

Abstract
RAG1 and RAG2 initiate V(D)J recombination, the process of rearranging the antigen-binding domain of immunoglobulins and T-cell receptors, by introducing site-specific double-strand breaks (DSB) in chromosomal DNA during lymphocyte development. These breaks are generated in two steps, nicking of one strand (hydrolysis), followed by hairpin formation (transesterification). The nature and location of the RAG active site(s) have remained unknown. Because acidic amino acids have a critical role in catalyzing DNA cleavage by nucleases and recombinases that require divalent metal ions as cofactors, we hypothesized that acidic active site residues are likewise essential for RAG-mediated DNA cleavage. We altered each conserved acidic amino acid in RAG1 and RAG2 by site-directed mutagenesis, and examined >100 mutants using a combination of in vivo and in vitro analyses. No conserved acidic amino acids in RAG2 were critical for catalysis; three RAG1 mutants retained normal DNA binding, but were catalytically inactive for both nicking and hairpin formation. These data argue that one active site in RAG1 performs both steps of the cleavage reaction. Amino acid substitution experiments that changed the metal ion specificity suggest that at least one of these three residues contacts the metal ion(s) directly. These data suggest that RAG-mediated DNA cleavage involves coordination of divalent metal ion(s) by RAG1.
Keywords: RAG1, RAG2, V(D)J recombination, immunoglobulin
V(D)J recombination is the process by which V (variable), D (diversity), and J (joining) gene segments are joined to form an exon that encodes the antigen-binding domain of immunoglobulins and T-cell receptors. These gene segments, termed coding segments, are flanked by recombination signal sequences (RSSs) that serve as recognition motifs for the recombinase machinery. The lymphoid-specific proteins RAG1 and RAG2 bind to the RSS and together constitute a site-specific endonuclease that introduces a double-strand break (DSB) between the RSS and the adjacent coding segment. DSB formation proceeds by two sequential single-strand cleavage events. In the first step of this reaction, hydrolysis, water is used as a nucleophile to attack a phosphodiester bond, introducing a nick precisely between the RSS and the coding segment. In the second step, transesterification, the newly formed 3′ OH is used as a nucleophile to attack the second phosphodiester bond, creating a covalently sealed hairpin coding end and a blunt, 5′-phosphorylated signal end (McBlane et al. 1995).
V(D)J recombination is central to a functional immune system. The activity of the RAG proteins must be carefully regulated, as inappropriate rearrangements catalyzed by this system can be oncogenic (Tycko and Sklar 1990; Korsmeyer 1992). Thus, it is critical to decipher the mechanism of catalysis to understand the multiple regulatory controls that guard against inappropriate recombination events. The nature and the location of the active site(s) responsible for hydrolysis and transesterification have not been established; consequently, it is not known whether a single active site carries out both reactions. Furthermore, whereas RAG1 alone can bind to the RSS (Difilippantonio et al. 1996; Spanopoulou et al. 1996; Akamatsu and Oettinger 1998; Nagawa et al. 1998), stable, efficient binding requires RAG2 (Hiom and Gellert 1997; Akamatsu and Oettinger 1998; Swanson and Desiderio 1998, 1999) and all known catalytic activities require the presence of both proteins (McBlane et al. 1995; Hiom and Gellert 1997; Agrawal et al. 1998; Besmer et al. 1998; Hiom et al. 1998; Melek et al. 1998; Shockett and Schatz 1999). Thus, it is not known whether RAG-1 or RAG-2 contains the active site(s) or whether these two proteins form one or more shared active sites.
Analysis of the predicted amino acid sequences of the RAG proteins has failed to reveal significant similarities with other recombinases that would provide hints about the location or nature of the active site. Neither the scant structural information nor the limited mutagenesis data currently available for the RAG proteins has yielded insight into the catalytic mechanism. Nevertheless, one important clue is provided by the observation that DNA cleavage by the RAG proteins requires the presence of divalent metal ions (van Gent et al. 1995). One feature common to nucleases and recombinases that use metal ions for catalysis of DNA cleavage is the presence of acidic amino acids in the active site that coordinate the divalent metal ion(s) (Vipond and Halford 1993; Grindley and Leschziner 1995).
Additional hints about the catalytic properties of the RAG proteins are provided by the functional similarities they share with members of the retroviral integrase superfamily (Craig 1996; Mizuuchi 1997; Roth and Craig 1998), which consists of several transposases and retroviral integrases (Grindley and Leschziner 1995; Polard and Chandler 1995). Shared features include the basic chemical mechanism of DNA cleavage (nicking by hydrolysis and strand transfer by transesterification) (Engelman et al. 1991; Mizuuchi and Adzuma 1991; Vink et al. 1991; van Gent et al. 1996a); a requirement for divalent metal ions (Mg2+ or Mn2+); the ability to use alternative nucleophiles such as alcohols (alcoholysis) (Engelman et al. 1991; Vink et al. 1991; van Gent et al. 1996a); the production of DNA hairpins (Roth et al. 1992; Mazumder et al. 1994; Van den Ent et al. 1994; Kennedy et al. 1998); and the ability to reverse the reaction (disintegration) (Chow et al. 1992; Engelman and Craigie 1992; Mazumder et al. 1994; Han et al. 1997; Melek et al. 1998).In fact, the RAG proteins have been shown recently to function as an authentic transposase in vitro (Agrawal et al. 1998; Hiom et al. 1998). These mechanistic similarities suggest that the active sites used by the RAG proteins for DNA cleavage might have critical features in common with those of the retroviral integrase superfamily.
The bacterial transposases MuA, Tn7, and Tn10, and the integrase proteins encoded by the HIV-1 and ASV retroviruses are the most thoroughly characterized members of the retroviral integrase superfamily. Each of these proteins contains a catalytic triad of two aspartic acids (D) and a glutamic acid (E), commonly referred to as the DDE motif (Kulkosky et al. 1992; Grindley and Leschziner 1995). The DDE motif is required for DNA cleavage: Mutation of any one of the three residues results in a severe (∼100-fold) decrease in activity (Engelman and Craigie 1992; Kulkosky et al. 1992; Baker and Luo 1994; Kim et al. 1995; Bolland and Kleckner 1996; Sarnovsky et al. 1996; Krementsova et al. 1998). Whereas the precise role of the DDE motif in phosphodiester bond cleavage has not been fully elucidated, crystallographic analysis of several superfamily members indicates that the two aspartic acids coordinate divalent metal ion(s) that are necessary for catalysis (Grindley and Leschziner 1995). Crystal structures of the HIV and ASV integrases in the presence of divalent metal ions reveal that the carboxylates of the two aspartates are close to one metal ion, suggesting that these residues are critical for positioning the metal ion to facilitate cleavage of the scissile phosphodiester bond (Bujacz et al. 1995, 1996; Goldgur et al. 1998; Maignan et al. 1998). The role of the glutamate in the DDE motif is less clear. Although required for catalysis, in every retroviral integrase superfamily member studied to date (Engelman and Craigie 1992; Kulkosky et al. 1992; Baker and Luo 1994; Sarnovsky et al. 1996; Kim et al. 1995; Bolland and Kleckner 1996; Krementsova et al. 1998), the available crystallographic data (HIV IN, ASV IN and the MuA transposase core domains) have not provided clear evidence that the carboxylate is positioned appropriately to interact with the metal ion (Bujacz et al. 1995, 1996; Rice and Mizuuchi 1995; Goldgur et al. 1998; Maignan et al. 1998).
Several approaches have been used to identify catalytic residues in retroviral integrase superfamily members, including primary sequence comparisons (Engelman and Craigie 1992; Kulkosky et al. 1992). The family members that have been crystallized, however, show <20% sequence identity in the region of the active site, even after structural alignment (Grindley and Leschziner 1995). Thus, functional analysis of site-directed mutants has been required to identify catalytic residues in several superfamily members (Engelman and Craigie 1992; Kulkosky et al. 1992; Baker and Luo 1994; Kim et al. 1995; Bolland and Kleckner 1996; Sarnovsky et al. 1996; Krementsova et al. 1998). Catalytic-deficient mutants fulfill the following criteria: (1) removal of the negative charge by substituting amino acids that lack the negative charge (alanine, asparagine, or glutamine) yields a severe defect in catalysis (≤1% activity) (Engelman and Craigie 1992; Kulkosky et al. 1992; Baker and Luo 1994; Kim et al. 1995; Bolland and Kleckner 1996; Sarnovsky et al. 1996; Krementsova et al. 1998); (2) activity is also lost in substitution mutants that retain the negative charge but alter the geometry of the side chain (changing D to E or E to D), indicating that precise positioning of the charge is important (Engelman and Craigie 1992; Kulkosky et al. 1992; Kim et al. 1995); and (3) catalytic mutants retain DNA-binding ability and interaction with protein partners, indicating that the mutations do not cause gross distortions in the overall protein structure (Baker and Luo 1994; Kim et al. 1995; Bolland and Kleckner 1996; Sarnovsky et al. 1996; Krementsova et al. 1998). An additional criterion that has been helpful in analyzing some family members is that cysteine substitution mutants, although catalytically inactive in Mg2+, show some activity when incubated in the presence of Mn2+ (Sarnovsky et al. 1996; Allingham et al. 1999). As Mn2+ is thiolphilic, this rescue is considered evidence for interaction between that cysteine and the divalent metal ion(s) (Dahm and Uhlenbeck 1991; Piccirilli et al. 1993; Sarnovsky et al. 1996; Allingham et al. 1999).
Given the numerous mechanistic similarities between V(D)J recombination and the activities of retroviral integrase superfamily members, as well as the common requirement for acidic amino acids in other nucleases and recombinases that catalyze metal-dependent DNA cleavage, we hypothesized that acidic amino acid residues play a critical role in catalysis of DNA cleavage by the RAG proteins. To test this hypothesis, we aligned the sequences of the RAG proteins from all available species and removed the negative charges from all conserved acidic amino acids in both RAG1 and RAG2 by site-directed mutagenesis. Of 109 mutants analyzed, 3 catalytic-deficient mutants, all in RAG1, were identified, affecting positions D600, D708, and E962. Biochemical analysis of these mutants indicates that these three acidic amino acids are critical for both nicking and hairpin formation, suggesting that one active site catalyzes both activities. A role for at least one of the critical acidic amino acids in metal ion binding is suggested by the rescue of a cysteine substitution mutant (D708C) in the presence of Mn2+.
Results
Experimental design
Our search for active site residues focused on the core regions of RAG1 [384–1008, truncated (t)RAG1] and RAG2 (1–387, tRAG2), as these are the minimal versions of the proteins capable of catalyzing V(D)J recombination (Sadofsky et al. 1993, 1994; Silver et al. 1993; Cuomo and Oettinger 1994). We identified conserved acidic amino acids by aligning the amino acid sequences of tRAG1 and tRAG2, individually, from all species for which sequence information was available (nine for tRAG1, seven for tRAG2), as summarized in Figure 1. All conserved D and E residues were mutated, as were positions in which either a D or an E is present in all species (conserved charge). Removal of the negative charge with minimal structural change was accomplished by changing D and E residues to N and Q, respectively. These substitutions have been shown to abolish activity in retroviral integrase superfamily members (Engelman and Craigie 1992; Kulkosky et al. 1992; Baker and Luo 1994; Kim et al. 1995; Sarnovsky et al. 1996; Krementsova et al. 1998).
Figure 1.

Targets for mutagenesis. Alignments of the predicted protein sequences of RAG1 (nine species) and RAG2 (seven species) were performed with ClustalW and used to determine conserved amino acids for mutagenesis. Accession nos. for RAG1 sequences are as follows: murine (P15919), human (P15918), rabbit (P34088), opossum (U51897), chicken (P24271), Xenopus (L19324.1), bull shark (U62645.1), rainbow trout (U15663), and zebrafish (U71093). Accession nos. for RAG2 sequences are as follows: murine (M64796), human (P55895), rabbit (M99311), chicken (M58531), Xenopus (L19325), rainbow trout (U31670), and zebrafish (U71094.1). Conserved aspartic acids (D) and glutamic acids (E) are shown as white on a black background. Positions of conserved charge (D or E), are black on a gray background. Nonconserved positions also chosen for mutagenesis are shown as white on gray.
Initially, we selected potential catalytic mutants by their inability to form signal joints in vivo using an established transient transfection assay (Steen et al. 1996; Han et al. 1999). Assaying for signal joint formation is preferable to testing for signal ends because the former involves less manipulation, thus facilitating the screening of a large number of mutants. Because cleavage is required for signal joint formation, this procedure identifies all cleavage-deficient mutants; however, other types of defects might also prevent signal joint formation. Therefore, mutants with severe defects in signal joint formation (∼100 fold, as expected for catalytic-deficient mutants) were also assayed for their ability to form signal ends in vivo with a standard semiquantitative ligation-mediated PCR (LMPCR) assay (Steen et al. 1996). Although mutations of DDE motif residues in other superfamily members profoundly impair cleavage efficiency (∼100-fold) (Engelman and Craigie 1992; Kulkosky et al. 1992; Baker and Luo 1994; Kim et al. 1995; Bolland and Kleckner 1996; Sarnovsky et al. 1996), we conservatively chose all mutants that decreased cleavage >10-fold for further biochemical analysis. Purified mutant proteins were tested for the properties expected of catalytic-deficient mutants, as described below.
No conserved acidic amino acids of RAG2 are essential for cleavage
We constructed 35 tRAG2 mutants (Fig. 1). Expression vectors encoding each mutant were cotransfected with wild-type tRAG1 and a recombination substrate, pJH290, into cultured fibroblasts. Plasmid DNA was harvested 48 hr after transfection and signal-joint formation was assessed. All mutant proteins catalyzed signal joint formation at or near wild-type levels (Fig. 2; data not shown), indicating that the mutated amino acids are not essential for recombination. These data demonstrate that conserved acidic residues in tRAG2 do not have a critical role in catalysis.
Figure 2.

RAG2 does not contribute conserved acidic amino acid residues to cleavage. Signal joints formed in vivo were detected by PCR and Southern blotting. The expected size of the signal joint-containing product is 220 bp. (1×) A PCR reaction containing one-thirtieth of the DNA recovered from a single transfection; (1:10, 1:100) PCR reactions containing the indicated dilutions. All mutants were assayed at the 1× concentration. The marker lane contains a radiolabeled 1-kb ladder (GIBCO/BRL). (Open triangle) 12-RSS; (solid triangle) 23-RSS; (rectangles) coding segments; (arrows) PCR primers; (SJ) signal joint; (WT) wild-type tRAG-1/tRAG-2.
Analysis of RAG1 mutants reveals several essential acidic amino acids
A total of 74 tRAG1 mutants were assayed for their ability to form signal joints in vivo. Of these, 15 mutants exhibited a severe (≥100-fold) decrease in signal joint formation. Because failure to form signal joints could reflect defects in either cleavage or joining, we assessed the ability of these 15 mutants to form signal ends in vivo. Eight of these mutants (E597Q, D600N, D708N, E719Q, D792N, E959Q, E962Q, and D986N) exhibited severe defects (≥100-fold) in cleavage (Fig. 3, lanes 4–6, 8, 9, 11–13); two mutants (E709Q and E811Q, lanes 7 and 10) yielded signal ends at a level ∼50-fold less than the wild-type control. These ten were considered candidates for catalytic residues. The remaining five mutants with much milder defects in cleavage (≤10-fold) were not studied further. Western blot analysis revealed that all 10 potential catalytic-deficient proteins were expressed at or near wild-type levels in transiently transfected mammalian cells (data not shown), indicating that the lack of cleavage was not due to effects of the mutations on protein expression. It should be noted that D546A and D560A are defective for DNA binding (Kim et al. 1999). Our corresponding mutants, D546N and D560N, showed only a 10-fold defect in cleavage (in accord with Kim et al.) and were therefore not studied in vitro.
Figure 3.

Several RAG1 mutants are severely impaired for cleavage. Signal ends formed in vivo were detected by ligation-mediated PCR for the 23 RSS. (Similar results were obtained in assays for the 12 RSS; data not shown.) (1X) Ligations containing one-fifteenth of the DNA recovered from a single transfection, diluted 1:100 then assayed by PCR. All mutants were assayed at the 1× concentration. The expected product for cleavage at the 23 RSS is 128 bp. (SE) Signal end; parallel lines denote ligation primers; other symbols are as in Fig. 2.
DNA-binding capabilities of mutant RAG1 proteins
Analysis of retroviral integrase superfamily members has shown that mutation of catalytic residues does not appreciably interfere with DNA-binding or protein–protein interactions (Baker and Luo 1994; Kim et al. 1995; Bolland and Kleckner 1996; Sarnovsky et al. 1996; Krementsova et al. 1998). One would thus expect catalytic-deficient RAG1 mutants to exhibit normal DNA-binding activity. We assessed the ability of purified mutant proteins (approximately equal amounts as judged by Coomassie-stained SDS–polyacrylamide gels) to bind to an oligonucleotide substrate containing a 12-RSS with a standard electrophoretic mobility shift assay (Hiom and Gellert 1997), which detects formation of a DNA–protein complex involving a dimer of RAG1 and one or two monomers of RAG2 (Hiom and Gellert 1997; Akamatsu and Oettinger 1998; Bailin et al. 1999; Rodgers et al. 1999; Swanson and Desiderio 1999). One mutant, E811Q, displayed only trace amounts of DNA binding (Fig. 4, lane 5). This result, along with the observation that this mutant protein was difficult to purify from baculovirus infected cells (data not shown), suggests that this mutation may interfere with proper protein folding. Therefore, this protein was not studied further in purified form (see below for additional analysis). Binding was reduced by no more than 10-fold for any of the other 9 mutants examined (Fig. 4, lanes 2–4, 6–11).
Figure 4.

Binding of RAG1 mutants to a 12 RSS. DNA binding was assayed by electrophoretic mobility shift with a radiolabeled oligonucleotide probe containing a 12 RSS. Proteins tested in lanes 1–5 were MBP fusions (of wild-type or mutant tRAG1 and wild-type tRAG2). In lanes 6–14, the RAG proteins do not contain the MBP fusion; hence, the mobility of the DNA–protein complex is faster than observed in lanes 1–5. Some mutants (D600N, D708N) were tested both as MBP and non-MBP forms with the same results.
DNA cleavage capabilities of mutant RAG-1 proteins
Next, we assessed the ability of the purified mutant proteins to cleave an RSS-containing oligonucleotide substrate in vitro. Cleavage occurs efficiently at a single RSS in the presence of Mn2+, allowing detection of both nicks and hairpins, the expected intermediates and products of the cleavage reaction (McBlane et al. 1995; van Gent et al. 1996b). Thus, in vitro analysis of single RSS cleavage allows us to focus directly on the effects of each mutation on the individual catalytic steps. Furthermore, the use of pre-nicked substrates in this assay allowed us to test the second catalytic step, hairpin formation, independently of nicking.
The results of in vitro cleavage analysis neatly divided the nine binding-proficient, cleavage-deficient mutants into two classes, those that retain some cleavage activity (class I) and those that do not (class II). Class-I mutants (E597Q, E709Q, E719Q, D792N, E959Q, and D986N) exhibited impaired, but detectable, nicking and hairpin formation. Each mutant nicked with efficiencies ≥10% of wild type (Fig. 5A, lanes 2–7). Although D986N nicks at an aberrant location, nicks were nonetheless produced and hairpins of normal size were generated (lane 4), indicating that this mutant retains catalytic activity. To analyze the hairpin formation step directly, we used prenicked substrates and found detectable hairpin formation with all class-I mutants (Fig. 5B, lanes 2–7). (Visualization of hairpins formed by D792N requires a long exposure; data not shown. This mutant is clearly capable of catalyzing hairpin formation in crude extracts; see below). Thus, class-I mutants are capable of carrying out both catalytic steps.
Figure 5.
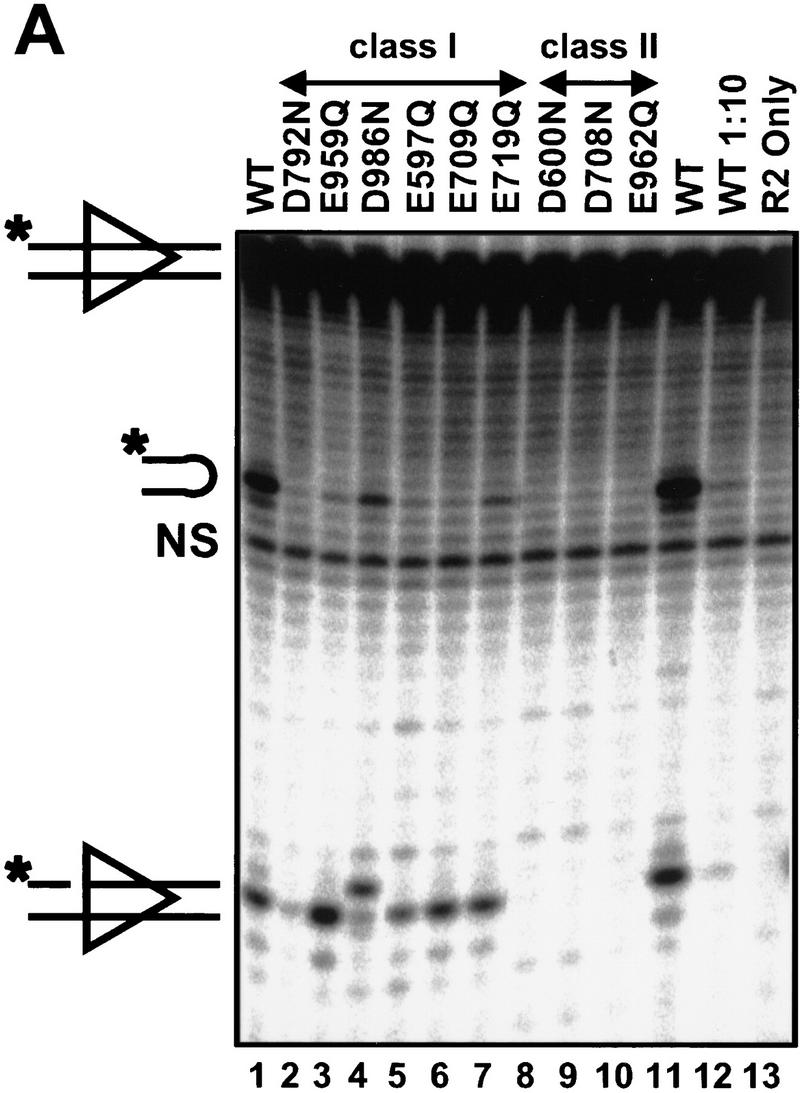

Class-II mutants are defective for both nicking and hairpin formation. Cleavage activity was tested in Mn2+ with the same protein preparations used in Fig. 4 with a radiolabeled oligonucleotide substrate containing a 12 RSS. The substrate is labeled on the 5′ end of the top strand, such that nicking produces a 16-nucleotide product and transesterification generates a 32-nucleotide hairpin. (A) A non-nicked substrate is used to assay nicking and hairpin formation. The species migrating slightly faster than the hairpin product (NS) does not depend on RAG-mediated cleavage (lane 13) and is a nonspecific band present in the substrate preparation. (B) A prenicked substrate is used to assay specifically for hairpin formation. (WT) Incubations with wild-type tRAG1 and tRAG2. (1:10) incubations done with a 1:10 dilution of wild-type tRAG1 and tRAG2. (R2 only) control incubations performed only with tRAG2.
All class-I mutants (E597Q, E709Q, E719Q, D792N, E959Q, and D986N) were further tested with crude extracts prepared from mammalian cells that express the appropriate mutant tRAG proteins. Crude extracts bypass potential problems introduced by overexpression and purification of mutant proteins from a heterologous system, which might cause reductions in specific activity of the mutants not directly related to specific catalytic defects. Furthermore, we have found that crude extracts catalyze more efficient cleavage than purified RAG proteins, presumably because the extracts contain cofactors that stimulate cleavage (L.E. Huye and D.B. Roth, unpubl.). Importantly, such cofactors should not stimulate cleavage by RAG mutants with alterations in amino acids that are critical for catalysis.
To assay for cleavage in crude extracts, a plasmid recombination substrate containing two RSS, pJH290, was incubated in the presence of extract and Mn2+. Deproteinized reaction products were then digested with PvuII, allowing the detection of cleaved molecules containing signal and coding ends by Southern blotting. When assayed in this system, the class-I mutants catalyzed cleavage at levels comparable with wild type (Fig. 6, lanes 2–7). Class-I mutants, therefore, created double-strand breaks, which require both nicking and hairpin formation; moreover, cleavage occurred at both RSS, yielding the normal excised linear fragment. Together, these data show that the class-I mutants carried out both steps of the cleavage reaction. Note that the E811Q mutant also catalyzed coupled cleavage at both RSS in the crude extract system (Fig. 6, lanes 12,17), indicating that this mutant is not completely defective for catalysis.
Figure 6.

Class-I, but not class-II, mutants are capable of DSB formation. Crude extracts containing RAG proteins were assayed with a plasmid substrate in the presence of Mn2+. Cleavage products were visualized by Southern blotting. Lanes 13–17 represent a longer exposure of lanes 8–12. Substrate and expected cleavage products are illustrated at right.
In contrast to the class-I mutants, the three class-II mutants (D600N, D708N, and E962Q) exhibited no detectable cleavage of oligonucleotide substrates in Mn2+ (Fig. 5A, lanes 8–10), did not form hairpins from pre-nicked substrates (Fig. 5B, lanes 8–10), and failed to catalyze detectable DSB formation in crude extracts (Fig. 6, lanes 8–10,13–15). Thus, these three mutants retained normal DNA-binding capability, yet displayed no detectable catalytic activity for either nicking or hairpin formation. The class-II mutants are catalysis deficient.
Side-chain geometry is critical for catalysis
As discussed above, a common feature of DDE motif amino acids is the dependence of catalytic activity, not just on the presence of a negative charge, but also on the configuration of the side chain. Changing a D to an E or an E to a D results in as severe a defect as mutating to an N, Q, or A, suggesting that precise positioning of the metal ion is critical for catalysis (Engelman and Craigie 1992; Kulkosky et al. 1992; Kim et al. 1995). We hypothesized that such charge-conserving mutations in the critical acidic residues in RAG1 would abolish catalytic activity. Such mutants were constructed for the three class-II mutants (D600E, D708E, and E962D), and at three other positions for which substitutions of N or Q substantially reduced cleavage (D792E, E811D, and E959D). We then assayed the mutants for signal end formation in vivo (the mutations did not affect protein expression as assessed by Western blotting; data not shown). As predicted, only the three class-II mutants failed to give detectable cleavage (Fig. 7, lanes 5–7). The remaining mutants formed signal ends (lanes 8–10). Notably, the E811D mutant generated signal ends at approximately one-half the wild-type level (cf. lanes 9 and 2). Together with the biochemical data described above, these observations lead us to conclude that E811 is not absolutely required for catalysis. Our observation that charge-preserving alterations at the three critical positions abolish cleavage strongly indicates that these three amino acids play essential roles in catalysis.
Figure 7.

Analysis of charge-conserving substitution mutants in vivo. Signal ends generated in vivo by the indicated RAG mutants were detected by ligation-mediated PCR as described in the legend to Fig. 3.
A cysteine-substitution mutant displays altered metal ion specificity
As mentioned above, Mn2+ is thiolphilic. Metal-binding amino acids in other nucleases have been identified by the ability of cysteine substitution mutants to alter the metal ion specificity of the reaction. The cysteine substitution mutants are catalytically inactive in Mg2+ (because the negative charge responsible for metal binding has been removed), but incubation in Mn2+ allows a partial rescue of activity, strongly suggesting that there is a direct interaction between this amino acid and the metal ion(s) (Piccirilli et al. 1993; Sarnovsky et al. 1996; Allingham et al. 1999). We constructed cysteine substitution versions of the three class-II mutants (D600C, D708C, and E962C) and tested them along with the N or Q substitution mutants in the crude extract system described above. As expected, none of the N or Q substitution mutants showed detectable activity in Mn2+ (Fig. 8, lanes 2–4) or Mg2+ (lanes 10–12). However, D708C formed DSB in Mn2+ (cf. lanes 6 and 14), demonstrating that catalytic activity is rescued in a Cys/Mn2+-dependent manner. Whereas neither D600C nor E962C exhibited Mn2+-dependent rescue (Fig. 8, lanes 5,7), these results do not rule out a role for these amino acids in metal binding (see Discussion).
Figure 8.

Mn2+ rescue of cysteine substitution mutants. The indicated mutants were tested in the crude extract system for their ability to catalyze cleavage of plasmid substrates in Mn2+ (left) and in Mg2+ (right). Substrate and expected cleavage products are illustrated at right.
Discussion
Our comprehensive mutational analysis of all conserved acidic amino acids in the core regions of RAG1 and RAG2 identified nine RAG1 mutants that retain normal RSS binding but exhibit specific defects in cleavage. Three of these mutants (class II), as well as charge-preserving alterations at these positions, are catalytically inactive under all conditions tested. The three amino acids identified by these experiments (D600, D708, and E962) are, therefore, required specifically for catalysis. Our results indicate that these three amino acids participate in the active site that catalyzes both nicking and hairpin formation. The three critical acidic amino acids and their neighbors are completely conserved among the RAG1 sequences from a variety of organisms (Fig. 9A), as expected for amino acids located in the region of the active site. Our data are in accord with previous mutational studies of RAG1, which showed that deletions encompassing D600 (Kirch et al. 1996) or E962 (Sadofsky et al. 1993) abolish recombination.
Figure 9.
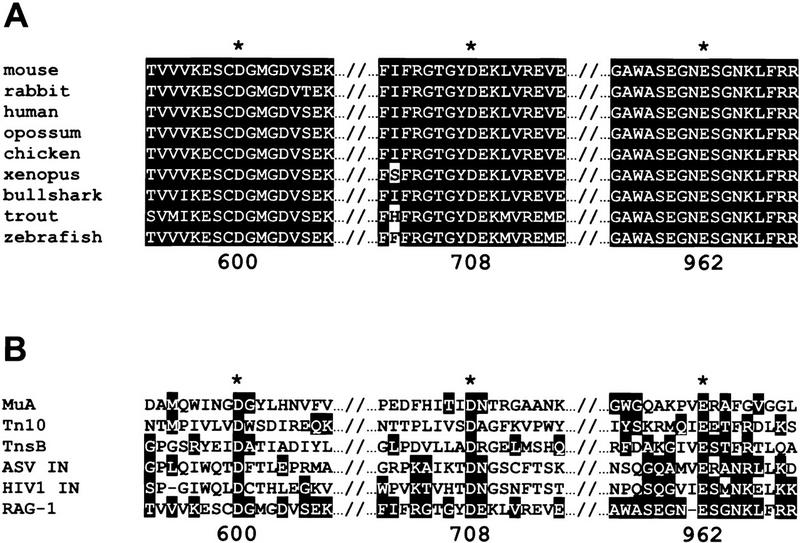
Sequence alignments of catalytic residues. Amino acid sequence alignments are shown for the relevant regions of RAG-1 from several organisms (A) and for selected retroviral integrase superfamily members and murine RAG-1 (B). (Asterisks) The amino acids defined by catalytic-deficient (class II) mutants. Highlighted amino acids were grouped as follows according to Engelman and Craigie (1992): (G,A,S,T,P); (L,I,V,M); (F,Y,W); (D,E,N,Q); (K,R,H); C. Amino acid numbers correspond to murine RAG-1.
One active site in RAG1 performs both hydrolysis and transesterification
RAG-mediated DNA cleavage occurs in two steps: hydrolysis (nicking) and transesterification (hairpin formation). In principle, these steps could involve the use of one active site, as in the Tn10 (Bolland and Kleckner 1996) and MuA (Namgoong and Harshey 1998; Williams et al. 1999) transposases, or use of two different active sites, as in the Tn7 transposase (Sarnovsky et al. 1996). In the case of multiple active sites, we would expect two types of catalytic-deficient mutants—those that would be able to nick at wild-type levels, but be severely defective for hairpin formation, and others that would be severely defective for nicking, but form hairpins at wild-type levels with prenicked substrates. We isolated no such mutants, which would have been identified as recombination-deficient mutants in our in vivo screen. Furthermore, the three class-II mutants we isolated are completely defective for both cleavage reactions. These data strongly suggest that these three amino acid residues contribute to the active site that performs both nicking and hairpin formation. This active site may also perform the other reactions carried out by the RAG proteins, transposition (Agrawal et al. 1998; Hiom et al. 1998), RSS-independent endonuclease activity (end processing) (Besmer et al. 1998), and hairpin opening (Besmer et al. 1998; Shockett and Schatz 1999). In support of this hypothesis, we have found that the three catalytic-deficient mutants are devoid of RSS-independent endonuclease activity (M. Landree and D. Roth, unpubl.). However, future experiments will be required to elucidate the contributions of each protomer's active site within the context of a RAG1 dimer.
RAG1 participates directly in catalysis
Although both RAG1 and RAG2 are required for all catalytic activities, the individual roles of these two proteins have remained unclear. Our data provide the first evidence that RAG1 participates directly in catalysis. Furthermore, our mutational analysis demonstrates that no acidic residues of RAG2 are required for cleavage. All catalytic steps might be carried out by RAG1. In this case, RAG2 could play a regulatory role, as in the Tn7 transposase, whose catalytic activity requires assembly of three proteins, TnsA, TnsB, and TnsC (Stellwagen and Craig 1997). Alternatively, RAG2 may contribute nonacidic amino acids to a shared active site, as observed in the Flp recombinase system (Lee et al. 1999). This possibility was suggested by recent DNA–protein cross-linking experiments, which localized RAG1 and RAG2 precisely to the site of DSB formation (Eastman et al. 1999).
Possible roles of the essential acidic amino acids
Several lines of evidence support the hypothesis that the class-II residues we have identified are involved in coordination of a divalent metal ion. First, rescue of the D708C mutant by incubation in Mn2+ provides a strong indication that this amino acid interacts directly with the metal ion(s). Rescue of D708C and weak rescue of D600C in Mn2+ have also been observed in the Oettinger laboratory (D.-R. Kim and M. Oettinger, pers. comm.), providing evidence that both of these essential amino acids participate in metal binding. This is further supported by Fe2+-induced protein cleavage data, which indicates that both D600 and D708 may bind to divalent metal ions (D.-R. Kim and M. Oettinger, pers. comm.). Together, these data strongly suggest that two of the three catalytic-deficient mutants we have isolated identify amino acids that coordinate divalent metal ion(s) required for DNA cleavage.
Whereas functional analysis of E962 mutants reveals a severe catalytic defect, so far there is no evidence directly implicating E962 in metal binding. The glutamate may have a somewhat different role in metal binding or in catalysis than the two aspartates. This hypothesis is supported by structural analysis of retroviral integrase superfamily members. In all cases examined to date, whereas the two aspartic acids in the DDE triad are located in positions appropriate for metal binding, there is no indication that the corresponding glutamates are capable of interacting directly with Mn2+ or Mg2+ (Bujacz et al. 1995, 1996; Rice and Mizuuchi 1995; Goldgur et al. 1998; Maignan et al. 1998). Thus, whereas the glutamate of the DDE motif is critical for catalysis, its precise role has not yet been fully delineated. The inability of the E962C mutant to be rescued in Mn2+ may also reflect the fact that, sterically, the cysteine side chain more closely resembles aspartate than glutamate.
Others have suggested that the ability of Mn2+ to rescue the activity of mutants (D to N or E to Q) may indicate a role for those amino acids in metal binding (Baker and Luo 1994; Sarnovsky et al. 1996). Does this indicate that our class-I mutants, which are active in the presence of Mn2+, affect metal binding residues? Mn2+ rescue (of amino acid substitutions other than cysteine) is not a universal characteristic of mutants that affect amino acids involved in metal binding, as it is not observed with the Tn10 transposase (Allingham et al. 1999) or the TnsA protein of Tn7 (Sarnovsky et al. 1996). In fact, in the two cases in which some Mn2+-dependent rescue was detected, the level of rescued activity was quite low (Baker and Luo 1994; Sarnovsky et al. 1996). Furthermore, in one case, prolonged incubation in very high concentrations (5- to 10-fold above the normal optimum) of Mn2+ was required to detect weak activity (Baker and Luo 1994). In contrast, our class-I mutants were highly active for both nicking and hairpin formation when assayed at Mn2+ concentrations that give optimum activity of the wild-type enzyme. Because Mn2+ can, by unknown mechanisms, relax the specificity of nucleases (Vermote and Halford 1992) and can rescue activities of integrase mutants not thought to affect metal binding (Blain and Goff 1996), we do not regard the stimulation of activity of class-I mutants by Mn2+ as evidence for involvement of these amino acids in protein–metal interactions. It should be stressed that the class-II mutants are catalytic-deficient in all assays, both in vivo and in vitro in the presence of Mg2+ or Mn2+. These mutants clearly identify residues that are critical for catalysis.
A DDE motif in RAG1?
Given the remarkable similarities between the properties of the RAG proteins and the members of the retroviral integrase superfamily, along with our identification of D600, D708, and E962 as the only essential acidic amino acids in RAG1, we must consider the possibility that these amino acids constitute a DDE motif. Early descriptions of the DDE motif noted that the spacing between the second aspartate and the glutamate, 35 amino acids, is conserved among the retroviral integrases (Kulkosky et al. 1992; Polard and Chandler 1995). This spacing is not conserved in other members of the superfamily; however, 56 and 131 amino acids separate the analogous residues in the MuA and Tn10 transposases, respectively (Baker and Luo 1994; Bolland and Kleckner 1996). Thus, it is not inconceivable that in RAG1, a substantially larger protein, this distance would also be larger (254 amino acids). A comparison of the amino acid sequences of the relevant regions of RAG1 and retroviral integrase superfamily members is shown in Figure 9B. Although some similarities are evident, this primary sequence comparison does not provide strong evidence that the three class-II residues of RAG1 constitute a DDE motif.
We cannot exclude the possibility that the essential acidic amino acids may participate in a structural element that does not resemble a DDE motif. Catalysis of DNA cleavage can use acidic amino acids arranged in a variety of structural motifs. For example, type-II restriction enzymes use acidic amino acids to coordinate metal ion(s), but the spatial arrangement of these amino acids is different from that of the DDE motif. Interestingly, crystallographic analysis has recently shown that acidic residues in the TnsA protein of Tn7, identified as catalytic residues by mutational analysis and Mn2+ rescue of cysteine mutants, are structurally similar to the metal-binding region of type-II restriction enzymes (F. Dyda, A. Hickman, and N. Craig, pers. comm.). Thus, a detailed understanding of how the essential acidic triad we have identified in RAG1 contributes to catalysis will require structural information.
The identification of three catalytic-deficient mutants of RAG1 provides a powerful new tool for dissecting the mechanism of V(D)J recombination. These mutants should facilitate more detailed analysis of the active site and its organization with respect to the RAG–RSS DNA–protein complex. The catalytic-deficient RAG1 mutants should also be useful for future investigations into the mechanisms and the regulation of DNA cleavage during V(D)J recombination.
Materials and methods
Plasmid constructs and mutagenesis
All 110 shaded amino acids in Figure 1 were mutated from D to N or E to Q, with the exception of E607, which has been shown previously to recombine at wild-type levels (Sadofsky et al. 1995; Sleckman et al. 1996; Swanson and Desiderio 1998). Single-strand DNA mutagenesis was performed according to the method of Kunkel et al. (1987). pMAL-2 encodes truncated (core) RAG-1 [amino acids 384–1008, derived from pMS127 (Sadofsky et al. 1993)] fused with three copies of the human c-myc epitope on the carboxyl terminus in a pcDNAI/Amp vector (Invitrogen). pMAL-1 encodes truncated (core) RAG2 (amino acids 1–387, derived from pMS216 (Sadofsky et al. 1994)) with a nine-histidine tag and three copies of the human c-myc epitope fused to its caboxyl terminus in a pcDNAI/Amp vector. Single-stranded DNA was prepared with CJ236 cells (dut−ung−). Mutagenic oligonucleotides were annealed to single-stranded template, serving as a primer for second strand synthesis, followed by ligation. The resulting double-stranded plasmid was transformed into ES1301 cells (mutS, dut+ung+). DNA preparations from individual colonies were screened by sequencing. Two independent isolates of each mutation were carried forward for analysis. Positive clones were transformed into DH5α cells and individual mutant colonies were identified by sequencing. Any cDNA encoding a RAG-1 mutant with a cleavage defect was sequenced in its entirety to rule out the presence of adventitious second site mutations.
Baculovirus transfer vectors encoding the mutant RAG proteins or their wild-type counterparts [all containing carboxy-terminal (his)9 (myc)3 tags] were made by subcloning into the pFastBac transfer vector (GIBCO/BRL). Some mutants (and wild-type controls) were also constructed as amino-terminal maltose-binding protein (MBP) fusions, as described previously (McBlane et al. 1995). These fusions retained the carboxy-terminal his and myc epitope tags.
Transfections
For transient transfection assays, Chinese hamster ovary fibroblasts (RMP41 cells) were transfected with 2.1 μg of wild-type or mutant RAG-1 expression vector (pMAL-2), 2.5 μg of wild-type or mutant RAG2 expression vector (pMAL-1), and 5 μg of pJH290 substrate as described previously (Steen et al. 1996, 1997). In control transfections lacking a RAG expression vector, 4.6 μg of the pcDNAI/Amp backbone vector was used. DNA was transfected with the Fugene-6 transfection reagent (Boehringer Mannheim). DNA was harvested 48 hr post-transfection by the method of Hirt (1967). The resulting DNA was resuspended in 30 μl of TE. All transfections were repeated at least four times, with two independent isolates of each mutant.
Signal joint assays
To detect signal joints, one-thirtieth of the total harvested DNA was assayed by PCR (24 cycles) with the DR55 and ML68 primers, as described previously (Steen et al. 1997). A total of 10 μl of each PCR reaction was loaded onto a 6% polyacrylamide gel, transferred to a membrane (Genescreen Plus), and hybridized with a radiolabeled oligonucleotide probe (DR55). All signal joint assays were repeated at least four times from independent transfections.
Signal end assays
To detect signal ends, one-fifteenth of the total harvested DNA was subjected to ligation-mediated PCR as described (Roth et al. 1993). For PCR assays, 1 μl of a 1:100 dilution of each ligation was amplified with the ML68 and DR20 primers. PCR products (10 μl) were separated by PAGE and detected by hybridization to an oligonucleotide probe (DR69) as described. All signal end assays were repeated at least four times, from independent transfections.
Purified proteins
The Bac-to-Bac protein expression system (GIBCO/BRL) was used for expression of recombinant RAG proteins as described previously (McBlane et al. 1995; van Gent et al. 1995; Akamatsu and Oettinger 1998; Kim and Oettinger 1998). Typically, ten 150-mm plates of Sf9 cells were infected with high-titer virus stocks (RAG1 and RAG2). Cells were harvested ∼60 hr postinfection and lysed in 10 ml of lysis buffer [20 mm Tris-Cl (pH 7.9) at 4°C, 0.5 m NaCl, 20% glycerol, 2 mm β-mercaptoethanol] plus 60 mm imidazole by Dounce homogenization (20 strokes, tight pestle). The resulting lysate was centrifuged at 100,000g for 30 min at 4°C. The supernatant was loaded onto a 0.5-ml metal chelating Sepharose column (Pharmacia) charged with NiSO4. The column was washed with 10 ml of lysis buffer containing 90 mm imidazole and eluted with lysis buffer containing 250 mm imidazole. Fractions containing the RAG proteins were dialyzed against 500 volumes of storage buffer (25 mm K-HEPES at pH 7.5, 150 mm potassium glutamate, 20% glycerol, 2 mm DTT) for 3 hrs at 4°C. Protein was aliquoted, flash frozen in liquid nitrogen, and stored at −80°C.
Crude extracts
RMP41 cells were transfected (21 μg each pMAL-1 and pMAL-2 per T25 flask) with the Fugene-6 transfection reagent. After 48 hr, cells were harvested, washed with PBS, and subjected to three cycles of freezing and thawing. The cells were then extracted for 2–3 hr at 4°C in 50 μl of extraction buffer (25 mm K-HEPES at pH 7.0, 260 mm KCl, 40 mm NaCl, 20% glycerol, 0.1% NP-40, 1 mm DTT, 0.5 mm PMSF). The lysate was then spun at 25,000g for 25 min at 4°C and the supernatant was frozen in liquid nitrogen and stored at −80°C.
Electrophoretic mobility shift assays
Purified RAG1 and RAG2 proteins (100 ng each as measured by Coomassie-stained gels) were incubated with 25 fmoles of the annealed oligonucleotide substrate, DAR39/40 (McBlane et al. 1995) in 10 μl of reaction buffer [37.8 mm HEPES-KOH at pH 7.5; 51 mm potassium glutamate, 10% glycerol; 3 mm DTT; 2.5 pmoles of the nonspecific competitor oligonucleotide, FM117 (McBlane et al. 1995); 1 mm MgCl2; 60 μg/ml BSA; 0.006% NP-40; 20% DMSO]. Incubations were at 30°C for 30 min, and were cross-linked by addition of glutaraldehyde (to 0.1%), with additional incubation for 10 min at 37°C, as described (Hiom and Gellert 1997; Akamatsu and Oettinger 1998). DNA binding was analyzed by nondenaturing electrophoresis through a 4%–20% polyacrylamide gel run in 1× TBE gel at 200 V for 1 hr at 4°C. Dried gels were visualized by autoradiography and/or PhosphorImager.
Oligonucleotide cleavage assays
Assays were performed as described previously (Kim and Oettinger 1998). RAG1 and RAG2 (100 ng each) were incubated with 0.25 pmole of annealed DAR39/40 (McBlane et al. 1995) (DAR 39 was 5′ 32P-endlabeled) in a 10-μl reaction (40 mm HEPES-KOH at pH 7.5, 60 mm potassium glutamate, 10% glycerol, 3 mm DTT, 1 mm MnCl2, 60 μg/ml BSA, 0.006% NP-40) at 30°C for 2 hr. The reaction was stopped by adding an equal volume of 94% formamide, 20 mm EDTA, and 0.05% bromophenol blue. Reaction products were separated by electrophoresis through a 10% acrylamide gel containing 30% formamide, 0.67× TBE, 7 m urea, and 12.5 mm HEPES-KOH at pH 7.5 for 2 hr at 75 W. Wet gels were visualized by autoradiography or PhosphorImager analysis.
Plasmid cleavage assays
RAG1 and RAG2 containing crude extracts (5 μl) were incubated with 100 ng of pJH290 for 3 hr at 30°C (38 mm HEPES-KOH at pH 8.0, 0.4 mm Tris-Cl at pH 8.0, 5 mm HEPES-KOH at pH 7.0, 68 mm KCl, 8 mm NaCl, 0.76 mm MgCl2 (or MnCl2), 0.76 mm ATP, 4% glycerol, 0.02% NP-40, 0.96 mm DTT, 0.04 mm EDTA, 0.1 mm PMSF). Cleavage reactions were stopped by the addition of 100 μl of stop buffer (100 mm Tris-Cl at pH 8.0, 0.2% SDS, 0.35 mg/ml proteinase K, 10 mm EDTA) and incubated at 55°C for 1 hr. The deproteinized cleavage products were extracted, EtOH precipitated, resuspended in 10 μl of TE and digested with PvuII (1 unit) for 1 hr at 37°C. One-half of the digested reaction products were then separated by electrophoresis through a 4.5% acrylamide gel, transferred to a solid support, and hybridized with a random primed 693-bp PvuII fragment from pJH290 that is complementary to all cleavage products.
Acknowledgments
We thank T. Palzkill and members of his laboratory for advice on site-directed mutagenesis and M. Estes and S. Crawford for advice on the baculovirus expression system. We are grateful to M. Oettinger and members of her laboratory (D.-R. Kim and C. Mundy) for advice on protein purification and DNA-binding assays, and for sharing information and reagents prior to publication. We thank L. Huye for assistance with crude extract experiments. T. Baker, M. Bogue, V. Brandt, N. Craig, L. Huye, S. Kale, S.-Y. Namgoong, and M. Purugganan provided comments on the manuscript. We thank S. Kale, L. Huye, J. Bryan, and F. Gimble, for helpful discussions. Mary Lowe provided excellent secretarial support. M. Calicchio, H. Kan, and J. Lin provided technical assistance. This work was supported by a grant from the National Institutes of Health (AI-36420). Early phases of this work were supported in part by the Charles E. Culpeper Foundation. M.A.L. was supported in early phases of this work by a predoctoral fellowship from the National Institutes of Health (GM-08231) and is currently supported by a predoctoral fellowship from the National Institutes of Health (AI-07495).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
Corresponding author.
E-MAIL davidbr@bcm.tmc.edu; FAX (713) 798-3033.
References
- Agrawal A, Eastman QM, Schatz DG. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 1998;394:744–751. doi: 10.1038/29457. [DOI] [PubMed] [Google Scholar]
- Akamatsu Y, Oettinger MA. Distinct roles of RAG1 and RAG2 in binding the V(D)J recombination signal sequences. Mol Cell Biol. 1998;18:4670–4678. doi: 10.1128/mcb.18.8.4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allingham JS, Pribil PA, Haniford DB. All three residues of the Tn10 transposase DDE catalytic triad function in divalent metal ion binding. J Mol Biol. 1999;289:1195–1206. doi: 10.1006/jmbi.1999.2837. [DOI] [PubMed] [Google Scholar]
- Bailin T, Mo X, Sadofsky MJ. A RAG1 and RAG2 tetramer complex is active in cleavage in V(D)J recombination. Mol Cell Biol. 1999;19:4664–4671. doi: 10.1128/mcb.19.7.4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, Luo L. Identification of residues in the Mu transposase essential for catalysis. Proc Natl Acad Sci. 1994;91:6654–6658. doi: 10.1073/pnas.91.14.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besmer E, Mansilia-Soto J, Cassard S, Sawchuk DJ, Brown G, Sadofsky M, Lewis SM, Nussenzweig MC, Cortes P. Hairpin coding end opening is mediated by RAG1 and RAG2 proteins. Mol Cell. 1998;2:817–828. doi: 10.1016/s1097-2765(00)80296-8. [DOI] [PubMed] [Google Scholar]
- Blain SW, Goff SP. Differential effects of moloney murine leukemia virus reverse transcriptase mutations on RNase H activity in Mg2+ and Mn2+ J Biol Chem. 1996;271:1448–1454. doi: 10.1074/jbc.271.3.1448. [DOI] [PubMed] [Google Scholar]
- Bolland S, Kleckner N. The three chemical steps of Tn10/IS10 transposition involve repeated utilization of a single active site. Cell. 1996;84:223–233. doi: 10.1016/s0092-8674(00)80977-0. [DOI] [PubMed] [Google Scholar]
- Bujacz G, Jaskólski M, Alexandratos J, Wlodawer A, Merkel G, Katz RA, Skalka AM. High-resolution structure of the catalytic domain of avian sarcoma virus integrase. J Mol Biol. 1995;253:333–346. doi: 10.1006/jmbi.1995.0556. [DOI] [PubMed] [Google Scholar]
- ————— The catalytic domain of avian sarcoma virus integrase: Conformation of the active-site residues in the presence of divalent cations. Structure. 1996;4:89–96. doi: 10.1016/s0969-2126(96)00012-3. [DOI] [PubMed] [Google Scholar]
- Chow SA, Vincent KA, Ellison V, Brown PO. Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science. 1992;255:723–726. doi: 10.1126/science.1738845. [DOI] [PubMed] [Google Scholar]
- Craig NL. V(D)J recombination and transposition: Closer than expected. Science. 1996;271:1512–1512. doi: 10.1126/science.271.5255.1512. [DOI] [PubMed] [Google Scholar]
- Cuomo CA, Oettinger MA. Analysis of regions of RAG-2 important for V(D)J recombination. Nucleic Acids Res. 1994;22:1810–1814. doi: 10.1093/nar/22.10.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahm SC, Uhlenbeck OC. Role of divalent metal ions in the hammerhead RNA cleavage reaction. Biochemistry. 1991;30:9464–9469. doi: 10.1021/bi00103a011. [DOI] [PubMed] [Google Scholar]
- Difilippantonio MJ, McMahan CJ, Eastman QM, Spanopoulou E, Schatz DG. RAG1 mediates signal sequence recognition and recruitment of RAG2 in V(D)J recombination. Cell. 1996;87:253–262. doi: 10.1016/s0092-8674(00)81343-4. [DOI] [PubMed] [Google Scholar]
- Eastman QM, Villey IJ, Schatz DG. Detection of RAG protein-V(D)J recombination signal interactions near the site of DNA cleavage by UV cross-linking. Mol Cell Biol. 1999;19:3788–3797. doi: 10.1128/mcb.19.5.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Craigie R. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J Virol. 1992;66:6361–6369. doi: 10.1128/jvi.66.11.6361-6369.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Mizuuchi K, Craigie R. HIV-1 DNA integration: Mechanism of viral cleavage and DNA strand transfer. Cell. 1991;67:1211–1221. doi: 10.1016/0092-8674(91)90297-c. [DOI] [PubMed] [Google Scholar]
- Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R. Three new structures of the core domain of HIV-1 integrase: An active site that binds magnesium. Proc Natl Acad Sci. 1998;95:9150–9154. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindley NDF, Leschziner AE. DNA transposition: From a black box to a color monitor. Cell. 1995;83:1063–1066. doi: 10.1016/0092-8674(95)90132-9. [DOI] [PubMed] [Google Scholar]
- Han J-O, Steen SB, Roth DB. Ku86 is not required for protection of signal ends or for formation of nonstandard V(D)J recombination products. Mol Cell Biol. 1997;17:2226–2234. doi: 10.1128/mcb.17.4.2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Intermolecular V(D)J recombination is prohibited specifically at the joining step. Mol Cell. 1999;3:331–338. doi: 10.1016/s1097-2765(00)80460-8. [DOI] [PubMed] [Google Scholar]
- Hiom K, Gellert M. A stable RAG1-RAG2-DNA complex that is active in V(D)J cleavage. Cell. 1997;88:65–72. doi: 10.1016/s0092-8674(00)81859-0. [DOI] [PubMed] [Google Scholar]
- Hiom K, Melek M, Gellert M. DNA transposition by the RAG1 and RAG2 proteins: A possible source of oncogenic translocations. Cell. 1998;94:463–470. doi: 10.1016/s0092-8674(00)81587-1. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Kennedy AK, Guhathakurta A, Kleckner N, Haniford DB. Tn10 transposition via a DNA hairpin intermediate. Cell. 1998;95:125–134. doi: 10.1016/s0092-8674(00)81788-2. [DOI] [PubMed] [Google Scholar]
- Kim D-R, Oettinger MA. Functional analysis of coordinated cleavage in V(D)J recombination. Mol Cell Biol. 1998;18:4679–4688. doi: 10.1128/mcb.18.8.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D.-R., Y. Dai, C.L. Mundy, W. Yang, and M.A. Oettinger. 1999. Mutations of acidic residues in RAG1 define the active site of the V(D)J recombinase. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Kim K, Namgoong S-Y, Jayaram M, Harshey RM. Step-arrest mutants of phage Mu transposase. J Biol Chem. 1995;270:1472–1479. doi: 10.1074/jbc.270.3.1472. [DOI] [PubMed] [Google Scholar]
- Kirch SA, Sudarsanam P, Oettinger MA. Regions of RAG1 protein critical for V(D)J recombination. Eur J Immunol. 1996;26:886–891. doi: 10.1002/eji.1830260425. [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ. Chromosomal translocations in lymphoid malignancies reveal novel proto-oncogenes. Annu Rev Immunol. 1992;10:785–807. doi: 10.1146/annurev.iy.10.040192.004033. [DOI] [PubMed] [Google Scholar]
- Krementsova E, Giffin MJ, Pincus D, Baker TA. Mutational analysis of the Mu transposase. J Biol Chem. 1998;273:31358–31365. doi: 10.1074/jbc.273.47.31358. [DOI] [PubMed] [Google Scholar]
- Kulkosky J, Jones KS, Katz RA, Mack JPG, Skalka AM. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol Cell Biol. 1992;12:2331–2338. doi: 10.1128/mcb.12.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- Lee J, Jayaram M, Grainge I. Wild-type Flp recombinase cleaves DNA in trans. EMBO J. 1999;18:784–791. doi: 10.1093/emboj/18.3.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maignan S, Guilloteau J-P, Zhou-Liu Q, Clément-Mella C, Mikol V. Crystal structures of the catalytic domain of HIV-1 integrase free and complexed with its metal cofactor: High level of similarity of the active site with other viral integrases. J Mol Biol. 1998;282:359–368. doi: 10.1006/jmbi.1998.2002. [DOI] [PubMed] [Google Scholar]
- Mazumder A, Engelman A, Craigie R, Fesen M, Pommier Y. Intermolecular disintegration and intramolecular strand transfer activities of wild-type and mutant HIV-1 integrase. Nucleic Acids Res. 1994;22:1037–1043. doi: 10.1093/nar/22.6.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBlane JF, van Gent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, Oettinger MA. Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell. 1995;83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- Melek M, Gellert M, van Gent DC. Rejoining of DNA by the RAG1 and RAG2 proteins. Science. 1998;280:301–303. doi: 10.1126/science.280.5361.301. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K. Polynucleotidyl transfer reactions in site-specific DNA recombination. Genes Cells. 1997;2:1–12. doi: 10.1046/j.1365-2443.1997.970297.x. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K, Adzuma K. Inversion of the phosphate chirality at the target site of Mu DNA strand transfer: Evidence for a one-step transesterification mechanism. Cell. 1991;66:129–140. doi: 10.1016/0092-8674(91)90145-o. [DOI] [PubMed] [Google Scholar]
- Nagawa F, Ishiguro K-I, Tsuboi A, Yoshida T, Ishikawa A, Takemori T, Otsuka AJ, Sakano H. Footprint analysis of the RAG protein recombination signal sequence complex for V(D)J type recombination. Mol Cell Biol. 1998;18:655–663. doi: 10.1128/mcb.18.1.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namgoong S-Y, Harshey RM. The same two monomers within a MuA tetramer provide the DDE domains for the strand cleavage and strand transfer steps of transposition. EMBO J. 1998;17:3775–3785. doi: 10.1093/emboj/17.13.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirilli JA, Vyle JS, Caruthers MH, Cech TR. Metal ion catalysis in the Tetrahymena ribozyme reaction. Nature. 1993;361:85–88. doi: 10.1038/361085a0. [DOI] [PubMed] [Google Scholar]
- Polard P, Chandler M. Bacterial transposases and retroviral integrases. Mol Microbiol. 1995;15:13–23. doi: 10.1111/j.1365-2958.1995.tb02217.x. [DOI] [PubMed] [Google Scholar]
- Rice P, Mizuuchi K. Structure of the bacteriophage Mu transposase core: A common structural motif for DNA transposition and retroviral integration. Cell. 1995;82:209–220. doi: 10.1016/0092-8674(95)90308-9. [DOI] [PubMed] [Google Scholar]
- Rodgers KK, Villey IJ, Ptaszek L, Corbett E, Schatz DG, Coleman JE. A dimer of the lymphoid protein RAG1 recognizes the recombination signal sequence and the complex stably incorporates the high mobility group protein HMG2. Nucleic Acids Res. 1999;27:2938–2946. doi: 10.1093/nar/27.14.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth DB, Craig NL. VDJ recombination: A transposase goes to work. Cell. 1998;94:411–414. doi: 10.1016/s0092-8674(00)81580-9. [DOI] [PubMed] [Google Scholar]
- Roth DB, Menetski JP, Nakajima PB, Bosma MJ, Gellert M. V(D)J recombination: Broken DNA molecules with covalently sealed (hairpin) coding ends in scid mouse thymocytes. Cell. 1992;70:983–991. doi: 10.1016/0092-8674(92)90248-b. [DOI] [PubMed] [Google Scholar]
- Roth DB, Zhu C, Gellert M. Characterization of broken DNA molecules associated with V(D)J recombination. Proc Natl Acad Sci. 1993;90:10788–10792. doi: 10.1073/pnas.90.22.10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadofsky MJ, Hesse JE, McBlane JF, Gellert M. Expression and V(D)J recombination activity of mutated RAG-1 proteins. Nucleic Acids Res. 1993;21:5644–5650. doi: 10.1093/nar/21.24.5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadofsky MJ, Hesse JE, Gellert M. Definition of a core region of RAG-2 that is functional in V(D)J recombination. Nucleic Acids Res. 1994;22:1805–1809. doi: 10.1093/nar/22.10.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadofsky MJ, Hesse JE, van Gent DC, Gellert M. RAG-1 mutations that affect the target specificity of V(D)J recombination: A possible direct role of RAG-1 in site recognition. Genes & Dev. 1995;9:2193–2199. doi: 10.1101/gad.9.17.2193. [DOI] [PubMed] [Google Scholar]
- Sarnovsky RJ, May EW, Craig NL. The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. EMBO J. 1996;15:6348–6361. [PMC free article] [PubMed] [Google Scholar]
- Shockett PE, Schatz DG. DNA hairpin opening mediated by the RAG1 and RAG2 proteins. Mol Cell Biol. 1999;19:4159–4166. doi: 10.1128/mcb.19.6.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver DP, Spanopoulou E, Mulligan RC, Baltimore D. Dispensable sequence motifs in the RAG-1 and RAG-2 genes for plasmid V(D)J recombination. Proc Natl Acad Sci. 1993;90:6100–6104. doi: 10.1073/pnas.90.13.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleckman BP, Gorman JR, Alt FW. Accessibility control of antigen-receptor variable-region gene assembly: Role of cis-acting elements. Annu Rev Immunol. 1996;14:459–481. doi: 10.1146/annurev.immunol.14.1.459. [DOI] [PubMed] [Google Scholar]
- Spanopoulou E, Zaitseva F, Wang F-H, Santagata S, Baltimore D, Panayotou G. The homeodomain region of Rag-1 reveals the parallel mechanisms of bacterial and V(D)J recombination. Cell. 1996;87:263–276. doi: 10.1016/s0092-8674(00)81344-6. [DOI] [PubMed] [Google Scholar]
- Steen SB, Gomelsky L, Roth DB. The 12/23 rule is enforced at the cleavage step of V(D)J recombination in vivo. Genes Cells. 1996;1:543–553. doi: 10.1046/j.1365-2443.1996.d01-259.x. [DOI] [PubMed] [Google Scholar]
- Steen SB, Gomelsky L, Speidel SL, Roth DB. Initiation of V(D)J recombination in vivo: Role of recombination signal sequences in formation of single and paired double-strand breaks. EMBO J. 1997;16:2656–2664. doi: 10.1093/emboj/16.10.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen AE, Craig NL. Avoiding self: Two Tn7-encoded proteins mediate target immunity in Tn7 transposition. EMBO J. 1997;16:6823–6834. doi: 10.1093/emboj/16.22.6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson PC, Desiderio S. V(D)J recombination signal recognition: Distinct, overlapping DNA-protein contacts in complexes containing RAG1 with and without RAG2. Immunity. 1998;9:115–125. doi: 10.1016/s1074-7613(00)80593-2. [DOI] [PubMed] [Google Scholar]
- ————— RAG–2 promotes heptamer occupancy by RAG-1 in the assembly of a V(D)J initiation complex. Mol Cell Biol. 1999;19:3674–3683. doi: 10.1128/mcb.19.5.3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko B, Sklar J. Chromosomal translocations in lymphoid neoplasia: A reappraisal of the recombinase model. Cancer Cells. 1990;2:1–8. [PubMed] [Google Scholar]
- Van den Ent FMI, Vink C, Plasterk RHA. DNA substrate requirements for different activities of the human immunodeficiency virus type I integrase protein. J Virol. 1994;68:7825–7832. doi: 10.1128/jvi.68.12.7825-7832.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gent DC, McBlane JF, Ramsden DA, Sadofsky MJ, Hesse JE, Gellert M. Initiation of V(D)J recombination in a cell-free system. Cell. 1995;81:925–934. doi: 10.1016/0092-8674(95)90012-8. [DOI] [PubMed] [Google Scholar]
- van Gent DC, Mizuuchi K, Gellert M. Similarities between initiation of V(D)J recombination and retroviral integration. Science. 1996a;271:1592–1594. doi: 10.1126/science.271.5255.1592. [DOI] [PubMed] [Google Scholar]
- van Gent DC, Ramsden DA, Gellert M. The RAG1 and RAG2 proteins establish the 12/23 rule in V(D)J recombination. Cell. 1996b;85:107–113. doi: 10.1016/s0092-8674(00)81086-7. [DOI] [PubMed] [Google Scholar]
- Vermote CLM, Halford SE. EcoRV restriction endonuclease: Communication between catalytic metal ions and DNA recognition. Biochemistry. 1992;31:6082–6089. doi: 10.1021/bi00141a018. [DOI] [PubMed] [Google Scholar]
- Vink C, Yeheskiely E, van der Marel GA, van Boom JH, Plasterk RHA. Site-specific hydrolysis and alcoholysis of human immunodeficiency virus DNA termini mediated by the viral integrase protein. Nucleic Acids Res. 1991;19:6691–6698. doi: 10.1093/nar/19.24.6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vipond IB, Halford SE. Structure-function correlation for the EcoRV restriction enzyme: From non-specific binding to specific DNA cleavage. Mol Microbiol. 1993;9:225–231. doi: 10.1111/j.1365-2958.1993.tb01685.x. [DOI] [PubMed] [Google Scholar]
- Williams TL, Jackson EL, Carritte A, Baker TA. Organization and dynamics of the Mu transpososome: Recombination by communication between two active sites. Genes & Dev. 1999;13:2725–2737. doi: 10.1101/gad.13.20.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]