

Abstract
A novel and simplified synthetic scaffold based on pladienolide was designed using a consensus pharmacophore hypothesis. An initial target was synthesized and evaluated to examine the role of the 3-hydroxy group and the methyl groups present at positions 10, 16, 20, 22 in 1, on biological activity. We report the first totally synthetic analog of this macrolide that shows biological activity. Our novel synthetic strategy enables the rapid synthesis of other new analogs of pladienolide in order to develop selective anticancer lead compounds.
Introduction
The pladienolides are 12-membered macrolides produced by Streptomyces platensis Mer 11107, some of which show remarkable antitumor activity.1–3 Among the seven pladienolides isolated, pladienolide B and D exhibit superior selective antitumor efficacy in vivo.4 These natural products have an antitumor mechanism of action that sets them apart from anticancer drugs currently in clinical use.4 In 2007 Mizui and co-workers identified the SF3b spliceosome subunit, a subcomplex of U2 SnRNP, as the molecular target of these natural products.5 Another microbial natural product (FR901464) and its methylated derivative (spliceostatin A) showed similar cellular effects to those induced by pladienolide.6 FR901464, a structurally distinct compound that had previously been isolated from a different bacterial genus, was reported to inhibit tumor growth in various xenograft models with a cytotoxicity profile similar to pladienolide.7–9 More recently spliceostatin (a partially stabilized derivative of FR901464) was also shown to interact with the SF3b subunit of the spliceosome.6 While this class of compounds were initially described as ‘inhibitors’ of splicing, very recent work has shown that this class of compounds act by modulation of alternate splicing, which may explain their selective toxicity to tumor cells versus normal tissue.10–12 We were motivated by the very similar (or possibly identical) mechanism of action of these two complex structurally distinct natural products to develop a hypothetical 3D pharmacophore model that allowed us to design analogs of FR901464 (sudemycins: 6–8),12 which showed significant antitumor activity in vitro and in vivo.13, 14 These compounds contain only three stereocenters, in contrast to the 9 stereocenters in FR901464 or the 10 stereocenters in pladienolide (see Figure 1).
Figure 1.

Natural and synthetic analogs of pladienolide and FR901464.
To date, only three approaches have been reported for the synthesis of these unique macrolides,3, 15, 16 including a total synthesis of pladienolide B and D.3 However, access to multi-gram quantities of totally synthetic pladienolides for in vivo studies remains a significant challenge, due to the synthetic complexity inherent to this class of compounds. Fortunately our pharmacophore design has allowed us to target simplified analogs of pladienolides that possess the potential to be potent and more drug-like than either FR901464 or pladienolide. As part of our ongoing efforts to extend our successful pladienolide–FR901464 consensus pharmacophore-based design approach to simplified synthetic analogs of pladienolides, we now report the synthesis and biological evaluation of our first synthetic pladienolide analogs 5 and E-26, as the starting point in our development of active simplified analogs of this natural product. The design of this scaffold is based on our published consensus-based pharmacophore (see Figure 2) that indicates that the C2 through C5 carbon atoms (highlighted in green) do not overlap with atoms in FR901464 analogs and are therefore not a likely component of the active pharmacophore.13, 14 In particular the C3 atom bearing the hydroxyl group does not appear to have an important interaction in this model, therefore this functionality is excluded from our design for the basic framework molecule 5 (see Scheme 1). However, inspection of the overlay in Figure 1 shows that the C6 OH group can readily overlay with the amide NH of FR901464 analogs, indicating that this feature could be a critical hydrogen-bond donor in the pharmacophore. In addition, from this overlay we assume that the methyl groups present at 10, 12, 16, 20, 22 positions of 1 are responsible for locking the free rotational bonds of the molecule. So, our preliminary hypothesis is that the side-chain chiral methyl centers may have limited importance in the activity of pladienolide. Our previous work that led to the development of the active analogs 6, 7, and 8, has already shown the critical importance of the carbonyloxy group (at the C7 position in pladienolide B) in the pharmacophore. 13, 14
Figure 2.
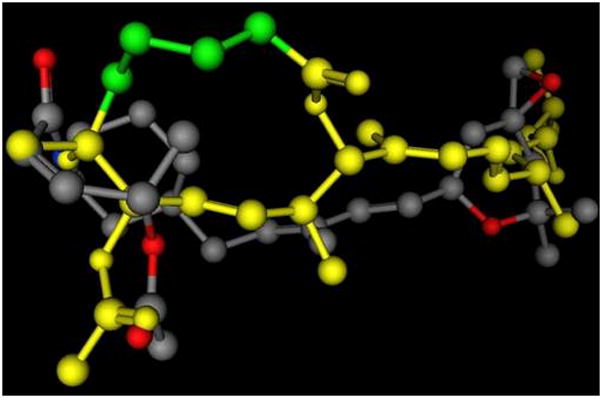
A 3D representation of an example of an overlay of the presumed key interaction groups in a low energy conformation of 3-deoxypladienolide (shown in yellow, some atoms are not shown) and compound 6, showing that the epoxy group and the carbonyloxy groups are the same distance in both molecules. In the pladienolide model the C2 through C5 carbons are highlighted in green. This alignment represents the best S value (S=167.18) for molecules matching the hypothetical pharmacophore, and the second best overall of 69 alignments from 500 iterations. The alignment was prepared using the Molecular Operating System (MOE 2007.09, Chemical Computing Group, Inc.) using the Flexible Alignment function with both molecules, following a conformational minimization using MOE default settings.14
Scheme 1.

The retrosynthetic analysis of simplified pladienolide analog scaffold 5. BT = Benzothiazole; PG = protecting group
Chemistry
We envisioned a novel synthetic strategy to rapidly access synthetic analogs by modification of the core macrolide unit 10 (for example the deletion of the 3-hydroxyl group of the macrolide) and/or the C15–C22 side chain unit 9 in regions that do not appear to be critical to the pharmacophore, based on our experience with the design of the active FR901464 analogs.13, 14 An efficient approach would allow for the preparation of both the modified fragments from commercially available starting materials and utilize an established late-stage coupling protocol in order to gain rapid access to numerous new analogs. Therefore our initial synthetic goal was the stereoselective construction of (±)-12 from a Mukaiyama aldol reaction, followed by a Baeyer–Villiger oxidation and subsequent lactone hydrolysis, starting from 2-methylcylcohexanone. The reported routes for the construction of the C1–C8 unit of the pladienolides require application of asymmetric dihydroxylation methods17 in order to install the C6–C7 diol moiety. We found that esterification of compound (±)-12 with allyl alcohol 11, followed by diastereoselective ring-closing metathesis, represents a practical strategy to rapidly access the desired 3-deoxy C1–C14 unit macrolide core.
Our approach to the synthesis of 5 began with a novel stereoselective approach to the C1–C9 fragment as shown in Scheme 2. The Mukaiyama aldol reaction18 between the 2,2-disubstituted silylenolether 13 (obtained by a regioselective silylation of 2-methylcyclohexanone)19 and 3-trimethylsilylpropynal provided the corresponding racemic aldol product (syn:anti 10:90) from which the pure anti isomer (±)-14 was isolated in 60% overall yield.20 It is worth noting that BF3·OEt2 afforded a cleaner reaction with better anti stereoselectivity among the Lewis acids that we explored: i.e. 1 eq. TiCl4 (syn:anti 23:77, 60% yield), 10 mol% TiCl4 (syn:anti 25:75, 40% yield) and 1 eq. SnCl4 (syn:anti 10:90, 32% yield).21 It can be noted that we explored conditions that could be expected to provide syn stereoselectivity by application of the Nicholas reaction with 13 (without success) using the dicobalt hexacarbonyl complex of 3-trimethylsilylpropynal20 and other conditions. The next step involved the inversion of C7 stereocenter by means of a Mitsunobu inversion-saponification protocol. Accordingly, the major anti aldol adduct (±)-14 was converted into its syn isomer (±)-15 in 40% yield using 4-nitrobenzoic acid under Mitsunobu conditions.22
Scheme 2.

Synthesis of C1–C9 unit. Reagents and conditions: a) 3-trimethylsilylpropynal, BF3·OEt2, CH2Cl2, 60%; b) i. 4-NO2C6H4CO2H, TPP, DIAD, 40%; ii. K2CO3, MeOH, 70%; C) m-CPBA, NaHCO3; 4:1 Hexane: EtOAc, 70%; d) i. Et3N, MeOH, 90 °C, quant; ii. Lindlar catalyst, EtOAc:pyridine:1-octene, 97%; e) i. 2,2-dimethoxypropane, PPTS, 90%; ii. LiOH, 2:2:1 THF:MeOH:H2O, quant.
As anticipated, the acetylenic TMS group was also cleaved during the hydrolysis of the ester group. Baeyer Villiger oxidation of (±)-15 then provided lactone (±)-16 in 70% yield as a single stereo- and regioisomer, as expected.23 The structure assignment, including the relative stereochemistry at the C6–C7 position, was confirmed by X-ray crystallographic analysis of lactone (±)-16 (see Scheme 2).24 Methanolysis of this lactone in the presence of Et3N, followed by controlled partial hydrogenation using Lindlar catalyst, gave diol (±)-17 in 97% yield.25 Installation of the acetonide diol protecting group and hydrolysis of the resulting methylester produced the acetonide acid (±)-12, in quantitative yield.
The synthesis of the C1–C14 unit was accomplished as shown in Scheme 3 giving aldehyde 18 in 4 steps, as previously reported.26 The Brown allylation of aldehyde 18 provided the enantioenriched (85% ee) alcohol 19 in 89% isolated yield as shown in Scheme 3.27, 28 The acid (±)-12 was then coupled with 19, using the Yamaguchi protocol, to furnish 20a and 20b as an inseparable mixture of diastereomers (1:1) in 88% yield.29 The ring-closing metathesis (RCM) of this diastereomeric pair, with the 2nd generation Hoveyda–Grubbs catalyst, smoothly afforded the macrolides 21a and 21b in 49% combined yield.30 As shown in Scheme 3, an interesting diastereoselection occurred during the RCM reaction, resulting in a 3:2 mixture of the desired 21a and the undesired 21b diastereomers respectively.31, 32 This diastereomeric mixture was separated by standard flash chromatography. The absolute configuration at the C7 carbinol center was determined by converting 21a into (R)- and (S)-MTP derivatives followed by a modified Mosher’s ester protocol (see Supporting Information).33, 34 The desired major diastereomer 21a was then further converted into the corresponding acetyl-aldehyde derivative 10. The deprotection of the acetonide then chemoselective acetylation of the C7 hydroxyl group followed by PMB-deprotection and finally Dess-Martin oxidation, furnished aldehyde 10 in excellent yields.
Scheme 3.

Synthesis of the C1–C14 unit. Reagents and conditions: a) (−)-Ipc2BOMe, Allylmagnesium bromide, diethyl ether, 89%; b) 19, 2,4,6-trichloro benzoylchloride, Et3N, DMAP, 88%, 1:1 diastereomers; c) 2nd generation Hoveyda-Grubbs catalyst, 49%, 3:2 diastereomers; d) i. PPTS, MeOH, 80 °C, 66%; ii. Et3N, Ac2O, DMAP, quant; e) i. DDQ, CH2Cl2, 70%; ii. Dess-Martin periodinane, CH2Cl2, 95%.
Next, the preparation of the C15–C22 side-chain unit was undertaken, as shown in Scheme 4. Commercially available 4-penten-1-ol was protected as the benzothiazole, followed by cross-metathesis with (S)-penten-2-ol smoothly afforded the desired olefin 24 as a 96:4 mixture of E:Z isomers respectively. Following the selective oxidation of 24, the resulting sulfone was subjected to Shi asymmetric epoxidation conditions to afford a 5:1 mixture of the β:α epoxides, from which the desired β-epoxide was isolated in 59% isolated yield. Silylation of the free hydroxy moiety furnished the side chain fragment 25.
Scheme 4.

The synthesis of C15–C22 unit, fragment coupling and the synthesis of simplified pladienolide analogs E-26 and 5. Reagents and conditions: a) i. 2-mercaptobenzothiazole, TPP, DIAD, 90%; ii. (S)-4-penten-2-ol, 2nd generation Grubbs catalyst, 60%; b) i. (NH3)6Mo7(H2O)4, H2O2, EtOH, 72%; ii. Shi epoxidation catalyst, oxone, K2CO3, 59%; iii. TBSOTf, 2,6-lutidine, 86% for compound 25; Ethyl vinyl ether, PPTS, CH2Cl2, 85% for compound 27; c) 10, NaHMDS, THF, E:Z 72:28, 54% for compound 26; mixture of stereoisomers, 38% for compound 28; d) SFC; e) 28, PPTS, MeOH, E:Z 72:28, 25%. (The reaction sequence with ethoxyethyl protecting group are presented in italics).
The final steps in the synthesis of the simplified pladienolide analog 5 entailed the coupling of sulfone 25 with aldehyde 10. This reaction provided compound 26 as a 72:28 mixture of E:Z isomers in 54% yield, which was quantitatively separated by Supercritical Fluid Chromatography (SFC) using an OD-H column to isolate major stereoisomer E-26. Unfortunately desilylation of compound E-26 under a variety of conditions produced inseparable complex mixtures. In order to circumvent this problem the alternate Julia coupling precursor 27 was prepared and used to successfully complete the synthesis of 5. Thus, employing a similar reaction sequence, compound 28 was prepared in 38% yield as an inseparable mixture of stereoisomers. Desilylation of 28 with PPTS/MeOH provided the target analog 5 as a 72:28 inseparable mixture of E:Z isomers in 25% yield. This inseparable mixture of cis and trans isomers (5) was submitted for cytotoxicity assay along with compound E-26 and with our previous FR active analogs 6 and 7 as positive controls.
Biology
As previously reported, the inhibition or knockdown of specific subunits of the spliceosome can induce changes in alternative splicing.11, 12 To investigate the ability of of our synthetic pladienolide analogs to modulate spliceosome function, we determined whether these compounds could alter the splicing of the MDM2 mRNA – the splicing of which has previously been shown to be altered by compound 7 and its analogs, and is now understood to be a defining property of this class of spliceosome modulators.10–12 We found that treatment of SK-MEL-2 melanoma cells with compound E-26 increased mRNA expression of the properly spliced form of MDM2 at lower compound concentrations, but resulted in the formation of two alternatively spliced variant MDM2 mRNAs at the higher concentration (Figure 3). The more potent FR901464 analog 7 induced only a minimal increase in properly spliced MDM2 at the lowest concentration tested but only the splice variant forms at higher concentrations, suggesting that SF3b spliceosome modulator compounds of differing classes and potencies may produce at least partially different profiles of alternatively spliced mRNA. Supporting a correlation between cytotoxicity and the formation of alternatively spliced mRNAs, the control compound 22 (IC50 >20 μM in the SK-MEL-2 cell line) only slightly increased levels of properly spliced MDM2 and failed to induce any splice variant forms of MDM2 and is therefore considered inactive, as expected.
Figure 3. Modulation of MDM2 mRNA splicing.
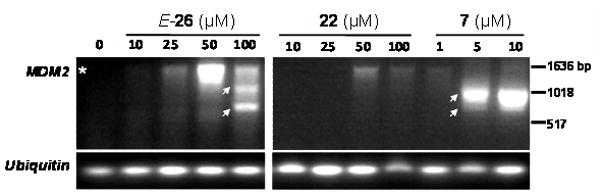
SK-MEL-2 melanoma cells were treated with vehicle (DMSO) or the designated concentrations of compounds E-26, 22, or 7 for 6 hours. Following RNA extraction and reverse transcription, cDNA was amplified by PCR using primers for MDM2 and the intronless gene ubiquitin. Due to the presence of endogenous, transcriptionally inactive, mutant p53 in the SK-MEL-2 cell line, basal levels of MDM2 are low-to-undetectable in the untreated cells.35,36 The asterisk (*) denotes the size of properly spliced MDM2; arrows indicate splice variants of MDM2.
Conclusion
We were gratified to see initial ‘hit-like’ activity with E-26 in a preliminary screen for cytotoxicity with compounds 5 and E-26 that also included the other macrolide intermediates. Initial assays examined the cytotoxicity of these compounds to the JeKo-1 and PC-3 cell lines, using our previous simplified FR901464 analogs (6–8) as standards.13, 14 The active compound (E-26) from this initial screen was then examined for cytotoxicity using a set of cancer cell lines that are sensitive to pladienolide5 and to our simplified FR901464 analogs14 (see Table 1). We were surprised that the TBS protected analog E-26 showed more activity in this assay than the corresponding alcohol 5, which implies a preference for hydrophobic groups at this position. Though this activity is much less potent than pladienolide B, we have succeeded in preparing the first active and totally synthetic analogs of the pladienolide framework that modulate mRNA splicing. These compounds contain only 6 chiral centers, compared to the 10 chiral centers in the natural products. We have now developed methods that will serve as a starting point in the elucidation of the structure-activity relationships for the refinement of the pladienolide splicing modulatory pharmacophore, which is an emerging area for the development of new chemotherapeutics for cancer..
Table 1.
In vitro cytotoxicity data as determined by XTT assay following a 72 h exposure. The data are expressed in μM as the mean ± standard error of 3 independent experiments. The IC50 is defined as the drug concentration that inhibits growth to 50% of the vehicle (DMSO)-treated control; IC50 values were calculated from sigmoidal analysis of the dose-response curves using Origin, version 7.5, software (OriginLab, Northampton, MA). ND, not determined. This assay was performed as previously reported.6
| Cell line | Cancer type | IC50 (μM)
|
|||
|---|---|---|---|---|---|
| E-26 | 5 | 6 | 7 | ||
| SK-MEL-2 | Melanoma | 10.71 ± 0.77 | >20 | 0.39 ± 0.11 | 0.70 ± 0.10 |
| JeKo-1 | Mantle cell lymphoma | 11.60 ± 0.27 | >20 | 0.11 ± 0.01 | 0.29 ± 0.02 |
| MOLT-4 | Acute lymphoblastic leukemia | 15.46 ± 0.76 | ND | 0.18 ± 0.01 | 0.66 ± 0.07 |
| PC-3 | Prostate | 18.03 ± 0.66 | >20 | 2.02 ± 0.39 | 4.16 ± 0.68 |
| MCF7 | Breast | 19.15 ± 0.55 | ND | 0.72 ± 0.02 | 1.69 ± 0.48 |
| NCI-H226 | Non-small cell lung cancer | >20 | ND | >10 | >10 |
Supplementary Material
Acknowledgments
This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital and by the National Cancer Institute Grant CA140474. We thank John Bollinger in the department of Structural Biology, St. Jude Children’s Research Hospital for the determination of the X-ray crystal structure of compound (±)-16.
Footnotes
Electronic Supplementary Information (ESI) available: X-ray crystallography data of compound (±)-16, modified Mosher ester protocol of compound 21a, experimental details and copies of 1H, 13C spectra for all new compounds. See DOI: xxxx
Author roles: M.K.G. performed all of the chemistry content of this work, collaborated on the design of targets with T.R.W., and wrote the initial draft manuscript including all supporting information; T.R.W. directed the project, designed the initial synthetic targets, direct the chemistry and drafted the final manuscript; A.P. and X.C. performed all of the biology studies described herein, with supervision from S.W.M.
References and notes
- 1.Sakai T, Sameshima T, Matsufuji M, Kawamura N, Dobashi K, Mizui Y. J Antibiot. 2004;57:173–179. doi: 10.7164/antibiotics.57.173. [DOI] [PubMed] [Google Scholar]
- 2.Asai N, Kotake Y, Niijima J, Fukuda Y, Uehara T, Sakai T. J Antibiot. 2007;60:364–369. doi: 10.1038/ja.2007.49. [DOI] [PubMed] [Google Scholar]
- 3.Kanada RM, Itoh D, Nagai M, Niijima J, Asai N, Mizui Y, Abe S, Kotake Y. Angev Chem (International ed. 2007;46:4350–4355. doi: 10.1002/anie.200604997. [DOI] [PubMed] [Google Scholar]
- 4.Mizui Y, Sakai T, Iwata M, Uenaka T, Okamoto K, Shimizu H, Yamori T, Yoshimatsu K, Asada M. J Antibiot. 2004;57:188–196. doi: 10.7164/antibiotics.57.188. [DOI] [PubMed] [Google Scholar]
- 5.Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y. Nat Chem Biol. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- 6.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, Tani T, Horinouchi S, Yoshida M. Nat Chem Biol. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, Shimomura K. J Antibiot. 1996;49:1204–1211. doi: 10.7164/antibiotics.49.1204. [DOI] [PubMed] [Google Scholar]
- 8.Nakajima H, Sato B, Fujita T, Takase S, Terano H, Okuhara M. J Antibiot. 1996;49:1196–1203. doi: 10.7164/antibiotics.49.1196. [DOI] [PubMed] [Google Scholar]
- 9.Nakajima H, Takase S, Terano H, Tanaka H. J Antibiot. 1997;50:96–99. doi: 10.7164/antibiotics.50.96. [DOI] [PubMed] [Google Scholar]
- 10.Folco EG, Coil KE, Reed R. Genes & development. 25:440–444. doi: 10.1101/gad.2009411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corrionero A, Minana B, Valcarcel J. Genes & development. 25:445–459. doi: 10.1101/gad.2014311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. ACS Chemical Biology. 2011 doi: 10.1021/cb100356k. Publication Date (Web): February 23, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagisetti C, Pourpak A, Jiang Q, Cui X, Goronga T, Morris SW, Webb TR. J Med Chem. 2008;51:6220–6224. doi: 10.1021/jm8006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lagisetti C, Pourpak A, Goronga T, Jiang Q, Cui X, Hyle J, Lahti JM, Morris SW, Webb TR. J Med Chem. 2009;52:6979–6990. doi: 10.1021/jm901215m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandel AL, Jones BD, La Clair JJ, Burkart MD. Bioorg Med Chem Lett. 2007;17:5159–5164. doi: 10.1016/j.bmcl.2007.06.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skaanderup PR, Jensen T. Organic letters. 2008;10:2821–2824. doi: 10.1021/ol800946x. [DOI] [PubMed] [Google Scholar]
- 17.Choudary BM, Chowdari NS, Jyothi K, Kantam ML. J Am Chem Soc. 2002;124:5341–5349. doi: 10.1021/ja017889j. [DOI] [PubMed] [Google Scholar]
- 18.Mukaiyama T, Banno K, Narasaka K. J Am Chem Soc. 1974;96:7503–7509. [Google Scholar]
- 19.Sarabèr FCE, Baranovsky A, Jansen BJM, Posthumus MA, de Groot A. Tetrahedron. 2006;62:1726–1742. [Google Scholar]
- 20.Mukai C, Nagami K, Hanaoka M. Tetrahedron Lett. 1989;30:5623–5626. [Google Scholar]
- 21.Mahrwald R. Chemical reviews. 1999;99:1095–1120. doi: 10.1021/cr980415r. [DOI] [PubMed] [Google Scholar]
- 22.Dodge JA, Nissan JS, Presnell M. Org Synth. 1996;73:110–115. [Google Scholar]
- 23.Kabat MM, Garofalo LM, Daniewski AR, Hutchings SD, Liu W, Okabe M, Radinov R, Zhou Y. J Org Chem. 2001;66:6141–6150. doi: 10.1021/jo015788y. [DOI] [PubMed] [Google Scholar]
- 24.CCDC 805182 contains the supplementary crystallographic data for compound (±)-16. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 25.Kumar P, Naidu SV, Gupta P. J Org Chem. 2005;70:2843–2846. doi: 10.1021/jo048087k. [DOI] [PubMed] [Google Scholar]
- 26.Curley RW, Jr, Ticoras CJ. Synth Commun. 1986;16:627–631. [Google Scholar]
- 27.Lafontaine JA, Provencal DP, Gardelli C, Leahy JW. J Org Chem. 2003;68:4215–4234. doi: 10.1021/jo034011x. [DOI] [PubMed] [Google Scholar]
- 28.Jadhav PK, Bhat KS, Perumal PT, Brown HC. J Org Chem. 1986;51:432–439. [Google Scholar]
- 29.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 30.Based on our investigation using diverse Grubbs catalysts and conditions, it appears that in addition to the relevant factors discussed by Kanada et al.(ref. 3), the C6–C7 relative stereochemistry may also play a key role in the efficiency of this RCM reaction.
- 31.Mehta G, Shinde HM. Tetrahedron Lett. 2005;46:6633–6636. [Google Scholar]
- 32.Bajwa N, Jennings MP. Tetrahedron Lett. 2008;49:390–393. [Google Scholar]
- 33.Dale JA, Mosher HS. J Amer Chem Soc. 1973;95:512–519. [Google Scholar]
- 34.Hoye TR, Jeffrey CS, Shao F. Nat Protoc. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 35.Haapajarvi T, Pitkanen K, Laiho M. Cell Growth Differ. 1999;3:163–71. [PubMed] [Google Scholar]
- 36.Landers JE, Cassel SL, George DL. Cancer Res. 1997;57:3562–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.