated transcription (Fig. 1B), and because Kss1 is concentrated in the nucleus (Ma et al. 1995), Kss1 has an opportunity to associate physically with and inhibit Ste12 and/or Tec1. Indeed, Kss1 interacts with Ste12 in the two-hybrid assay (Printen and Sprague 1994; Cook et al. 1996). Moreover, Kss1- and Ste12-containing complexes (Madhani et al. 1997; L. Bardwell, unpubl.), and Fus3- and Ste12-containing complexes (Elion et al. 1993; Tedford et al. 1997), have been recovered from cell extracts. However, neither of these assays demonstrates that the interaction between a MAPK and Ste12 is direct. It has been proposed (Tedford et al. 1997) that the Dig1/Rst1 and Dig2/Rst2 proteins, which bind directly to both Ste12 and Kss1 (Cook et al. 1996), may bridge the interaction between the MAPKs and Ste12. To determine whether Kss1 can bind directly to full-length Ste12 in the absence of any other yeast protein, radiolabeled versions of these proteins were prepared by in vitro translation in rabbit reticulocyte lysates, and their interactions were assessed by coimmunoprecipitation (Fig. 2). For this purpose, Ste12 was fused to an amino-terminal c-Myc epitope tag (see Material and Methods for details). Kss1 and Ste12 were efficiently produced in radiolabeled form under the conditions used (Fig. 2, Input). As expected, tagged Ste12 was immunoprecipitated by the monoclonal antibody recognizing the c-Myc epitope (Fig. 2, lane 8). When Kss1 and Ste12 were present in the same reaction, Kss1 was coimmunoprecipitated (Fig. 2, lane 5) at a level reproducibly three- to fivefold above both the nonspecific association of Kss1 with the antibody (Fig. 2, lane 4) and the background displayed by Kss1Y231C, a mutant specifically defective in Ste12 binding (described in greater detail below) (Fig. 2, lanes 6,7). When determined as described (Bardwell et al. 1996), the Kd of the Ste12–Kss1 interaction was ∼400 nm. For comparison, the Ste7–Kss1 interaction measured by the same methodology had a Kd of ∼5 nm (Bardwell et al. 1996).
Figure 2.

Direct binding of Kss1 to Ste12 in vitro. Radiolabeled Kss1, Kss1Y231C, and myc epitope-tagged Ste12 (meSte12) were prepared by in vitro translation, partially purified by ammonium sulfate precipitation, and portions [for Kss1 and Kss1Y231C, 5% of the amount added in the immunoprecipitation (pptn) reactions, and for meSte12, 20%; Input] were resolved on a 10% SDS–polyacrylamide gel. Samples (2 pmoles) of Kss1 and Kss1Y231C, each accompanied by ∼200 μg of total protein from the rabbit reticulocyte lysate, were immunoprecipitated with the anti-c-Myc mAb 9E10 either in the absence (lanes 4,6) or presence (lanes 5,7,8) of 2 pmoles meSte12 protein, and the resulting immunoprecipitates were analyzed on the same gel. The percentage of the input Kss1 derivatives bound in the reactions corresponding to lanes 4–7 was, respectively, 0.6, 1.7, 0.5, and 0.7; and of meSte12 in lanes 5,7,8: 37.2, 30.7, and 35.1.
To delineate the Kss1-binding domain of the 688-residue Ste12 protein, we used Kss1 as bait to screen a random library and recovered seven different Ste12 isolates, including amino- and/or carboxy-terminal truncations (Cook et al. 1996). These isolates shared a common segment (residues 298–473). This portion of Ste12 lacks both the amino-terminal DNA-binding domain and the carboxy-terminal element that mediates cooperativity with the Mcm1 transcription factor, yet contains part of the transactivating region and a segment implicated in pheromone inducibility (Kirkman-Correia et al. 1993). We verified that this region of Ste12 is sufficient for MAPK association by constructing and purifying a GST–Ste12298–473 fusion protein, and demonstrating that this fusion (but not GST alone) specifically retains Kss1 (see below; Figs. 4B and 6).
Figure 4.
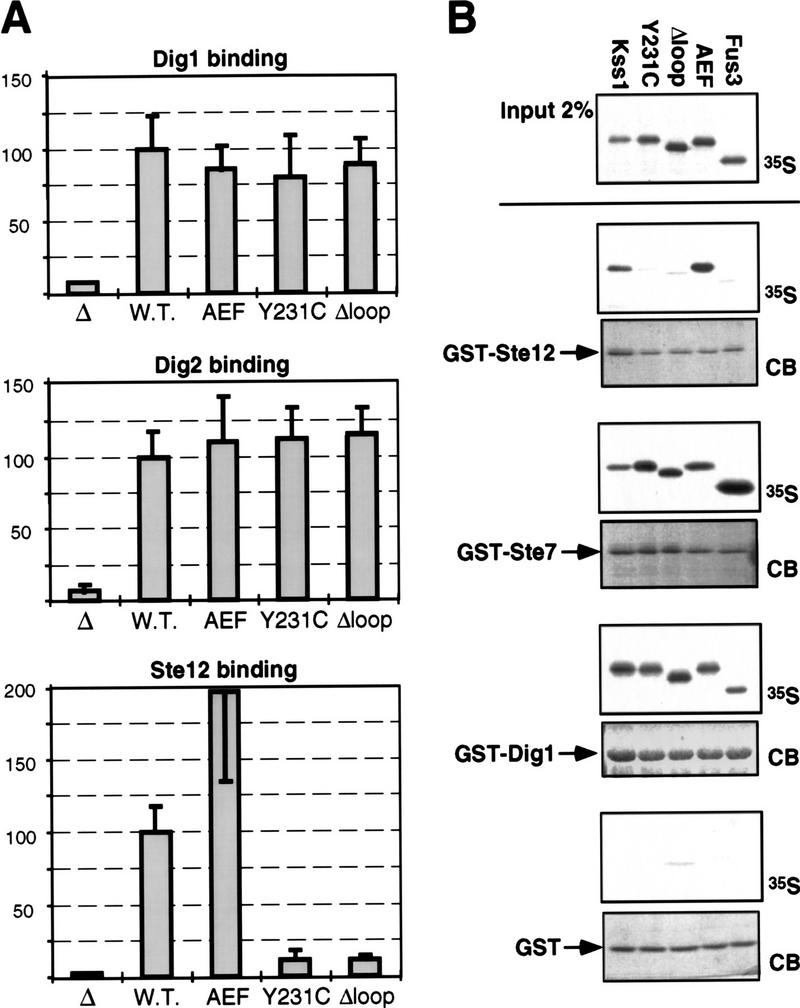
Kss1Δloop and Kss1Y231C are specifically defective in binding to Ste12. (A) Two-hybrid interaction of alleles of KSS1 with DIG1, DIG2, and STE12. Strain MaV103a was cotransformed with pKSS1–GDB (W.T.), pkss1(AEF)–GDB (encoding the T183A, Y185F derivative), pkss1(Y231C)–GDB, pkss1(Δloop)-GDB, or the plasmid encoding the GDB domain only (Δ); and with pGAD–DIG1(7–452), pGAD–DIG2(56–323), or pGAD–STE12(191–478); and β-galactosidase-specific activity was measured as described in the legend to Fig. 1B. Values are normalized to that observed for the interaction of wild-type KSS1 with DIG1, DIG2, or STE12 (41, 49 and 52 nmoles/min per milligram of protein, respectively). None of the KSS1–GDB alleles displayed an appreciable two-hybrid interaction with an empty GAD plasmid (data not shown). (B) In vitro binding of Kss1, Kss1 mutants, and Fus3 to Ste12, Ste7, and Dig1. Radiolabeled (35S) Kss1, Kss1Y231C, Kss1Δloop, Kss1AEF, and Fus3 proteins were prepared by in vitro translation, partially purified by ammonium sulfate precipitation, and portions (2% of the amount added in the binding reactions; Input) were resolved on a 10% SDS-polyacrylamide gel. Samples (1 pmole) of the same proteins were incubated with ∼2 μg of GST–Ste12298–473, GST–Ste71–172, GST–Dig1213–452, or GST bound to glutathione–Sepharose beads, and the resulting bead-bound protein complexes were isolated, resolved by 10% SDS-PAGE, and analyzed by staining with Coomassie blue (CB) for visualization of the bound GST fusion protein, and by autoradiography for visualization of the bound radiolabeled (35S) protein. GST, GST–Ste71–172, and GST–Dig1213–452 were purified from E. coli; GST–Ste12298–473 was purified from yeast (see Materials and Methods for details). The percentage of input Kss1, Kss1Y231C, Kss1Δloop, Kss1AEF, and Fus3 proteins bound to GST–Ste12 was, respectively: 1.6, 0.1, 0.1, 2.2, and 0.1; to GST–Ste7: 8.3, 13.6, 4.4, 6.1, and 30.4; to GST–Dig1: 9.1, 7.8, 7.4, 7.2, and 5.1; and to GST: 0.1 for all five proteins.
Figure 6.

Phosphorylation of Kss1 reduces its affinity for Ste12. Cultures of strain JCY100 (W.T.) or JCY107 (ste7Δ), expressing Kss1 from a multicopy plasmid (YEpT–KSS1), were grown, treated (+), or not (−) with α-factor (αF) mating pheromone as indicated, and harvested. Cell extracts were prepared, and portions (5% of the amount added in the binding reactions; input) were resolved on a 10% SDS–polyacrylamide gel. Samples (1 mg) of the same extracts were incubated with ∼2 μg of GST–Ste12298–473, GST–Ste71–172, or GST bound to glutathione–Sepharose beads, and the resulting bead-bound protein complexes were isolated, resolved on the same gel, and analyzed by immunoblot analysis with anti-phospho–MAPK antiserum (middle) followed by stripping and reprobing with anti-Kss1 (top) and anti-GST (bottom) antiserum.
Kss1 mutants specifically defective in Ste12 binding
To provide a critical in vivo test of the importance of Kss1 association with Ste12 in the mechanism of Kss1-mediated inhibition of invasive growth, we sought to generate Kss1 derivatives specifically defective in binding to Ste12. Two such mutants were identified. The first derivative, Kss1Δloop (Fig. 3), was constructed by site-directed mutagenesis (Bardwell et al. 1996). In Kss1Δloop, a sequence designated the activation loop—also called the activation segment (Johnson et al. 1996), phosphorylation lip (Cobb and Goldsmith 1995), or T-loop (Morgan and De Bondt 1994)—has been deleted and replaced with a shorter segment from an unrelated protein kinase (yeast Tpk3). The activation loop lies along the bottom of the active site cleft in the crystal structure of unphosphorylated Erk2 (Zhang et al. 1994, 1995), and contains the target phosphoacceptor residues for MEK-mediated phosphorylation (Fig. 3). In Erk2, this loop refolds upon phosphorylation, leading to activation of the enzyme (Canagarajah et al. 1997).
Figure 3.

Structure and properties of the Kss1Δloop and Kss1Y231C derivatives. Amino acid sequence of Kss1 and homology to yeast Fus3 and rat Erk2 in the affected regions. Dashes (−) indicate single-residue gaps; identities are boxed. In Kss1Δloop protein, 21 residues of Kss1 (indicated by brackets) have been replaced with the unrelated 11-residue activation segment from S. cerevisiae Tpk3 (GenBank accession no. M17074), a homolog of the catalytic subunit of mammalian cyclic AMP-dependent protein kinase. Y231 is indicated by a carat (^). The target phosphoacceptor residues for MEK-mediated phosphorylation are indicated by asterisks (*). A hydrogen bond between Y231 and E184 in Erk2 is indicated by colons (:).
We isolated the second Kss1 mutant using a reverse two-hybrid screen (see Materials and Methods for details). This derivative, Kss1Y231C, contains a single-residue substitution (to cysteine) at a conserved tyrosine that, in unphosphorylated Erk2 (Zhang et al. 1995), forms a hydrogen bond with the glutamate in the TEY phosphoacceptor motif (Fig. 3). Hence, alteration of this residue in Kss1Y231C may disrupt this hydrogen bond (and/or cause other perturbations resulting from the chemical differences between tyrosine and cysteine), thereby affecting the conformation of the activation loop region. Consistent with this conclusion, when tested in an in vitro protein kinase assay (Bardwell et al. 1996), Kss1Y231C was a much less efficient phosphoacceptor substrate for Ste7 than Kss1 (data not shown).
The fact that the mutations in Kss1Δloop and Kss1Y231C specifically affect the ability of Kss1 to interact with Ste12 was demonstrated in several ways. First, Kss1, Kss1AEF, Kss1Δloop, and Kss1Y231C were expressed as Gal4 DNA-binding domain (GDB) fusions and tested for interaction with either Ste12, Dig1, or Dig2 expressed as Gal4 activation domain (GAD) fusions by use of the two-hybrid method in a yeast strain containing a GAL promoter-driven reporter gene (Escherichia coli lacZ). Interaction was measured by determination of β-galactosidase-specific activity (Fig. 4A). The Kss1AEF, Kss1Δloop, and Kss1Y231C fusions exhibited near wild-type two-hybrid interactions with Dig1 and Dig2 fusions. In contrast, the Kss1Δloop and Kss1Y231C fusions showed a severely reduced two-hybrid interaction (∼8% of wild type) with the Ste12 fusion, whereas the Kss1AEF fusion reproducibly displayed an enhanced interaction relative to wild type.
To confirm these binding properties, Kss1, Kss1Y231C, Kss1Δloop, Kss1AEF, as well as Fus3, were radiolabeled by translation in vitro in rabbit reticulocyte lysate, and tested for binding to purified GST–Ste12298–473, GST–Ste71–172, GST–Dig1213–452, or GST alone. Kss1 and Kss1AEF bound specifically to GST–Ste12 (but not to GST), whereas Kss1Y231C and Kss1Δloop did not bind to GST–Ste12 at a level above background binding to GST (Fig. 4B). In contrast, all four of the Kss1 derivatives bound with approximately equal efficiency to both GST–Ste7 and GST–Dig1 at a level well above nonspecific binding to GST (Fig. 4B). Fus3 did not bind detectably to purified GST–Ste12298–473 in this assay, although it bound with about the same efficiency as Kss1 to GST–Dig1213-452, and bound with greater efficiency than Kss1 to GST–Ste71–172 (Fig. 4B). Compared with Kss1, Fus3 also exhibited a weaker association with Ste12 in yeast cell extracts (data not shown).
Finally, as judged by coimmunoprecipitation, radiolabeled Kss1Y231C (Fig. 2) and Kss1Δloop (data not shown) were unable to interact detectably with full-length, epitope-tagged and radiolabeled Ste12.
Binding of Kss1 to Ste12 is required for Kss1 to inhibit Ste12
Having demonstrated that Kss1Δloop and Kss1Y231C are specifically defective in binding to Ste12 (but fully able to interact with Ste7, Dig1, and Dig2) allowed us to assess whether direct association between Kss1 and Ste12 is involved in Kss1-mediated inhibition of invasive growth. For this purpose, alleles of KSS1 (Fig. 5A, a) carried on low copy (CEN) plasmids were introduced into a haploid strain JCY137 (ste7Δ kss1Δ fus3Δ). In this strain, even normal Kss1 remains unphosphorylated as a result of the absence of Ste7. Hence, wild-type, catalytically inactive and unactivatable Kss1 derivatives all behaved as potent inhibitors of invasive growth (Fig. 5A, c) and of FRE-mediated transcription (Fig. 5B). In marked contrast, both Kss1Δloop and Kss1Y231C permitted a substantial degree of invasive growth (Fig. 5A, c) and a measurably elevated level of FRE-mediated transcription (Fig. 5B). All of these Kss1 derivatives were produced at about the same level as endogenous Kss1 (Fig. 5C).
Figure 5.

Ste12 binding contributes to Kss1-mediated inhibition of invasive growth. (A) Strain JCY137 (MATa ste7Δ kss1Δ fus3Δ) was transformed with plasmid YCpU (empty vector), YCpU–KSS1 (W.T.), YCpU–kss1(Y24F), YCpU–kss1(T183A), YCpU–kss1(Y185F), YCpU–kss1(AEF), YCpU–kss1(Δloop), or YCpU–kss1(Y231C) (a). The resulting transformants were streaked onto a plate selective for plasmid maintenance. After 2 days at 30°C, plates were replica-plated onto rich medium (YPD) plates and scored for surface growth (b), or invasive growth after 30 hr at 30°C (c). (B) Expression of the FRETy1–lacZ reporter gene. The strains described in A were transformed with plasmid YEpL–FTyZ, grown to mid-exponential phase in liquid medium, and β-galactosidase specific activity was measured as detailed in the legend to Fig. 1. Values are normalized to that observed for JCY137 carrying YCpU (3450 nmoles/min per milligram of protein). (C) Portions (20 μg) of the protein extracts used in B, plus an additional extract (lane 1) prepared from strain JCY100 (MATa KSS1+), were resolved on a 10% SDS-polyacrylamide gel and analyzed by immunoblotting with anti-Kss1 antiserum.
When introduced into a ste7Δ/ste7Δ kss1Δ/kss1Δ diploid strain, wild-type KSS1, as well as the catalytically inactive (Y24F) and unactivatable (AEF) alleles potently inhibited formation of filaments (pseudohyphae), whereas the Y231C and Δloop alleles permitted at least some filamentation (data not shown). These findings suggest that Kss1 binding to Ste12 mediates Kss1-imposed inhibition of invasive growth in diploids, as in haploids. However, in diploids expressing Kss1Y24F, reintroduction of Ste7 only weakly stimulated filamentation (data not shown). Because Kss1Y24F is phosphorylatable, but nonfunctional (Ma et al. 1995), this result suggests that Kss1 catalytic activity is required for efficient filamentation. Nonetheless, vigorous filament formation is observed in diploids when Kss1 is absent, even if Ste7 is also absent (Cook et al. 1997). Apparently, therefore, the catalytic activity of phosphorylated Kss1 is required in diploids to overcome the inhibitory function of those Kss1 molecules that remain unphosphorylated.
Phosphorylation of Kss1 reduces its affinity for Ste12
The preceding results provide strong support for the conclusion that direct physical interaction between Kss1 and Ste12 is required for Kss1-mediated inhibition of invasive growth. Moreover, the findings presented above establish that Ste7 relieves this inhibition via phosphorylation of Kss1. These observations predict that phosphorylation of Kss1 decreases its affinity for Ste12. To test this prediction, the ability of phosphorylated and unphosphorylated Kss1 to bind in vitro to Ste12 was examined.
To obtain extracts containing roughly equivalent amounts of total Kss1, but different amounts of phosphorylated Kss1, lysates were prepared from three different Σ1278b-derived strains: wild-type cells overproducing Kss1 that contain a readily detectable level of phosphorylated Kss1 (Fig. 6, lane 1); the same cells treated with α-factor mating pheromone that contain an increased level of phosphorylated Kss1 (Fig. 6, lane 2); and, an otherwise isogenic ste7Δ strain overexpressing Kss1 from the same plasmid that contains no detectable phosphorylated Kss1 (Fig. 6, lane 3). Because Kss1 is overproduced, the bulk of the Kss1 pool remains unphosphorylated, even in pheromone-treated cells (L. Bardwell and J.G. Cook, unpubl.). Total and phosphorylated Kss1 were detected as in Figure 1, quantitated by immunoblotting of serial dilutions of the samples, and normalized to the input level of Kss1 (which was used as the standard).
As predicted, phosphorylated Kss1 clearly bound less well to GST–Ste12 than the bulk unphosphorylated Kss1 (Fig. 6, lanes 7–9). The amount of unphosphorylated Kss1 bound was ∼25% of input, whereas only 1%–2% of the input phosphorylated Kss1 bound. The >10-fold difference in relative retention between phosphorylated and unphosphorylated Kss1 indicates that the phosphorylated form of Kss1 must have a correspondingly weaker affinity for Ste12. This effect was specific to the Kss1–Ste12 interaction because phosphorylated and unphosphorylated Kss1 associated with GST–Ste7 at the same efficiency (∼25% of input) (Fig. 6, lanes 10–12), consistent with our previous finding that Kss1–Ste7 interaction is unaffected by pheromone treatment (Bardwell et al. 1996). As expected, neither phosphorylated nor unphosphorylated Kss1 bound to GST alone (Fig. 6, lanes 4–6).
Discussion
Modulation of transcription factor function by protein kinase-mediated phosphorylation is a ubiquitous regulatory strategy. Phosphorylation-independent modes of protein kinase-mediated regulation are feasible because protein kinases often form relatively stable complexes with their substrates.
This investigation addressed two main mechanistic questions. First, how does unactivated Kss1 MAPK inhibit invasive growth? Second, how do upstream signals relieve this inhibition? We found that Kss1 binds directly to the Ste12 transcription factor and thereby represses its function (Fig. 7A). Phosphorylation of the activation loop of Kss1 by the Ste7 MEK weakens Kss1–Ste12 interaction, and is sufficient to relieve Kss1-mediated repression (Fig. 7B). Thus, we have demonstrated that an unactivated kinase isoform potently inhibits a transcription factor, and that activation of the kinase lifts this inhibition.
Figure 7.
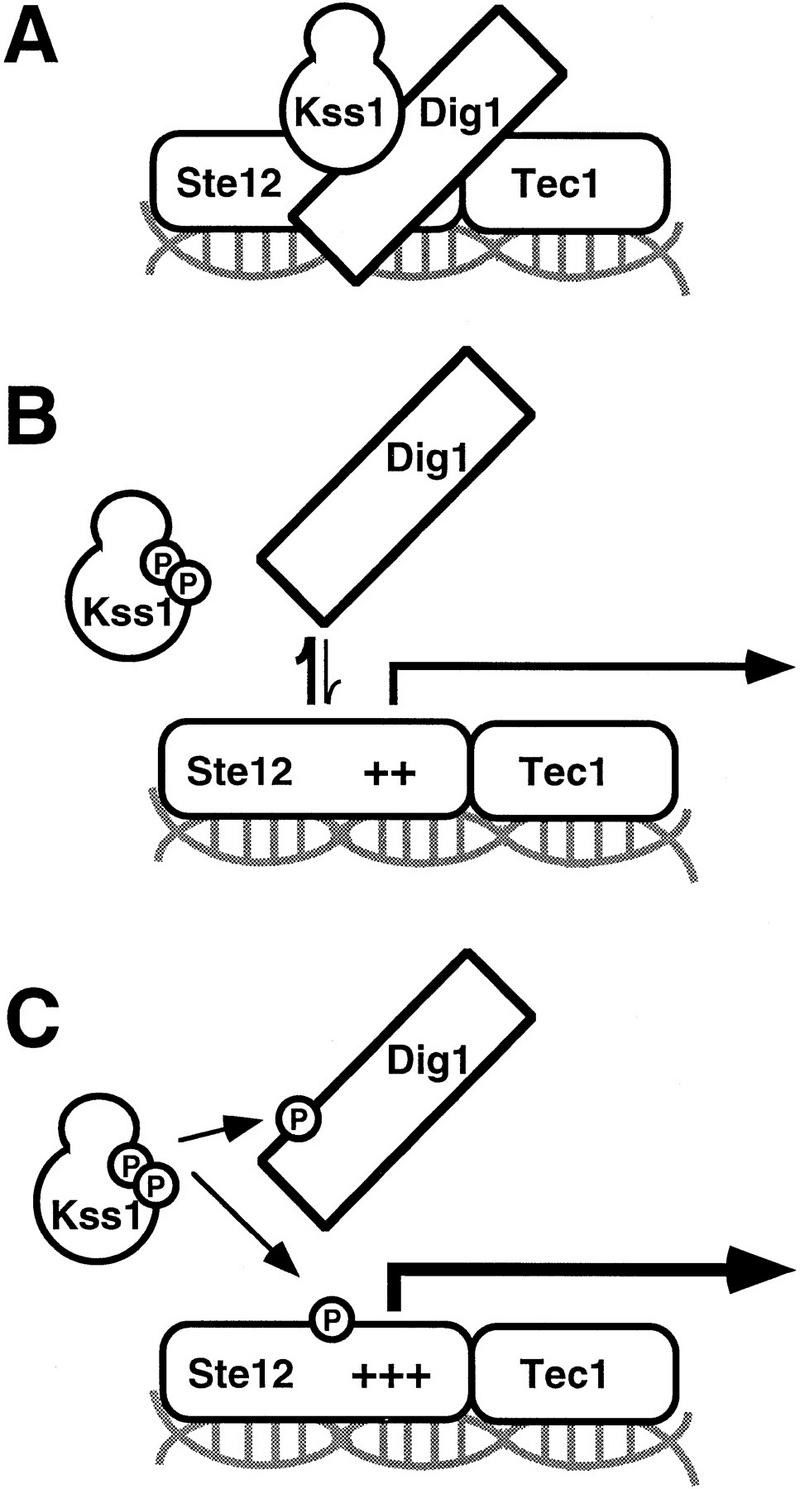
Model for Kss1-mediated regulation of Ste12 in invasive growth. (A) Unphosphorylated Kss1 binds directly to Ste12, and to Dig1 and Dig2, thereby stabilizing Dig1/2–Ste12 complexes and potentiating Dig-mediated repression of Ste12. (B) Phosphorylation of Kss1 by Ste7, in response to upstream signals, causes a conformational change in Kss1 that weakens its association with Ste12, and consequently reduces Dig1/2 interaction with Ste12–Tec1. (C) Phosphorylated and activated Kss1 further reinforces the transition, presumably by phosphorylating Dig1/2, Ste12, and/or Tec1, permitting full derepression.
Two nuclear proteins—Dig1 and Dig2—bind to Kss1 (and Fus3) and to Ste12 (Cook et al. 1996; Tedford et al. 1997). In addition, Dig1 and Dig2 show an interaction with Tec1 in the two-hybrid assay (M. Lorenz and J. Heitman, pers. comm.). Although it has been suggested that the Dig proteins bridge the interaction between the MAPKs and Ste12, we have shown here that Kss1 can bind directly to Ste12 in the absence of any other yeast proteins. Moreover, our recent results (L. Bardwell, J.G. Cook, J.X. Zhu-Shimoni and J. Thorner, in prep.) suggest that the Kss1–Ste12 interaction promotes the recruitment of Dig1 and Dig2, which act as negative regulators of Ste12 function (Cook et al. 1996; Tedford et al. 1997), because, for example, Kss1-mediated repression is defective in the absence of Dig1 and Dig2. Hence, the Dig proteins may constitute (or attract) factors that directly block Ste12 (and/or Tec1) action, whereas Kss1 serves primarily to stabilize the Dig–Ste12–Tec1 complex (Fig. 7A).
It seems likely, therefore, that phosphorylation of Kss1 relieves repression by a dual mechanism. First, as we have demonstrated, phosphorylation of Kss1 by Ste7 weakens Kss1–Ste12 interaction and presumably, therefore, weakens Dig binding to (or Dig action on) Ste12 and/or Tec1 (Fig. 7B). Second, derepression of Ste12 may be further reinforced by phosphorylation of Ste12, Tec1, and/or Dig1/Dig2 by activated Kss1 (Fig. 7C). Phosphorylation of these proteins may further weaken these complexes and promote interaction of Ste12/Tec1 with the transcription machinery. It has been shown that both Ste12 and Dig1 can be phosphorylated in a Kss1-dependent manner in immune complexes (Cook et al. 1996; Madhani et al. 1997) and that both GST–Ste12298–473 (containing the Kss1-binding site) and GST–Dig1213–452 (containing the Kss1- and Ste12-binding sites) serve as efficient in vitro substrates for purified Kss1 (L. Bardwell, unpubl.). In fact, Kss1 catalytic activity is required for optimal invasive growth in haploids, in which repression by Fus3 must also be prevented, and for efficient filamentous growth in diploids, in which the ratio of unphosphorylated Kss1 (and the Dig1 and Dig2 proteins) to Ste12 is perhaps higher than in haploids because of the reduced expression of STE12 in diploids (Fields and Herskowitz 1987). In toto, these events provide a switch-like mechanism to achieve commitment to a differentiated state.
Ste12 is the target of Kss1-mediated inhibition of invasive growth
The conclusion that Ste12 is the direct target of Kss1-mediated inhibition of invasive growth is supported by multiple observations. First, both here and elsewhere (Cook et al. 1997), we found a remarkable correlation between the extent of inhibition caused by alleles of Kss1 (and by Fus3) and the level of expression of a Ste12-dependent reporter gene (FRETy1–lacZ). Second, as we and others have found, Kss1 associates with Ste12 in cell extracts (Madhani et al. 1997) and in the two-hybrid assay (Printen and Sprague 1994; Cook et al. 1996). This interaction is direct because, as we showed here, Kss1 and Ste12 translated in vitro can associate. Third, Kss1 mutants specifically deficient in binding to Ste12 that we generated exhibited greatly reduced inhibition of invasive growth and FRE-mediated transcription in vivo. In a complementary approach, Madhani et al. (1997) isolated Kss1 mutants that displayed hyperfilamentation and found that some were defective in formation of Ste12-containing complexes. [Because, under some in vitro conditions, association of Kss1 with Ste12 is dependent on the presence of the Dig proteins (Tedford et al. 1997; Mike Tyers, pers. comm.), the alleles identified by Madhani et al. (1997) could be defective in Kss1–Dig binding, rather than in Kss1–Ste12 binding per se, or both.] Fourth, we found that Fus3, which is a weaker inhibitor of invasive growth than Kss1 (Cook et al. 1997), displays a correspondingly weaker binding to Ste12. Fifth, Ste7-mediated phosphorylation of Kss1 on T183 and Y185 was both necessary and sufficient to prevent Kss1-mediated inhibition and, correspondingly, reduced the affinity of Kss1 for Ste12.
Phosphorylated Kss1 exhibits substantially reduced, yet still detectable, affinity for Ste12. The residual binding of activated Kss1 to Ste12 may expedite Kss1-mediated phosphorylation of Ste12, Tec1, and/or Dig1 and Dig2. Such a docking function has been suggested for the Ste7–Kss1, Jnk–Jun, and Erk–Elk interactions (Bardwell and Thorner 1996; Bardwell et al. 1996; Kallunki et al. 1996; Yang et al. 1998).
The activation loop of Kss1 controls Ste12 binding and inhibition
The activation loop of MAPKs is flexible, and its conformation dictates catalytic activity and contributes to substrate recognition (Cobb and Goldsmith 1995). Our results indicate that this segment of Kss1 controls its ability to repress Ste12. Binding of unphosphorylated Kss1 to Ste12 requires integrity of the activation loop. The Kss1Δloop mutant, in which this segment was removed and replaced with an unrelated sequence, exhibited substantially reduced binding to Ste12 (but undiminished binding to Dig1, Dig2, and Ste7) and correspondingly, reduced ability to repress Ste12. A substitution (Y231C) in a residue that is conserved in most MAPKs caused the same reduction in specific binding and repression. This residue contacts the activation loop in unphosphorylated Erk2; but in phosphorylated Erk2, this contact has been broken (Zhang et al. 1995; Canagarajah et al. 1997).
The activation loop of rat Erk2 refolds following MEK-mediated phosphorylation (Canagarajah et al. 1997). Thus, Ste7-mediated phosphorylation of the activation loop of Kss1 provides a simple mechanism to relieve Kss1-imposed repression. We demonstrated here that phosphorylation of T183 and Y185 in Kss1 is necessary for Ste7 to relieve Kss1-mediated inhibition of invasive growth in vivo, and weakens Kss1–Ste12 binding in vitro. This dual phosphorylation is sufficient to relieve Kss1-mediated inhibition of haploid invasive growth, even when Kss1 is not catalytically active. Thus, residues in the activation loop of Kss1 may be in direct contact with Ste12. However, in Erk2, the state of the activation loop affects the conformation of the active site, the P+1 specificity pocket, the extended carboxyl terminus, and the so-called MAPK insertion (Zhang et al. 1995; Canagarajah et al. 1997). Hence, other regions of Kss1 could participate in Ste12 recognition. In this regard, two of the mutations (D249G and E260G) isolated by Madhani et al. (1997) are located in the MAPK insertion region.
Phosphorylation-induced refolding of an activation segment situated between conserved subdomains VII and VIII causes conformational changes required for catalysis and for access of substrates to the active site in many protein kinases (Johnson et al. 1996; Canagarajah et al. 1997). Our results argue that such structural rearrangements can also diminish the binding of a kinase to a target, with important physiological consequences.
Generality of MAPK-mediated inhibition
Our work demonstrates that regulation of a transcription factor by a MAPK involves direct protein kinase-transcription factor binding (and recruitment by the bound kinase of other regulatory factors). How general is this mechanism? Relatively stable interactions between mammalian MAPKs and transcription factors have been observed (Gupta et al. 1996; Kallunki et al. 1996; Yang et al. 1998). It was proposed that binding of SAPK/Jnk may inhibit c-Jun function directly (Gupta et al. 1996), although this suggestion is at odds with some recent results (May et al. 1998). Nonetheless, the inhibitory role of Kss1 was revealed initially by use of genetic approaches that are more difficult in organisms less tractable than S. cerevisiae. It seems likely, therefore, that the mechanism characterized here will be found in other protein kinase signaling pathways that act as developmental switches, but may not have been readily discernible heretofore.
Materials and methods
Yeast strains and media
Yeast strains used in this work are shown in Table 1. Standard yeast media were prepared as described (Bardwell et al. 1996), except that, in synthetic complete medium, twice the recommended level of nutritional supplements was used.
Table 1.
S. cerevisiae strains used in this study
| Strain
|
Genotype
|
Source or reference
|
|---|---|---|
| Haploid strains derived from the Σ1278b lineage | ||
| JCY100 | MATa his3Δ∷hisG leu2Δ∷hisG trp1Δ∷hisG ura3-52 | Cook et al. (1997),Madhani and Fink (1997) |
| JCY107 | JCY100 ste7Δ∷ura3 | Cook et al. (1997) |
| Madhani and Fink (1997) | ||
| JCY130 | JCY100 kss1Δ∷hisG fus3Δ∷TRP1 | Cook et al. (1997) |
| Madhani and Fink (1997) | ||
| JCY137 | JCY100 ste7Δ∷ura3 kss1Δ∷hisG fus3Δ∷TRP1 | Cook et al. (1997) |
| Other strains | ||
| DC17 | MATα his1 | J.B. Hicks (Cold Spring Harbor Laboratory) |
| BJ2168 | MATa leu2 trp1 ura3-52 prb1-1112 pep4-3 prc1-407 gal2 | Jones (1991) |
| MaV103a | MATa leu2-3,112 trp1-901 his3Δ200 ade2-101 gal4Δ gal80Δ SPAL10∷URA3 GAL1∷lacZ GAL1∷HIS3@lys2 can1R cyh2R | Vidal et al. (1996) |
Plasmid constructions and recombinant DNA methods
Plasmids YCpU (Cook et al. 1997) and YEpT–KSS1 (Bardwell et al. 1996) are described in the citations given. YEpU–FTyZ, containing the FRETy1–lacZ reporter gene (and the URA3 gene) on a 2 μm DNA plasmid, has been described elsewhere (Cook et al. 1997). The URA3 gene was excised from YEpU–FTyZ by digesting with Sse8387 I and XmaI and replaced with the LEU2 gene on a PstI–XmaI fragment excised from pJJ252 (Jones and Prakash 1990), to yield YEpL–FTyZ.
To construct YCpU–KSS1, an EcoRI–SphI fragment containing KSS1 and its promoter was excised from plasmid YEp–KSS1 (Ma et al. 1995) and inserted into the corresponding sites of YCpU. YCpU–kss1(K42R Q45P), YCpU–kss1(Y24F), YCpU–kss1(T183A), YCpU–kss1(Y185F), and YCpU–kss1(AEF) were constructed in a similar fashion with EcoRI–SphI fragments excised from the corresponding YEp-based plasmids carrying these mutant alleles (Ma et al. 1995). Plasmid pRS316–KSS1 (gift of Doreen Ma, this laboratory), contains KSS1 and its promoter on an EcoRI–BglII fragment in the corresponding sites of pRS316 (Sikorski and Hieter 1989). To facilitate efficient subcloning of KSS1 alleles obtained from the reverse two-hybrid screen (see below) into this vector, the 0.96-kb BsiWI–BspEI fragment of KSS1 was replaced with oligonucleotide BXB1 (5′-CCGGAGAACCCCTTCC-3′) annealed to BXB2 (5′-GTACGGAAGGGGTTCT-3′), yielding pRS316–kss1ΔBXB. YCpU–kss1(Δloop) was generated by use of the BsiWI–BspEI fragment from YEpU–kss1(Δloop) (Bardwell et al. 1996) to replace the oligonucleotide fragment of pRS316–kss1ΔBXB. YCpU–kss1(Y231C) was generated in a similar fashion by the BsiWI–BspEI fragment from pkss1(Y231C)–GDB (see below).
pKSS1–GDB (formerly pJGC2), encoding a Kss1–GDB domain fusion protein, has been described elsewhere (Cook et al. 1996). To facilitate the subcloning of other KSS1 alleles into this vector, the BsiWI–BspEI fragment of KSS1 was replaced with oligonucleotide BXB1 annealed to BXB2, yielding pkss1ΔBXB–GDB. pkss1(AEF)–GDB (encoding the T183A Y185F derivative) was generated by use of the BsiWI–BspEI fragment from YEp-T183A Y185F (Ma et al. 1995) to replace the oligonucleotide fragment of pkss1ΔBXB–GDB. pkss1(Δloop)–GDB and pkss1(Y231C)–GDB were generated in a similar fashion with BsiWI–BspEI fragments from YEpU–kss1(Δloop) (Bardwell et al. 1996) and p141 (see Reverse two-hybrid screen, below), respectively. pGAD–DIG1(7–452), pGAD–DIG2(56–323), pGAD–STE12(191–478), and pGAD–STE12(298–534) have been described elsewhere (Cook et al. 1996). To construct pEG-105, a BamHI fragment containing STE12(298–473) was excised from pGAD–STE12(298–534) and inserted into the BamHI site of pEG(KT) (Mitchell et al. 1993).
To fuse Ste12 to an amino-terminal c-Myc epitope tag, an EcoRI fragment containing the STE12 ORF was excised from plasmid pGAD–STE12 (gift of Stan Fields, University of Washington, Seattle) and inserted into the EcoRI site of pGEM4Z–9E10 (Bardwell et al. 1994a), yielding pGEM4Z–meSTE12, in which the amino-terminal sequence is MEQKLISEEDLEFHM- (epitope for mAb 9E10 underlined, native Ste12 initiator Met in boldface). pGEM4Z–kss1(Y231C) was generated by use of the BsiWI–BspEI fragment from pkss1(Y231C)–GDB to replace the corresponding fragment of pGEM4Z–KSS1 (Bardwell et al. 1996).
Reverse two-hybrid screen
The kss1(Y231C) allele was isolated in a reverse two-hybrid screen by essentially the method described by Vidal and colleagues (Vidal et al. 1996). Briefly, a library of mutagenized pKSS1–GDB plasmids was created (Greener et al. 1996) by growing this plasmid for ∼25 generations in E. coli mutator strain XL-1 Red (Stratagene), using carbenicillin to select for the plasmid. Plasmid DNA was prepared and used to transform yeast strain MaV103a carrying plasmid pGAD–STE12(191–478). MaV103a contains three GAL promoter-driven reporter genes (URA3, HIS3, and E. coli lacZ). Transformants (19,000) were screened for their ability to grow on agar medium containing 0.1% 5-FOA, thus selecting for loss of URA3 expression, and thereby for drastic reduction of the KSS1–STE12 two-hybrid interaction. The 218 colonies so obtained were then tested for those that could grow on agar medium containing 20 mm 3-amino-1H-1,2,4-triazole (3-AT), but not on 50 mm 3-AT, thus screening for clones that have diminished, but not completely abolished, HIS3 expression (and thereby a greatly reduced, but not completely null, KSS1–STE12 two-hybrid interaction). From the resulting five clones, the pKSS1–GDB plasmids were rescued, retransformed into MaV103a along with pGAD–DIG1(7–452), and scored for lacZ expression to identify Kss1 mutants that fully retained the ability to interact with Dig1. Only two plasmids (designated p141 and p242) met all three criteria. Nucleotide sequence analysis revealed that both of these plasmids (most likely siblings from the mutagenized DNA library) contained a single A-to-G transition at nucleotide 692 of the KSS1 ORF, resulting in a tyrosine-to-cysteine substitution at residue 231.
Transcription and translation in vitro
Transcription and translation reactions in vitro , and the partial purification of translation products by ammonium sulfate precipitation, were performed as described previously (Bardwell et al. 1992, 1996). Preparation of in vitro-translated Kss1, Kss1Δloop, Kss1AEF, and Fus3 has been described elsewhere (Bardwell et al. 1996). The kss1(Y231C) mRNA was transcribed from plasmid pGEM4Z–kss1(Y231C) linearized with NdeI. The meSTE12 mRNA was transcribed from plasmid pGEM4Z–meSTE12 linearized with SalI.
GST-fusion protein production and binding assays
GST–Ste71–172 and GST–Dig1213–452 were prepared from E. coli as described elsewhere (Bardwell et al. 1996; Cook et al. 1996). GST–Ste12298–473 was expressed from plasmid pEG-105 in yeast strain BJ2168 and purified as follows: Cultures (250 ml) were grown overnight in selective medium containing 2% galactose and 0.2% sucrose, and harvested. Cell pellets were resuspended in buffer B (Bardwell et al. 1996) containing 5 mm dithiothreitol (DTT), and extracts were prepared as described previously (Bardwell et al. 1996). The extracts were adjusted to a final concentration of 1.5 % (wt/vol) N-lauroyl-sarcosine (sarcosyl) and rocked at 4°C for 15 min. Triton X-100 was then added to a final concentration of 3.0% (vol/vol), followed by a further 15 min of rocking. The lysate was centrifuged for 5 min at 13,000g at 4°C to remove insoluble material. To the resulting clarified extract, 0.1 ml of a 50% slurry of glutathione–Sepharose (Pharmacia) was added, and the mixture was rotated for 1 hr at 4°C. The glutathione–Sepharose beads were collected, and washed thoroughly with buffer B containing 5 mm DTT and 0.75 m potassium acetate, followed by buffer B containing 5 mm DTT. Bound proteins were eluted with three 10-min incubations at room temperature in 50 μl of freshly prepared 10 mm reduced glutathione, 50 mm Tris-HCl (pH 8.0). The eluate fractions were pooled (150 μl total) and frozen at −70°C. Alternatively, fusion proteins bound to glutathione–Sepharose beads were stored on ice in buffer B containing 5 mm DTT and 1 mg/ml BSA until needed. GST–Ste12298–473 protein purified from yeast by this method was judged to be >90% pure by Coomassie blue staining and to be free of contaminating Dig1, Dig2, Kss1, or Fus3 by immunoblotting. GST was purified from both yeast and E. coli. No difference in the binding properties of GST purified from these two sources was found.
Binding assays using GST fusions and in vitro-translated proteins were as described elsewhere (Cook et al. 1996). Binding assays using GST fusions and yeast cell extracts were performed by our published procedure for immunoprecipitation from yeast cell extracts (Bardwell et al. 1996), except that glutathione–Sepharose-bound fusion proteins were used in place of Protein A/G-antibody beads, and 1 mg/ml BSA and 5 mm DTT were present in all buffers.
Other methods and reagents
Expression of the FRETy1–lacZ reporter gene was determined as described elsewhere (Cook et al. 1997). The bioassay for haploid invasive growth has been described (Roberts and Fink 1994). Growth and harvesting of yeast cultures for biochemical analysis, preparation of cell extracts, and coimmunoprecipitation assays were performed exactly as described previously (Bardwell et al. 1996). Anti-GST antisera was purchased from Santa Cruz Biotechnologies. The rabbit polyclonal antisera used for analysis of Kss1 (Ma et al. 1995) has been described. Anti-phospho-MAPK antisera (Khokhlatchev et al. 1997), initially a gift of Erik Schaefer and thereafter obtained from a commercial source (anti-ACTIVE-MAPK antisera; Promega), was used at a dilution of 1:1300 with overnight incubation at 4°C.
Acknowledgments
We thank Brad Cairns, Bob Deschenes, Beverly Errede, Stan Fields, Gerald Fink, Ira Herskowitz, Erik Schaefer, Mike Tyers, and Marc Vidal for generous gifts of research materials, and also Mike Tyers and Joe Heitman for communication of unpublished results. We thank Mike Lorenz and Tom Burke, and especially the members of our own laboratory, for critical discussion. This work was supported by Special Senior Fellow Award 3754-98 from the Leukemia Society of America (to L.B.), by National Institutes of Health–National Cancer Institute (NIH–NCI) Postdoctoral Traineeship CA09041 (to J.G.C.), by NIH–National Institute of General Medical Sciences (NIGMS) Predoctoral Traineeship GM07232 (to D.M.B.), by funds from the Berkeley campus Summer Research Opportunities Program (to A.R.M.), by NIH research grant GM21841 (to J.T.), and by resources provided by the Berkeley campus Cancer Research Laboratory.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jeremy@socrates.berkeley.edu: FAX (510) 643-5035.
References
- Bardwell L, Thorner J. A conserved motif at the amino termini of MEKs might mediate high-affinity interaction with the cognate MAPKs. Trends Biochem Sci. 1996;1:373–374. [PubMed] [Google Scholar]
- Bardwell L, Cooper AJ, Friedberg EC. Stable and specific association between the yeast recombination and DNA repair proteins RAD1 and RAD10 in vitro. Mol Cell Biol. 1992;12:3041–3049. doi: 10.1128/mcb.12.7.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell AJ, Bardwell L, Iyer N, Svejstrup JQ, Feaver WJ, Kornberg RD, Friedberg EC. Yeast nucleotide excision repair proteins Rad2 and Rad4 interact with RNA polymerase II basal transcription factor b (TFIIH) Mol Cell Biol. 1994a;14:3569–3576. doi: 10.1128/mcb.14.6.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell L, Cook JG, Inouye CJ, Thorner J. Signal propagation and regulation in the mating pheromone response pathway of the yeast Saccharomyces cerevisiae. Devel Biol. 1994b;166:363–379. doi: 10.1006/dbio.1994.1323. [DOI] [PubMed] [Google Scholar]
- Bardwell L, Cook JG, Chang EC, Cairns BR, Thorner J. Signaling in the yeast pheromone response pathway: Specific and high-affinity interaction of the mitogen-activated protein (MAP) kinases Kss1 and Fus3 with the upstream MAP kinase kinase Ste7. Mol Cell Biol. 1996;16:3637–3650. doi: 10.1128/mcb.16.7.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur M, Esch RK, Errede B. Cooperative binding interactions required for function of the Ty1 sterile responsive element. Mol Cell Biol. 1997;17:4330–4337. doi: 10.1128/mcb.17.8.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns BR, Ramer SW, Kornberg RD. Order of action of components in the yeast pheromone response pathway revealed with a dominant allele of the STE11 kinase and the multiple phosphorylation of the STE7 kinase. Genes & Dev. 1992;6:1305–1318. doi: 10.1101/gad.6.7.1305. [DOI] [PubMed] [Google Scholar]
- Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- Chandarlapaty S, Errede B. Ash1, a daughter cell-specific protein, is required for pseudohyphal growth of Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:2884–2891. doi: 10.1128/mcb.18.5.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, van de Wetering M. TCF/LEF factors earn their wings. Trends Genet. 1997;13:485–489. doi: 10.1016/s0168-9525(97)01305-x. [DOI] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- Cook JG, Bardwell L, Kron SJ, Thorner J. Two novel targets of the MAP kinase Kss1 are negative regulators of invasive growth in the yeast Saccharomyces cerevisiae. Genes & Dev. 1996;10:2831–2848. doi: 10.1101/gad.10.22.2831. [DOI] [PubMed] [Google Scholar]
- Cook JG, Bardwell L, Thorner J. Inhibitory and activating functions for MAPK Kss1 in the S. cerevisiae filamentous growth signaling pathway. Nature. 1997;390:85–88. doi: 10.1038/36355. [DOI] [PubMed] [Google Scholar]
- DePinho R. Transcriptional repression: The cancer-chromatin connection. Nature. 1998;391:533. doi: 10.1038/35257. [DOI] [PubMed] [Google Scholar]
- Elion EA, Brill JA, Fink GR. FUS3 represses CLN1 and CLN2 and in concert with KSS1 promotes signal transduction. Proc Natl Acad Sci. 1991;88:9392–9396. doi: 10.1073/pnas.88.21.9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion E, Satterberg B, Kranz J. Fus3 phosphorylates multiple components of the mating signal transduction cascade: Evidence for Ste12 and Far1. Mol Biol Cell. 1993;4:495–510. doi: 10.1091/mbc.4.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdman S, Lin L, Malczynski M, Snyder M. Pheromone-regulated genes required for yeast mating differentiation. J Cell Biol. 1998;140:461–483. doi: 10.1083/jcb.140.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errede B, Gartner A, Zhou Z, Naysmith K, Ammerer G. MAP kinase-related FUS3 from S. cerevisiae is activated by STE7 in vitro. Nature. 1993;362:261–264. doi: 10.1038/362261a0. [DOI] [PubMed] [Google Scholar]
- Fields S, Herskowitz I. Regulation by the yeast mating type locus of STE12, a gene required for expression of two sets of cell-type-specific genes. Mol Cell Biol. 1987;7:3818–3821. doi: 10.1128/mcb.7.10.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrias V, Andrianopoulos A, Gimeno CJ, Timberlake WE. Saccharomyces cerevisiae TEC1 is required for pseudohyphal growth. Mol Microbiol. 1996;19:1255–1263. doi: 10.1111/j.1365-2958.1996.tb02470.x. [DOI] [PubMed] [Google Scholar]
- Gimeno CJ, Ljungdahl PO, Styles CA, Fink GR. Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: Regulation by starvation and RAS. Cell. 1992;68:1077–1090. doi: 10.1016/0092-8674(92)90079-r. [DOI] [PubMed] [Google Scholar]
- Goldman P, Tran V, Goodman R. The multifunctional role of the co-activator CBP in transcriptional regulation. Recent Prog Horm Res. 1997;52:103–119. [PubMed] [Google Scholar]
- Greener A, Callahan M, Jerpseth B. An efficient random mutagenesis technique using an E. coli mutator strain. In: Trower MK, editor. Methods in molecular biology. Tolowa, NJ: Humana Press; 1996. pp. 375–385. [DOI] [PubMed] [Google Scholar]
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HL, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- Heldin C, Miyazono K, ten Dijke P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Hill CS, Treisman R. Transcriptional regulation by extracellular signals: Mechanisms and specificity. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- Hunter T, Plowman GD. The protein kinases of budding yeast: Six score and more. Trends Biochem Sci. 1997;22:18–22. doi: 10.1016/s0968-0004(96)10068-2. [DOI] [PubMed] [Google Scholar]
- Johnson LN, Noble MEM, Owen DJ. Active and inactive protein kinases: Structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Jones EW. Tackling the protease problem in yeast. Methods Enzymol. 1991;194:428–453. doi: 10.1016/0076-6879(91)94034-a. [DOI] [PubMed] [Google Scholar]
- Jones JS, Prakash L. Yeast Saccharomyces cerevisiae selectable markers in pUC18 polylinkers. Yeast. 1990;6:363–366. doi: 10.1002/yea.320060502. [DOI] [PubMed] [Google Scholar]
- Kallunki T, Deng T, Hibi M, Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- Karin M. Signal transduction from the cell surface to the nucleus through the phosphorylation of transcription factors. Curr Opin Cell Biol. 1994;6:415–424. doi: 10.1016/0955-0674(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Khokhlatchev A, Xu S, English J, Wu P, Schaefer E, Cobb MH. Reconstitution of mitogen-activated protein kinase phosphorylation cascades in bacteria. J Biol Chem. 1997;272:11057–11062. doi: 10.1074/jbc.272.17.11057. [DOI] [PubMed] [Google Scholar]
- Kirkman-Correia C, Stroke IL, Fields S. Functional domains of the yeast STE12 protein, a pheromone-responsive transcriptional activator. Mol Cell Biol. 1993;13:3765–3772. doi: 10.1128/mcb.13.6.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron SJ, Styles CA, Fink GR. Symmetric cell division in pseudohyphae of the yeast Saccharomyces cerevisiae. Mol Biol Cell. 1994;5:1003–1022. doi: 10.1091/mbc.5.9.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kübler E, Mösch H-U, Rupp S, Lisanti MP. Gpa2p, a G-protein α-subunit, regulates growth and pseudohyphal development in Saccharomyces cerevisiae via a cAMP-dependent mechanism. J Biol Chem. 1997;272:20321–20323. doi: 10.1074/jbc.272.33.20321. [DOI] [PubMed] [Google Scholar]
- Laloux I, Jacobs E, Dubois E. Involvement of SRE element of Ty1 transposon in TEC1-dependent transcriptional activation. Nucleic Acids Res. 1994;22:999–1005. doi: 10.1093/nar/22.6.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberer E, Thomas D, Whiteway M. Pheromone signalling and polarized morphogenesis in yeast. Curr Opin Genet Dev. 1997;7:59–66. doi: 10.1016/s0959-437x(97)80110-4. [DOI] [PubMed] [Google Scholar]
- Liu H, Styles CA, Fink GR. Elements of the yeast pheromone response pathway required for filamentous growth of diploids. Science. 1993;262:1741. doi: 10.1126/science.8259520. [DOI] [PubMed] [Google Scholar]
- Lo HJ, Köhler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, Fink GR. Nonfilamentous C. albicans are avirulent. Cell. 1997;90:939–949. doi: 10.1016/s0092-8674(00)80358-x. [DOI] [PubMed] [Google Scholar]
- Lorenz MC, Heitman J. The MEP2 ammonium permease regulates pseudohyphal differentiation in Saccharomyces cerevisiae. EMBO J. 1998;17:1236–1247. doi: 10.1093/emboj/17.5.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Cook JG, Thorner J. Phosphorylation and localization of Kss1, a MAP kinase of the Saccharomyces cerevisiae pheromone response pathway. Mol Biol Cell. 1995;6:889–909. doi: 10.1091/mbc.6.7.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Fink GR. Combinatorial control required for the specificity of yeast MAPK signaling. Science. 1997;275:1314–1317. doi: 10.1126/science.275.5304.1314. [DOI] [PubMed] [Google Scholar]
- Madhani HD, Styles CA, Fink GR. MAP kinases with distinct inhibitory functions impart signaling specificity during yeast differentiation. Cell. 1997;91:673–684. doi: 10.1016/s0092-8674(00)80454-7. [DOI] [PubMed] [Google Scholar]
- May GHW, Funk M, Black EJ, Clark W, Hussain S, Woodgett JR, Gillespie DAF. An oncogenic mutation uncouples the v-Jun oncoprotein from positive regulation by the SAPK/JNK pathway in vivo. Curr Biol. 1998;8:117–120. doi: 10.1016/s0960-9822(98)70043-0. [DOI] [PubMed] [Google Scholar]
- Mitchell D, Marshall T, Deschenes R. Vectors for the inducible overexpression of glutathione S-transferase fusion proteins in yeast. Yeast. 1993;9:715–722. doi: 10.1002/yea.320090705. [DOI] [PubMed] [Google Scholar]
- Morgan DO, De Bondt HL. Protein kinase regulation: Insights from crystal structure analysis. Curr Opin Cell Biol. 1994;6:239–246. doi: 10.1016/0955-0674(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Mösch H-U, Roberts RL, Fink GR. Ras2 signals via the Cdc42/Ste20/mitogen-activated protein kinase module to induce filamentous growth in Saccharomyces cerevisiae. Proc Natl Acad Sci. 1996;93:5352–5356. doi: 10.1073/pnas.93.11.5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman AM, Herskowitz I. Reconstitution of a yeast protein kinase cascade in vitro: Activation of the yeast MEK homolog STE7 by STE11. Proc Natl Acad Sci. 1994;91:3398–3402. doi: 10.1073/pnas.91.8.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printen JA, Sprague GF., Jr Protein-protein interactions in the yeast pheromone response pathway: Ste5p interacts with all members of the MAP kinase cascade. Genetics. 1994;138:609–619. doi: 10.1093/genetics/138.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes N, Connell L, Errede B. STE11 is a protein kinase required for cell-type-specific transcription and signal transduction in yeast. Genes & Dev. 1990;4:1862–1874. doi: 10.1101/gad.4.11.1862. [DOI] [PubMed] [Google Scholar]
- Roberts RL, Fink GR. Elements of a single MAP kinase cascade in Saccharomyces cerevisiae mediate two developmental programs in the same cell type: Mating and invasive growth. Genes & Dev. 1994;8:2974–2985. doi: 10.1101/gad.8.24.2974. [DOI] [PubMed] [Google Scholar]
- Roberts RL, Mösch H-U, Fink GR. 14-3-3 proteins are essential for RAS/MAPK cascade signaling during pseudohyphal development in S. cerevisiae. Cell. 1997;89:1055–1065. doi: 10.1016/s0092-8674(00)80293-7. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedford K, Kim S, Sa D, Stevens K, Tyers M. Regulation of the mating pheromone and invasive growth responses in yeast by two MAP kinase substrates. Curr Biol. 1997;7:228–238. doi: 10.1016/s0960-9822(06)00118-7. [DOI] [PubMed] [Google Scholar]
- Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- Vandromme M, Gautheir-Rouviere C, Lamb N, Fernandez A. Regulation of transcription factor localization: Fine-tuning of gene expression. Trends Biochem Sci. 1996;21:59–64. [PubMed] [Google Scholar]
- Vidal M, Brachmann R, Fattaey A, Harlow E, Boeke J. Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc Natl Acad Sci. 1996;93:10315–10320. doi: 10.1073/pnas.93.19.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S-H, Yates PR, Whitmarsh AJ, Davis RJ, Sharrocks AD. The Elk-1 ETS domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol Cell Biol. 1998;18:710–720. doi: 10.1128/mcb.18.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2.3 Å resolution. Nature. 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang F, Ebert D, Cobb MH, Goldsmith EJ. Activity of the MAP kinase ERK2 is controlled by a flexible surface loop. Structure. 1995;3:299–307. doi: 10.1016/s0969-2126(01)00160-5. [DOI] [PubMed] [Google Scholar]