

Abstract
Peroxisome proliferator-activated receptors (PPARs), members of the nuclear hormone receptor superfamily, function as transcription factors and modulators of gene expression. These actions allow PPARs to regulate a variety of biological processes and to play a significant role in several diseases and conditions. The current literature describes frequently opposing and paradoxical roles for the three PPAR isotypes, PPARα, PPARβ/δ and PPARγ, in cancer. While some studies have implicated PPARs in the promotion and development of cancer, others, in contrast, have presented evidence for a protective role for these receptors against cancer. In some tissues, the expression level of these receptors and/or their activation correlates with a positive outcome against cancer, while, in other tissue types, their expression and activation have the opposite effect. These disparate findings raise the possibility of (i) PPAR receptor-independent effects, including effects on receptors other than PPARs by the utilized ligands; (ii) cancer stage-specific effect; and/or (iii) differences in essential ligand-related pharmacokinetic considerations. In this review, we highlight the latest available studies on the role of the various PPAR isotypes in cancer in several major organs and present challenges as well as promising opportunities in the field.
Keywords: nuclear receptors, PPARs, cancer
Peroxisome proliferator-activated receptors
Peroxisome proliferator-activated receptors (PPARs) are nuclear transcription factors that were discovered in 1990 (Issemann and Green, 1990) and are classified as members of the steroid hormone receptor superfamily. To date, three related PPAR isotypes have been identified: PPARα, PPARβ/δ and PPARγ (Figure 1). The three isotypes share a high degree of homology but differ in tissue distribution and ligand specificity (Berger and Moller, 2002). These receptors bind to and are activated by fatty acids, eicosanoids and numerous xenobiotics (Figure 2) some of which have therapeutic value (Forman et al., 1997; Kliewer et al., 1997; Lalloyer and Staels, 2010). Prior to ligand binding, however, PPARs heterodimerize with retinoid X receptor (RXR), forming a complex. This complex is required for binding to specific DNA sequences, known as PPAR response elements, in the promoter region of target genes (Figure 3). Upon binding to their ligands, PPARs undergo conformational changes allowing release of co-repressors, and recruitment of coactivators, followed by the activation of transcription (Berger and Moller, 2002; Feige et al., 2006).
Figure 1.
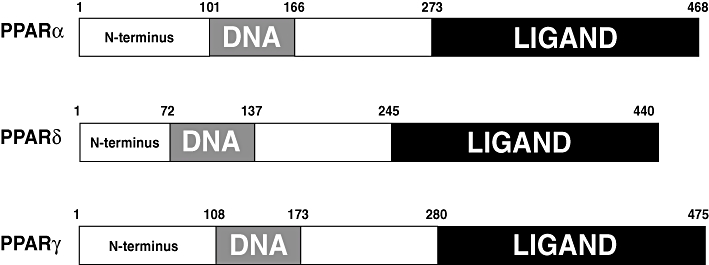
Functional domains of mouse PPARα, PPARβ/δ and PPARγ. PPAR, peroxisome proliferator-activated receptor.
Figure 2.
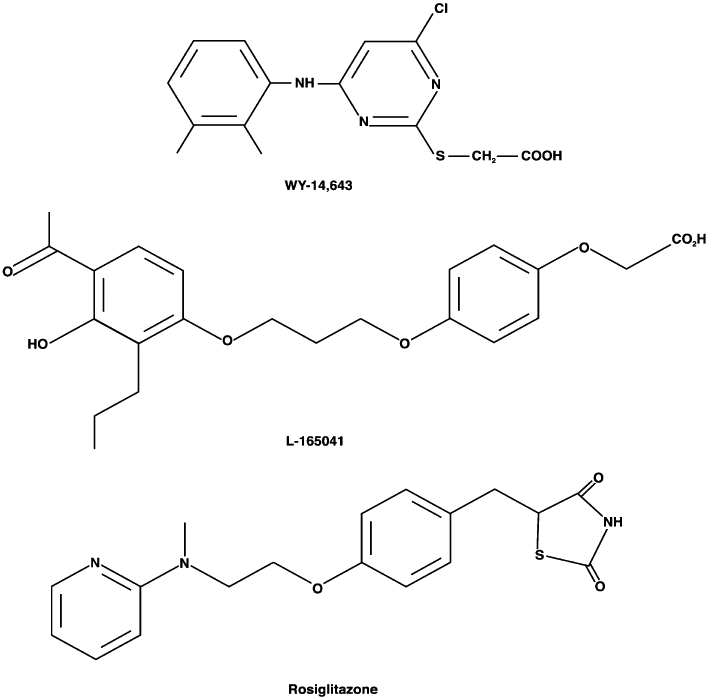
Chemical structures of representative PPAR agonists. PPARα, WY 14643; PPARβ/δ, L-165041; PPARγ, rosiglitazone. PPAR, peroxisome proliferator-activated receptor.
Figure 3.
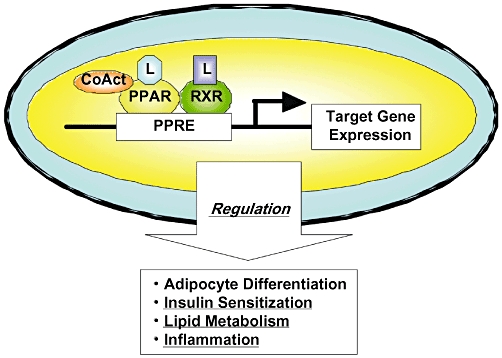
Transcription regulation of target genes by PPARs [reproduced from Shimizu and Moriwaki (2008) with permission from Dr Masahito Shimizu]. PPAR, peroxisome proliferator-activated receptor; PPRE, PPAR response element; RXR, retinoid X receptors.
Peroxisome proliferator-activated receptors have been implicated, in a subtype-specific manner, in several important diseases and pathological conditions such as senescence and senescence-related diseases (Masters and Crane, 1995; Youssef and Badr, 1999; Youssef and Badr, 2001; Han et al., 2010), inflammation (Chinetti et al., 2000; Delerive et al., 2001; Guri et al., 2010), immunity (Spiegelman, 1998; Michalik and Wahli, 1999; Peyrin-Biroulet et al., 2010), obesity (Brun et al., 1996; Spiegelman and Flier, 1996; Vidal-Puig et al., 1997; Lefebvre et al., 1998a; Gregoire et al., 2007; Zhang et al., 2007), diabetes (Lefebvre et al., 1998a; Allen et al., 2006), and in the regulation of male and female fertility (Lim et al., 1999; Barak et al., 2002; Froment, 2008). In addition, a large body of literature is available on the role of these receptors in various cancers (Badr, 2004; Panigrahy et al., 2008).
Role of PPARs in brain tumorigenesis
Peroxisome proliferator-activated receptors are expressed in several cell types of the brain including microglia, astrocytes, oligodendrocytes and neurons (Heneka and Landreth, 2007). These receptors are thought to play a role in controlling brain cell growth and differentiation. Studies have shown that PPARγ agonists interfere with glioblastoma growth and malignancy (Grommes et al., 2006), as well as inhibit growth and expansion of brain tumour stem cells (Chearwae and Bright, 2008). Using glioma cell lines and a murine glioma model, research has shown that pioglitazone, a PPARγ agonist, in combination with RXRγ agonist, is effective in reducing proliferation and invasion of glioblastoma (Papi et al., 2009). This finding provides the basis for the current clinical use of PPARγ agonists against this aggressive and currently incurable disease (Papi et al., 2009). Indeed, clinical studies have revealed that pioglitazone is beneficial in some patients with high-grade glioma treated with cyclooxygenase-2 (COX-2) inhibitors and low-dose chemotherapy (Hau et al., 2007), and showed a synergistic anti-tumour effect when given with immunotherapeutic agents (Lichtor et al., 2008). However, despite the fact that rosiglitazone, another PPARγ agonist, was effective in inhibiting neuroblastoma in vitro, this drug has a marginal effect in vivo (Krieger-Hinck et al., 2010). In this regard, pharmacokinetic issues encountered in vivo, but not in vitro, such as absorption, distribution, metabolism and excretion, should not be overlooked.
As whole-brain irradiation represents the primary mode of treatment for brain metastases, the role of PPARs in combating brain injury in response to radiation has been explored. This strategy is based on the fact that radiation causes inflammation and that PPARs are known to control inflammatory responses. Indeed, activation of PPARα by fenofibrate did confer neuroprotection against radiation-induced brain proinflammatory responses in vitro as well as in vivo (Ramanan et al., 2010), and activation of PPARγ by pioglitazone did ameliorate the severity of radiation-induced cognitive impairment (Ramanan et al., 2010).
PPARs and lung cancer
Studies in different models suggest that PPARβ/δ activation could attenuate lung cancer. A synthetic high-affinity PPARβ/δ ligand, L165041, inhibited human lung adenocarcinoma cell proliferation (Fukumoto et al., 2005) and elimination of expression of the PPARβ/δ gene in a transgenic mouse model was associated with exacerbation of lung cancer (Muller-Brusselbach et al., 2007). Conversely, it has recently been suggested that growth of two human lung cancer cell lines was stimulated by PPARβ/δ activation (Han et al., 2008; Pedchenko et al., 2008). Indeed, a more recent report by Han et al. (2009) showed that GW501516, a selective PPARβ/δ agonist, stimulates human lung carcinoma cell proliferation. A reason for these conflicting results may lie in the facts that the reported studies were performed utilizing different lung carcinoma cell lines (Fukumoto et al., 2005; Han et al., 2009). In addition, while the study by Han et al. used GW501516 as a model PPARδ agonist (Han et al., 2009), L-165041 was used in the study by Fukumoto et al. (2005). As the latter agonist is known to activate both PPARδ and PPARγ (Han et al., 2009), it remained possible that the inhibition of lung tumour cell proliferation by L-165041 was mediated by PPARγ under the experimental conditions used (Fukumoto et al., 2005). This notion is refuted, however, by the assertion made by Fukumoto and colleagues indicating that their unpublished data showed that L-165041 did not activate PPARγ at the concentrations they employed in their study (Fukumoto et al., 2005), leaving the differences in the cell type used as the only potential explanation for these conflicting findings.
In contrast to the conflicting data on the role of PPARβ/δ in cancer prevention and enhancement, the evidence is more uniform and compelling in favour of a role for PPARγ in the treatment of lung cancer. In this regard, decreased expression of PPARγ has been associated with poor prognosis in lung cancer patients (Sasaki et al., 2002) and activating PPARγ by either endogenous or synthetic agonists was found to inhibit growth of human lung cancer cells (Tsubouchi et al., 2000). Transgenic mice that over-expressed PPARγ in their lungs were less susceptible to the development of lung tumours (Bren-Mattison et al., 2008). This receptor isotype may also mediate selective inhibition of invasive metastasis and activates pathways such as those involved in the anti-tumour effect of prostacyclin (Nemenoff et al., 2008) and COX-2 down-regulation (Hazra et al., 2008), which promote a more differentiated epithelial phenotype (Bren-Mattison et al., 2005).
In vitro treatment of human non-small-cell lung cancer cells with PPARγ activators induced differentiation and apoptosis (Chang and Szabo, 2000; Inoue et al., 2001; Satoh et al., 2002), as well as potentiated the inhibitory effects of cisplatin and paclitaxel (Reddy et al., 2008). In vivo experiments using a xenograft model showed similar results (Keshamouni et al., 2005). Other studies demonstrate that inhibition of angiogenesis contributes to the inhibitory effects of pioglitazone and troglitazone on primary tumour growth (Keshamouni et al., 2005) and that ciglitazone suppressed A-549-induced tumours in nude mice (Zhang et al., 2006). In addition, patients receiving thiazolidinedione PPARγ agonists for treatment of diabetes exhibited a significant lower risk for developing lung cancer (Govindarajan et al., 2007), suggesting a protective role for PPARγ ligands against this disease (Girnun et al., 2008; Roman, 2008).
PPARs in stomach and intestinal tumour formation
Expression of both PPARα and PPARγ has been consistently detected in normal colonic mucosal human biopsies, but PPARδ expression has not been detected (Matthiessen et al., 2005). While activation of PPARα had no effect on colonocyte proliferation, activation of PPARγ significantly decreased proliferation of these cells (Matthiessen et al., 2005). Surprisingly, however, a PPARδ ligand also significantly decreased cell proliferation, despite the absence of PPARδ expression in these cells (Matthiessen et al., 2005), suggesting PPAR receptor-independent effects.
In human colonic polyps, mRNA and protein expression of PPARα were significantly lower compared with normal colonic mucosa (Jackson et al., 2003; Matthiessen et al., 2005), while no difference was observed with regard to either PPARδ or PPARγ (Matthiessen et al., 2005) Investigations using two different colorectal cancer models suggest that PPARβ/δ expression attenuated colon carcinogenesis (Harman et al., 2004), while other studies show that PPARβ/δ activation promoted the growth of intestinal adenomas (Gupta et al., 2004). In the first study (Harman et al., 2004), it was shown that colon polyp formation was significantly greater in mice nullizygous for PPARδ than in control mice, while the latter study (Gupta et al., 2004) documented that exposure to the PPARδ ligand GW50156 resulted in a significant increase in the number and size of intestinal polyps in control mice compared with the nullizygous group. Resolution of this discrepancy with regard to the role of PPARβ/δ in colon cancer will require determination of whether the synthetic PPARδ ligand GW50156 has PPARδ-dependent and/or independent effects that are different from those expressed by putative endogenous PPARδ ligands.
Anti-cancer effects of PPARγ ligands have been reported in several gastric cancer cell lines, an effect attributed to induction of apoptosis and to G1 cell cycle arrest (Takahashi et al., 1999; Sato et al., 2000; Chen et al., 2003). Studies have also shown that PPARγ activation suppresses gastric carcinogenesis in mice, suggesting that PPARγ ligands may act as chemopreventive agents in human gastric carcinogenesis (Lu et al., 2005). However, recent investigations suggest that anti-proliferative effect of ciglitazone and troglitazone in stomach cancer could proceed via a PPARγ-independent pathway, as studies examining GW9662, a PPARγ antagonist, did not report a growth suppressant effect exerted by either of the two receptor activators (Cheon et al., 2009). Epidemiological studies associate PPARγ Pro12Ala polymorphism with gastric cancer and peptic ulcer disease (Tahara et al., 2007; Prasad et al., 2008).
It is well documented that PPARγ exerts both common and tissue-specific genomic and physiologic effects in the proximal and distal colon (Su et al., 2007) and regulates proliferation and motility of intestinal epithelial cells (Chen et al., 2006). Further studies are needed, however, to identify the exact role of PPARγ activation on colon tumour behaviour.
Although PPARγ ligands have been shown to inhibit proliferation and to induce differentiation of human colon cancer cells (Sarraf et al., 1998; Kopelovich et al., 2002; Ban et al., 2010), the growth inhibiting effect of PPARγ agonists shown in cellular studies was not evident in most studies performed in intact animals. Indeed, in vivo studies suggest that activation of PPARγ promotes colon tumours in animal models (Saez et al., 1998; Lefebvre et al., 1998b). In an attempt to explain this paradox, it was suggested that the anti-proliferative effects of PPARγ ligands may depend on the level of cellular differentiation: well-differentiated cancer may lose sensitivity to, or become deficient in factors involved in, PPARγ activation (Sato et al., 2000). Raising the possibility of species-specific differences, a recent clinical investigation demonstrates that PPARγ expression is associated with good prognosis of colorectal cancer (Ogino et al., 2009). Data have suggested that in humans, PPARγ acts as colon cancer suppressor and that decreased expression of this receptor may increase colon cancer risk (Chen et al., 2006; Necela et al., 2008; Ogino et al., 2009).
Paradoxical roles of liver PPARs in hepatic carcinogenesis
Peroxisome proliferator-activated receptor α has been implicated as a key mediator responsible for non-genotoxic hepatocarcinogensis in rodents. Chronic treatment of these animals with PPARα agonists results in increased incidence of liver tumours through a PPARα-mediated mechanism, which may include induction of cell proliferation and oxidative stress (Peters et al., 1997; Pyper et al., 2010).
Potential involvement of non-cancer cells in the mechanism through which PPAR agonists cause cancer has been extensively evaluated. Specifically, much attention has been given to Kupffer cells, the resident liver macrophages, with results suggesting a role for these cells in liver cancer caused by PPARα agonists in rodents (Marsman et al., 1988; Bojes et al., 1997; Rose et al., 1997). Evidence in support of this proposed role includes: (i) inactivation of Kupffer cells prevented the mitogenic effect of the PPARα agonist WY 14643 (Rose et al., 1997); (ii) replicative DNA synthesis in hepatocytes cultured in the presence of WY 14643 was dependent on the presence of non-parenchymal cells (Karam and Ghanayem, 1997); (iii) antibodies against tumour necrosis factor-α (TNF-α), presumably released from Kupffer cells upon their activation by PPARα agonists, blocked the increase in liver cell replication in response to WY 14643 (Bojes et al., 1997); and (iv) induction of hepatic DNA synthesis and suppression of liver cell apoptosis, effects that are produced by PPAR activators, were mimicked by TNF-α (Rolfe et al., 1997).
In contrast to the aforementioned assertion in favour of a role for Kupffer cells in mediating PPARα-induced hepatocellular proliferation and liver cancer, results from our laboratory (Youssef and Badr, 1997; Alsarra et al., 2006) and others (Uchimura et al., 2001; Woods et al., 2007) do not support the existence of such a role. These studies showed: (i) perfluorooctanoic acid, a PPARα agonist, caused a remarkable increase in liver cell proliferation in vivo in the absence of measurable changes in reliable markers of Kupffer cell activation (Youssef and Badr, 1997; Alsarra et al., 2006); (ii) activating RXR, the obligatory heterodimer of PPAR, did indeed inhibit, rather than stimulate TNF-α production by isolated Kupffer cells (Uchimura et al., 2001); and (iii) pathway mapping of genes that respond to WY 14643 in a time- and dose-dependent manner strongly demonstrated that Kupffer cells do not appear to play a role in chronic hepatic effects of PPARα agonists (Woods et al., 2007). In addition, it has been shown that Kupffer cells do not express PPARα receptors (Peters et al., 2000), and that PPARα agonists were able to stimulate hepatocellular proliferation in both TNF-α- and TNF-α-receptor-null mice (Anderson et al., 2001; Lawrence et al., 2001). Thus, participation of non-cancer cells in PPARα agonist-induced cancer remains controversial, necessitating further evaluation before a final conclusion can be reached.
Importantly, however, human subjects receiving fibrates for treatment of hyperlipidaemia are resistant to the carcinogenic effects of these drugs, suggesting significant differences between human PPARα and rodents PPARα-dependent pathways (Mukherjee et al., 1994). Species-specific effects of fibrates are likely due to differences in the level of receptor expression (Palmer et al., 1998), ligand affinity and/or other factors involved in PPARα activation (Gonzalez and Shah, 2008), as well as the profile of genes activated by mouse PPARα versus human PPARα following treatment with the fibrate drugs (Morimura et al., 2006; Yang et al., 2008). In order to delineate the mechanisms involved in human lack of susceptibility to the heptocarcinogenic effect of PPARα activation, attempts are underway to identify specific factors involved in receptor regulation in each species. The availability of PPARα-humanized mice (Yang et al., 2008) may be beneficial in that regard.
The role of PPARβ/δ in liver cancer is controversial. While some cellular studies show that PPARβ/δ activation promote proliferation and growth of human hepatic cancer cell lines through up-regulation of COX-2 gene expression and PGE2 production (Glinghammar et al., 2003; Hellemans et al., 2003), other studies demonstrate that COX-2 expression does not change when the same liver cancer cell lines are treated with PPARβ/δ ligands. No cell growth or increase in proliferation is reported by these investigators (Lollingshead et al., 2007). Therefore, the role of PPARβ/δ in liver cancer is uncertain and further studies using different models and various experimental approaches are still needed before reaching a final conclusion regarding this matter.
Several reports suggest a role for PPARγ in prevention and treatment of hepatocellular carcinoma, where increasing evidence suggests a potential role for the PPARγ agonists thiazolidinediones as anti-proliferative agents (Borbath and Horsmans, 2008). Studies show that PPARγ ligands inhibit proliferation of human liver cancer cells and induce cell cycle arrest (Toyoda et al., 2002; Hsu et al., 2008; Zhou et al., 2008; Yu et al., 2010). Induction of apoptosis through caspase 3-activation is proposed to be another mechanism for growth inhibition of human liver cancer cells by troglitazone (Toyoda et al., 2002), which was also found to modulate the expression of several cell cycle regulating proteins (Yu et al., 2010). Another PPARγ ligand, rosiglitazone, is also suggested to be beneficial in liver cancer therapy due to its ability to induce apoptosis (Cao et al., 2007). In addition, the PPARγ agonist pioglitazone was found to inhibit early carcinogenic transformation in rat liver (Borbath et al., 2007).
Paradoxically, studies suggest that PPARγ antagonists may provide greater hepatic anti-tumour effects than the receptor agonists. Specific PPARγ antagonists were found to reduce adhesion of hepatocellular carcinoma cells to extracellular matrix leading to inhibition of cell growth and migration (Schaefer et al., 2005; Kim et al., 2007). To explain this apparent paradox, it is suggested that inducing cell death by anoikis, caused by PPARγ antagonists, may be a more effective mechanism to control tumour growth and invasion compared with that caused by cell cycle arrest caused by PPARγ agonists (Schaefer et al., 2005).
Pancreatic PPARs and pancreatic cancer
Several cellular studies demonstrate that PPARγ activation decreases pancreatic cancer cell growth and attenuates their migration and invasive capacity (Motomura et al., 2000; Toyota et al., 2002; Tsujie et al., 2003; Motomura et al., 2004; Adrian et al., 2008; Kumei et al., 2009). Using a pancreatic carcinoma xenograft model of nude mice, it was reported that PPARγ activation inhibited pancreatic cancer growth and suppressed tumour angiogenesis (Dong et al., 2009). However, like in other types of cancers, the role PPARγ in pancreatic cancer remains controversial (Eibl, 2008). In contrast to the above studies, PPARγ expression in pancreatic cancer was correlated with shorter patient survival suggesting a role for PPARγ in tumour progression (Kristiansen et al., 2006). Although species-related differences, mice versus human, may be of important consequences in explaining the paradox at hand, further investigations are still needed to clarify the role of PPARγ and its ligands in pancreatic cancer.
PPAR in cancer of the urinary system
Although some studies claim that increased expression of PPARγ protein is positively correlated with increasing grade and stage of bladder cancer (Mansure et al., 2009), others report that PPARγ is expressed in normal urothelium and is associated with lower incidence of tumour progression (Myloma et al., 2009).
Again, paradoxically, it has been demonstrated that PPARγ activation is associated with the opposing actions of inducing cell differentiation on one hand and cancer on the other, in the urinary tract (Lubet et al., 2008; Oleksiewicz et al., 2008; Lee et al., 2010. Regrettably, some dual-acting PPARα and γ agonists exhibit carcinogenic effects in rats and mice bladder urothelium raising concerns for safety issues regarding the clinical use of these drugs (Rubenstrunk et al., 2007). In this regard, it is hypothesized that simultaneous activation of PPARα and PPARγ could modulate the proliferation/differentiation balance contributing to carcinogenesis of PPARα + γ dual agonists (Oleksiewicz et al., 2008).
Although a new class of PPARγ agonists, methylene-substituted diindolylmethanes (C-DIMs), which are more potent than the previous generation of agonists, exhibits anti-tumour activity in bladder cancer cells (Lubet et al., 2008), subsequent studies revealed that these chemicals exert their anti-tumour activity through PPARγ-independent mechanisms, involving activation of pro-apoptotic proteins (Kassouf et al., 2006).
PPARs in other cancers
Although this review focuses on the role of PPARs in cancer in selected major organs, it should be recognized that these receptors also play an important role in cancer of numerous other organs (Figures 4 and 5). For example, PPARγ expression was found to be higher in human testicular cancer cells than in their normal counterparts, and PPARγ agonists exerted an anti-proliferative effect in testicular cancer cells (Hase et al., 2002). In addition, it is suggested that PPARγ agonists could be beneficial in preventing as well as treating osteosarcoma, by promoting osteoplastic terminal differentiation (Wagner et al., 2010). Furthermore, PPARγ ligands have anti-proliferative, prodifferentiative, anti-metastatic and pro-apopototic effects on several hematological malignancies (Figure 4), making PPARs a promising target in therapeutic regimens designed to combat these types of cancer (Garcia-Bates et al., 2008).
Figure 4.
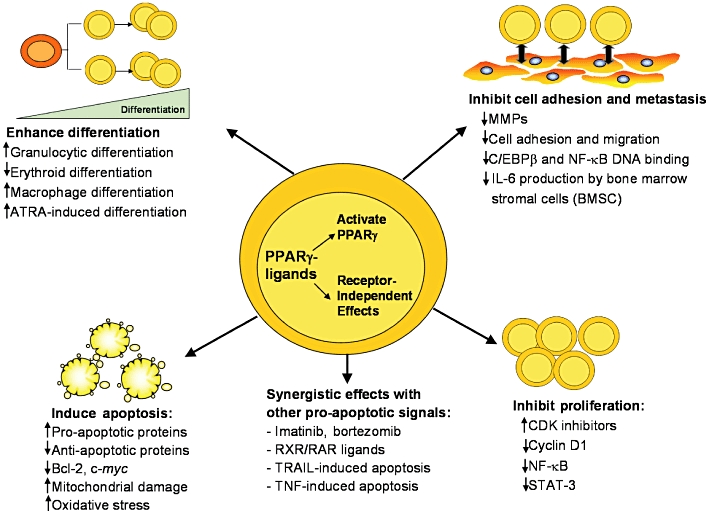
Mechanism of action of PPARγ ligands in hematological malignancies [reproduced from Garcia-Bates et al. (2008) with permission from Dr Richard Phipps]. PPAR, peroxisome proliferator-activated receptor; RXR, retinoid X receptors.
Figure 5.
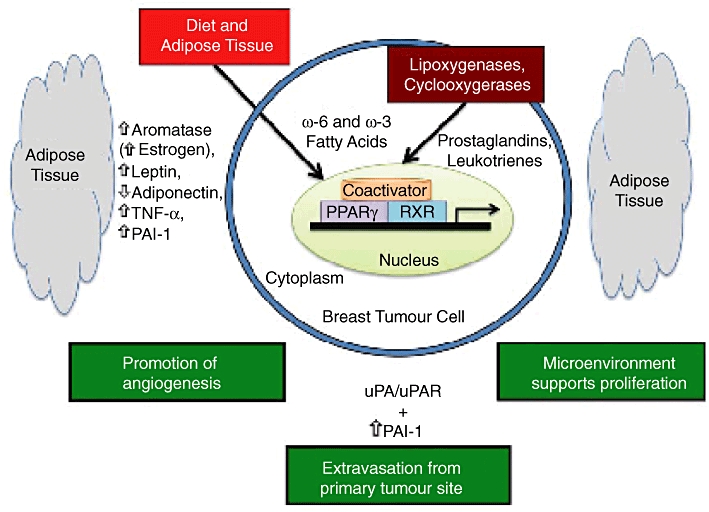
Potential role of PPARγ, fatty acid ligands, adipose tissue and the plasminogen activator system in breast cancer [reproduced from Carter and Church (2009) with permission from Dr Frank C. Church]. PPAR, peroxisome proliferator-activated receptor.
In addition to the above, PPARγ is up-regulated in breast cancer cells (Badawi and Badr, 2003), and several PPARγ ligands have been shown to exert anti-proliferative effects in these cells (Bonofiglio et al., 2009). PPARγ ligands also significantly delayed tumour formation onset in vivo, when given simultaneously with celecoxib, a COX-2 inhibitor (Mustafa and Kruger, 2008). It is noteworthy, however, that the PPARγ agonist troglitazone suppressed telomerase activity in breast cancer cells independently of PPARγ (Rashid-Kolvear et al., 2010). Furthermore, while rosiglitazone, another PPARγ agonist, had a pro-apoptotic effect in breast cancer cells (Bonofiglio et al., 2009), troglitazone did not influence apoptosis, casting doubt on the role of this receptor isotype in controlling breast cancer. Indeed, evidence has been presented showing that activating PPARγ promoted tumour growth in vivo (Saez et al., 2004), possibly by enhancing angiogenesis (Tian et al., 2009). Although the underlying reasons for these discrepancies remain to be delineated, differences in experimental models, such as cell type used, as well as PPARγ ligands utilized and duration of treatment as well as dosage, are among candidate factors.
Proposed mechanisms of action
Because of the conflicting evidence with regard to the role PPARs play in cancer, there has not been a unified, universally accepted mechanism of action to describe such a role. Therefore, the following is an overview of the various hypotheses put forth in an attempt to describe and explain the observed effects.
It has been reported that PPARγ activation inhibits cell growth (Garcia-Bates et al., 2008), as well as causes both differentiation and apoptosis in a variety of cancer cell types (Bonofiglio et al., 2009). Activation of PPARγ by rosiglitazone has been shown to change mitochondrial membrane permeability, an important step in the induction of cellular apoptosis (Bonofiglio et al., 2009). This effect was blocked in the presence of specific antagonists of PPARγ, as well as in cells pretreated with antisense for the tumour suppressor P53 (Bonofiglio et al., 2009). The latter effect, coupled with the observation that PPARγ activation resulted in the up-regulation of P53 mRNA and protein levels, suggests a crucial potential role of P53 in PPARγ-mediated apoptosis in MCF-7, a human breast cancer cells line. Adding to the potential complexity of this system, another study showed that troglitazone-induced enhancement of apoptosis was caspase-3-dependent (Yamakawa-Karakida et al., 2002); treatment with a caspase-3 inhibitor completely abolished troglitazone-induced cell death (Yamakawa-Karakida et al., 2002). PPARγ agonists also increased the levels of the pro-apoptotic proteins, Bcl-xl and Mcl-1 Bax and down-regulated the anti-apoptotic protein Bcl-2 (Liu et al., 2005; Liu et al., 2007). In contrast, troglitazone did not induce apoptosis in the breast cancer cell line, MDA-MB-231 (Rashid-Kolvear et al., 2010).
Although it is not feasible to directly compare the effect of PPARγ agonists with that of a PPARγ mutant that mimics the ligand-activated state of the receptor, it is noteworthy to mention here that a recent study suggests that non-ligand-activated PPARγ can actually enhance, rather than reduce, mammary tumour growth in vivo, potentially through enhancing angiogenesis (Tian et al., 2009).
The action of PPARγ agonists and antagonists is by no means straightforward and likely involves multiple cellular systems. It has been reported that troglitazone suppresses telomerase activity in the breast cancer cell line, MDA-MB-231 via a PPARγ-independent mechanism (Rashid-Kolvear et al., 2010). This conclusion was based on the findings that PPARγ-specific antagonists were unable to block the observed effect of troglitazone on telomerase, as well as on the fact that troglitazone suppressed telomerase activity even in the absence of PPARγ (Rashid-Kolvear et al., 2010). Troglitazone has also been proposed to inhibit cell growth by inducing a G1 cell cycle arrest (Fujimura et al., 1998), and to dramatically reduce the expression levels of the proto-oncogene product c-myc (Yamakawa-Karakida et al., 2002). In a human eosinophilic leukaemia cell line, treatment with troglitazone caused a G0/G1 cell cycle arrest that correlated with increased mRNA levels of the cyclin-dependent kinase inhibitor p21WAF1/CIP1 (Sugimura et al., 1999). A novel PPARγ agonist, DIM#34, has been recently shown to induce apoptosis and inhibit cell growth through both, PPARγ-dependent and -independent mechanisms (Contractor et al., 2005).
Clinical application of PPAR agonists in cancer
It is noteworthy that various problems have been encountered with some clinically approved PPAR agonists and their use has been consequently restricted or discontinued (Krishnaswami et al., 2010). Nevertheless, clinical trials continue to assess the impact of PPAR agonists, particularly those that activate PPARγ, on various types of cancer, with trials completed to date producing conflicting outcomes.
In patients treated with the PPARγ agonist rosiglitazone or troglitazone, there was no objective tumour response noted in the overall incidence of malignancy or in several specific tumour types including breast, bladder, colon and liposarcomas (Kulke et al., 2002; Burstein et al., 2003; Debrock et al., 2003; Home et al., 2009. Home et al., 2010). However, in one of these studies there was a significant reduction in the incidence of pancreatic cancer in patients receiving rosiglitazone compared with the control group (Home et al., 2009), and another study reported a positive response, albeit modest, to the same drug against thyroid cancer (Kebebew et al., 2006). Furthermore, a meta-analysis of 80 randomized clinical studies comprising close to 30 000 patients, reported an overall incidence of malignancy that was significantly lower in patients treated for diabetes with rosiglitazone compared with control groups (Monami et al., 2008).
Conclusions
In the last 20 years, PPARs have gone from virtually unknown receptors to being major players in numerous physiological functions and pathological conditions. Among the most consequential involvements of these receptors is their role in cell differentiation and cancer.
The literature is replete with contradictory evidence implicating PPARs in the promotion and development of cancer, as well as for a protective role against cancer. While numerous studies report that the expression level of these receptors and/or their activation correlates with a positive outcome against cancer, this does not appear to be a universal phenomenon, raising the possibility of complexity arising from (i) cell type- and organ-specific effects; (ii) receptor-independent effects by the utilized PPAR ligands; (iii) pharmacokinetic considerations; and/or (iv) the stage of cancer formation at the time of PPAR ligand exposure.
The availability of three different PPAR isotypes with common as well as a number of isotype-specific ligands presents both opportunities and challenges for the goal of targeting these receptors as a means to combat cancer. The complementary action of simultaneous activation of more than one PPAR isotype has lead early on to pharmacological strategies focused on the development of agonists targeting more than one receptor isotype (Rubenstrunk et al., 2007). However, these strategies were soon challenged by emerging evidence showing, for example, that administering bezafibrate, a PPARα selective agonist, simultaneously with pioglitazone, a PPARγ selective agonist, prevented the beneficial effect of the latter on liver fat content (Balasubramanian et al., 2010). More relevant to the focus of this review, it was found that while agonists of PPARα or PPARγ have been shown to exert anti-proliferative effect, in various tissues, when used separately, dual activation of these two receptor isotypes was found to promote cancer in the bladder (Oleksiewicz et al., 2008). More recent strategies therefore have aimed at the identification and development of selective PPAR modulators (Rubenstrunk et al., 2007). These strategies are based on the hypothesis that optimizing the selectivity ratio between the different PPAR isotypes allows the selection of new PPAR agonists with improved efficacy and/or safety profiles (Rubenstrunk et al., 2007).
Emerging evidence points to the potential of PPAR isotype-specific agonists as anti-cancer therapies when administered in combination with conventional chemotherapeutic agents and/or radiation treatment in many types of malignancies (Robbins et al., 2010; Simpson-Haidaris et al., 2010). The synergistic effects of PPARγ agonists with other agents have been reported. For example, it has been found that PPARγ activation synergistically increases the growth inhibitory effect of platinum-based anti-cancer drugs such as cisplatin, both in vivo and in vitro (Shimizu and Moriwaki, 2008).
Because of the central permissive effect RXR plays in the activation of PPARs (Figure 6), it is reasonable to predict that the combination of PPARγ and RXR agonists may offer a new therapeutic strategy to combat various types of human malignancies (Shimizu and Moriwaki, 2008). Indeed, it has been reported that in human colon cancer cells, the combination of ciglitazone and 9-cis RA, agonists of PPARγ and RXR, respectively, produces greater efficacy in inhibiting cell growth than does either agonist alone (Shimizu and Moriwaki, 2008). This strategy has also proven successful in other types of malignancies, as well (Shimizu and Moriwaki, 2008).
Figure 6.
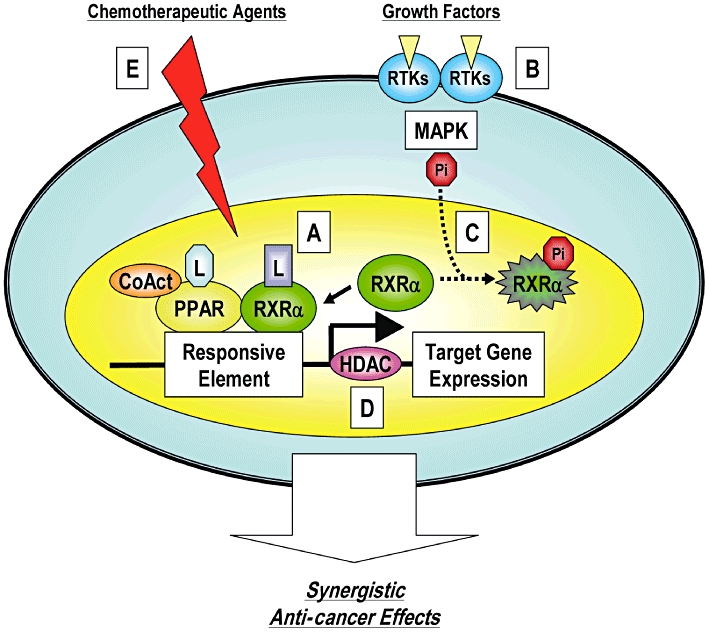
A hypothetical schematic representation of the synergistic anti-cancer effects of the combination of PPAR ligands plus other agents [reproduced from Shimizu and Moriwaki (2008) with permission from Dr Masahito Shimizu]. PPAR, peroxisome proliferator-activated receptor; RXR, retinoid X receptors.
Acknowledgments
Research in the laboratory of Dr Badr has been funded, in part, by the University of Missouri Research Board. The authors would like to express their gratitude to Dr Barbour Warren for his valuable comments on the manuscript.
Glossary
Abbreviations
- COX-2
cyclooxygenase-2
- PPARs
peroxisome proliferator-activated receptors
- RXR
retinoid X receptors
Future directions
Using androgen receptors as a model, McDonnell et al. have recently identified over 150 proteins/polypeptides whose ability to interact with full-length receptor was influenced by which ligand was bound to this nuclear receptor (Norris et al., 2009). According to these investigators, the nature of the bound ligand determines the overall conformation of the receptor, influencing the receptor's ability to recruit specific functionally distinct coactivators (Norris et al., 2009). Accordingly, the ability of the target cell to distinguish between receptors, presented to it, in different conformations would dictate the resulting cellular response (Norris et al., 2009).
Based on available experimental evidence and an understanding of the molecular actions of nuclear receptors, we present a hypothesis (Figure 7) to explain the paradoxical involvement of PPARs in cancer. According to this hypothesis, different cell types contain different metabolic pathways that produce quantitatively and/or qualitatively different chemical moieties from various PPAR ligands. The resulting metabolites/ligands induce a range of receptor conformational changes. These ‘ligand’-induced specific conformational changes lead to the recruitment of specific coactivators and subsequently produce a specific profile of gene transcription associated with either enhanced or diminished cancer (Figure 7). Essential to this hypothesis is the notion that different types of cells may vary in their levels, types and/or functionality of coactivators involved in PPAR activity, as well as in their ability to recognize various receptor conformations.
Figure 7.
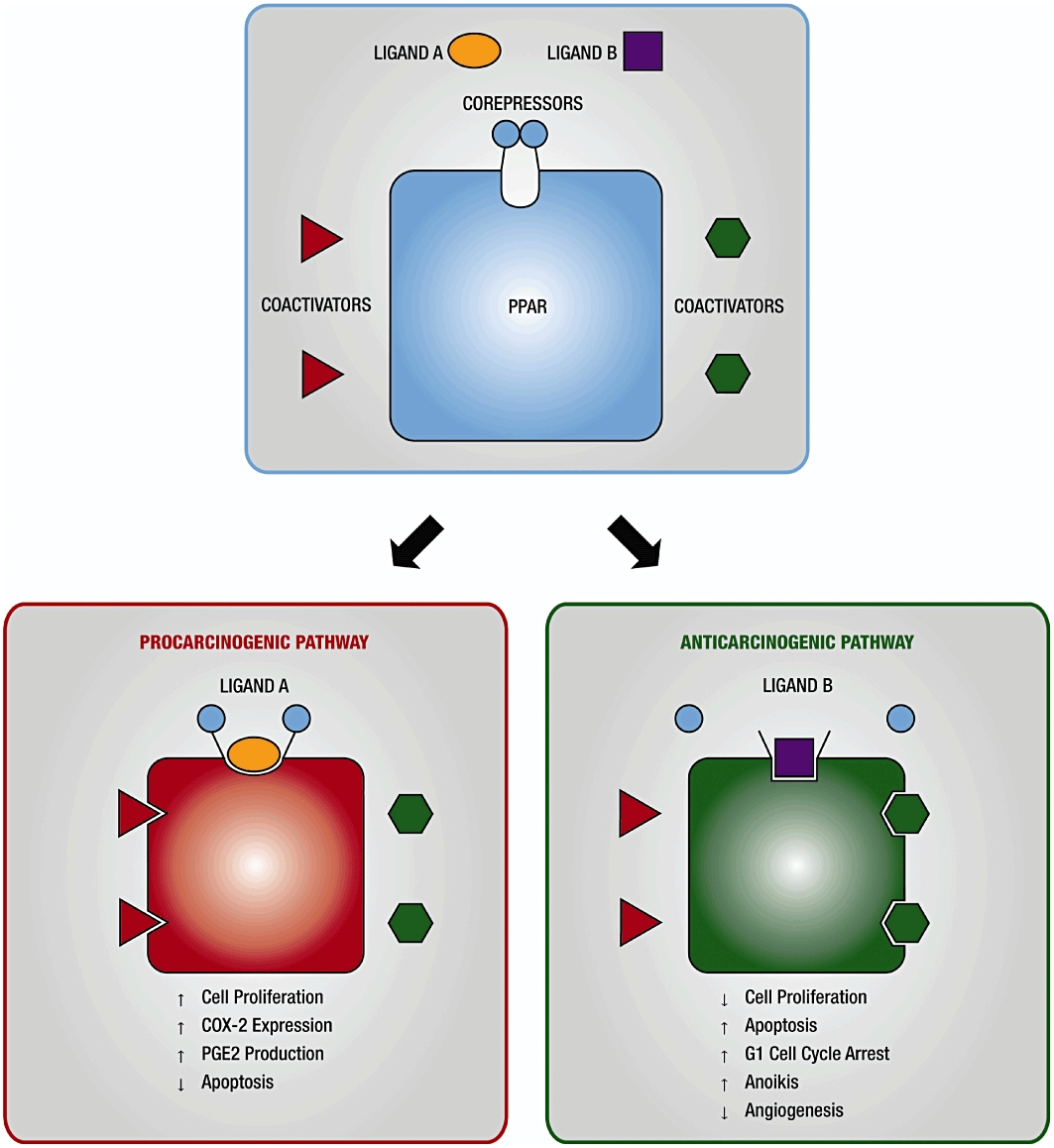
A schematic representation depicting a hypothesis explaining the paradoxical, anti-carcinogenic/pro-carcinogenic, effect of PPAR agonists. COX-2, cyclooxygenase-2; PPAR, peroxisome proliferator-activated receptor.
In conclusion, targeting PPARs in cancer treatment remains a valuable goal of researchers in the field, as evidenced by the ongoing numerous experimental as well as clinical trials. As this review has delineated, the agonists and antagonists of these receptors have a wide yet varied activity profile against cancer, providing a great opportunity for the development of new therapies for the disease.
Conflict of interest
The authors declare no conflict of interest.
References
- Adrian TE, Hennig R, Friess H, Ding X. The role of PPARgamma receptors and leukotriene B(4) receptors in mediating the effects of LY293111 in pancreatic cancer. PPAR Res. 2008;2008:827096. doi: 10.1155/2008/827096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen T, Zhang F, Moodie SA, Clemens LE, Smith A, Gregoire F, et al. Halofenate is a selective peroxisome proliferator-activated receptor gamma modulator with antidiabetic activity. Diabetes. 2006;55:2523–2533. doi: 10.2337/db06-0618. [DOI] [PubMed] [Google Scholar]
- Alsarra I, Brockmann W, Cunningham M, Badr M. Hepatocellular proliferation in response to agonists of peroxisome proliferator-activated receptor alpha: a role for kupffer cells? J Carcinogenesis. 2006;5:26. doi: 10.1186/1477-3163-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Dunn C, Cattley R, Corton JC. Hepatocellular proliferation in response to a peroxisome proliferators does not require TNFalpha signaling. Carcinogenesis. 2001;22:1843–1851. doi: 10.1093/carcin/22.11.1843. [DOI] [PubMed] [Google Scholar]
- Badawi AF, Badr MZ. Expression of cyclooxygenase-2 and peroxisome proliferators-activated receptor-gamma and levels of prostaglandin E2 and 15-deoxy-delta 12, 14-prostaglandin J2 in human breast cancer and metastasis. Int J Cancer. 2003;103:84–90. doi: 10.1002/ijc.10770. [DOI] [PubMed] [Google Scholar]
- Badr M. Peroxisome proliferator-activated receptor alpha and cancer: friends or foes? Int J Cancer Prev. 2004;1:77–87. [Google Scholar]
- Balasubramanian R, Gerrard J, Dalla Man C, Firbank MJ, Lane A, English PT, et al. Combination peroxisome proliferator-activated receptor gamma and alpha agonist treatment in Type 2 diabetes prevents the beneficial pioglitazone effect on liver fat content. Diabet Med. 2010;27:150–156. doi: 10.1111/j.1464-5491.2009.02906.x. [DOI] [PubMed] [Google Scholar]
- Ban JO, Kwak DH, Oh JH, Park EJ, Cho MC, Song HS, et al. Suppression of NF-kappaB and GSK-3beta is involved in colon cancer cell growth inhibition by the PPAR agonist troglitazone. Chem Biol Interact. 2010;188:75–85. doi: 10.1016/j.cbi.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Barak Y, Liao D, He W, Ong ES, Nelson MC, Olefsky JM, et al. Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proc Natl Acad Sci U S A. 2002;99:303–308. doi: 10.1073/pnas.012610299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger J, Moller DF. The mechanisms of action of PPARs. Ann Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Bojes H, Germolec D, Simoeonova P, Bruccoleri A, Schoonhoven R, Luster M, et al. Antibodies to tumor necrosis factor α prevent increases in cell replication in liver due to the potent peroxisome proliferator, Wy-14,643. Carcinogenesis. 1997;18:669–674. doi: 10.1093/carcin/18.4.669. [DOI] [PubMed] [Google Scholar]
- Bonofiglio D, Cione E, Qi H, Pingitore A, Perri M, Catalano S, et al. Combined low doses of PPARgamma and RXR ligands trigger an intrinsic apoptotic pathway in human breast cancer cells. Am J Pathol. 2009;175:1270–1280. doi: 10.2353/ajpath.2009.081078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbath I, Horsmans Y. The role of PPARgamma in hepatocellular carcinoma. PPAR Res. 2008;2008:209520. doi: 10.1155/2008/209520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbath I, Leclercq I, Moulin P, Sempoux C, Horsmans Y. The PPARgamma agonist pioglitazone inhibits early neoplastic occurrence in the rat liver. Eur J Cancer. 2007;43:1755–1763. doi: 10.1016/j.ejca.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Bren-Mattison Y, Van Putten V, Chan D, Winn R, Geraci MW, Nemenoff RA. Peroxisome proliferator-activated receptor-gamma (PPARgamma) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC) Oncogene. 2005;24:1412–1422. doi: 10.1038/sj.onc.1208333. [DOI] [PubMed] [Google Scholar]
- Bren-Mattison Y, Meyer AM, Van Putten V, Li H, Kuhn K, Stearman R, et al. Antitumorigenic effects of peroxisome proliferator-activated receptor-gamma in non-small-cell lung cancer cells are mediated by suppression of cyclooxygenase-2 via inhibition of nuclear factor-kappaB. Mol Pharmacol. 2008;37:709–717. doi: 10.1124/mol.107.042002. [DOI] [PubMed] [Google Scholar]
- Brun R, Tontonoz P, Forman B, Ellis R, Chen J, Evans R, et al. Differential activation of adipogenesis by multiple PPAR isoforms. Gene Dev. 1996;10:974–984. doi: 10.1101/gad.10.8.974. [DOI] [PubMed] [Google Scholar]
- Burstein H, Demetri G, Mueller E, Sarraf P, Spiegelman B, Winer E. Use of peroxisone proliferator-activated receptor (PPAR) γ ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat. 2003;79:391–397. doi: 10.1023/a:1024038127156. [DOI] [PubMed] [Google Scholar]
- Cao LQ, Chen XL, Wang Q, Huang XH, Zhen MC, Zhang LJ, et al. Upregulation of PTEN involved in rosiglitazone-induced apoptosis in human hepatocellular carcinoma cells. Acta Pharmacol Sin. 2007;28:879–887. doi: 10.1111/j.1745-7254.2007.00571.x. [DOI] [PubMed] [Google Scholar]
- Carter JC, Church FC. Obesity and breast cancer: the roles of peroxisome proliferator-activated receptor-gamma and plasminogen activator inhibitor-1. PPAR Res. 2009;2009:345320. doi: 10.1155/2009/345320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TH, Szabo E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator-activated receptor gamma in non-small cell lung cancer. Cancer Res. 2000;60:1129–1138. [PubMed] [Google Scholar]
- Chearwae W, Bright JJ. PPARgamma agonists inhibit growth and expansion of CD133+ brain tumour stem cells. Br J Cancer. 2008;99:2044–2053. doi: 10.1038/sj.bjc.6604786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YX, Zhong XY, Qin YF, Bing W, He LZ. 15d-PGJ2 inhibits cell growth and induces apoptosis of MCG-803 human gastric cancer cell line. World J Gastroenterol. 2003;9:2149–2153. doi: 10.3748/wjg.v9.i10.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Bush CR, Necela BM, Su W, Yanagisawa M, Anastasiadis PZ, et al. RS5444, a novel PPARgamma agonist, regulates aspects of the differentiated phenotype in nontransformed intestinal epithelial cells. Mol Cell Endocrinol. 2006;251:17–32. doi: 10.1016/j.mce.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Cheon CW, Kim DH, Kim DH, Cho YH, Kim JH. Effects of ciglitazone and troglitazone on the proliferation of human stomach cancer cells. World J Gastroenterol. 2009;15:310–320. doi: 10.3748/wjg.15.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res. 2000;49:497–505. doi: 10.1007/s000110050622. [DOI] [PubMed] [Google Scholar]
- Contractor R, Samudio IJ, Estrov Z, Harris D, McCubrey JA, Safe SH, et al. A novel ring-substituted diindolylmethane,1,1-bis[3′-(5-methoxyindolyl)]-1-(p-t-butylphenyl) methane, inhibits extracellular signal-regulated kinase activation and induces apoptosis in acute myelogenous leukemia. Cancer Res. 2005;65:2890–2898. doi: 10.1158/0008-5472.CAN-04-3781. [DOI] [PubMed] [Google Scholar]
- Debrock G, Vanhentenrijk V, Sciot R, Debiec-Rychter M, Oyen R, Van Oosterom A. A phase II trial with rosiglitazone in liposarcoma patients. Br J Cancer. 2003;89:1409–1412. doi: 10.1038/sj.bjc.6601306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol. 2001;169:453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- Dong YW, Wang XP, Wu K. Suppression of pancreatic carcinoma growth by activating peroxisome proliferator-activated receptor gamma involves angiogenesis inhibition. World J Gastroenterol. 2009;15:441–448. doi: 10.3748/wjg.15.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eibl G. The role of PPAR-gamma and its interaction with COX-2 in pancreatic cancer. PPAR Res. 2008;2008:326915. doi: 10.1155/2008/326915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige NJ, Gelman L, Michalik L, Desvergne B, Wahli W. From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog Lipid Res. 2006;45:120–159. doi: 10.1016/j.plipres.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci (USA) 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froment P. PPARs and RXRs in male and female fertility and reproduction. PPAR Res. 2008;2008:637490. doi: 10.1155/2008/637490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura S, Suzumiya J, Nakamura K, Ono J. Effects of troglitazone on the growth and differentiation of hematopoietic cell lines. Int J Oncol. 1998;13:1263–1267. doi: 10.3892/ijo.13.6.1263. [DOI] [PubMed] [Google Scholar]
- Fukumoto K, Yano Y, Virgona N, Hagiwara H, Sato H, Senba H, et al. Peroxisome proliferator-activated receptor delta as a molecular target to regulate lung cancer cell growth. FEBS Lett. 2005;579:3829–3836. doi: 10.1016/j.febslet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Garcia-Bates TM, Lehmann GM, Simpson-Haidaris PJ, Bernstein SH, Sime PJ, Phipps RP. Role of peroxisome proliferator-activated receptor gamma and its ligands in the treatment of hematological malignancies. PPAR Res. 2008;2008:834612. doi: 10.1155/2008/834612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girnun GD, Chen L, Silvaggi J, Drapkin R, Chirieac LR, Padera RF, et al. Regression of drug-resistant lung cancer by the combination of rosiglitazone and carboplatin. Clin Cancer Res. 2008;14:6478–6486. doi: 10.1158/1078-0432.CCR-08-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinghammar B, Skogsberg J, Hamsten A, Ehrenborg E. PPARdelta activation induces COX-2 gene expression and cell proliferation in human hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2003;308:361–368. doi: 10.1016/s0006-291x(03)01384-6. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ, Shah YM. PPARalpha: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology. 2008;246:2–8. doi: 10.1016/j.tox.2007.09.030. [DOI] [PubMed] [Google Scholar]
- Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER, Midathada MV, Kim L, et al. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J Clin Oncology. 2007;25:1476–1481. doi: 10.1200/JCO.2006.07.2777. [DOI] [PubMed] [Google Scholar]
- Gregoire FM, Kersten S, Harrington W. PPARS and obesity. PPAR Res. 2007;2007:78475. doi: 10.1155/2007/78475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grommes C, Landreth GE, Sastre M, Beck M, Feinstein DL, Jacobs AH, et al. Inhibition of in vivo glioma growth and invasion by peroxisome proliferator-activated receptor gamma agonist treatment. Mol Pharmacol. 2006;70:1524–1533. doi: 10.1124/mol.106.022194. [DOI] [PubMed] [Google Scholar]
- Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-delta accelerates intestinal adenoma growth. Nature Medicine. 2004;10:245–247. doi: 10.1038/nm993. [DOI] [PubMed] [Google Scholar]
- Guri AJ, Mohapatra SK, Horne WT, 2nd, Hontecillas R, Bassaganya-Riera J. The role of T cell PPAR gamma in mice with experimental inflammatory bowel disease. BMC Gastroenterol. 2010;10:60. doi: 10.1186/1471-230X-10-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Ritzenthaler JD, Zheng Y, Roman J. PPARbeta/delta agonist stimulates human lung carcinoma cell growth through inhibition of PTEN expression: the involvement of PI3K and NF-kappaB signals. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1238–L1249. doi: 10.1152/ajplung.00017.2008. [DOI] [PubMed] [Google Scholar]
- Han S, Ritzenthaler JD, Sun X, Zheng Y, Roman J. Activation of peroxisome proliferator-activated receptor beta/delta induces lung cancer growth via peroxisome proliferator-activated receptor coactivator gamma-1alpha. Am J Respir Cell Mol Biol. 2009;40:325–331. doi: 10.1165/rcmb.2008-0197OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Han L, Zhou R, Niu J, McNutt MA, Wang P, Tong T. SIRT1 is regulated by a PPAR-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010;38:7458–7471. doi: 10.1093/nar/gkq609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman FS, Nicol CJ, Marin HE, Ward JM, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-delta attenuates colon carcinogenesis. Nat Med. 2004;10:481–483. doi: 10.1038/nm1026. [DOI] [PubMed] [Google Scholar]
- Hase T, Yoshimura R, Mitsuhashi M, Segawa Y, Kawahito Y, Wada S, et al. Expression of peroxisome proliferator-activated receptors in human testicular cancer and growth inhibition by its agonists. Urology. 2002;60:542–547. doi: 10.1016/s0090-4295(02)01747-8. [DOI] [PubMed] [Google Scholar]
- Hau P, Kunz-Schughart L, Bogdahn U, Baumgart U, Hirschmann B, Weimann E, et al. Low-dose chemotherapy in combination with COX-2 inhibitors and PPAR-gamma agonists in recurrent high-grade gliomas – a phase II study. Oncology. 2007;73:21–25. doi: 10.1159/000120028. [DOI] [PubMed] [Google Scholar]
- Hazra S, Peebles KA, Sharma S, Mao JT, Dubinett SM. The role of PPARgamma in the cyclooxygenase pathway in lung cancer. PPAR Res. 2008;2008:790568. doi: 10.1155/2008/790568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans K, Michalik L, Dittie A, Knorr A, Rombouts K, De Jong J, et al. Peroxisome proliferator-activated receptor-beta signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology. 2003;124:184–201. doi: 10.1053/gast.2003.50015. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Landreth GE. PPARs in the brain. Biochim Biophys Acta. 2007;1771:1031–1045. doi: 10.1016/j.bbalip.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Home P, Pocock S, Beck-Nielsen H, Curtis P, Gomis R, Hanefeld M, et al. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicenter, randomised, open-label trial. Lancet. 2009;373:2125–2135. doi: 10.1016/S0140-6736(09)60953-3. [DOI] [PubMed] [Google Scholar]
- Home P, Hahn S, Jones N, Noronha D, Beck-Nielsen H, Viberti G. Experience of malignancies with oral glucone-lowering drugs in the randomized controlled ADOPT (A Diabetes Outcome Progression Trial) and RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycaemia in Diabetes) clinical trials. Diabetologia. 2010;53:1838–18845. doi: 10.1007/s00125-010-1804-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu MC, Huang CC, Chang HC, Hu TH, Hung WC. Overexpression of Jab1 in hepatocellular carcinoma and its inhibition by peroxisome proliferator-activated receptor ligands in vitro and in vivo. Clin Cancer Res. 2008;14:4045–4052. doi: 10.1158/1078-0432.CCR-07-5040. [DOI] [PubMed] [Google Scholar]
- Inoue K, Kawahito Y, Tsubouchi Y, Yamada R, Kohno M, Hosokawa Y, et al. Expression of peroxisome proliferator-activated receptor (PPAR)-gamma in human lung cancer. Anticancer Res. 2001;21:2471–2476. [PubMed] [Google Scholar]
- Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- Jackson L, Wahli W, Michalik L, Watson SA, Morris T, Anderton K, et al. Potential role for peroxisome proliferator activated receptor (PPAR) in preventing colon cancer. Gut. 2003;52:1317–1322. doi: 10.1136/gut.52.9.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam WG, Ghanayem BI. Induction of replicative DNA Synthesis and PPAR alpha-dependent gene transcription by Wy-14,643 in primary rat hepatocyte and non-parenchymal cell co-cultures. Carcinogenesis. 1997;18:2077–2083. doi: 10.1093/carcin/18.11.2077. [DOI] [PubMed] [Google Scholar]
- Kassouf W, Chintharlapalli S, Abdelrahim M, Nelkin G, Safe S, Kamat AM. Inhibition of bladder tumor growth by 1,1-bis(3′-indolyl)-1-(p-substitutedphenyl)methanes: a new class of peroxisome proliferator-activated receptor gamma agonists. Cancer Res. 2006;66:412–418. doi: 10.1158/0008-5472.CAN-05-2755. [DOI] [PubMed] [Google Scholar]
- Kebebew E, Peng M, Reiff E, Treseler P, Woeber K, Clark O, et al. A phase II trial of rosiglitazone in patients with thyroglobulin-positive and radioiodine-negative differentiated thyroid cancer. Surgery. 2006;140:960–967. doi: 10.1016/j.surg.2006.07.038. [DOI] [PubMed] [Google Scholar]
- Keshamouni VG, Arenberg DA, Reddy RC, Newstead MJ, Anthwal S, Standiford TJ. PPAR-gamma activation inhibits angiogenesis by blocking ELR+CXC chemokine production in non-small cell lung cancer. Neoplasia. 2005;7:294–301. doi: 10.1593/neo.04601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KR, Choi HN, Lee HJ, Baek HA, Park HS, Jang KY, et al. A peroxisome proliferator-activated receptor gamma antagonist induces vimentin cleavage and inhibits invasion in high-grade hepatocellular carcinoma. Oncol Rep. 2007;18:825–832. [PubMed] [Google Scholar]
- Kliewer S, Sundseth S, Jones S, Brown P, Wisely GB, Koble C, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc Natl Acad Sci (USA) 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopelovich L, Fay JR, Glazer RI, Crowell JA. Peroxisome proliferator-activated receptor modulators as potential chemopreventive agents. Mol Cancer Ther. 2002;1:357–363. [PubMed] [Google Scholar]
- Krieger-Hinck N, Schumacher U, Müller A, Valentiner U. The effect of the PPAR-gamma agonist rosiglitazone on neuroblastoma SK-N-SH cells in a metastatic xenograft mouse model. Oncol Res. 2010;18:387–393. doi: 10.3727/096504010x12644422320708. [DOI] [PubMed] [Google Scholar]
- Krishnaswami A, Ravi-Kumar S, Lewis J. Thiazolidinediones: a 2010 perspective. Perm J. 2010;14:64–72. doi: 10.7812/tpp/09-052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen G, Jacob J, Buckendahl AC, Grützmann R, Alldinger I, Sipos B, et al. Peroxisome proliferator-activated receptor gamma is highly expressed in pancreatic cancer and is associated with shorter overall survival times. Clin Cancer Res. 2006;12:6444–6451. doi: 10.1158/1078-0432.CCR-06-0834. [DOI] [PubMed] [Google Scholar]
- Kulke M, Sharpless N, Ryan D, Shivdasani R, Clark J, Spiegelman B, et al. A phase II study of troglitazone, an activator of the PPARgamma receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. Cancer J. 2002;8:395–399. doi: 10.1097/00130404-200209000-00010. [DOI] [PubMed] [Google Scholar]
- Kumei S, Motomura W, Yoshizaki T, Takakusaki K, Okumura T. Troglitazone increases expression of E-cadherin and claudin 4 in human pancreatic cancer cells. Biochem Biophys Res Commun. 2009;380:414–419. doi: 10.1016/j.bbrc.2009.01.134. [DOI] [PubMed] [Google Scholar]
- Lalloyer F, Staels B. Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2010;30:894–899. doi: 10.1161/ATVBAHA.108.179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence J, Wollengerg G, DeLuca J. Tumor necrosis factor alpha is not required for Wy-14,643-induced cell proliferation. Carcinogenesis. 2001;22:381–386. doi: 10.1093/carcin/22.3.381. [DOI] [PubMed] [Google Scholar]
- Lee JY, Kim JK, Shin S, Yoon DY, Heo YS, Kim Y. Cytotoxic flavonoids on agonists of peroxisome proliferator-activated receptor gamma on human cervical and prostate cancer cells. J Nat Prod. 2010;73:1261–1265. doi: 10.1021/np100148m. [DOI] [PubMed] [Google Scholar]
- Lefebvre A-M, Laville M, Vega N, Riou J, Gaal L, Auwerx J, et al. Depot-specific differences in adipose tissue gene expression in lean and obese subjects. Diabetes. 1998a;47:98–103. doi: 10.2337/diab.47.1.98. [DOI] [PubMed] [Google Scholar]
- Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, et al. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998b;4:1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- Lichtor T, Spagnolo A, Glick RP, Feinstein DL. PPAR-gamma thiazolidinedione agonists and immunotherapy in the treatment of brain tumors. PPAR Res. 2008;2008:547470. doi: 10.1155/2008/547470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, Gupta R, Ma W-G, Paria B, Moller D, Morrow J, et al. Cyclooxygenase-2-derived prostacyclin mediates embryo implantation in the mouse via PPARγ. Gene Dev. 1999;13:1561–1574. doi: 10.1101/gad.13.12.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Huang RW, Lin DJ, Peng J, Wu XY, Lin Q, et al. Expression of survivin and bax/bcl-2 in peroxisome proliferator activated receptor-gamma ligands induces apoptosis on human myeloid leukemia cells in vitro. Ann Oncol. 2005;16:455–459. doi: 10.1093/annonc/mdi077. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Liu PQ, Lin DJ, Xiao RZ, Huang M, Li XD, et al. Downregulation of cyclooxygenase-2 expression and activation of caspase-3 are involved in peroxisome proliferator-activated receptor-gamma agonists induced apoptosis in human monocyte leukemia cells in vitro. Ann Hematol. 2007;86:173–183. doi: 10.1007/s00277-006-0205-2. [DOI] [PubMed] [Google Scholar]
- Lollingshead HE, Killins RL, Borland MG, Girroir EE, Billin AN, Willson TM, et al. Peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) ligands do not potentiate growth of human cancer cell lines. Carcinogenesis. 2007;28:2641–2649. doi: 10.1093/carcin/bgm183. [DOI] [PubMed] [Google Scholar]
- Lu J, Imamura K, Nomura S, Mafune K, Nakajima A, Kadowaki T, et al. Chemopreventive effect of peroxisome proliferator-activated receptor gamma on gastric carcinogenesis in mice. Cancer Res. 2005;65:4769–4774. doi: 10.1158/0008-5472.CAN-04-2293. [DOI] [PubMed] [Google Scholar]
- Lubet RA, Fischer SM, Steele VE, Juliana MM, Desmond R, Grubbs CJ. Rosiglitazone, a PPAR gamma agonist: potent promoter of hydroxybutyl(butyl)nitrosamine-induced urinary bladder cancers. Int J Cancer. 2008;123:2254–2259. doi: 10.1002/ijc.23765. [DOI] [PubMed] [Google Scholar]
- Mansure JJ, Nassim R, Kassouf W. Peroxisome proliferator-activated receptor gamma in bladder cancer: a promising therapeutic target. Cancer Biol Ther. 2009;8:6–15. doi: 10.4161/cbt.8.7.7853. [DOI] [PubMed] [Google Scholar]
- Marsman D, Cattley R, Conway J, Popp JA. Relationship of hepatic peroxisome proliferation and replicative DNA synthesis to the hepatocarinogenicity of the peroxisome proliferators di(2-ethylhexyl) phthalate and [4-Chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid (Wy-14,643) in rats. Cancer Res. 1988;48:6739–6744. [PubMed] [Google Scholar]
- Masters C, Crane D. On the role of the peroxisome in ontogeny, aging and degenerative disease. Mech Ageing Dev. 1995;80:69–83. doi: 10.1016/0047-6374(94)01563-2. [DOI] [PubMed] [Google Scholar]
- Matthiessen MW, Pedersen G, Albrektsen T, Adamsen S, Fleckner J, Brynskov J. Peroxisome proliferator-activated receptor expression and activation in normal human colonic epithelial cells and tubular adenomas. Scand J Gastroenterol. 2005;40:198–205. doi: 10.1080/00365520410009573. [DOI] [PubMed] [Google Scholar]
- Michalik L, Wahli W. Peroxisome proliferator-activated receptors: three isotypes for a multitude of functions. Curr Opin Biotechnol. 1999;10:564–570. doi: 10.1016/s0958-1669(99)00030-0. [DOI] [PubMed] [Google Scholar]
- Monami M, Lamanna C, Marchionni N, Mannucci E. Rosiglitazone and risk of cancer. A meta-analysis of randomized clinical trials. Diabetes Care. 2008;31:1455–1460. doi: 10.2337/dc07-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura K, Cheung C, Ward JM, Reddy JK, Gonzalez FJ. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor alpha to Wy-14,643-induced liver tumorigenesis. Carcinogenesis. 2006;27:1074–1080. doi: 10.1093/carcin/bgi329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motomura W, Okumura T, Takahashi N, Obara T, Kohgo Y. Activation of peroxisome proliferator-activated receptor gamma by troglitazone inhibits cell growth through the increase of p27KiP1 in human. Pancreatic carcinoma cells. Cancer Res. 2000;60:5558–5564. [PubMed] [Google Scholar]
- Motomura W, Nagamine M, Tanno S, Sawamukai M, Takahashi N, Kohgo Y, et al. Inhibition of cell invasion and morphological change by troglitazone in human pancreatic cancer cells. J Gastroenterol. 2004;39:461–468. doi: 10.1007/s00535-003-1324-3. [DOI] [PubMed] [Google Scholar]
- Mukherjee R, Jow L, Noonan D, McDonnell DP. Human and rat peroxisome proliferator activated receptors (PPARs) demonstrate similar tissue distribution but different responsiveness to PPAR activators. J Steroid Biochem Mol Biol. 1994;51:157–166. doi: 10.1016/0960-0760(94)90089-2. [DOI] [PubMed] [Google Scholar]
- Muller-Brusselbach S, Ebrahimsade S, Jäkel J, Eckhardt J, Rapp UR, Peters JM, et al. Growth of transgenic RAF-induced lung adenomas is increased in mice with a disrupted PPARbeta/delta gene. Int J Oncol. 2007;31:607–611. [PubMed] [Google Scholar]
- Mustafa A, Kruger WD. Suppresion of tumor formation by a cyclooxygenase-2 inhibitor and a peroxisome proliferator-activated receptor gamma agonist in an in vivo mouse model of spontaneous breast cancer. Clin Cancer Res. 2008;14:4935–4949. doi: 10.1158/1078-0432.CCR-08-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myloma E, Giannopoulou I, Diamantopoulou K, Bakarakos P, Nomikos A, Zervas A, et al. Peroxisome proliferator-activated receptor gamma expression in urothelial carcinomas of the bladder: association with differentiation, proliferation and clinical outcome. Eur J Surg Oncol. 2009;35:197–201. doi: 10.1016/j.ejso.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Necela BM, Su W, Thompson EA. Peroxisome proliferator-activated receptor gamma down-regulates follistatin in intestinal epithelial cells through SP1. J Biol Chem. 2008;283:29784–29794. doi: 10.1074/jbc.M804481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemenoff R, Meyer AM, Hudish TM, Mozer AB, Snee A, Narumiya S, et al. Prostacyclin prevents murine lung cancer independent of the membrane receptor by activation of peroxisomal proliferator-activated receptor gamma. Cancer Prev Res. 2008;1:349–356. doi: 10.1158/1940-6207.CAPR-08-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JD, Joseph JD, Sherk AB, Juzumiene D, Turnbull PS, Rafferty SW, et al. Differential presentation of protein interaction surfaces on the androgen receptor defines the pharmacological actions of bound ligands. Chem & Biol. 2009;16:452–460. doi: 10.1016/j.chembiol.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino S, Shima K, Baba Y, Nosho K, Irahara N, Kure S, et al. Colorectal cancer expression of peroxisome proliferator-activated receptor gamma (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology. 2009;136:1242–1250. doi: 10.1053/j.gastro.2008.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiewicz MB, Southgate J, Iversen L, Egerod FL. Rat urinary bladder carcinogenesis by dual-acting PPARalpha + gamma agonists. PPAR Res. 2008;2008:103167. doi: 10.1155/2008/103167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CN, Hsu MH, Griffin KJ, Raucy JL, Johnson EF. Peroxisome proliferator activated receptor-alpha expression in human liver. Mol Pharmacol. 1998;53:14–22. [PubMed] [Google Scholar]
- Panigrahy D, Kaipainen A, Kieran MW. Huang, S. PPARs: a double-edged sword in cancer therapy? PPAR Res. 2008;2008:350351. doi: 10.1155/2008/350351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papi A, Tatenhorst L, Terwel D, Hermes M, Kummer MP, Orlandi M, et al. PPARgamma and RXRgamma ligands act synergistically as potent antineoplastic agents in vitro and in vivo glioma models. J Neurochem. 2009;109:1779–1790. doi: 10.1111/j.1471-4159.2009.06111.x. [DOI] [PubMed] [Google Scholar]
- Pedchenko TV, Gonzalez AL, Wang D, DuBois RN, Massion PP. Peroxisome proliferator-activated receptor beta/delta expression and activation in lung cancer. Am J Respir Cell Mol Biol. 2008;39:689–696. doi: 10.1165/rcmb.2007-0426OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Cattley RC, Gonzalez FG. E mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis. 1997;18:2029–2033. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- Peters J, Rusyn I, Rose M, Gonzalez F, Thurman RG. Peroxisome proliferator-activated receptor α is restricted to hepatic parenchymal cells, not kupffer cells: implications for the mechanism of action of peroxisome proliferators in hepatocarcinogenesis. Carcinogenesis. 2000;21:823–826. doi: 10.1093/carcin/21.4.823. [DOI] [PubMed] [Google Scholar]
- Peyrin-Biroulet L, Beisner J, Wang G, Nuding S, Oommen ST, Kelly D, et al. Peroxisome proliferator-activated receptor gamma activation is required for maintenance of innate antimicrobial immunity in the colon. Proc Natl Acad Sci U S A. 2010;107:8772–8777. doi: 10.1073/pnas.0905745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad KN, Saxena A, Ghoshal UC, Bhagat MR, Krishnani N. Analysis of Pro12Ala PPAR gamma polymorphism and Helicobacter pylori infection in gastric adenocarcinoma and peptic ulcer disease. Ann Oncol. 2008;19:1299–1303. doi: 10.1093/annonc/mdn055. [DOI] [PubMed] [Google Scholar]
- Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nucl Recept Signal. 2010;8:e002. doi: 10.1621/nrs.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan S, Zhao W, Riddle DR, Robbins ME. Role of PPARs in radiation-induced brain injury. PPAR Res. 2010;2010:234975. doi: 10.1155/2010/234975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid-Kolvear F, Taboski MA, Nguyen J, Wang DY, Harrington LA, Done SJ. Troglitazone suppresses telomerase activity independently of PPAR gamma in estrogen-receptor negative breast cancer cells. BMC Cancer. 2010;10:390. doi: 10.1186/1471-2407-10-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy RC, Srirangam A, Reddy K, Chen J, Gangireddy S, Kalemkerian GP, et al. Chemotherapeutic drugs induce PPAR-gamma expression and show sequence-specific synergy with PPAR-gamma ligands in inhibition of non-small cell lung cancer. Neoplasia. 2008;10:597–603. doi: 10.1593/neo.08134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins M, Linard C, Panigrahy D. PPARs and anticancer therapies. PPAR Res. 2010;2010:536415. doi: 10.1155/2010/536415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfe M, James N, Roberts R. Tumor necrosis factor α (TNFα) suppresses apoptosis and induces DNA synthesis in rodent hepatocytes: a mediator of the hepatocarcinogenicity of peroxisome proliferators? Carcinogenesis. 1997;18:2277–2280. doi: 10.1093/carcin/18.11.2277. [DOI] [PubMed] [Google Scholar]
- Roman J. Peroxisome proliferator-activated receptor gamma and lung cancer biology: implications for therapy. J Invest Med. 2008;56:528–533. doi: 10.2310/JIM.0b013e3181659932. [DOI] [PubMed] [Google Scholar]
- Rose M, Germolec DR, Schoonhoven R, Thurman RG. Kupffer cells are casually responsible for mitogenic effect of peroxisome proliferators. Carcinogenesis. 1997;18:1453–1456. doi: 10.1093/carcin/18.8.1453. [DOI] [PubMed] [Google Scholar]
- Rubenstrunk A, Hanf R, Hum DW, Fruchart JC, Staels B. Safety issues and prospects for future generations of PPAR modulators. Biochimica Biophysica Acta. 2007;1771:1065–1081. doi: 10.1016/j.bbalip.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Saez E, Saez E, Tontonoz P, Nelson MC, Alvarez JG, Ming UT, et al. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat Med. 1998;4:1058–1061. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- Saez E, Rosenfeld J, Livolsi A, Olson P, Lombardo E, Nelson M, et al. PPAR gamma signaling exacerbates mammary gland tumor development. Genes Dev. 2004;18:528–540. doi: 10.1101/gad.1167804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nature Med. 1998;4:1046–1052. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Tanahashi M, Yukiue H, Moiriyama S, Kobayashi Y, Nakashima Y, et al. Decreased peroxisome proliferator-activated receptor gamma gene expression was correlated with poor prognosis in patients with lung cancer. Lung Cancer. 2002;36:71–76. doi: 10.1016/s0169-5002(01)00449-4. [DOI] [PubMed] [Google Scholar]
- Sato H, Ishihara S, Kawashima K, Moriyama N, Suetsugu H, Kazumori H, et al. Expression of peroxisome proliferator-activated receptor (PPAR)gamma in gastric cancer and inhibitory effects of PPARgamma agonists. Br J Cancer. 2000;83:1394–1400. doi: 10.1054/bjoc.2000.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Toyoda M, Hoshino H, Monden T, Yamada M, Shimizu H, et al. Activation of peroxisome proliferator-activated receptor-gamma stimulates the growth arrest and DNA-damage inducible 153 gene in non-small cell lung carcinoma cells. Oncogene. 2002;21:2171–2180. doi: 10.1038/sj.onc.1205279. [DOI] [PubMed] [Google Scholar]
- Schaefer KL, Wada K, Takahashi H, Matsuhashi N, Ohnishi S, Wolfe MM, et al. Peroxisome proliferator-activated receptor gamma inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Res. 2005;65:2251–2259. doi: 10.1158/0008-5472.CAN-04-3037. [DOI] [PubMed] [Google Scholar]
- Shimizu M, Moriwaki H. Synergistic effects of PPARgamma ligands and retinoids in cancer treatment. PPAR Res. 2008;2008:181047. doi: 10.1155/2008/181047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson-Haidaris PJ, Pollock SJ, Ramon S, Guo N, Woeller CF, Feldon SE, et al. Anticancer role of PPARgamma agonists in hematological malignancies found in the vasculature, marrow, and eyes. PPAR Res. 2010;2010:814609. doi: 10.1155/2010/814609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman B. PPARγ in monocytes: less pain, any gain. Cell. 1998;93:153–155. doi: 10.1016/s0092-8674(00)81567-6. [DOI] [PubMed] [Google Scholar]
- Spiegelman B, Flier J. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87:377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- Su W, Bush CR, Necela BM, Calcagno SR, Murray NR, Fields AP, et al. Differential expression, distribution, and function of PPAR-gamma in the proximal and distal colon. Physiol Genomics. 2007;30:342–353. doi: 10.1152/physiolgenomics.00042.2007. [DOI] [PubMed] [Google Scholar]
- Sugimura A, Kiriyama Y, Nochi H, Tsuchiya H, Tamoto K, Sakurada Y, et al. Troglitazone suppresses cell growth of myeloid leukemia cell lines by induction of p21WAF1/CIP1 cyclin-dependent kinase inhibitor. Biochem Biophys Res Commun. 1999;261:833–837. doi: 10.1006/bbrc.1999.1049. [DOI] [PubMed] [Google Scholar]
- Tahara T, Arisawa T, Shibata T, Nakamura M, Wang F, Maruyama N, et al. Influence of peroxisome proliferator-activated receptor (PPAR)gamma Plo12Ala polymorphism as a shared risk marker for both gastric cancer and impaired fasting glucose (IFG) in Japanese. Dig Dis Sci. 2007;53:614–621. doi: 10.1007/s10620-007-9944-8. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Okumura T, Motomura W, Fujimoto Y, Kawabata I, Kohgo Y. Activation of PPARgamma inhibits cell growth and induces apoptosis in human gastric cancer cells. FEBS Lett. 1999;455:135–139. doi: 10.1016/s0014-5793(99)00871-6. [DOI] [PubMed] [Google Scholar]
- Tian L, Zhou J, Casimiro MC, Liang B, Ojeifo JO, Wang M, et al. Activating peroxisome proliferator-activated receptor gamma mutant promotes tumor growth in vivo by enhancing angiogenesis. Cancer Res. 2009;69:9236–9244. doi: 10.1158/0008-5472.CAN-09-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda M, Takagi H, Horiguchi N, Kakizaki S, Sato K, Takayama H, et al. A ligand for peroxisome proliferator activated receptor gamma inhibits cell growth and induces apoptosis in human liver cancer cells. Gut. 2002;50:563–567. doi: 10.1136/gut.50.4.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M, Miyazaki Y, Kitamura S, Nagasawa Y, Kiyohara T, Shinomura Y, et al. Peroxisome proliferator-activated receptor gamma reduces the growth rate of pancreatic cancer cells through the reduction of cyclin D1. Life Sci. 2002;70:1565–1575. doi: 10.1016/s0024-3205(01)01524-7. [DOI] [PubMed] [Google Scholar]
- Tsubouchi Y, Sano H, Kawahito Y, Mukai S, Yamada R, Kohno M, et al. Inhibition of human lung cancer cell growth by the peroxisome proliferator-activated receptor-gamma agonists through induction of apoptosis. Biochem Biophys Res Commun. 2000;270:400–405. doi: 10.1006/bbrc.2000.2436. [DOI] [PubMed] [Google Scholar]
- Tsujie M, Nakamori S, Okami J, Hayashi N, Hiraoka N, Nagano H, et al. Thiazolidinediones inhibit growth of gastrointestinal, biliary, and pancreatic adenocarcinoma cells through activation of the peroxisome proliferator-activated receptor gamma/retinoid X receptor alpha pathway. Exp Cell Res. 2003;289:143–151. doi: 10.1016/s0014-4827(03)00263-5. [DOI] [PubMed] [Google Scholar]
- Uchimura K, Nakamuta M, Enjoji M, Irie T, Sugimoto R, Muta T, et al. Activation of retinoic X receptor and peroxisome proliferator-activated receptor-γ inhibits nitric oxide and tumor necrosis factor-α production in rat kupffer cells. Hepatology. 2001;33:91–99. doi: 10.1053/jhep.2001.21145. [DOI] [PubMed] [Google Scholar]
- Vidal-Puig A, Considine R, Jimenez-Linan M, Werman A, Porries W, Caro J, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss and regulation by insulin and glucocorticoids. J Clin Invest. 1997;99:2416–2422. doi: 10.1172/JCI119424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner ER, He BC, Chen L, Zuo GW, Zhang W, Shi Q, et al. Therapeutic implications of PPARgamma in human osteosarcoma. PPAR Res. 2010;2010:956427. doi: 10.1155/2010/956427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C, Kosyk O, Bradford B, Ross P, Burns A, Cunningham M, et al. Time-course investigation of PPARα- and Kupffer cell-dependent effects of WY 14643 in mouse liver using micrparray gene expression. Toxicol Appl Pharmacol. 2007;225:267–277. doi: 10.1016/j.taap.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa-Karakida N, Sugita K, Inukai T, Goi K, Nakamura M, Uno K, et al. Ligand activation of peroxisome proliferator-activated receptor gamma induces apoptosis of leukemia cells by down-regulating the c-myc gene expression via blockade of the Tcf-4 activity. Cell Death Differ. 2002;9:513–526. doi: 10.1038/sj.cdd.4401000. [DOI] [PubMed] [Google Scholar]
- Yang Q, Nagano T, Shah Y, Cheung C, Ito S, Gonzalez FJ. The PPAR alpha-humanized mouse: a model to investigate species differences in liver toxicity mediated by PPAR alpha. Toxicol Sci. 2008;101:132–139. doi: 10.1093/toxsci/kfm206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef J, Badr M. Activated Kupper cells attenuate the liver response to the peroxisome proliferator perfluoroocatanoic acid. Mol Cell Biochem. 1997;169:143–147. doi: 10.1023/a:1006806820951. [DOI] [PubMed] [Google Scholar]
- Youssef J, Badr M. Biology of senescent liver peroxisomes: role in hepatocellular aging and disease. Environ Health Perspect. 1999;107:791–797. doi: 10.1289/ehp.99107791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef J, Badr M. Peroxisomal alterations in aging and disease. In: Mattson M, editor. Advances in Cell Aging and Gerontology Vol. 7, Interorganellar Signaling in Age-Related Disease. Amsterdam, the Netherlands: Elsevier Press; 2001. pp. 1–28. [Google Scholar]
- Yu J, Shen B, Chu ES, Teoh N, Cheung KF, Wu CW, et al. Inhibitory role of peroxisome proliferator-activated receptor gamma in hepatocarcinogenesis in mice and in vitro. Hepatology. 2010;51:2008–2019. doi: 10.1002/hep.23550. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhang H, Xing L. Influence of ciglitazone on A549 cells growth in vitro and in vivo and mechanism. J Huazhong Univ Sci Technol Med Sci. 2006;26:36–39. doi: 10.1007/BF02828033. [DOI] [PubMed] [Google Scholar]
- Zhang F, Lavan BE, Gregoire FM. Selective modulators of PPAR-gamma activity: molecular aspects related to obesity and side-effects. PPAR Res. 2007;2007:32696. doi: 10.1155/2007/32696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YM, Wen YH, Kang XY, Qian HH, Yang JM, Yin ZF. Troglitazone, a peroxisome proliferator-activated receptor gamma ligand, induces growth inhibition and apoptosis of HepG2 human liver cancer cells. World J Gastroenterol. 2008;14:2168–2173. doi: 10.3748/wjg.14.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]