

Abstract
The RAG1 and RAG2 proteins collaborate to initiate V(D)J recombination by binding recombination signal sequences (RSSs) and making a double-strand break between the RSS and adjacent coding DNA. Like the reactions of their biochemical cousins, the bacterial transposases and retroviral integrases, cleavage by the RAG proteins requires a divalent metal ion but does not involve a covalent protein/DNA intermediate. In the transposase/integrase family, a triplet of acidic residues, commonly called a DDE motif, is often found to coordinate the metal ion used for catalysis. We show here that mutations in each of three acidic residues in RAG1 result in mutant derivatives that can bind the RSS but whose ability to catalyze either of the two chemical steps of V(D)J cleavage (nicking and hairpin formation) is severely impaired. Because both chemical steps are affected by the same mutations, a single active site appears responsible for both reactions. Two independent lines of evidence demonstrate that at least two of these acidic residues are directly involved in coordinating a divalent metal ion: The substitution of Cys for Asp allows rescue of some catalytic function, whereas an alanine substitution is no longer subject to iron-induced hydroxyl radical cleavage. Our results support a model in which the RAG1 protein contains the active site of the V(D)J recombinase and are interpreted in light of predictions about the structure of RAG1.
Keywords: RAG1, RAG2; V(D)J recombination; T-cell receptor; recombination signal sequence
A series of site-specific somatic DNA rearrangements is required for the assembly of the immunoglobulin and T-cell receptor (TCR) genes of the vertebrate immune system (for reviews, see Lewis 1994; Gellert 1997; Oettinger 1999). These V(D)J recombination events are initiated by the combined action of two lymphoid-specific proteins, RAG1 and RAG2. Together, these proteins recognize recombination signal sequences (RSSs) that flank immunoglobulin and TCR gene segments and introduce a double-strand break (DSB) at the border of the coding gene segment and the RSS. The RSS is comprised of conserved heptamer and nonamer elements separated by a spacer region of conserved length (12 or 23 bp) but variable sequence. The DSB is generated in two steps (McBlane et al. 1995). In the first step, a nick is introduced at the 5′ end of the heptamer at the signal/coding boundary. In the second step, the newly liberated hydroxyl on the coding DNA attacks the phosphodiester bond of the opposing strand to form a hairpinned coding end and a blunt signal end. Subsequent processing and rejoining of the cleaved ends to form the mature coding segment and a signal junction requires the action of a number of proteins, including several involved in the repair of X-ray-induced damage (Jeggo 1998).
The reactions carried out by the RAG proteins are chemically similar to those carried out by the transposase proteins of bacterial transposons such as Mu or Tn10 or by the integrase proteins of retroviruses such as HIV (Plasterk 1998; Roth and Craig 1998). In addition to carrying out the two steps of V(D)J cleavage, the RAG proteins can mediate transpositional recombination in vitro, joining the 3′ OH of a cleaved signal end to an unrelated target sequence (Agrawal et al. 1998; Hiom et al. 1998). A shared feature of the strand transfer reactions of these mobile elements and the RAG recombinase is that they occur in the absence of an external energy source via a one-step transesterification reaction with no protein–DNA intermediate (van Gent et al. 1996). One distinction between the RAG proteins and other recombinases is the reversal of the initial cleavage site relative to the DNA recognition sequence. Specifically, RAG proteins nick the top strand (the 5′ end of the heptamer at the signal/coding boundary), whereas MuA and Tn10 transposases, for example, nick the bottom strand (the 3′ end of the mobile element).
A large number of enzymes that catalyze phosphoryl-transfer reactions, including all members of the transposase/integrase family, the type II restriction enzymes, DNA polymerases, and the RAG proteins, are metal dependent (McBlane et al. 1995; Rice et al. 1996; Pingoud and Jeltsch 1997; Steitz 1999). Extensive mutational, biochemical, and structural studies of these enzymes have established several sequence motifs, all of which have a common requirement for conserved carboxylates to chelate metal ions for catalysis. The metal ions are not only required for the phosphoryl-transfer reaction, they also play a role in substrate binding. Often metal ions are utilized as a means to bridge close and specific contacts between protein and nucleic acid (Beese and Steitz 1991; Roberts and Halford 1993; Cao 1999). By identifying metal-chelating carboxylates, the catalytic sites can essentially be defined.
Members of the bacterial transposase/retroviral integrase family share a common folding topology in addition to a conserved carboxylate triad, the so-called DDE motif (Kulkosky and Skalka 1994; Polard and Chandler 1995; Rice et al. 1996). Substitution of Ala, Asn, or Gln for residues of the catalytic triad substantially reduces the activity of these proteins, often to undetectable levels. In some cases, when cysteine is substituted for one of the acidic residues, an alteration in the metal binding preference from Mg2+ to Mn2+ is observed, supporting the conclusion that the residue is involved in chelating a metal ion (Sarnovsky et al. 1996; Allingham et al. 1999; T.A. Baker, pers. comm.). This use of cysteine substitutions for defining metal ion binding sites within a protein is based on the observation that although oxygen binds well to both Mg2+ and Mn2+, a sulfur group serves well as a ligand only for Mn2+ (Pecoraro et al. 1984). Recent high-resolution structural studies of ASV and HIV1 integrase proteins in the presence of Mg2+ have allowed direct detection of the interaction between the acidic residues in the active site with the metal ion (Bujacz et al. 1996; Goldgur et al. 1998; Maignan et al. 1998). The similarity in reaction mechanism of the transposase/integrase family with that of the RAG proteins suggests that the RAG proteins might share this phylogenetically conserved catalytic triad and that the catalytic mechanism of the two systems may be similar.
In addition to mutational analysis as a means to identify residues involved in metal binding and catalysis, the location of a metal binding site can be physically mapped by taking advantage of iron-induced hydroxyl radical protein cleavage. The highly reactive hydroxyl radicals generated by chelated Fe2+ can cause localized cleavage of nearby sites. This method, often used for DNA footprinting, can also be applied to proteins, in which the hydroxyl radicals cleave peptide bonds in the immediate vicinity of the chelated iron. Fe2+ cleavage has been used successfully as a tool to map the metal binding sites in several proteins (Wei et al. 1994; Ettner et al. 1995; Zaychikov et al. 1996; Lykke-Andersen et al. 1997).
Here, we report the results of applying a combination of these methods to the identification and characterization of residues in RAG1 that are involved in catalysis. We have identified two acidic residues that appear to have direct roles in metal binding based on iron cleavage and cysteine substitution criteria. A third acidic residue is identified that also appears to have a critical role in catalysis. These results argue strongly that RAG1 contains most, if not all, of the active site of the RAG1/RAG2 recombinase.
Results
Purification of active RAG1 protein from Escherichia coli
Although previous biochemical analysis of RAG1 was carried out with protein produced from recombinant baculoviral expression in insect cells or from GST fusions expressed in mammalian cells, the work presented here was greatly facilitated by our development of expression vectors and purification conditions that allow functional RAG1 protein to be purified from Escherichia coli. Two bacterial expression constructs were generated. The first, pDRK534, contains a truncated form of the RAG1 gene encoding amino acids 352–1040 fused in-frame with a carboxy-terminal epitope tag of 8 histidines, under the control of the PA1 promoter (see Materials and Methods). The second derivative (pDRK573) contains the maltose binding protein (MBP) fused to the amino terminus of RAG1 (amino acids 352–1040) and a carboxy-terminal 8 histidine epitope tag (see Fig. 1A). Proteins were purified to near homogeneity using Ni-chelating chromatography. Using comparable amounts of protein, as determined by Coomassie blue staining, the proteins purified from E. coli were consistently 8- to 10-fold more active in an oligonucleotide cleavage assay than our standard protein purified from insect cells (data not shown). [Successful purification of E. coli RAG1 has also been reported recently by others (Rodgers et al. 1999)]. The pDRK534 expression plasmid formed the basis for the mutational studies described below, whereas pDRK537, generating the MBP:RAG1 fusion protein, was used for Fe2+-induced cleavage assays.
Figure 1.

Iron-induced hydroxyl radical cleavage of RAG1. (A) Diagram of the MBP–RAG1 fusion protein used for this analysis, with positions of the MBP and 8 histidine tag indicated. (B) Western blot with anti-MBP antibody reveals a ∼70-kD iron-dependent cleavage product. (C) Western blot with anti-His tag antibody reveals a ∼47-kD carboxy-terminal cleavage product. The position of the cleaved products is indicated by an arrow; the presence (+) or absence (−) of Fe2+ in the reaction is indicated.
Fe2+-induced cleavage of RAG1 protein
Previous mutational studies of the RAG proteins revealed that all residues essential for V(D)J cleavage lie between amino acids 384 and 1008 of RAG1 and 1 and 383 of RAG2, indicating that any crucial metal binding site must lie within these regions (Sadofsky et al. 1993; Silver et al. 1993; Cuomo and Oettinger 1994; Sadofsky et al. 1994; Kirch et al. 1996). To further narrow down the region responsible for metal ion binding and to develop a physical basis for assigning the residues involved in metal binding, we sought to make use of iron-induced hydroxyl radical protein cleavage. Both the nicking and hairpinning steps of DNA cleavage by RAG1/2 protein can occur with Fe2+ as a cofactor (data not shown), suggesting that Fe2+ can bind to the same active site without significantly distorting the structure. Thus, the RAG proteins appeared to be good candidates for Fe2+-mediated protein cleavage.
Purified RAG1 protein alone or in combination with RAG2 protein was incubated with an oligonucleotide substrate containing a 12 RSS in the presence of Fe2+, ascorbic acid, and hydrogen peroxide. Reaction products were analyzed by Western blot following SDS-PAGE. RAG1 cleavage products were visualized with antibodies directed against MBP (Fig. 1B) or a RAG1 epitope (RAG1 amino acids 511–530; data not shown). Both antibodies revealed the presence of a novel species of ∼70 kD following incubation. Maximum formation of this product required both hydrogen peroxide and ferrous ion (although low levels of cleavage products were occasionally observed when Ca2+ was substituted for iron; Figs. 1B and 6A, below; data not shown). The same 70-kD product was observed either in the presence or absence of RAG2 protein (data not shown). Given that MBP contributes 40 kD to the observed product, the approximate location of the Fe2+-induced cleavage was 30 kD in from the amino terminus of truncated RAG1, around amino acid 630. Using antibodies to the carboxy-terminal histidine tag, a doublet of ∼47 kD was observed (Fig. 1C). A species of this size would be expected for the reciprocal cleavage product, suggesting that there is only one region in the protein in which Fe2+-induced cleavage occurs. (In some gels, a smaller carboxy-terminal fragment of ∼35 kD was observed, corresponding to a cleavage near amino acid 720.)
Figure 6.

The D600A/D708A double mutant is not subject to iron-induced cleavage. (A) Cleavage products were visualized with anti-MBP antibodies. Cleavage reactions were carried out in the absence of metal (−) or in the presence of iron (Fe2+) or calcium (Ca2+). (B) Carboxy-terminal cleavage products of RAG1 derivatives were visualized with anti-histidine tag antibody. The presence (+) or absence (−) of Fe2+ is indicated. (WT) Wild-type RAG1.
Single alanine substitutions at each of three acidic residues yields mutant proteins that are catalytically impaired
Having observed one or two Fe2+-induced cleavage sites in the vicinity of amino acids 630–720, we made a series of alanine substitutions of the acidic residues between amino acids 530–780. If the three-dimensional structure of the metal binding pocket brought a more distant residue into close proximity, additional cleaved species might have been expected. We further limited our initial selection of mutations as follows. First, we focused on aspartate and glutamate residues that are evolutionarily conserved in the RAG genes of humans, mouse, rabbit, chicken, Xenopus, and salmon. Second, mutations shown previously to support at least 25% of the wild-type level of recombination in vivo were not analyzed further. Third, we included two additional residues outside of this region, E811 and E962, that supported little or no V(D)J cleavage in vivo when changed to glutamine (M.A. Landree and D. Roth, pers. comm.). Residues mutated for this study are indicated in Figure 7 (below) and all mutant derivatives listed in Materials and Methods.
Figure 7.

Predicted structure of the core domain of RAG1. The structure predicted by the PSIpred program for RAG1 amino acids 481–1020 is shown above the murine RAG1 sequence. (H) Helix; (C) coil; (E) sheet. Structural predictions with a high confidence level (7–10) are indicated with uppercase letters; lower confidence levels are shown in lowercase. RAG1 sequences conserved between humans, mouse, chicken, Xenopus, and salmon are shown in uppercase letters; nonconserved residues in lowercase. Positions where either a D or E is observed are underlined. The positions of all residues mutagenized for this study are indicated with a black diamond; arrows and boldface type indicate the position of D600, D708, and E962.
By analogy with active site mutants in the transposase/integrase family, RAG mutants defective in the binding of a metal ion at the active site might be expected to be catalytically impaired while retaining their DNA-binding activity (though as discussed above, the metal ion may also play a role in DNA binding). As an initial screen, all mutant proteins were tested for their ability to cleave a 12-RSS in a standard oligonucleotide-based cleavage assay (Fig. 2; Table 1; data not shown) and for their ability to bind to DNA (see below). Equivalent amounts of protein, as determined by Coomassie blue staining following SDS-PAGE, were used for each assay. Reactions were carried out in Mn2+ to permit both nick and hairpin formation and also in Mg2+ in which, on a single RSS, only nicking occurs with wild-type RAG proteins. (Hairpin formation in Mg2+ requires a synapsed 12/23 signal pair, in which coupled cleavage can occur at both RSSs).
Figure 2.
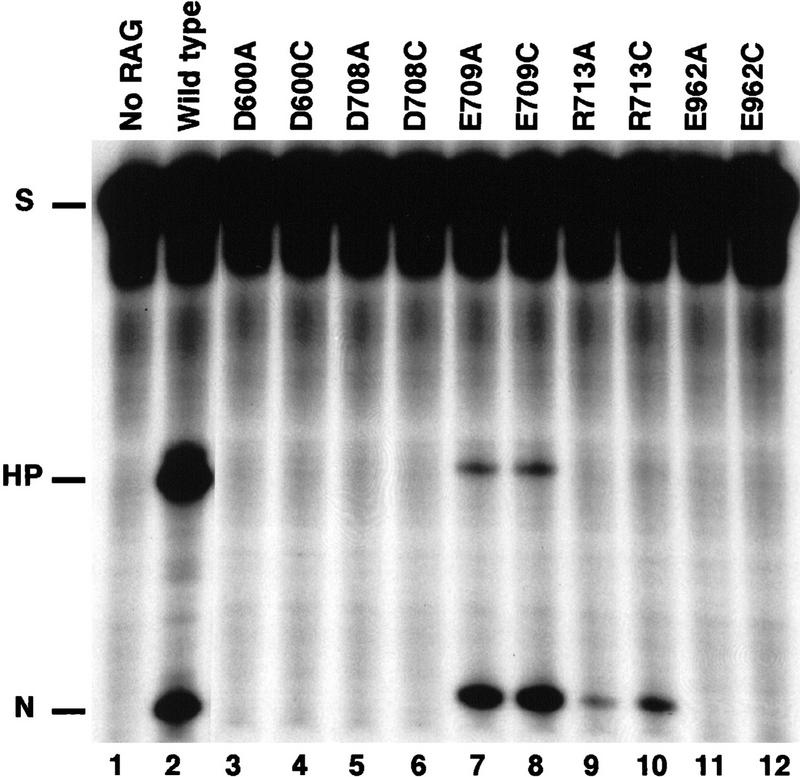
RAG1 mutant proteins D600A, D708A, and E962A are defective in V(D)J cleavage. Single site cleavage of a 32P-labeled 12-RSS oligonucleotide substrate (see Materials and Methods) is shown. Lane 1 contains no RAG protein; all other lanes contain the indicated RAG1 protein in addition to RAG2. Reactions were carried out in 0.1 mm Mn2+ for 30 min. The positions of the substrate (S) and nicked (N) and hairpin (HP) cleavage products are marked.
Table 1.
The effects of mutations in RAG1 on DNA binding and cleavage
| Mutant
|
RAG 1/2 bindinga
|
Nickingb
|
HPc
|
HP with prenickd
|
Coupled cleavagee
|
Tnf
|
|---|---|---|---|---|---|---|
| D546A | + | + | + | N.T. | + | N.T. |
| D600A | +++ | − | − | − | − | + |
| D600C | +++ | − | − | +/− | − | + |
| D708A | +++ | − | − | − | − | − |
| D708C | +++ | −(+/−) | −(+) | ++ | +/−:− | ++ |
| E709A | +++ | +++ | + {+++} | + {+++} | +++ | N.T. |
| E709C | +++ | +++ | + {+++} | + {+++} | +++ | N.T. |
| R713A | +++ | + | − | N.T. | +:− | N.T. |
| R713C | +++ | ++ | − | N.T. | ++:− | N.T. |
| E811A | + | + | + | N.T. | N.T. | |
| E962A | +++ | − | − | +/− | − | +/− |
| E962C | +++ | − | − | +/− | − | +/− |
| K966A | +++ | +++ | +++ | N.T. | N.T. | N.T. |
| D600A/D708A | +++ | − | − | − | − | N.T. |
| D600C/D708C | +++ | − | − | − | − | N.T. |
| D708A/E709A | +++ | − | − | − | − | N.T. |
| D708C/E709A | +++ | −(+/−) | −(+) | ++ | − | N.T. |
| E962A/D708C | +++ | − | − | − | − | N.T. |
| E962A/D708A | +++ | − | − | − | − | N.T. |
| E962A/D600A | +++ | − | − | − | − | N.T. |
(N.T.) Not tested.
DNA-binding levels indicated. Results listed reflect a summary of single and paired complex formation; these were similar in all cases.
The levels of nicking on a 12-RSS substrate relative to wild type are shown for standard reaction conditions; results from a longer incubation (2–4 hr) are shown in parentheses.
The levels of hairpinning observed following cleavage of an intact 12-RSS substrate are shown. Results from longer incubation (2–4) are shown in parentheses; brackets indicate results at higher Mn2+ concentration (1 mm).
The levels of hairpin formation on a prenicked 12-RSS substrate was shown. Brackets are as in c.
The levels of coupled cleavage in Mn2+ and Mg2+ are indicated relative to wild type. Only one level is indicated when the results were the same in both metal ions. Where the results differ, those in Mn2+ are shown before the colon; Mg2+ after the colon.
The levels of transposition with precleaved donor DNA is shown (see also Fig. 5).
All results are shown relative to wild-type RAG1 protein (+++).
Seven mutants exhibited <20% of the wild-type level of cleavage activity (D546A, D560A, D600A, D708A, E709A, E811A, E962A). No cleavage was detected with D600A, D708A, or E962A, whereas D546A and D560A showed residual activity at ∼5%–10% of wild type (Fig. 2, lanes 3,5,11; Table 1; data not shown). E709A produced a normal level of nicked product, but was severely defective in hairpin formation (Fig. 2, lane 7). However, an increase in Mn2+ concentration from the standard used here of 0.1 to 1 mm restored the level of hairpin formation for E709A to wild-type levels (Table 1). Increases in Mn2+ concentration (up to 10 mm) had no effect on the cleavage activity of the other mutants (data not shown). E811A, a poorly expressed and unstable derivative, appeared to have a decreased level of cleavage activity, but the levels were normal when normalized for the amount of protein present that could bind DNA (data not shown; see below).
It was necessary to distinguish catalytically impaired mutants from those defective in DNA binding. RAG1 can bind to a single RSS on its own or in conjunction with RAG2. When RAG1 and RAG2 bind together to a single RSS, two complexes (SC1 andSC2) are formed that differ in the amount of RAG2 present (C. Mundy and M. Oettinger, in prep.). When a pair of RSSs (one with a 12- and one with a 23-bp spacer) is available, a third (paired) complex containing RAG1 and RAG2 and the two signals is formed (Hiom and Gellert 1998). All three complexes can be distinguished by their differing mobilities in a gel retardation assay. The ability of the seven mutants described above to form these individual complexes was assessed. D546A and D560A were found to be defective in DNA binding and were therefore not considered further (Table 1; data not shown). E811A formed the expected complexes, but in very low abundance, making a detailed assessment of this protein's activity impractical (data not shown). D600A, D708A, E709A, and E962A formed all of the expected complexes at normal levels (Fig. 3, A, lanes 4, 6, 8, and 10, and B, lanes 5, 6, 9, 10, 17, 18), indicating that the defect in these mutants was not an obvious failure to make appropriate protein–protein contacts or a failure to complex with DNA. These mutations were thus suspected to involve the active site.
Figure 3.

DNA binding and synaptic complex formation. (A) Bandshift assays in the presence of a labeled 12 RSS are shown for the indicated RAG1 derivatives. The presence (+) or absence (−) of RAG1 and RAG2 in the reactions is indicated. (B) Bandshifts assays were carried out as in A with the addition of HMG1 and an unlabeled 23-RSS oligonucleotide (see Materials and Methods; Hiom and Gellert 1998). The presence (+) or absence (−) of the 23 RSS is noted. Lanes 3–20 contain RAG2 protein. The positions of the RAG1 alone (R1), single complex (SC1 and SC2), and paired complex (PC) bands, as well as the position of the labeled substrate (S), are indicated.
The catalytic properties of RAG1 derivatives carrying mutations at D600, D708, E709, and E962 were explored in greater detail to determine if their phenotypes were consistent with those expected of an active site mutant. (E709 was included in these analyses because hairpin formation with the E709A derivative showed an unusual response to metal ion concentration). The ability to carry out coupled cleavage with a 12/23 RSS pair was measured to exclude mutations that could be rescued by assembly into a synaptic complex (Fig. 4A). Coupled cleavage was assessed using an oligonucleotide substrate with duplex 12 and 23 RSS elements held together by a single-stranded tether, as described previously (Kim and Oettinger 1998). Although all four mutants could form a paired complex as determined by gel shift analysis, only E709A could support coupled cleavage, allowing both nick and hairpin formation in either Mn2+ or Mg2+ (see Fig. 4A, lanes 7 and 18). No nick or hairpin formation was observed for the other three.
Figure 4.
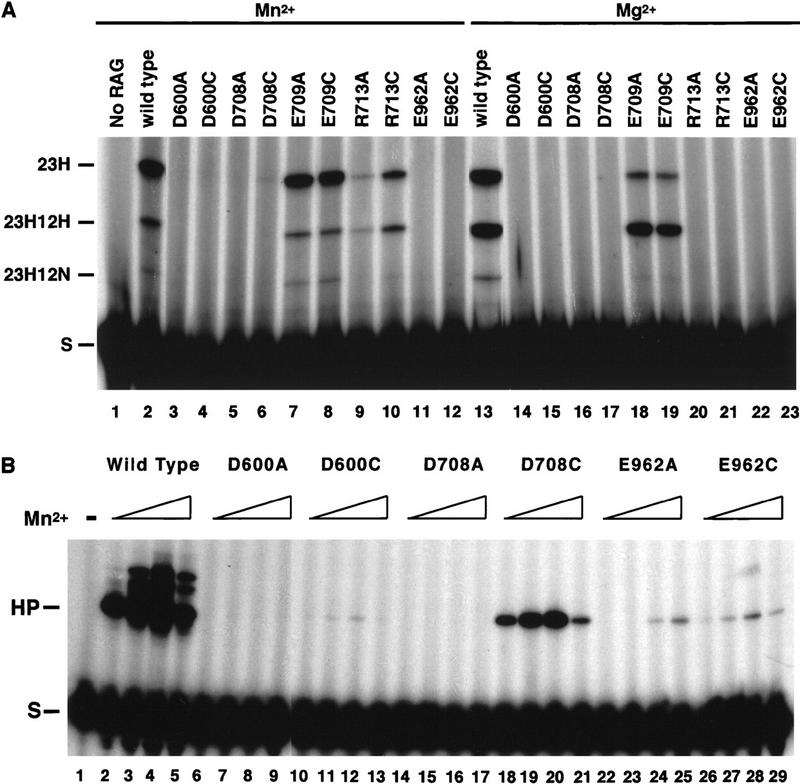
Effects of RAG1 mutations on coupled cleavage and hairpin formation. (A) Coupled cleavage of a tethered substrate by the indicated RAG1 derivatives in reaction buffer containing Mn2+ or Mg2+ is shown. The substrate (S) is labeled at the 23 RSS and the positions of cleavage products with a hairpin only at the 23 RSS (23H), a hairpin at the 23 RSS, and a nick at the 12 RSS (23H12N) or a hairpin at both signals (23H12H) are marked. (B) Hairpin formation on a prenicked substrate following a 2-hr incubation is shown for either the wild-type RAG1 protein or the indicated mutant derivatives. The presence of increasing concentrations of Mn2+ (0.05, 0.2, 2, 10 mm) is indicated by an open wedge.
A mutation in the metal binding site might have a differential effect on the two steps of V(D)J cleavage. Therefore, the ability of the mutant proteins to catalyze hairpin formation independently of the nicking step was assessed. A radioactively labeled oligonucleotide containing a 12RSS with a preformed nick at the 5′ end of the heptamer was subjected to cleavage with each of the mutants. No hairpin product was observed following incubation with D600A, D708A, or E962A (Table 1; Fig. 4B). Following a prolonged incubation of 2 hr instead of 30 min, E962A exhibited a very low but detectable level of hairpinning activity (Fig. 4B, lanes 24,25). E709A again showed a sensitivity to Mn2+ concentration, supporting some hairpinning at low Mn2+ concentration and nearly normal levels at higher Mn2+ concentrations (data not shown).
Cysteine substitution can rescue cleavage in Mn2+
To assess the roles of the acidic residues identified above in metal binding, we tested whether the substitution of cysteine for aspartate or glutamate could rescue activity. Mutants with individual cysteine substitutions at D600, D708, E709, and E962 were expressed and purified from E. coli, as were double mutants combining alanine and cysteine substitutions (see Table 1). Mutants with cysteine substitutions were subjected to the same series of experiments described above and the behavior of these mutants compared to their alanine counterparts. All the individual cysteine mutants as well as the double mutants (including D600A/D708A, Fig. 3A, lane 12) bound to RSS substrates and formed the expected complexes (see Table 1; Fig. 3).
Under standard reaction conditions, no rescue of cleavage (defined as increased activity when compared to the corresponding alanine substitution) was seen for any of the mutants on a single RSS over a range of Mn2+ concentration from 0.01 to 20 mmm (Fig. 2 shows results for 0.1 mm Mn2+). However, after prolonged incubation (2–4 hr), some rescue of nicking activity was detectable for D708C at 2 mm Mn2+, suggesting that this mutant protein was catalytically active but only weakly (data not shown). A far more striking rescue was observed when D708C was incubated with a prenicked substrate (Fig. 4B). In this case, hairpin activity was restored to near normal levels. A range of Mn2+ concentrations was tested and maximal activity was observed at a concentration of 2 mm for D708C (see Fig. 4B, lanes 18–21). Hairpin formation on a prenicked substrate was also detected for D600C, though at much lower levels (Fig. 4B, lanes 10–13), and for E962C at levels comparable to those seen with E962A (Fig. 4B, cf. lanes 23–25 and lanes 26–29). No cleavage was observed for the double mutants D600CD708C, D600CE962C, or D708CE962C (Table 1; data not shown).
When wild-type RAG proteins are incubated with RSS substrates in the presence of Mg2+, the formation of a synaptic complex containing an RSS pair is required to permit cleavage at either signal. We asked whether the catalytic activity of proteins carrying Cys substitutions might be affected under conditions in which the presence of a second signal would permit paired complex formation. Twelve and 23 RSSs were presented in cis on a tethered substrate or in trans on separate oligonucleotides, and cleavage by the mutant proteins assayed over a range of Mg2+ and Mn2+ concentrations (Fig. 4A; data not shown). Of the Cys substitution mutants, only E709C supported coupled cleavage, as expected from the results with a single RSS.
Because cysteine substitution affected the two steps of V(D)J cleavage differentially, we asked what effect this substitution would have on another RAG-mediated activity, transposition. To test the target DNA capture and strand transfer steps of transposition without the need to cleave the RSS, radioactively labeled precleaved 12 and 23 signals were incubated with a plasmid target in the presence of RAG2 and either wild-type RAG1 or the alanine or cysteine substitution mutants for D600, D708, and E962 (see Materials and Methods). Single ended transposition results in the formation of a labeled plasmid migrating as a nicked circle, whereas double-ended events yield a linear molecule (see Hiom et al. 1998; Materials and Methods). Results of transposition reactions carried out with Ca2+, Mn2+, or Mg2+ as the cofactor are shown (Fig. 5). Both E962A and E962C supported a very low level of transposition in Mn2+, reminiscent of the low level of hairpin formation observed on a prenicked substrate (Fig. 5, lanes 17,20). However, in keeping with the observed differential effect on first strand nicking versus strand transfer, D708C supported substantial levels of transposition, but only in Mn2+ (Fig. 5, lane 13). In addition, D600C, which showed minimal hairpin activity on a prenicked substrate, carried out transposition at levels only slightly lower than D708C. Interestingly, D600A could also mediate some transposition.
Figure 5.

Effects of RAG1 mutation on transposition. Transposition of 32P-labeled precleaved 12/23 RSS donors into a target plasmid is shown for wild-type (WT) and mutant RAG1 proteins in the presence of the indicated divalent metal ion. Arrows indicate the position of linearized and nicked target DNA. Lanes 14–20 come from the same exposure of the same gel, spliced to remove intervening lanes.
The double mutant D600A/D708A is resistant to Fe2+-induced hydroxyl radical cleavage
The observation of catalytic activity following substitution of Cys but not Ala at D600 and D708 strongly suggested that these residues are directly involved in metal binding and catalysis. The Fe2+-induced cleavage assay provides a second, independent criterion to assess the roles of these residues. Single alanine substitutions and double mutations were tested for their ability to undergo Fe2+-induced cleavage, and the pattern of cleavage products was compared to wild-type RAG1 protein. Following Western blot analysis with antibodies directed to the amino-terminal MBP moiety, the 70-kD product, comigrating with the hydrogen peroxide-dependent cleavage product seen with the wild-type protein, was observed for D600A, D708A, and E962A (Fig. 6A, lanes 5,8; data not shown). However, in a mutant where both D600 and D708 were simultaneously substituted with Ala, no cleavage was seen (Fig. 6A, lane 11). This observation strongly suggests that Fe2+ could no longer be bound by this derivative and suggests that D600 and D708 are directly involved in binding to the same divalent metal ion. As expected, the reciprocal carboxy-terminal fragments generated by iron cleavage were not observed for the D600A/D708A double mutant (Fig. 6B, lane 8). The other double mutant derivatives tested, E962A/D600A and E962A/D708A, continued to be subject to iron-induced cleavage (Fig. 6B, lanes 4,6), suggesting that E962A is not responsible for binding the same metal ion as D600 and D708 or is not accessible to iron-induced cleavage.
Discussion
Identification of active site residues in RAG1
V(D)J cleavage requires the combined action of the RAG1 and RAG2 proteins. Prior to this work, no information has been available about the nature of the active site of the V(D)J recombinase or even in which protein this activity resides. The active sites of other metal-dependent proteins that catalyze phosphoryl-transfer reactions have been identified by defining the metal-chelating carboxylates. Here we show that amino acid residues that coordinate a metal ion (as discussed below) lie within RAG1. Moreover, we show that mutations in these two acidic residues (and one additional Glu) result in RAG proteins that bind to DNA and assemble into a synaptic complex but that are catalytically impaired at both steps of V(D)J cleavage and in the strand transfer step of transposition. In addition, mutational analysis of RAG2 has revealed that no acidic residues in RAG2 are required for catalysis (Landree et al. 1999; M.A. Landree and D. Roth, pers. comm.).Taken together, these results strongly suggest that RAG1 contains most, if not all, of the active site for V(D)J cleavage.
Amino acid residues D600 and D708 coordinate a metal ion
Several lines of evidence support the conclusion that residues D600 and D708 coordinate a metal ion at the active site of RAG1. First, substitution of either residue with alanine completely abrogates cleavage in vitro, while leaving DNA binding, RAG1–RAG2 interaction and synaptic complex formation intact. Second, substitution with cysteine allows the restoration of some catalytic activity, permitting the strand transfer reactions of hairpin formation and transposition to proceed. This strongly argues that the sulfur group of cysteine is permitting metal ion coordination where the alanine mutant cannot. The rescue of activity with Cys substitution suggests that the coordinations of this metal ion by these residues are rather flexible. Third, iron-induced hydroxyl radical cleavage of RAG1 is inhibited when alanine substitutions are present at both residues indicating that the iron molecule can no longer be bound or is no longer present in a fashion that permits peptide bond cleavage. Fourth, the unusual behavior of the neighboring residue, E709, with respect to hairpin formation and sensitivity to metal ion concentration, while not supporting a direct role for this residue in metal binding, suggests that it lies near the active site.
The precise role of E962 in RAG1 function is less clear. E962 is required for catalytic activity, because substitution of either alanine or cysteine for this residue reduces both cleavage steps to barely detectable levels. The failure to support catalysis cannot simply be the result of some gross alteration in protein conformation, as DNA binding and synapsis are unaffected. In addition, an E962Q mutant of RAG1 is inactive in vivo and in vitro (M.A. Landree and D. Roth, pers. comm.), suggesting that the carboxylate oxygen must be the functional group of E962. Thus, although we do not have direct evidence for metal binding, such a role remains likely. It is possible that the location of E962 renders it insusceptible to iron cleavage or that a cleavage product from this site went undetected. In addition, a strict requirement for ligand coordination could prevent rescue of a Cys substitution in Mn2+ (in some metal binding sites even a D cannot substitute for an E) (Kanaya et al. 1990).
We cannot formally rule out a role for D546, D560, and E811 in metal binding or catalysis due to the abnormal DNA-binding properties of D546 and D560 and the expression and purification problems observed with E811. Experiments in vivo suggest that proteins carrying mutations at these three residues can support at least some level of V(D)J recombination, in contrast to derivatives carrying D600N, D708N, or E962Q, which cannot (M.A. Landree and D. Roth, pers. comm.).
A single active site
Mutations of D600, D708, and E962 all affect both steps of chemical cleavage (nick and hairpin), as well as inhibiting transposition. An additional residue (R713) that may also be involved in catalysis (see below) also abrogates both steps of cleavage. These results strongly argue that a single active site is involved in all of these steps, though each chemical step may require distinct additional residues or rely on different protomers. In the case of other well-characterized transposases/integrases, a single active site is also used repeatedly to carry out the distinct chemical steps of transposition/integration (Mizuuchi 1997).
Although mutations of D600 and D708 affect both steps of V(D)J cleavage, we observe differential effects of cysteine substitutions. Cysteine substitution at D600 or D708 provides little or no rescue of the nicking step under the conditions of our assay. In contrast, the strand transfer reactions required for hairpin formation and for transposition are partially restored (most notably for D708C). The nick and strand transfer reactions differ in a number of ways, including the use of an activated water molecule as the nucleophile for the first strand nick compared to the use of the 3′ hydroxyl on the nicked DNA strand for the hairpin step. Differential effects of mutations on the sequential chemical steps of transposition have been seen for bacterial transposons and retroviral integrases (e.g., Leavitt et al. 1993; Kennedy and Haniford 1996).
A mechanism involving at least two metal ions at a single active site has been proposed to explain how a single active site can catalyze two phosphoryl transfer steps (for discussion, see Steitz and Steitz 1993). According to the proposed model, metal ion A forms a metal OH− ion for nucleophilic attack of the scissile bond, whereas metal ion B stabilizes the pentacovalent cleavage intermediate and acts as a Lewis acid to facilitate the leaving of the 3′ oxyanion. Although our results do not permit a determination of the number of metal ions involved, the distinct behaviors of the mutant derivatives of E962 in comparison to those of D600 and D708 are compatible with the two-metal ion model. D600 and D708 most likely chelate the same metal, as evidenced by their similar response to Cys substitution, the catalytic inactivity of the D600C/D708C double mutant, and the inhibition of Fe2+-induced cleavage for the double but not single alanine substitution mutants. Assuming that E962 is also involved in chelation, the lack of an effect of E962A mutants on iron cleavage and the lack of Cys rescue suggests interaction with a different metal ion.
Is RAG1 a three-carboxylate motif protein?
There are two well-characterized families of endonucleases that require metal ions for catalysis. The first family includes the bacterial transposase/retroviral integrases RNase H and RuvC recombinase (Rice et al. 1996). Despite a lack of sequence similarity, all of these nucleotidyltransferases exhibit striking structural similarities within their active site domains and in the relative positioning of the catalytic triad of acidic residues (the so-called DDE motif). The second family includes the type II restriction enzymes and structurally homologous endonucleases. All of the restriction enzymes characterized contain a conserved structural domain and two carboxylates in a largely conserved sequence motif, DEK (Ban and Yang 1998).
A comparison of the structures of members of the DDE superfamily shows that the two aspartates are located on two adjacent β-strands and that one of these two is usually at the end of a strand (Yang and Steitz 1995). The third carboxylate, usually glutamate, is most often located at the amino terminus of an α helix, which may be disordered in the absence of substrate or metal ion. In some cases, the Glu residue is situated between the two aspartate residues in the primary structure (as in E. coli RNase H), whereas in others it is carboxy-terminal to the Ds (as in transposases and integrases) (Yang and Steitz 1995). In the tertiary structures, the two Asp residues along with the β strands superimpose well among members of the DDE motif family, whereas the Glu residue is present on opposite sides of the β strands in RNase H compared to the integrase/transposases. Finally, despite the motif name, the D and E residues are sometimes interchanged, as family members with DED motifs have been identified (Yang and Steitz 1995).
In the absence of structural information, the data presented in this paper are not sufficient to distinguish between these models and to determine into what structural family RAG1 might fall. However, considering the postulated evolutionary relationship of RAG proteins to transposable elements and the closely related stereochemical reaction catalyzed by RAG proteins and transposases, it is worth considering how closely our data, in conjunction with computer-generated predictions of RAG1 structure, would fit to the DDE model.
We used the PSIpred program to predict the secondary structure of RAG1 (Fig. 7) (Jones 1999). This program was first tested for its ability to predict the secondary structure of several nucleotidyl transferases of known structure (including Tn5 transposase and HIV integrase) and succeeded in accurately predicting these structures (W. Yang, unpubl.). According to the predicted structure, D600 is at the end of a β strand, D708 at the amino terminus of an α helix, and E962 lies within an unstructured coil between two α helices. Given the characteristic proximity of a basic residue to the carboxylate in the helical conformation of the triad (Jenkins et al. 1997), we made RAG1 mutants carrying alanine or cysteine substitutions at R713 and K966 and tested them in the binding and cleavage assays described above. No defect was seen for K966A (Table 1; data not shown). In contrast, R713A and R713C were severely defective: Less than one-tenth of the normal level of nicking and barely detectable levels of hairpin formation on a single RSS were observed, although DNA binding appeared normal (see Figs. 2, lanes 9 and 10, and 3B, lanes 13–16). Some activity was restored, primarily for R713C, when cleavage was carried out on a tethered substrate in Mn2+, but no cleavage was observed for either mutant in Mg2+ (Fig. 4, lanes 9,10,20,21). These results suggest that R713 may have a role in catalysis.
The evident ability of D708 to bind metal, its predicted position on an α helix, and the existence of the neighboring basic residue, R713, that is important for catalytic activity suggests that D708 could be analogous to the E of the DDE motif. D600 would then be equivalent to one of the two Ds in the DDE motif. Where would the third carboxylate come from? One obvious choice is E962. The order of these three acidic residues in the primary sequence would be unusual, but such positioning is possible, as evidenced by the structure of RNase H (see above). The distance of E962 from D600 and D708 is also unusual, with classical DDE and DEK domains usually contained within 200 residues. However, E962 could fold back to be adjacent to D600 and D708 in the tertiary structure.
Other candidates for the third residue remain formally possible. A role for D546, D600, or E811 in metal binding has not been ruled out (see above). Other residues in RAG1 or RAG2 that were not tested here could have a role, but mutagenesis of all evolutionarily conserved acidic residues in RAG1 and RAG2 has failed to identify any additional candidates (Landree et al. 1999; M.A. Landree and D.B. Roth, pers. comm.). Finally, RAG1 might not contain a DDE or DEK motif, thereby relying on some alternative method of metal binding. Given the reversal of cleavage evidenced by the RAG recombinase as compared to other transposases (initial nicking of the top rather than bottom strand), some deviation from the classical DDE motif might be expected. Resolution of these issues awaits structural information.
Materials and methods
Plasmid construction and site-directed mutagenesis
DNA manipulations were carried out according to standard protocols. To generate pDRK534, a RAG1 fragment containing a 5′ Shine–Dalgarno site, RAG1 nucleotides encoding amino acids 352–1040, and a carboxy-terminal 8 histidine tag was generated by Pfu PCR from LG6 (Cuomo et al. 1994), subcloned into pDRK-N(M) (Kim and McHenry 1996), and the fidelity of the PCR confirmed by DNA sequencing. For pDRK573, a fragment containing a PA1 promoter was introduced upstream of the MBP gene in pMal-c2x (NEB) and the resulting NdeI–KpnI fragment carrying the PA1 promoter and MBP was subcloned in-frame into pDRK534 with an adaptor oligonucleotide. A complete sequence of plasmids is available on request.
All RAG1 mutants are derived from pDRK534 using QuickChange site-directed mutagenesis kit (Stratagene) following the manufacturer's protocols. Plasmids were transformed in E. coli DH5α and the resulting mutants screened by sequencing. Constructs encoding MBP fusion proteins carrying these same mutations were generated by subcloning the RAG1 mutant derivative from the pDRK534 backbone into pDRK573. A complete list of mutants tested follows: D536A, D546A, D546C, E547A, E547C, D551A, D560A, D569A, D572A/E573A, D582A, D584A/D585A, E643A, D600A, D600C, D604A, D661A, D664A, D708A, D708C, E709A, E709C, D600A/D708A, D600C/D708C, D708A/E709A, D708C/E709C, K710A, R713A, R713C, E716A, E716K, E719A, E719C, D731A, E736A, E811A, E811C, E962A, E962C, E962A/D708C, E962A/D708A, E962A/D600A, K966A, K966C.
Proteins
HMG1 protein was a generous gift from T. Paull (NIH). Recombinant RAG2 from vaccinia infection of HeLa cells was produced as described previously. Recombinant RAG1 was generated as follows. Overnight cultures of E. coli DH5α containing the appropriate RAG1 expression plasmid were diluted 40-fold into 1 liter of LB supplemented with 100 μg/ml Amp. Cells were grown at 37°C to an OD600 of 0.7–0.8, induced with IPTG (1 mm final concentration), and incubated at room temperature for 3 hr with the addition of 50 μg/ml amp. Cells were harvested at 4200 rpm for 15 min, resuspended in 20 ml of Ni-binding buffer (20 mm Tris at pH 7.9, 0.5 m NaCl, 10% glycerol, 2 mm β-mercaptoethanol, and 1 mm PMSF) containing 5 mm imidazole, and frozen in liquid nitrogen. Frozen cells were thawed and brought to a final volume of 35 ml with Ni-binding buffer containing 5 mm imidazole. Cells were lysed by sonication (four to six times at 80% power, Fisher Scientific) in the presence of 0.2 mg/ml lysozyme, subject to centrifugation in a Beckman SW28 rotor, at 25,000 rpm for 30 min at 4°C, and the resulting supernatant added to 0.5 ml of pre-equilibrated Ni–Sepharose resin (Pharmacia) and incubated at 4°C for 1 hr with gentle shaking. Purification by Ni-affinity (elution at ∼200 mm imidazaole) was as described (Kim and Oettinger 1998).
Fe-induced RAG1 cleavage
MBP RAG1 (from pDRK573, 16 pmoles) with or without RAG2 protein (16 pmoles) was mixed with 12 RSS substrate (28 pmoles, VDJ100/101, Fe(NH4)SO4 (final concentration 0.02 mm), and ascorbic acid (final concentration 20 mm), and incubated at room temperature for 30 min. Hydrogen peroxide (final 0.2%) was added to the reaction for 10 sec, and the reaction was stopped by adding SDS gel-loading solution. Proteins were separated by 7.5% SDS-PAGE and transferred to nitrocellulose membrane. RAG1 fragments were identified by Western blot using anti-MBP (NEB), anti-RAG1 (Santa Cruz Biotech) or anti-His (Santa Cruz Biotech) antibodies. All data shown are from experiments without RAG2 protein but were indistinguishable from those containing RAG2 protein.
Cleavage and binding assays
Cleavage reactions were performed as described previously (Cuomo et al. 1996; Kim and Oettinger 1998) except as noted. Cleavage reactions were carried out in 0.1 mm Mn2+ at 30°C for 30 min, except where indicated. Binding reactions to detect single RSS complexes (SC1 and SC2), were carried out in 1 mm CaCl2, 25 mm K-MOPS (pH 7.0), 50 mm K-Glu, 2 mm DTT, and 100 μg/ml BSA. Each 10-μl reaction contained 20 fmoles of radiolabeled V(D)J 100/101, 100 ng of RAG1 protein, 50 ng of RAG2 protein, and 2 pmoles of cold, single-stranded, nonspecific competitor DNA. Paired complex formation was carried out after the method described in Hiom and Gellert (1998), except that 200 fmoles of unlabeled V(D)J 132/133 (Cuomo et al. 1996) and 50 ng of HMG-1 were added to each reaction. All binding reactions took place at 37°C for 30 min and were stopped by adding 2 μl of 50% glycerol loading dye. Reactions were analyzed without cross-linking in either a 4%–20% precast TBE gel (NOVEX) or in a 4% TBE gel.
Transposition
Transposition reactions were carried out after the method described in Hiom et al. (1998). Precleaved donor DNAs were constructed by annealing VDJ106 (5′-CACAGTGCTACAGACTGGAACAAAAACCCAGGTCTC) and VDJ104 (5′-TGAGACCTGGTTTTTGTTCCAGTCTGTAGCACTGTG) for the 12 RSS and VDJ134 (5′-CACAGTGGTAGTACTCCACTGtCTGGCTGTACAAAAACCCAGGT) and YD24 (5′-TACCTGGGTTTTTGTACAGCCAGACAGTGGAGTACTACCACTGTG) for the 23 RSS in 25 mm MOPS (pH 7.0). pUC19 DNA was used as the target sequence. Donor DNAs were 3′end-labeled on the top strand (nonamer 5′-ACAAAAACC-3′) with 32P-labeled cordycepin (NEN) using recombinant terminal deoxynucleotidyl transferase.
Transposition assays were initiated by the addition of RAG1 (∼80 ng), RAG2 (∼40 ng), and HMG1 (∼30 ng) proteins to a 10-μl reaction mixture containing 0.02 pmole of 32P-labeled, precleaved donor DNA in 25 mm KMOPS (pH 7.0), 5 mm CaCl2, 75 mm potassium acetate, 1 mm DTT, and 100 μg/ml BSA. After a 30-min incubation at 37°C, pUC19 target DNA (200 ng) was added along with PEG 8000 (10% final concentration) and, for Figure 5, either 5 mm MgCl2 or 2 mm MnCl2 was added (a range of concentrations from 0.1 to 50 mm was tested, and the optimal conditions are presented). Following a 1-hr incubation at 37°C, reactions were treated with SDS and proteinase K, and reaction products visualized by autoradiography of samples separated by electrophoresis through a 1.2% agarose gel.
Acknowledgments
We gratefully acknowledge the technical assistance provided by Doug Chan and thank Mark Landree and David Roth for their generous communication of results prior to publication. We thank T. Paull (NIH) for the gift of HMG1 protein and members of the Oettinger laboratory for helpful discussions. This work was supported by a postdoctoral fellowship from the Cancer Research Institute (D.R.K.) and by NIH grant GM58026 (M.A.O.), the Leukemia Society Scholars Program (M.A.O.), and Hoechst Ag (M.A.O.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Oettinger@frodo.mgh.harvard.edu; FAX (617) 726-5949.
References
- Agrawal A, Eastman QM, Schatz DG. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 1998;394:744–751. doi: 10.1038/29457. [DOI] [PubMed] [Google Scholar]
- Allingham JS, Pribil PA, Haniford DB. All three residues of the Tn 10 transposase DDE catalytic triad function in divalent metal ion binding. J Mol Biol. 1999;289:1195–1206. doi: 10.1006/jmbi.1999.2837. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York, NY: Greene Publishing Associates and Wiley-Interscience; 1989. [Google Scholar]
- Ban C, Yang W. Structural basis for MutH activation in E. coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J. 1998;17:1526–1534. doi: 10.1093/emboj/17.5.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese LS, Steitz TA. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: A two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujacz G, Jaskolski M, Alexandratos J, Wlodawer A, Merkel G, Katz RA, Skalka AM. The catalytic domain of avian sarcoma virus integrase: Conformation of the active-site residues in the presence of divalent cations. Structure. 1996;4:89–96. doi: 10.1016/s0969-2126(96)00012-3. [DOI] [PubMed] [Google Scholar]
- Cao W. Binding kinetics and footprinting of TaqI endonuclease: Effects of metal cofactors on sequence-specific interactions. Biochemistry. 1999;38:8080–8087. doi: 10.1021/bi9903796. [DOI] [PubMed] [Google Scholar]
- Cuomo CA, Oettinger MA. Analysis of regions of RAG-2 important for V(D)J recombination. Nucleic Acids Res. 1994;22:1810–1814. doi: 10.1093/nar/22.10.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuomo CA, Kirch SA, Gyuris J, Brent R, Oettinger MA. Rch1, a protein that specfically interacts with the recombination activating gene, RAG-1. Proc Natl Acad Sci. 1994;91:6156–6160. doi: 10.1073/pnas.91.13.6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuomo CA, Mundy CL, Oettinger MA. DNA sequence and structure requirements for cleavage of V(D)J recombination signal sequences. Mol Cell Biol. 1996;16:5683–5690. doi: 10.1128/mcb.16.10.5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettner N, Metzger JW, Lederer T, Hulmes JD, Kisker C, Hinrichs W, Ellestad GA, Hillen W. Proximity mapping of the Tet repressor-tetracycline-Fe2+ complex by hydrogen peroxide mediated protein cleavage. Biochemistry. 1995;34:22–31. doi: 10.1021/bi00001a004. [DOI] [PubMed] [Google Scholar]
- Gellert M. Recent advances in understanding V(D)J recombination. Adv Immunol. 1997;64:39–64. doi: 10.1016/s0065-2776(08)60886-x. [DOI] [PubMed] [Google Scholar]
- Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R, Davies DR. Three new structures of the core domain of HIV-1 integrase: An active site that binds magnesium. Proc Natl Acad Sci. 1998;95:9150–9154. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiom K, Gellert M. Assembly of a 12/23 paired signal complex: A critical control point in V(D)J recombination. Mol Cell. 1998;1:1011–1019. doi: 10.1016/s1097-2765(00)80101-x. [DOI] [PubMed] [Google Scholar]
- Hiom K, Melek M, Gellert M. DNA transposition by the RAG1 and RAG2 proteins: A possible source of oncogenic translocations. Cell. 1998;94:463–470. doi: 10.1016/s0092-8674(00)81587-1. [DOI] [PubMed] [Google Scholar]
- Jeggo PA. Identification of genes involved in repair of DNA double-strand breaks in mammalian cells. Radiat Res. 1998;150:S80–S91. [PubMed] [Google Scholar]
- Jenkins TM, Esposito D, Engelman A, Craigie R. Critical contacts between HIV-1 integrase and viral DNA identified by structure-based analysis and photo-crosslinking. EMBO J. 1997;16:6849–6859. doi: 10.1093/emboj/16.22.6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- Kanaya S, Kohara A, Miura Y, Sekiguchi A, Iwai S, Inoue H, Ohtsuka E, Ikehara M. Identification of the amino acid residues involved in an active site of Escherichia coli ribonuclease H by site-directed mutagenesis. J Biol Chem. 1990;265:4615–4621. [PubMed] [Google Scholar]
- Kennedy AK, Haniford DB. Isolation and characterization of IS10 transposase separation of function mutants: Identification of amino acid residues in transposase that are important for active site function and the stability of transposition intermediates. J Mol Biol. 1996;256:533–547. doi: 10.1006/jmbi.1996.0106. [DOI] [PubMed] [Google Scholar]
- Kim DR, McHenry CS. Biotin tagging deletion analysis of domain limits involved in protein-macromolecular interactions. Mapping the tau binding domain of the DNA polymerase III alpha subunit. J Biol Chem. 1996;271:20690–20698. doi: 10.1074/jbc.271.34.20690. [DOI] [PubMed] [Google Scholar]
- Kim DR, Oettinger MA. Functional analysis of coordinated cleavage in V(D)J recombination. Mol Cell Biol. 1998;18:4679–4688. doi: 10.1128/mcb.18.8.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirch SA, Sudarsanam P, Oettinger MA. Regions of RAG1 protein critical for V(D)J recombination. Eur J Immunol. 1996;26:886–891. doi: 10.1002/eji.1830260425. [DOI] [PubMed] [Google Scholar]
- Kulkosky J, Skalka AM. Molecular mechanism of retroviral DNA integration. Pharmacol Ther. 1994;61:185–203. doi: 10.1016/0163-7258(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Landree, M.A., J.A. Wobbenmeyer, and D.B. Roth. 1999. Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination. Gene & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Leavitt AD, Shiue L, Varmus HE. Site-directed mutagenesis of HIV-1 integrase demonstrates differential effects on integrase functions in vitro. J Biol Chem. 1993;268:2113–2119. [PubMed] [Google Scholar]
- Lewis SM. The mechanism of V(D)J joining: Lessons from molecular, immunological, and comparative analyses. Adv Immunol. 1994;56:27–150. doi: 10.1016/s0065-2776(08)60450-2. [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen J, Garrett RA, Kjems J. Mapping metal ions at the catalytic centres of two intron-encoded endonucleases. EMBO J. 1997;16:3272–3281. doi: 10.1093/emboj/16.11.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maignan S, Guilloteau JP, Zhou-Liu Q, Clement-Mella C, Mikol V. Crystal structures of the catalytic domain of HIV-1 integrase free and complexed with its metal cofactor: High level of similarity of the active site with other viral integrases. J Mol Biol. 1998;282:359–368. doi: 10.1006/jmbi.1998.2002. [DOI] [PubMed] [Google Scholar]
- McBlane JF, van Gent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, Oettinger MA. Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell. 1995;83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K. Polynucleotidyl transfer reactions in site-specific DNA recombination. Genes Cells. 1997;2:1–12. doi: 10.1046/j.1365-2443.1997.970297.x. [DOI] [PubMed] [Google Scholar]
- Oettinger MA. V(D)J recombination: on the cutting edge. Curr Opin Cell Biol. 1999;11:325–329. doi: 10.1016/S0955-0674(99)80044-1. [DOI] [PubMed] [Google Scholar]
- Pecoraro VL, Hermes JD, Cleland WW. Stability constants of Mg2+ and Cd2+ complexes of adenine nucleotides and thionucleotides and rate constants for formation and dissociation of MgATP and MgADP. Biochemistry. 1984;23:5262–5271. doi: 10.1021/bi00317a026. [DOI] [PubMed] [Google Scholar]
- Pingoud A, Jeltsch A. Recognition and cleavage of DNA by type-II restriction endonucleases. Eur J Biochem. 1997;246:1–22. doi: 10.1111/j.1432-1033.1997.t01-6-00001.x. [DOI] [PubMed] [Google Scholar]
- Plasterk R. V(D)J recombination. Ragtime jumping. Nature. 1998;394:718–719. doi: 10.1038/29389. [DOI] [PubMed] [Google Scholar]
- Polard P, Chandler M. Bacterial transposases and retroviral integrases. Mol Microbiol. 1995;15:13–23. doi: 10.1111/j.1365-2958.1995.tb02217.x. [DOI] [PubMed] [Google Scholar]
- Rice P, Craigie R, Davies DR. Retroviral integrases and their cousins. Curr Opin Struct Biol. 1996;6:76–83. doi: 10.1016/s0959-440x(96)80098-4. [DOI] [PubMed] [Google Scholar]
- Roberts RJ, Halford SE. Recognition sequences and cleavage properties. In: Linn SM, LLoyd RS, Roberts RJ, editors. Nucleases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 35–88. [Google Scholar]
- Rodgers KK, Villey IJ, Ptaszek L, Corbett E, Schatz DG, Coleman JE. A dimer of the lymphoid protein RAG1 recognizes the recombination signal sequence and the complex stably incorporates the high mobility group protein HMG2. Nucleic Acids Res. 1999;27:2938–2946. doi: 10.1093/nar/27.14.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth DB, Craig NL. VDJ recombination: A transposase goes to work. Cell. 1998;94:411–414. doi: 10.1016/s0092-8674(00)81580-9. [DOI] [PubMed] [Google Scholar]
- Sadofsky MJ, Hesse JE, McBlane JF, Gellert M. Expression and V(D)J recombination activity of mutated RAG-1 proteins. Nucleic Acids Res. 1993;21:5644–5650. doi: 10.1093/nar/21.24.5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadofsky MJ, Hesse JE, Gellert M. Definition of a core region of RAG-2 that is functional in V(D)J recombination. Nucleic Acids Res. 1994;22:1805–1809. doi: 10.1093/nar/22.10.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnovsky RJ, May EW, Craig NL. The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. EMBO J. 1996;15:6348–6361. [PMC free article] [PubMed] [Google Scholar]
- Silver DP, Spanopolou E, Mulligan RC, Baltimore D. Dispensable sequence motifs in the RAG-1 and RAG-2 genes for plasmid V(D)J recombination. Proc Natl Acad Sci. 1993;90:6100–6104. doi: 10.1073/pnas.90.13.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz TA. DNA polymerases: Structural diversity and common mechanisms. J Biol Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gent DC, Mizuuchi K, Gellert M. Similarities between initiation of V(D)J recombination and retroviral integration. Science. 1996;271:1592–1594. doi: 10.1126/science.271.5255.1592. [DOI] [PubMed] [Google Scholar]
- Wei CH, Chou WY, Huang SM, Lin CC, Chang GG. Affinity cleavage at the putative metal-binding site of pigeon liver malic enzyme by the Fe(2+)-ascorbate system. Biochemistry. 1994;33:7931–7936. doi: 10.1021/bi00191a021. [DOI] [PubMed] [Google Scholar]
- Yang W, Steitz TA. Recombining the structures of HIV integrase, RuvC and RNase H. Structure. 1995;3:131–134. doi: 10.1016/s0969-2126(01)00142-3. [DOI] [PubMed] [Google Scholar]
- Zaychikov E, Martin E, Denissova L, Kozlov M, Markovtsov V, Kashlev M, Heumann H, Nikiforov V, Goldfarb A, Mustaev A. Mapping of catalytic residues in the RNA polymerase active center. Science. 1996;273:107–109. doi: 10.1126/science.273.5271.107. [DOI] [PubMed] [Google Scholar]