

Abstract
In mammals, the first branchial arch (BA1) develops into a number of craniofacial skeletal elements including the jaws and teeth. Outgrowth and patterning of BA1 during early embryogenesis is thought to be controlled by signals from its covering ectoderm. Here we used Cre/loxP technology to inactivate the mouse Fgf8 gene in this ectoderm and have obtained genetic evidence that FGF8 has a dual function in BA1: it promotes mesenchymal cell survival and induces a developmental program required for BA1 morphogenesis. Newborn mutants lack most BA1-derived structures except those that develop from the distal-most region of BA1, including lower incisors. The data suggest that the BA1 primordium is specified into a large proximal region that is controlled by FGF8, and a small distal region that depends on other signaling molecules for its outgrowth and patterning. Because the mutant mice resemble humans with first arch syndromes that include agnathia, our results raise the possibility that some of these syndromes are caused by mutations that affect FGF8 signaling in BA1 ectoderm.
Keywords: Agnathia, Barx1, BMP4, endothelin-1, FGF8, first branchial arch
In vertebrates, many structures including parts of the face develop from small primordia or “buds” consisting of undifferentiated mesenchymal cells covered by a layer of epithelium. One of these primordia is the first branchial arch (BA1), which in mammals develops into teeth, skeletal elements of the jaws, lateral skull wall, and middle ear, as well as part of the tongue and other soft tissue derivatives. In the mouse embryo, BA1 first becomes apparent at the six- to eight-somite stage [approximately embryonic day (E) 8.25] as a small swelling on the side of the head. This bud rapidly increases in size as cranial neural crest cells migrate into and proliferate within the arch. This neural crest-derived mesenchyme, which is termed ectomesenchyme and is localized immediately subadjacent to the covering epithelium, differentiates into cartilagenous (chondrocranial) and osseous (dermatocranial) structures. The central core of BA1 mesenchyme is derived from somitomeres and forms craniofacial muscle and vascular tissue. At ∼E9.5 the outgrowing BA1 on each side of the head develops into the primordia of the mandibular and maxillary arches, which grow toward the ventral midline. Subsequently, multiple fusions involving the paired mandibular arches, maxillary arches, and the frontonasal process establish the basic form of the face. Errors in these complex morphogenetic events cause craniofacial anomalies including defects of the mandible, which are among the most common malformations in humans. More than 130 human syndromes appear to involve incorrect development of the BA1. The most severe of these syndromes are associated with agnathia, a condition in which the lower jaw and other BA1-derived structures are absent (for review, see Bixler et al. 1985; Escobar 1993).
Classic experimental embryological studies have suggested that development of BA1 mesenchyme is controlled by signals from its covering ectoderm, which regulate cell proliferation, survival, patterning, and differentiation. In turn, signals from the mesenchyme may influence development of the ectoderm. The molecular basis of these epithelial–mesenchymal interactions is not yet well understood. Genetic analysis in mice has provided evidence that members of several homeobox gene families are necessary for normal BA1 development (for review, see Francis-West et al. 1998). However, relatively little is known about the precise role of the epithelium and the signaling molecules it produces in the regulation of outgrowth and patterning at early stages of BA1 development.
Members of the FGF family, particularly FGF8, have been implicated as epithelial signals that regulate gene expression during BA1 development. When beads soaked in FGF8 (FGF8-beads) were inserted into isolated mandibular mesenchymal explants, expression of several genes could be induced (for review, see Peters and Balling 1999). Although such bead implantation studies provide valuable clues to the identity of signaling molecules and potential target genes, the long-term consequences of altering gene expression on skeletal patterning cannot be addressed in organ culture. Furthermore, firm conclusions about gene function, particularly when several members of a multigene family are coexpressed, require loss-of-function studies. However, standard genetic approaches cannot be used to analyze FGF8 function in craniofacial development because Fgf8−/− embryos die during gastrulation (Sun et al. 1999). Here we have used Cre/loxP technology to circumvent this problem and to generate mutant embryos in which the Fgf8 gene is inactivated in BA1 ectoderm. Our analysis identifies FGF8 as an epithelial signal essential for the outgrowth and patterning of BA1 from the earliest stages of its development.
Results
Cre-mediated inactivation of Fgf8 in BA1 ectoderm
Inactivation of a particular gene in a specific tissue can be achieved by mating mice carrying a mutant allele in which essential regions of the gene are flanked by loxP sites (the recognition sequence for the site-specific DNA recombinase, Cre) with mice that express the cre gene in the tissue of interest (Gu et al. 1994; Tsien et al. 1996; Kulkarni et al. 1999). To inactivate Fgf8 in BA1, we used mice carrying Fgf8flox, an allele in which vital coding exons are flanked by loxP sites. Fgf8flox has wild-type Fgf8 activity but can be converted to a null allele (Fgf8Δ2,3) by Cre-mediated recombination (Meyers et al. 1998). Using the mating scheme outlined in Figure 1A we produced animals that were compound heterozygotes for Fgf8flox and a null allele of Fgf8, and which also carried Nes–cre1, a transgene containing a modified cre gene (Lewandoski et al. 1997) under the control of the rat Nestin promoter and intron-2 enhancer (Zimmerman et al. 1994; A. Trumpp, G.R. Martin, and J.M. Bishop, unpubl.). In Fgf8flox/Fgf8 null;Nes–cre1 embryos, hereafter referred to as Fgf8;Nes–cre or mutant embryos, Cre-mediated conversion of Fgf8flox to a null allele occurs only in cells that express Nes–cre1, resulting in complete loss of Fgf8 gene function in those cells and their descendants. Their Fgf8flox/Fgf8 null littermates that did not inherit Nes–cre1 are phenotypically normal and served as controls.
Figure 1.
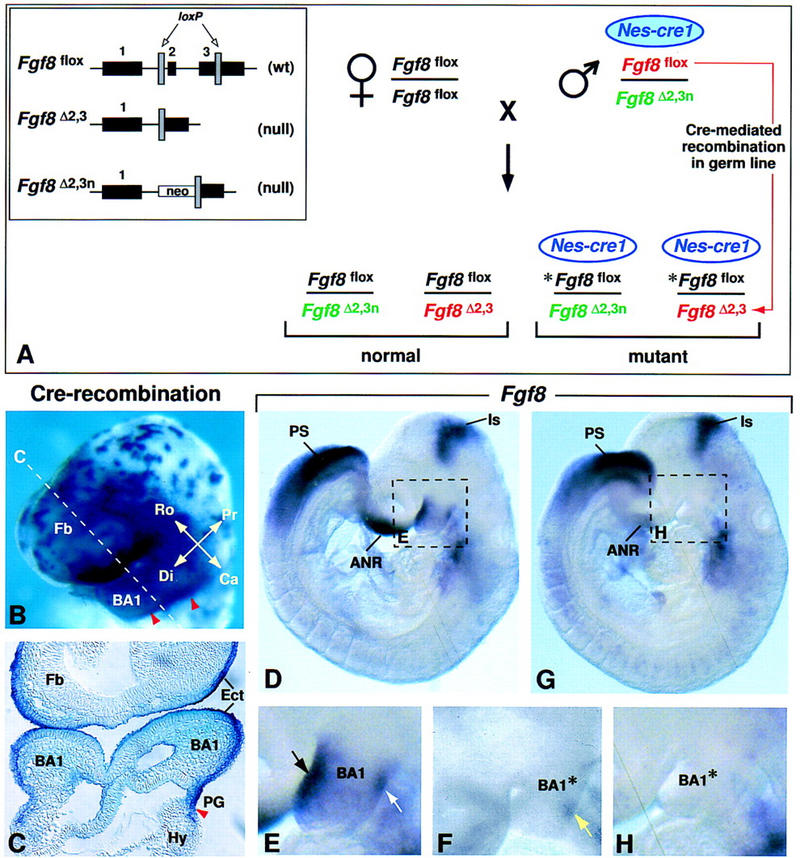
Inactivation of the Fgf8 gene in BA1 ectoderm. (A) Breeding scheme used to generate 50% mutant offspring. The three different Fgf8 alleles present in this cross are illustrated in the box on the left. The Fgf8flox allele has wild-type (wt) activity and can be converted to a null allele (Fgf8Δ2,3) by Cre-mediated recombination (Meyers et al. 1998). The Nes–cre1 gene used in this cross is imprinted, and is active in somatic tissues as described (see text) only when it is paternally inherited. However, it is active in the germ line irrespective of whether it is paternally or maternally inherited (A. Trumpp, B. Bates, R. Jaenisch, G.R. Martin, and J.M. Bishop, unpubl.). In the cross performed, the parental male has a normal phenotype because he carries a maternally inherited Nes–cre1 transgene (blue filled oval), which has little activity in somatic cells. When this transgene is transmitted to his offspring (unfilled oval), it is active in somatic tissues. In addition, this male transmits two different Fgf8 null alleles: Fgf8Δ2,3n (shown in green) and Fgf8Δ2,3 (shown in red), which is generated by Cre-mediated recombination in his germ line of the Fgf8flox allele he carries (also shown in red). Thus, all offspring carry a paternally inherited Fgf8 null allele, as well as a maternally inherited Fgf8flox allele (shown in black). In the 50% of offspring that inherit Nes–cre1, the floxed allele (*Fgf8flox) undergoes Cre-mediated recombination, resulting in tissue-specific inactivation of Fgf8. (B,C) Assay for Cre-mediated recombination using the Z/AP reporter gene (Lobe et al. 1999). Cells in which the Nes–cre1 gene was active produce human alkaline phosphatase (hAP). (B) Nes–cre1;Z/AP embryo at E9.0 (16 somites) stained for hAP activity in whole mount. The dashed line indicates the level of the section shown in C. The caudal limit of recombination (marked by red arrowheads) is located in the first pharyngeal groove. Note that recombination occurs in BA1 ectoderm. (D–H) Fgf8 expression as detected by whole mount RNA in situ hybridization in D and E control and F to H Fgf8;Nes–cre mutant embryos at E9.0 (14 somites). The dashed boxes in D and G indicate the regions shown at higher magnification in E and H, respectively. In the mutant embryo, the levels of Fgf8 RNA in most expression domains are similar to those in the normal embryo (D). However, they appear to be reduced in the front of the face/anterior neural ridge, and, significantly, no Fgf8 RNA is detected in BA1. Black and white arrows in E point to the normal Fgf8 expression domains on the rostral and caudal side of BA1, respectively. (F) Fgf8 RNA is detected on the caudal side of BA1 (yellow arrow) in a mutant embryo at a slightly earlier stage. (ANR) Anterior neural ridge; (BA1) first branchial arch; (BA1*) mutant BA1; (Ca) caudal; (Di) distal; (Ect) ectoderm; (Fb) forebrain; (Hy) hyoid arch (second branchial arch); (Is) isthmic constriction; (PG) pharyngeal groove; (Pr) proximal; (PS) primitive streak; (Ro) rostral.
The stage and tissue specificity of Nes–cre1 activity was determined by crossing males carrying Nes–cre1 to females carrying the Z/AP reporter gene, which produces human alkaline phosphatase (hAP) only after it has undergone Cre-mediated recombination (Lobe et al. 1999). A detailed description of the results of that analysis will be reported elsewhere. Here we focused on an assessment of cre activity in the head. In Nes–cre1;Z/AP embryos assayed at ∼E9.0 (16-somite stage), little hAP activity was detected in the brain, but strong activity was detected in surface ectoderm, particularly that covering BA1 (Fig. 1B,C). The domain of Nes–cre1 activity in BA1 ectoderm had a sharp caudal limit at the level of the first pharyngeal groove (red arrowheads in Fig. 1B,C). Analysis at earlier stages suggested that Cre-mediated recombination first occurred in the cranial surface ectoderm at ∼E7.75, and that subsequently, recombination occurred in cells on the rostral side of BA1 before it occurred in those on the caudal side (data not shown; see below).
During BA1 development, Fgf8 is expressed in a dynamic pattern in the surface ectoderm (Crossley and Martin 1995; Mahmood et al. 1995; Kettunen and Thesleff 1998). At E8.5, when the nascent arch is a discrete bud, Fgf8 expression is detected at low levels throughout the ectoderm, but subsequently becomes more robust in the rostral- and caudal-most aspects of BA1 (Fig. 1D,E; data not shown). By E9.5, Fgf8 expression becomes restricted to the ectoderm on the rostral side. By E10.5, when the maxillary and mandibular primordia have expanded, Fgf8 is abundant in the so-called “oral ectoderm,” which covers the caudal side of the maxillary and the rostral side of the mandibular arches (see Fig. 6O).
Figure 6.
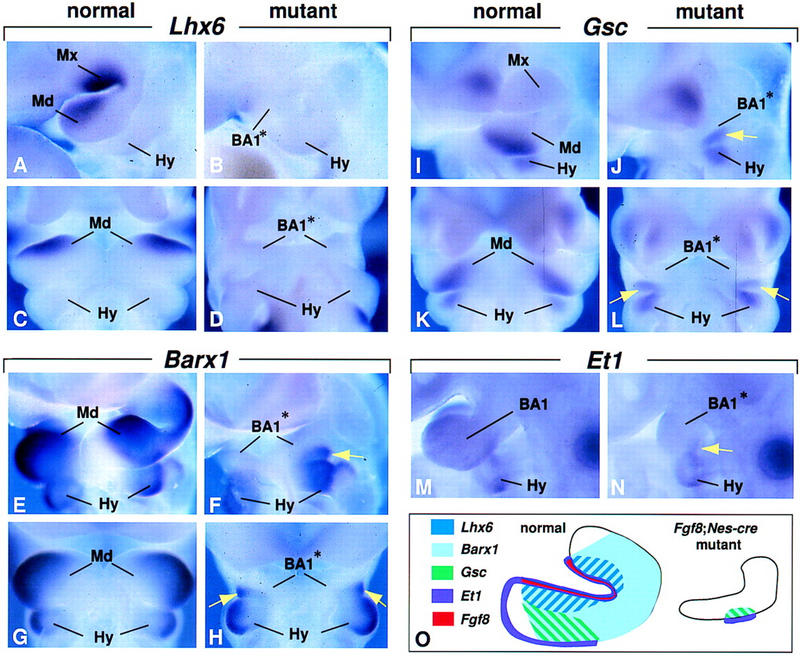
Gene expression regulated by FGF8 signaling in BA1. (A–D) Expression of Lhx6 at E10.5, (E–H) Barx1 at E10.0, (I–L) Gsc at E10.5, and (M,N) Et1 at E9.5. Lateral views (A,B,E,F,I,J,M,N) and ventral views (C,D,G,H,K,L) of embryos assayed for expression of the genes indicated. The yellow arrows point to gene expression in the region in which Fgf8 was expressed transiently before E9.0. (O) Schematic diagram summarizing gene expression patterns in the normal and mutant BA1 at E10.5.
In Fgf8;Nes–cre embryos, Fgf8 is inactivated in the surface ectoderm of prospective BA1. By E9.0 (14–18 somites), no Fgf8 RNA was detected in BA1, although it was detected in all other normal Fgf8 expression domains (n = 5; Fig. 1G,H; data not shown). Significantly, in two slightly younger embryos a small patch of Fgf8-expressing cells was detected on the caudal side of BA1 (yellow arrow, Fig. 1F). This patch appeared to be a subset of the normal caudal Fgf8 expression domain. The presence of this patch confirms that inactivation does not occur synchronously across the arch, and shows that some cells on the caudoproximal side transiently express Fgf8.
Mutant embryos survive to birth but lack most BA1-derived structures
The swelling that heralds BA1 development is evident in Fgf8;Nes–cre embryos, but by E9.0 it is clearly smaller than normal (Figs. 1E,H and 2A,B). As the embryos mature, there is relatively little expansion of either the mandibular or maxillary primordia, whereas development in neighboring regions, such as the hyoid arch (second branchial arch), appears normal (Fig. 2A–D; data not shown). It is important to note, however, that outgrowth of the mutant BA1 is not completely arrested, as the mandibular primordia extend distally and meet at the midline (white arrowhead in Fig. 2D; see also Fig. 6D,H,L). Furthermore, at E11.25 a distinct hillock (yellow arrow in Fig. 2D) forms in caudoproximal BA1, immediately adjacent to the external acoustic meatus (EAM). This hillock appears to form in the region in which Fgf8 expression was detected transiently at E9.0 (yellow arrow in 1F).
Figure 2.
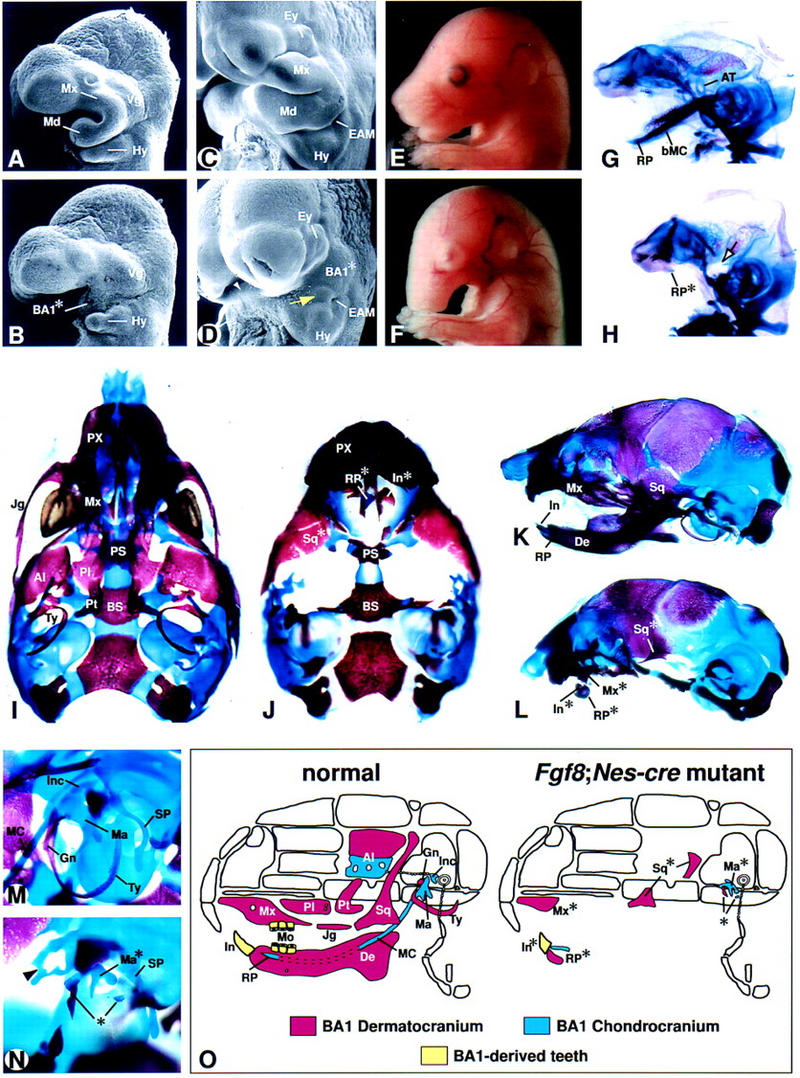
Phenotypic analysis of Fgf8;Nes–cre embryos. (A–D) Scanning electron microscope images of control (A,C) and mutant embryos (B,D) at E9.5 (A,B) and E11.25 (C,D). The yellow arrow in D points to a small outgrowth in the mutant BA1 primordium (BA1*) adjacent to the external acoustic meatus (EAM). The white arrowhead points to the region in which the distal ends of the paired BA1* meet. (E,F) Lateral views of control (E) and mutant (F) embryos at E16.5. (G–N) Skeletal preparations (cartilage stained blue and bone stained red) of control (G,I,K,M) and mutant (H,J,L,N) embryos. (G,H) Lateral view of E14.5 chondrocranium. Note the absence of the body of Meckel's cartilage (bMC) and ala temporalis (AT) in the mutant embryo. The arrow points to the region in which the AT is normally found. A vestigial rostral process (RP*) is present. (I–L) Newborn skulls. (I,J) Ventral views in which the hyoid and roof bones have been removed from both control and mutant, and the dentary has been removed from the control skull. (K,L) Lateral views. Note that in the mutant the dentary (De) is absent, except for the RP* with vestigial incisors (In*) and associated alveolar bone. The distal maxilla (Mx*) and a vestigial squamosal (Sq*) are present. (M,N) Lateral view of E16.5 embryos showing the region in which the ear is developing. The arrowhead in N points to ectopic cartilage of unknown identity found in association with the tegmen tympani. Ossified elements in the region where the gonial and tympanic normally form are indicated by an asterisk. (O) Schematic diagram depicting BA1-derived craniofacial elements of normal and mutant skulls. Although the origin of the squamosal is controversial, it is shown here as being BA1 derived. Abbreviations as in Fig. 1. (Al) Alisphenoid; (BS) basisphenoid; (Ey) eye; (Gn) gonial; (Hy) hyoid arch; (Inc) incus; (Jg) jugal; (Ma) malleus; (MC) Meckel's cartilage; (Mo) molars; (Mx) maxillary process; (Pl) palatine; (PS) presphenoid; (Pt) pterygoid; (PX) premaxilla; (SP) styloid process; (Ty) tympanic; (Vg) swelling over trigeminal ganglion.
Fgf8;Nes–cre mutants die shortly after birth. Newborns appear grossly normal except for severe craniofacial defects and the presence of an ectodermal covering over the prospective mouth (Fig. 2E,F; data not shown). The lungs do not inflate suggesting that lethality is caused by anoxia. Characterization of the craniofacial defects in Fgf8;Nes–cre embryos revealed that cartilagenous elements thought to develop from BA1 by E14.5, such as the ala temporalis and incus (maxillary arch-derived), and body of Meckel's cartilage (mandibular arch-derived) fail to develop. However mutant embryos have a vestigial malleus (Ma*) and rostral process (RP*), the distal symphyseal end of Meckel's cartilage (Fig. 2G–N).
Normally, between E14.5 and birth, a number of bones develop from BA1 neural crest-derived mesenchyme (summarized in Fig. 2O). In Fgf8;Nes–cre newborns, most of these bones are absent. A few mandibular arch-derived elements remain, including alveolar bone associated with RP* (Fig. 2L), Ma*, and some small ossified elements in the general region of the gonial and tympanic bones (asterisk in Fig. 2N). Of the maxillary arch-derived dermal elements, the mutants contain the distal maxilla (Mx*) and portions of the squamosal (Fig. 2L). Molars are absent, but vestigial lower incisors are observed in association with RP* (Figs. 2L and 5H). A summary of skeletal development in Fgf8;Nes–cre mutants is presented in Figure 2O.
Figure 5.
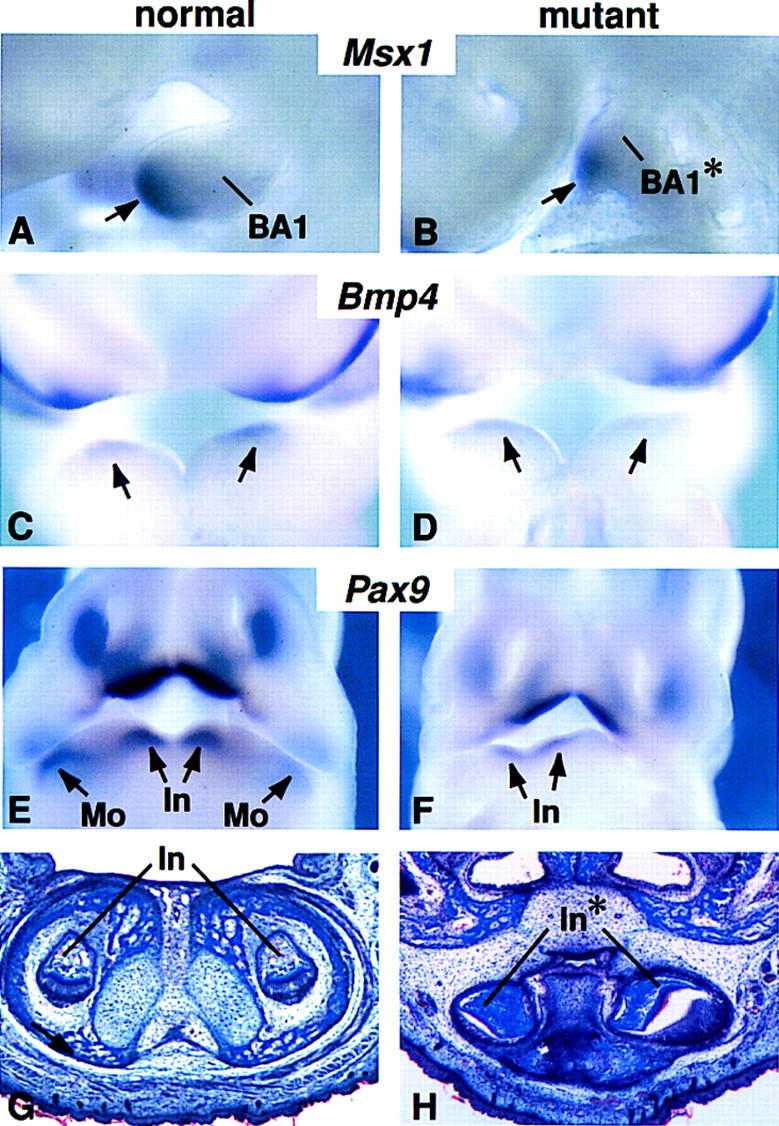
Tooth formation and expression of patterning genes in distal BA1 of Fgf8;Nes–cre embryos. (A–F) Gene expression at E9.5 (A,B) E10.0 (C,D) E11.5 (E,F). Arrows point to expression domains of (A,B) Msx1 in the mesenchyme; (C,D) Bmp4 in the ectoderm; and (E,F) Pax9 in the mandibular mesenchyme. Note that in the mutant embryo (F) Pax9 expression is detected in the presumptive incisor but not in the presumptive molar domain. (G,H) Frontal sections through the incisor domain of newborn pups.
Newborn mutants had other abnormalities consistent with a failure of BA1 development, including a small disorganized tongue (microglossia) and a truncated and misrouted mandibular division of the trigeminal nerve (data not shown). There was some variation in the mutant phenotype. The majority (28 of 42) displayed the phenotype described above, whereas the remaining mutants showed a slightly less severe phenotype, with a small increase in the extent of dermal bone development. In many cases, this less severe phenotype was observed on only one side of the head, which is reminiscent of asymmetries found in a variety of human congenital syndromes (for review, see Escobar 1993).
FGF8 is necessary for survival of mesenchymal cells in BA1
Analysis of Fgf8-null mutant homozygotes has shown that Fgf8 is required for cell migration at primitive streak stages of development (Sun et al. 1999). Therefore, we speculated that the early failure of BA1 outgrowth in mutant embryos might be due to a lack of neural crest cell migration into the BA1 primordium. To investigate this possibility we assayed for Cad6, Crabp1, and Ap2.2 expression, which marks migrating neural crest cells (Mitchell et al. 1991; Maden et al. 1992; Ruberte et al. 1992; Inoue et al. 1997). Expression of these markers was similar in mutant and normal embryos (Fig. 3A,B; data not shown), indicating that despite loss of Fgf8 function in BA1, the neural crest cell population follows its normal migration path and appears normal in size.
Figure 3.
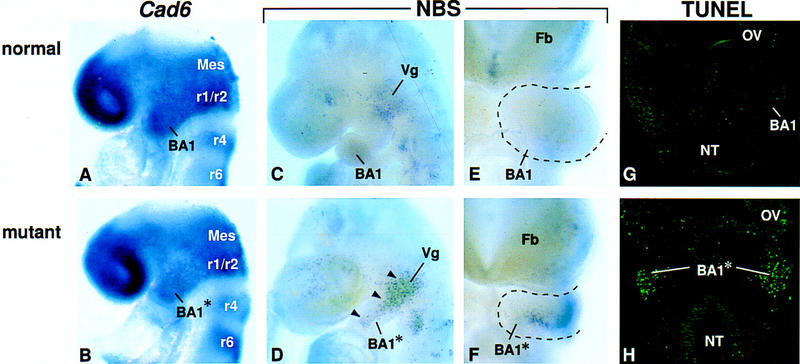
Analysis of neural crest cell migration and cell death in Fgf8;Nes–cre mutant embryos. (A,B) Expression of Cad6 at E8.5. (C–F) Cell death as assayed by Nile blue sulfate (NBS) staining in whole mount. Lateral (C,D) and frontal (E,F) views of E9.5 embryos. Black arrowheads point to areas of cell death in the mutant BA1. (G,H) Frontal sections of E9.5 embryos at the level of the maxillary primordium, stained for the presence of apoptotic cells using TUNEL. Abbreviations as in Figs. 1 and 2. (Mes) Mesencephalon; (NT) neural tube; (OV) optic vesicle; (r) rhombomere.
To determine why the mutant BA1 is hypoplastic, we then studied cell proliferation by assaying for BrdU incorporation. BrdU-labeled cells were detected in the mutant BA1 at E9.5, at least 12 hr after Cre-mediated inactivation of Fgf8, and the ratio of BrdU-labeled to unlabeled cells appeared roughly the same as in the normal arch (data not shown). This result suggests that FGF8 is not required for cell proliferation in BA1. We then assayed for cell survival by staining in whole mount for Nile blue sulfate (NBS) uptake, which marks dying and dead cells (Bowen 1981), and by performing TUNEL assays on tissue sections to detect apoptosis. In the BA1 region of control embryos at E8.5–E10.0, NBS staining was observed only proximally (Fig. 3C,E), where trigeminal ganglion cells die (Noden 1983; Davies and Lumsden 1984). In Fgf8;Nes–cre embryos, however, extensive cell death was detected at E8.75–E10.0 (Fig. 3D,F; data not shown). At E9.5, intense NBS staining was detected throughout the BA1 primordium (Fig. 3D) except at the extreme distal tip near the ventral midline (Fig. 3F). The area in which dying cells were detected stretched proximally to the trigeminal swelling and included the region forming the maxillary arch (Fig. 3D,H). The results of the TUNEL analysis indicated that the observed abnormal cell death, which was detected exclusively in the mesenchyme of BA1, is due to apoptosis (Fig. 3G,H). Together these data suggest that the lack of BA1 development in Fgf8;Nes–cre embryos is due, at least in part, to apoptosis of a substantial proportion of the cells that normally populate the arch, and therefore, that FGF8 produced in the surface ectoderm is essential for their survival.
Patterned expression of some regulatory genes is maintained in the hypoplastic BA1 of Fgf8;Nes–cre mutants
Because a proportion of mutant BA1 mesenchyme survives and forms a hypoplastic arch, we performed an analysis to identify genes that require FGF8 signaling and to determine the extent of mesenchymal patterning in the absence of FGF8. Pitx1, a bicoid-related homeobox gene (Lanctot et al. 1997; Szeto et al. 1999) is normally coexpressed with Fgf8 in the oral ectoderm of the mandibular and maxillary primordia, and is also expressed in the underlying mesenchyme (Fig. 4A). The oral mesenchyme also expresses Lhx7, a LIM homeodomain encoding gene (Grigoriou et al. 1998; Tucker et al. 1999) (Fig. 4C,E). Expression of both genes was clearly detected in the mutant BA1, but the signals were less extensive than normal (Fig. 4A–F), presumably because BA1 mesenchyme is hypocellular. These results show that mutant BA1 is sufficiently healthy to express at least some of the genes that mark the oral side of BA1, and suggest that some aspects of oral–aboral patterning are intact in the absence of Fgf8.
Figure 4.
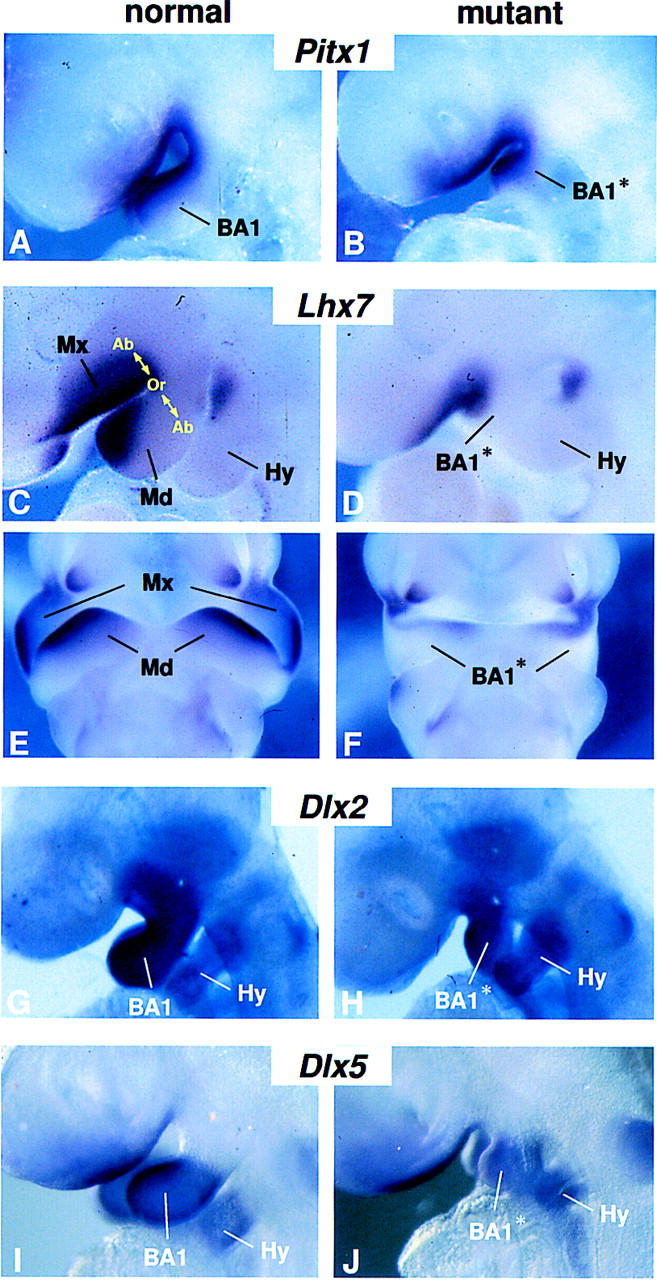
Patterning genes with normal expression domains in the Fgf8;Nes–cre BA1. (A–J) Gene expression in BA1 at E9.5 (A,B,G–J) and E10.5 (C–F) embryos. (A–D,G–J) Lateral views. (E,F) Ventral views. Abbreviations as in Fig. 1. (Ab) Aboral; (Md) mandibular primordium of BA1; (Mx) maxillary primordium of BA1; (Or) oral.
Dlx1, Dlx2, and Dlx5 are three members of the Dlx multigene family related to the Drosophila distalless gene, which are thought to regulate morphogenesis along the P–D axis of BA1 (Qiu et al. 1995, 1997; Depew et al. 1999). Dlx1 and Dlx2 are expressed in the ectomesenchyme along most of the P–D length of BA1, including the maxillary primordium (Fig. 4G; data not shown). In contrast, Dlx5 expression is not detected in the maxillary primordium and is restricted within the distal two-thirds of the mandibular primordium (Qiu et al. 1997; Depew et al. 1999) (Fig. 4I). Expression of all three genes was detected in BA1 of mutant embryos in roughly their normal domains (Fig. 4H,J). This suggests that cells in the mutant arch have sufficient P–D positional information to maintain differential expression of these genes.
To further investigate the extent to which distal BA1 is patterned, we assayed for Msx1 and Bmp4 expression. Between E9.5 and E10.5, Msx1 expression is normally restricted to the distal (medial) ectomesenchyme and Bmp4 expression is detected in distal BA1 ectoderm overlying the Msx1 expression domain (Tucker et al. 1998a) (Fig. 5A,C). Both Msx1 and Bmp4 expression appeared relatively normal in mutant BA1 (Fig. 5B,D). Similar results (not shown) were obtained with probes for Msx2, dHand, and eHand, other genes that are normally expressed in distal BA1 (Thomas et al. 1998). Pax9, which is required for tooth development, is normally detected in the mandibular arches at E11.5 in four spots, two distal and two proximal, representing the prospective incisor and molar domains, respectively (Neubuser et al. 1997; Peters et al. 1998) (Fig. 5E). In the mutant embryos, Pax9 was detected in only two spots near the ventral midline (Fig 5F), which presumably mark the regions in which the incisors will form. These early gene expression patterns are consistent with the observation that the distal-most portions of BA1 are the least affected: in newborn Fgf8;Nes–cre mutants the distal part of the maxilla and the distal mandible, including truncated lower incisors, are present (Fig. 5G,H; summarized in Fig. 2O). These data suggest that FGF8 is not required for development of distal BA1.
Identification of FGF8-responsive genes
FGF8-bead implantation experiments have identified Lhx7, and the closely related gene Lhx6, which are coexpressed in oral mesenchyme of mandibular and maxillary arches at E10.5 (Figs. 4C,E; 6A,C), as potential targets of FGF8 signaling in BA1 (Grigoriou et al. 1998). Interestingly, although we found that Lhx7, is expressed in its normal domain in Fgf8;Nes–cre mutants (Fig. 4D,F), Lhx6 expression was not detected in mutant BA1 (Fig. 6B,D). Thus, our data suggest that they are independently regulated in vivo.
Barx1 is another homeobox gene previously identified as inducible by FGF8 (Tucker et al. 1998b). Beginning at E9.5 it is expressed in mesenchyme throughout the proximal but not in the distal portion of BA1 (Fig. 6E,G; data not shown). In E9.5–E11.5 Fgf8;Nes–cre embryos, Barx1 RNA was not detectable, except in a small patch of cells on the caudal side of the mandibular arch (yellow arrows in Fig. 6F,H; data not shown). Significantly, this patch of Barx1 expression was localized in mesenchyme that appeared to underlie the region in which Fgf8 expression was detected transiently at E9.0 (cf. Figs. 1F and 6F). At E9.5, the mutant ectoderm that appears to overlie the patch of Barx1-expressing cells expresses Endothelin-1 (Et1; yellow arrow, Fig. 6N). Et1 RNA was not detected elsewhere in mutant BA1 ectoderm, but was detected in the second and third branchial arch epithelium (Fig. 6N; data not shown). In contrast, in control embryos, Et1 RNA was detected at low levels throughout the epithelium of branchial arches 1, 2, and 3 (Clouthier et al. 1998) (Fig. 6M). By E10.5, Goosecoid (Gsc) expression, which is normally detected throughout the caudal half of BA1 (Rivera-Perez et al. 1995; Yamada et al. 1995) (Fig. 6I,K) is restricted in the mutants to the region in which Barx1- and Et1-expressing cells are found (yellow arrow, Fig. 6J,L). These results, summarized in Figure 6O, show that expression of Lhx6, Barx1, Et1, and Gsc in BA1 are dependent, directly or indirectly, on FGF8 signaling. Furthermore, they suggest that transient expression of Fgf8 on the caudal side of BA1 at E9.0 is sufficient to induce local expression of Barx1, Et1, and Gsc, but that continued expression of Fgf8 is not required to maintain it.
Discussion
Inactivation of Fgf8 by Cre-mediated recombination in the ectoderm of the nascent first branchial arch severely impairs development of the BA1 primordium. This appears to be due to both apoptosis of the mesenchyme and failure to express a set of genes essential for BA1 morphogenesis. Newborn mutants lack most BA1-derived structures, except those formed from the distal-most region. These results provide genetic evidence that the ectoderm produces factors essential for BA1 outgrowth and patterning at a very early stage. They further suggest a model in which BA1 is specified into two domains at early stages in its development, a proximal region that is dependent on FGF8 signaling for its outgrowth and patterning, and a distal domain that is controlled by other signaling molecules (Fig. 7).
Figure 7.
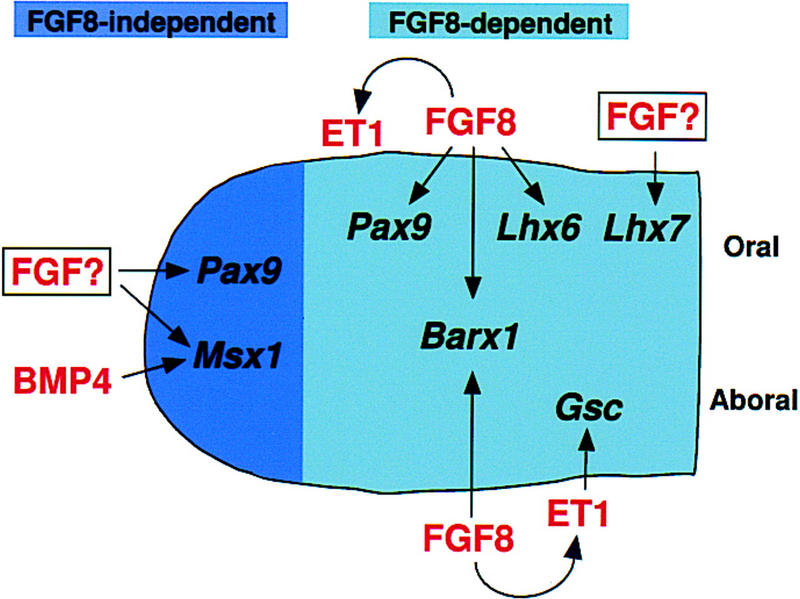
Inductive activities of signaling molecules produced in BA1 ectoderm. Schematic diagram illustrating effects of FGF8 signaling in BA1. According to this model, FGF8 signaling is not required to induce gene expression in the distal region. However, in the proximal region, expression of Barx1, Pax9, Lhx6, and Gsc in the mesenchyme is directly or indirectly dependent on FGF8, which is also required for Et1 expression in the ectoderm. Signaling molecules produced in the ectoderm are shown in red; transcription factors expressed in the mesenchyme are shown in black.
FGF8 is an essential signal for cell survival in proximal BA1
Shortly after Fgf8 is inactivated in the nascent BA1 epithelium, there is a brief period of cell death that peaks at ∼E9.5, during which a large proportion of proximal but not distal BA1 mesenchyme undergoes apoptosis. This cell death is presumably responsible for the small size of BA1 from an early stage of its development in the mutant embryos, and most likely contributes to the final mutant phenotype. Thus, one important conclusion from our study is that FGF8 is required, directly or indirectly, for cell survival in proximal BA1. Preliminary data suggest that reduction in the level of FGF8 also causes cell death in the developing limb and brain (M. Lewandoski, E. Storm, and G.R. Martin, unpubl.). In contrast, analysis of Fgf8−/− embryos indicates that during gastrulation FGF8 is not required for cell proliferation or survival, but instead is necessary for cell migration at the primitive streak stage (Sun et al. 1999). Thus, it appears that FGF8 performs different functions in different developmental contexts.
There is a substantial body of evidence from experimental studies in vitro showing that FGFs, particularly FGF1 and FGF2, can function as survival factors for a wide variety of cell types including neurons, glia, endothelial cells, and smooth muscle cells (for review, see Szebenyi and Fallon 1999). For example, it has been found that addition of FGF2 to explants of trunk neural crest enhances cell survival without stimulating mitosis (Kalcheim 1989), and local application of beads containing FGF4 prevents apoptosis in dental mesenchyme isolated from the mandibular arch at E13 (Vaahtokari et al. 1996). Despite the wealth of data from such in vitro studies, previous genetic analysis has provided little evidence that FGFs are required for cell survival. Null mutations have been generated in Fgf2 and at least 10 other mouse FGF genes, as well as all four FGF receptor (FGFR) genes (Goldfarb 1996; Floss et al. 1997; Dono et al. 1998; Min et al. 1998; Ortega et al. 1998; Weinstein et al. 1998; Zhou et al. 1998; Sekine et al. 1999; Sun et al. 1999; D. Ornitz, pers. comm.; C. Basilico, pers. comm.), but to our knowledge, effects specifically on cell survival have been described only in Fgf4−/− (Feldman et al. 1995) and Fgfr2−/− (Arman et al. 1998) embryos. These mutants display a similar early postimplantation lethal phenotype in which the inner cell mass dies.
One interesting question is why a proportion of the cells in proximal BA1 survive. One possibility is that their survival is dependent on other FGF family members, such as Fgf9, which is detected at low levels in both wild-type (Kettunen and Thesleff 1998) and mutant BA1 epithelium (data not shown), or on other types of signals. Another intriguing possibility is based on the premise that FGFs and other survival factors that signal through receptor tyrosine kinases (RTKs) function to prevent apoptosis by stimulating the antiapoptotic activity of Ras (for review, see Downward 1998), and the observation that FGF signaling can induce the expression of Sprouty (Spry) genes, which encode inhibitors of RTK signaling (Hacohen et al. 1998; Casci et al. 1999; Kramer et al. 1999; Minowada et al. 1999; Reich et al. 1999). Thus, the extent of cell survival in a given tissue may be determined by the balance between factors that stimulate and inhibit Ras activity. We have found that the expression of at least one member of the Sprouty gene family, Spry2, is greatly diminished in BA1 of Fgf8;Nes–cre embryos (data not shown), supporting the hypothesis based on FGF-bead implantation experiments that FGF8 positively regulates Sprouty gene expression in BA1 (Minowada et al. 1999). The reduced level of Spry2 in mutant BA1 mesenchyme might compensate partially for the reduced Ras activity caused by the inactivation of Fgf8 by derepressing other RTK pathways normally inhibited by Sprouty, and thereby promote cell survival.
FGF8 induces gene expression necessary for proximal BA1 morphogenesis
Fgf8;Nes–cre embryos lack most of the chondrocranial and dermatocranial elements that form the jaws, the lateral skull wall, and middle ear (Fig. 2O). It is possible that their failure to develop is due solely to the reduction in cell number that occurs between E8.75 and E10. However, it seems likely that the failure of surviving cells to express genes downstream of FGF8 also contributes to the final phenotype. One genetic pathway that is affected in Fgf8;Nes–cre mutants involves Endothelin-1 (ET1) (Levin 1995). Mice in which the genes encoding ET1 or its receptor (ETA) have been inactivated display a pleiotropic phenotype, including severe effects on the lower jaw and other BA1-derived structures (Kurihara et al. 1994; Clouthier et al. 1998). Although ET1 signaling is not required for the early stages of arch outgrowth and BA1 appears grossly normal until E10.5 in Et1−/− mutants (Kurihara et al. 1994), it is required for expression of Gsc (Clouthier et al. 1998), which is necessary for formation of some craniofacial skeletal elements (Rivera-Perez et al. 1995; Yamada et al. 1995). Our results show that Et1 and Gsc expression are dependent on FGF8 signaling, indicating that FGF8 is upstream of this pathway (Fig. 7). Consistent with this hypothesis, the BA1 structures that are missing or malformed in Et1−/− and EtA−/− mice are also affected in Fgf8;Nes–cre embryos. However, loss of Fgf8 function in BA1 results in a more severe phenotype. This could be explained either by the hypocellularity of the Fgf8;Nes–cre BA1 or by the lack of expression of genes that are downstream of FGF8 but not ET1 signaling.
One good candidate for such a gene is Barx1 (Tissier-Seta et al. 1995). Previous studies have shown that Barx1 expression, which is normally restricted to the region that gives rise to the same elements that fail to develop in Fgf8;Nes–cre embryos, can be induced in explants of mandibular arch mesenchyme by FGF8 (Tucker et al. 1998b). Our data show that FGF8 is required on both the rostral and caudal side of BA1 to establish the Barx1 expression domain. We found that Lhx6 is also downstream of FGF8. Surprisingly, Lhx7 expression does not require FGF8, despite the fact that Lhx6 and Lhx7, which appear to be expressed in the same cells on the oral side of the arch, can both be induced by placing FGF8-beads in isolated mandibular arch mesenchyme (Grigoriou et al. 1998). A possible explanation for these findings is that Lhx7 expression is induced by an FGF family member other than FGF8.
FGF8 as a switch that induces but does not maintain the BA1 developmental program
An important question is whether FGF8 functions solely to induce gene expression or whether it is also necessary to maintain it. We were able to address this question because Fgf8 is expressed transiently in a few epithelial cells on the caudoproximal side before it is inactivated. After Fgf8 RNA is no longer detected, Et1 is expressed in what appears to be the same caudal cell population. Moreover Barx1 and subsequently Gsc are induced in the adjacent underlying ectomesenchyme. The finding that expression of Et1, Barx1, and Gsc in the mutant BA1 is restricted to the region in which Fgf8 is expressed transiently suggests that FGF8 is necessary and sufficient to induce expression of these genes in vivo. Furthermore, because Barx1 and Gsc expression persists in this region through E11.5, at least 60 hr after Fgf8 has been inactivated, we propose that FGF8 can function as a switch to induce a gene expression cascade that rapidly becomes independent of FGF8 signaling. Thus, Barx1 and Gsc expression may be maintained by other signals produced in the epithelium, possibly ET1.
Analysis of the skeletons of Fgf8;Nes–cre newborns indicated that the only proximal BA1-derived skeletal elements present are a malleus-like middle ear element and nearby amorphous cartilagenous and bony fragments (Fig. 2O). It seems likely that they develop from cells on the caudal side of the mutant BA1 where outgrowth is observed at early stages in close proximity to a structure (the EAM) that will form the outer ear canal. Cells in this outgrowth apparently express Gsc, which is required for malleus development (Rivera-Perez et al. 1995; Yamada et al. 1995). Because this outgrowth develops in the region that was exposed transiently to FGF8 signaling, the presence of a vestigial malleus in the Fgf8;Nes–cre newborns does not contradict the hypothesis that FGF8 is required for development of the entire proximal domain of BA1.
Control of distal BA1 development
In contrast to the lack of development of skeletal elements derived from proximal BA1, structures derived from the distal portion of the arch are invariably present in Fgf8;Nes–cre newborns (Fig. 2O). There is genetic evidence that development of the distal region of BA1 is dependent on Msx1, as the distal maxilla and mandible (including the incisors), the very structures that are present in Fgf8;Nes–cre pups, fail to form in Msx1 null embryos (Satokata and Maas 1994). As expected, Msx1 expression was detected in its normal domain in distal BA1 of Fgf8;Nes–cre embryos. Although it is formally possible that the distal domain develops because Fgf8 is expressed transiently in distal BA1 ectoderm, we think this unlikely because no such Fgf8 expression was detected in the mutant embryos. Instead, we suggest that development of distal BA1 is not dependent on FGF8. This leaves open the question of what controls Msx1 expression? It is possible that FGF signaling is involved, as several FGFs have been found to induce Msx1 in dental mesenchyme (Bei and Maas 1998; Kettunen and Thesleff 1998), at the border of the neural plate (Streit and Stern 1999), and in the limb (Wang and Sassoon 1995). Other than Fgf8, the only FGF family member presently known to be expressed in BA1 epithelium before E12.5 is Fgf9 (Kettunen and Thesleff 1998; http://honeybee.helsinki.fi/toothexp/index.htm). Thus, FGF9 or some other FGF that has yet to be identified may provide the signal necessary for outgrowth and patterning of the distal domain of BA1. The hypothesis that another FGF in addition to FGF8 is required for BA1 development could also explain the pattern of expression we observed for Pax9, a marker for the prospective tooth-forming domains in the oral mesenchyme, which can be induced in isolated mesenchyme by any of several different FGFs (Neubuser et al. 1997). In the mutant BA1, Pax9 expression is absent in the proximal, molar domain because it is dependent on FGF8, whereas Pax9 expression is detected in the distal, incisor domain, perhaps because it is induced by an FGF signal other than FGF8.
Another factor that may play a role in the development of the distal domain is BMP4, as it too is capable of inducing Msx1 expression in BA1 (Vainio et al. 1993; Tucker et al. 1998a,b), limb (Wang and Sassoon 1995), and brain (Furuta et al. 1997), and is expressed in distal BA1 of Fgf8;Nes–cre mutants. In view of the evidence that BMP signaling plays a role in determining tooth identity (Tucker et al. 1998b), and that FGF and BMP signaling pathways function antagonistically to regulate tooth induction (Neubuser et al. 1997), it seems likely that interactions between the two types of signals control development of the distal-most BA1 structures.
Concluding remarks
The Fgf8;Nes–cre mutants resemble humans with first arch syndromes that include agnathia. Agnathia alone occurs very rarely, and is often associated with holoprosencephaly and sometimes with situs inversus totalis, or both (Pauli et al. 1983; Bixler et al. 1985; Leech et al. 1988; Escobar 1993). Significantly, embryos that are compound heterozygous for a null and a hypomorphic allele of Fgf8 show forebrain defects including holoprosencephaly (E. Meyers, E. Storm, and G.R. Martin, unpubl.) and display abnormalities in left-right asymmetry determination (Meyers and Martin 1999). Therefore, it is tempting to speculate that mutations in Fgf8 or in genes directly upstream or downstream of it might cause some of the human syndromes characterized by agnathia/micrognathia.
Materials and methods
Production and analysis of mutant embryos
Production and full characterization of the Nes–cre1 transgenic mouse line will be reported elsewhere. The Fgf8flox and Fgf8Δ2,3n alleles (Meyers et al. 1998) were maintained on a mixed genetic background. Fgf8;Nes–cre mutants were produced using the breeding scheme outlined in Figure 1A and genotyped using previously described primers (Lewandoski et al. 1997; Meyers et al. 1998; Sun et al. 1999). Histological analysis of embryos, scanning electron microscopy, and skeletal preparations were carried out as described by Depew et al. (1999). Nes–cre1;Z/AP double hemizygotes (E7.5–E11.5) were stained for alkaline phosphatase activity essentially as described by Lobe et al. (1999).
Cell death analysis
For whole mount NBS staining, E8.0–E10.5 embryos were dissected, washed in PBS, and incubated for 30–45 min at 37°C in filtered NBS solution [10 mg/ml NBS (Sigma N-5632) in PBS containing 0.1% Tween 20]. Embryos were then washed several times in PBS at room temperature and photographed immediately. TUNEL analysis was performed on paraffin sections using the In Situ Cell Death Detection kit (Boehringer-Mannheim) following the manufacturer's protocol.
RNA in situ hybridization
Whole mount RNA in situ hybridization analysis was carried out as previously described (Neubuser et al. 1997) using riboprobes prepared from plasmids described in references cited for each gene. Fgf8 RNA was detected using a probe for sequences in exons 2 and 3, which are deleted in the Fgf8Δ2,3 and the Fgf8Δ2,3n mutant alleles. The expression of each gene was analyzed in at least three Fgf8;Nes–cre embryos at each stage.
Acknowledgments
We are very grateful to Andras Nagy for providing the Z/AP reporter line, and thank the following for providing plasmids used to prepare probes used in this study: J.F. Brunet (Barx1); E. deRobertis (Gsc); J. Drouin (Pitx1); B. Hogan (Bmp4); V. Pachnis (Lhx6, Lhx7); P. Sharpe (Msx1); M. Takeichi (Cad6); and M. Yanagisawa (Et1). We are grateful to A. Gannon and D. Trail for excellent technical assistance. We also thank our colleagues in the Martin and Bishop laboratories for helpful discussion and critical readings of the manuscript. A.T. was the recipient of postdoctoral fellowships from the Deutsche Forschungsgemeinschaft, Human Frontiers Science Program, and the California Division of the American Cancer Society. This work was supported by grants from the March of Dimes and Nina Ireland (to J.L.R.R.), the Howard Hughes Medical Institute Research Resources Program grant (76296-549901) to the UCSF School of Medicine, and National Institutes of Health grants KO2 MH01046 (to J.L.R.R.), RO1 CA44338 (to J.M.B.), and RO1 HD34380 (to G.R.M.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gmartin@itsa.ucsf.edu; FAX (415) 476-3493
References
- Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci. 1998;95:5082–5087. doi: 10.1073/pnas.95.9.5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei M, Maas R. FGFs and BMP4 induce both Msx1-independent and Msx1-dependent signaling pathways in early tooth development. Development. 1998;125:4325–4333. doi: 10.1242/dev.125.21.4325. [DOI] [PubMed] [Google Scholar]
- Bixler D, Ward R, Gale DD. Agnathia-holoprosencephaly: A developmental field complex involving face and brain. Report of 3 cases. J Craniofac Genet Dev Biol (Suppl.) 1985;1:241–249. [PubMed] [Google Scholar]
- Bowen ID. Techniques for demonstrating cell death. In: Bowen ID, Lockshin RA, editors. Cell death in biology and pathology. London, UK: Chapman and Hall; 1981. pp. 379–444. [Google Scholar]
- Casci T, Vinos J, Freeman M. Sprouty, an intracellular inhibitor of Ras signaling. Cell. 1999;96:655–665. doi: 10.1016/s0092-8674(00)80576-0. [DOI] [PubMed] [Google Scholar]
- Clouthier DE, Hosoda K, Richardson JA, Williams SC, Yanagisawa H, Kuwaki T, Kumada M, Hammer RE, Yanagisawa M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125:813–824. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Martin GR. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development. 1995;121:439–451. doi: 10.1242/dev.121.2.439. [DOI] [PubMed] [Google Scholar]
- Davies A, Lumsden A. Relation of target encounter and neuronal death to nerve growth factor responsiveness in the developing mouse trigeminal ganglion. J Comp Neurol. 1984;223:124–137. doi: 10.1002/cne.902230110. [DOI] [PubMed] [Google Scholar]
- Depew MJ, Liu JK, Long JE, Presley R, Meneses JJ, Pedersen RA, Rubenstein JLR. Dlx5 regulates regional development of the branchial arches and sensory capsules. Development. 1999;126:3831–3846. doi: 10.1242/dev.126.17.3831. [DOI] [PubMed] [Google Scholar]
- Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Ras signalling and apoptosis. Curr Opin Genet Dev. 1998;8:49–54. doi: 10.1016/s0959-437x(98)80061-0. [DOI] [PubMed] [Google Scholar]
- Escobar LF. Facial bones. In: Stevenson RE, Hall JG, Goodman RM, editors. Human malformations and related anomalies. 1993. , II, pp. 629–653. Oxford University Press, New York, NY. [Google Scholar]
- Feldman B, Poueymirou W, Papaioannou VE, DeChiara TM, Goldfarb M. Requirement of FGF-4 for postimplantation mouse development. Science. 1995;267:246–249. doi: 10.1126/science.7809630. [DOI] [PubMed] [Google Scholar]
- Floss T, Arnold HH, Braun T. A role for FGF-6 in skeletal muscle regeneration. Genes & Dev. 1997;11:2040–2051. doi: 10.1101/gad.11.16.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis-West P, Ladher R, Barlow A, Graveson A. Signalling interactions during facial development. Mech Dev. 1998;75:3–28. doi: 10.1016/s0925-4773(98)00082-3. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Piston DW, Hogan BL. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development. 1997;124:2203–2212. doi: 10.1242/dev.124.11.2203. [DOI] [PubMed] [Google Scholar]
- Goldfarb M. Functions of fibroblast growth factors in vertebrate development. Cytokine Growth Factor Rev. 1996;7:311–325. doi: 10.1016/s1359-6101(96)00039-1. [DOI] [PubMed] [Google Scholar]
- Grigoriou M, Tucker AS, Sharpe PT, Pachnis V. Expression and regulation of Lhx6 and Lhx7, a novel subfamily of LIM homeodomain encoding genes, suggests a role in mammalian head development. Development. 1998;125:2063–2074. doi: 10.1242/dev.125.11.2063. [DOI] [PubMed] [Google Scholar]
- Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA. sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell. 1998;92:253–263. doi: 10.1016/s0092-8674(00)80919-8. [DOI] [PubMed] [Google Scholar]
- Inoue T, Chisaka O, Matsunami H, Takeichi M. Cadherin-6 expression transiently delineates specific rhombomeres, other neural tube subdivisions, and neural crest subpopulations in mouse embryos. Dev Biol. 1997;183:183–194. doi: 10.1006/dbio.1996.8501. [DOI] [PubMed] [Google Scholar]
- Kalcheim C. Basic fibroblast growth factor stimulates survival of nonneuronal cells developing from trunk neural crest. Dev Biol. 1989;134:1–10. doi: 10.1016/0012-1606(89)90072-9. [DOI] [PubMed] [Google Scholar]
- Kettunen P, Thesleff I. Expression and function of FGFs-4, -8, and -9 suggest functional redundancy and repetitive use as epithelial signals during tooth morphogenesis. Dev Dynamics. 1998;211:256–268. doi: 10.1002/(SICI)1097-0177(199803)211:3<256::AID-AJA7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Kramer S, Hacohen N, Okabe M, Krasnow MA, Hiromi Y. Sprouty: A common antagonist of FGF and EGF signaling pathways in Drosophila. Development. 1999;126:2515–2525. doi: 10.1242/dev.126.11.2515. [DOI] [PubMed] [Google Scholar]
- Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96:329–339. doi: 10.1016/s0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, Oda H, Kuwaki T, Cao WH, Kamada N, et al. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368:703–710. doi: 10.1038/368703a0. [DOI] [PubMed] [Google Scholar]
- Lanctot C, Lamolet B, Drouin J. The bicoid-related homeoprotein Ptx1 defines the most anterior domain of the embryo and differentiates posterior from anterior lateral mesoderm. Development. 1997;124:2807–2817. doi: 10.1242/dev.124.14.2807. [DOI] [PubMed] [Google Scholar]
- Leech RW, Bowlby LS, Brumback RA, Schaefer GB., Jr Agnathia, holoprosencephaly, and situs inversus: Report of a case. Am J Med Genet. 1988;29:483–490. doi: 10.1002/ajmg.1320290303. [DOI] [PubMed] [Google Scholar]
- Levin ER. Endothelins. N Engl J Med. 1995;333:356–363. doi: 10.1056/NEJM199508103330607. [DOI] [PubMed] [Google Scholar]
- Lewandoski M, Wassarman KM, Martin GR. Zp3-cre, a transgenic mouse line for the activation or inactivation of loxP-flanked target genes specifically in the female germ line. Curr Biol. 1997;7:148–151. doi: 10.1016/s0960-9822(06)00059-5. [DOI] [PubMed] [Google Scholar]
- Lobe CG, Koop KE, Kreppner W, Lomeli H, Gertsenstein M, Nagy A. Z/AP, a double reporter for Cre-mediated recombination. Dev Biol. 1999;208:281–292. doi: 10.1006/dbio.1999.9209. [DOI] [PubMed] [Google Scholar]
- Maden M, Horton C, Graham A, Leonard L, Pizzey J, Siegenthaler G, Lumsden A, Eriksson U. Domains of cellular retinoic acid-binding protein I (CRABP I) expression in the hindbrain and neural crest of the mouse embryo. Mech Dev. 1992;37:13–23. doi: 10.1016/0925-4773(92)90011-8. [DOI] [PubMed] [Google Scholar]
- Mahmood R, Bresnick J, Hornbruch A, Mahony C, Morton N, Colquhoun K, Martin P, Lumsden A, Dickson C, Mason I. A role for FGF-8 in the initiation and maintenance of vertebrate limb bud outgrowth. Curr Biol. 1995;5:797–806. doi: 10.1016/s0960-9822(95)00157-6. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nature Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Martin GR. Differences in left-right axis pathways in mouse and chick: Functions of FGF8 and SHH. Science. 1999;285:403–406. doi: 10.1126/science.285.5426.403. [DOI] [PubMed] [Google Scholar]
- Min H, Danilenko DM, Scully SA, Bolan B, Ring BD, Tarpley JE, DeRose M, Simonet WS. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes & Dev. 1998;12:3156–3161. doi: 10.1101/gad.12.20.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowada G, Jarvis LA, Chi CL, Neubüser A, Sun X, Hacohen N, Krasnow MA, Martin GR. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development. 1999;126:4465–4475. doi: 10.1242/dev.126.20.4465. [DOI] [PubMed] [Google Scholar]
- Mitchell PJ, Timmons PM, Hebert JM, Rigby PW, Tjian R. Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes & Dev. 1991;5:105–119. doi: 10.1101/gad.5.1.105. [DOI] [PubMed] [Google Scholar]
- Neubuser A, Peters H, Balling R, Martin GR. Antagonistic interactions between FGF and BMP signaling pathways: A mechanism for positioning the sites of tooth formation. Cell. 1997;90:247–255. doi: 10.1016/s0092-8674(00)80333-5. [DOI] [PubMed] [Google Scholar]
- Noden DM. The role of the neural crest in patterning of avian cranial skeletal, connective, and muscle tissues. Dev Biol. 1983;96:144–165. doi: 10.1016/0012-1606(83)90318-4. [DOI] [PubMed] [Google Scholar]
- Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. Neuronal defects and delayed wound healing in mice lacking FGF2. Proc Natl Acad Sci. 1998;95:5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli RM, Pettersen JC, Arya S, Gilbert EF. Familial agnathia-holoprosencephaly. Am J Med Genet. 1983;14:677–698. doi: 10.1002/ajmg.1320140411. [DOI] [PubMed] [Google Scholar]
- Peters H, Balling R. Teeth. Where and how to make them. Trends Genet. 1999;15:59–65. doi: 10.1016/s0168-9525(98)01662-x. [DOI] [PubMed] [Google Scholar]
- Peters H, Neubuser A, Kratochwil K, Balling R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes & Dev. 1998;12:2735–2747. doi: 10.1101/gad.12.17.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu M, Bulfone A, Martinez S, Meneses JJ, Shimamura K, Pedersen RA, Rubenstein JL. Null mutation of Dlx-2 results in abnormal morphogenesis of proximal first and second branchial arch derivatives and abnormal differentiation in the forebrain. Genes & Dev. 1995;9:2523–2538. doi: 10.1101/gad.9.20.2523. [DOI] [PubMed] [Google Scholar]
- Qiu M, Bulfone A, Ghattas I, Meneses JJ, Christensen L, Sharpe PT, Presley R, Pedersen RA, Rubenstein JL. Role of the Dlx homeobox genes in proximodistal patterning of the branchial arches: Mutations of Dlx-1, Dlx-2, and Dlx-1 and -2 alter morphogenesis of proximal skeletal and soft tissue structures derived from the first and second arches. Dev Biol. 1997;185:165–184. doi: 10.1006/dbio.1997.8556. [DOI] [PubMed] [Google Scholar]
- Reich A, Sapir A, Shilo B. Sprouty is a general inhibitor of receptor tyrosine kinase signaling. Development. 1999;126:4139–4147. doi: 10.1242/dev.126.18.4139. [DOI] [PubMed] [Google Scholar]
- Rivera-Perez JA, Mallo M, Gendron-Maguire M, Gridley T, Behringer RR. Goosecoid is not an essential component of the mouse gastrula organizer but is required for craniofacial and rib development. Development. 1995;121:3005–3012. doi: 10.1242/dev.121.9.3005. [DOI] [PubMed] [Google Scholar]
- Ruberte E, Friederich V, Morriss-Kay G, Chambon P. Differential distribution patterns of CRABP I and CRABP II transcripts during mouse embryogenesis. Development. 1992;115:973–987. doi: 10.1242/dev.115.4.973. [DOI] [PubMed] [Google Scholar]
- Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nature Genet. 1994;6:348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S. Fgf10 is essential for limb and lung formation. Nature Genet. 1999;21:138–141. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- Streit A, Stern CD. Establishment and maintenance of the border of the neural plate in the chick: involvement of FGF and BMP activity. Mech Dev. 1999;82:51–66. doi: 10.1016/s0925-4773(99)00013-1. [DOI] [PubMed] [Google Scholar]
- Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes & Dev. 1999;13:1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebenyi G, Fallon JF. Fibroblast growth factors as multifunctional signaling factors. Int Rev Cytol. 1999;185:45–106. doi: 10.1016/s0074-7696(08)60149-7. [DOI] [PubMed] [Google Scholar]
- Szeto DP, Rodriguez-Esteban C, Ryan AK, O'Connell SM, Liu F, Kioussi C, Gleiberman AS, Izpisua-Belmonte JC, Rosenfeld MG. Role of the Bicoid-related homeodomain factor Pitx1 in specifying hindlimb morphogenesis and pituitary development. Genes & Dev. 1999;13:484–494. doi: 10.1101/gad.13.4.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Kurihara H, Yamagishi H, Kurihara Y, Yazaki Y, Olson EN, Srivastava D. A signaling cascade involving endothelin-1, dHAND and msx1 regulates development of neural-crest-derived branchial arch mesenchyme. Development. 1998;125:3005–3014. doi: 10.1242/dev.125.16.3005. [DOI] [PubMed] [Google Scholar]
- Tissier-Seta JP, Mucchielli ML, Mark M, Mattei MG, Goridis C, Brunet JF. Barx1, a new mouse homeodomain transcription factor expressed in cranio-facial ectomesenchyme and the stomach. Mech Dev. 1995;51:3–15. doi: 10.1016/0925-4773(94)00343-l. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Tucker AS, Al Khamis A, Sharpe PT. Interactions between Bmp-4 and Msx-1 act to restrict gene expression to odontogenic mesenchyme. Dev Dynamics. 1998a;212:533–539. doi: 10.1002/(SICI)1097-0177(199808)212:4<533::AID-AJA6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Tucker AS, Matthews KL, Sharpe PT. Transformation of tooth type induced by inhibition of BMP signaling. Science. 1998b;282:1136–1138. doi: 10.1126/science.282.5391.1136. [DOI] [PubMed] [Google Scholar]
- Tucker AS, Yamada G, Grigoriou M, Pachnis V, Sharpe PT. Fgf-8 determines rostral-caudal polarity in the first branchial arch. Development. 1999;126:51–61. doi: 10.1242/dev.126.1.51. [DOI] [PubMed] [Google Scholar]
- Vaahtokari A, Aberg T, Thesleff I. Apoptosis in the developing tooth: Association with an embryonic signaling center and suppression by EGF and FGF-4. Development. 1996;122:121–129. doi: 10.1242/dev.122.1.121. [DOI] [PubMed] [Google Scholar]
- Vainio S, Karavanova I, Jowett A, Thesleff I. Identification of BMP-4 as a signal mediating secondary induction between epithelial and mesenchymal tissues during early tooth development. Cell. 1993;75:45–58. [PubMed] [Google Scholar]
- Wang Y, Sassoon D. Ectoderm–mesenchyme and mesenchyme–mesenchyme interactions regulate Msx-1 expression and cellular differentiation in the murine limb bud. Dev Biol. 1995;168:374–382. doi: 10.1006/dbio.1995.1087. [DOI] [PubMed] [Google Scholar]
- Weinstein M, Xu X, Ohyama K, Deng C-X. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development. 1998;125:3615–3623. doi: 10.1242/dev.125.18.3615. [DOI] [PubMed] [Google Scholar]
- Yamada G, Mansouri A, Torres M, Stuart ET, Blum M, Schultz M, De Robertis EM, Gruss P. Targeted mutation of the murine goosecoid gene results in craniofacial defects and neonatal death. Development. 1995;121:2917–2922. doi: 10.1242/dev.121.9.2917. [DOI] [PubMed] [Google Scholar]
- Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, Boivin GP, Duffy JJ, Pawlowski SA, Doetschman R. Fibroblast growth factor 2 control of vascular tone. Nature Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G, McMahon A. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron. 1994;12:11–24. doi: 10.1016/0896-6273(94)90148-1. [DOI] [PubMed] [Google Scholar]