

Abstract
BACKGROUND AND PURPOSE
Up-regulation of thioredoxin interacting protein (TXNIP), an endogenous inhibitor of thioredoxin (Trx), compromises cellular antioxidant and anti-apoptotic defences and stimulates pro-inflammatory cytokines expression, implying a role for TXNIP in apoptosis. Here we have examined the causal role of TXNIP expression in mediating retinal neurotoxicity and assessed the neuroprotective actions of verapamil, a calcium channel blocker and an inhibitor of TXNIP expression.
EXPERIMENTAL APPROACH
Retinal neurotoxicity was induced by intravitreal injection of NMDA in Sprague–Dawley rats, which received verapamil (10 mg·kg−1, p.o.) or vehicle. Neurotoxicity was examined by terminal dUTP nick-end labelling assay and ganglion cell count. Expression of TXNIP, apoptosis signal-regulating kinase 1 (ASK-1), NF-κB, p38 MAPK, JNK, cleaved poly-ADP-ribose polymerase (PARP), caspase-3, nitrotyrosine and 4-hydroxy-nonenal were examined by Western and slot-blot analysis. Release of TNF-α and IL-1β was examined by elisa.
KEY RESULTS
NMDA injection enhanced TXNIP expression, decreased Trx activity, causing increased oxidative stress, glial activation and release of TNF-α and IL-1β. Enhanced TXNIP expression disrupted Trx/ASK-1 inhibitory complex leading to release of ASK-1 and activation of the pro-apoptotic p38 MAPK/JNK pathway, as indicated by cleaved PARP and caspase-3 expression. Treatment with verapamil blocked these effects.
CONCLUSION AND IMPLICATIONS
Elevated TXNIP expression contributed to retinal neurotoxicity by three different mechanisms, inducing release of inflammatory mediators such as TNF-α and IL-1β, altering antioxidant status and disrupting the Trx-ASK-1 inhibitory complex leading to activation of the p38 MAPK/JNK apoptotic pathway. Targeting TXNIP expression is a potential therapeutic target for retinal neurodegenerative disease.
Keywords: Thioredoxin, TXNIP, verapamil, neuroprotection, peroxynitrite, retinal ganglion cells, apoptosis, NMDA, neurodegeneration, retina
Introduction
Retinal ganglion cell (RGC) death plays a critical role in the pathogenesis of many retinal neurodegenerative disorders, including models of glaucoma, diabetic retinopathy, traumatic optic neuropathy, uveitis and retinal ischaemia (El-Remessy et al., 2003; 2008; Zheng et al., 2007; Ali et al., 2008; 2011; Schmidt et al., 2008). Understanding the molecular mechanism of neuronal cell death in such retinal diseases is of great clinical significance for devising new treatments. We and others have demonstrated that the application of NMDA, an agonist at the major excitatory receptors in the CNS and retina, generated a reliable model of retinal neurotoxicity with which to screen neuroprotective agents (Lam et al., 1999; El-Remessy et al., 2003; Goebel and Winkler, 2006; Al-Gayyar et al., 2010; Bessero et al., 2010).
Thioredoxin interacting protein (TXNIP) is an endogenous inhibitor of the thioredoxin (Trx) system, a major cellular thiol-reducing and antioxidant system. Recently, it has been demonstrated that over-expression of TXNIP up-regulates the transcription factor NF-κB in retinal endothelial cells (Perrone et al., 2009). Under basal conditions, NF-κB is found in the cytosol bound to its inhibitor, but upon activation NF-κB is allowed to enter the nucleus, where it induces transcription of pro-inflammatory cytokines such as TNF-α and IL-1β (Rutledge and Adeli, 2007). In addition, previous studies demonstrated that over-expression of TXNIP can augment the inflammatory actions of TNF-α-mediated activation of p38MAPK and apoptosis in vascular endothelial cells (Yamawaki et al., 2005; Chen et al., 2008). We have previously shown that enhanced TXNIP expression was associated with increases in Müller cell activation in the NMDA model (Al-gayyar et al., 2010). As a consequence of Müller cell activation, retinal NF-κB expression and inflammation can be triggered in retinal neurodegenerative diseases (Laabich et al., 2002; Wang et al., 2002; Lebrun-Julien et al., 2009). However, the causal role and molecular events by which TXNIP causes retinal neuroinflammation and cell death have not been elucidated.
Protection from reactive oxygen species is mediated by many systems including the Trx system, which consists of Trx, TXNIP, Trx reductase and NADPH (Holmgren, 1995). TXNIP modulates the cellular redox state by inhibiting the Trx system leading to increased reactive oxygen species and oxidative stress (Junn et al., 2000). Trx is not only an antioxidant, but it can also regulate cell survival by binding and inhibiting the activity of apoptosis signal-regulating kinase 1 (ASK-1), a member of the MAP kinase kinase kinase family (Ichijo et al., 1997). As such, TXNIP plays a critical role in stress-induced cellular apoptosis as it binds reduced-Trx and inhibits its activity releasing free ASK-1, leading to activation of the p38 MAPK pathway and cell death (Ichijo et al., 1997; Nishiyama et al., 1999; Yamawaki et al., 2005; Chen et al., 2008). So far, the research exploring the causal role of TXNIP in mediating cell death has been hampered by the lack of a specific pharmacological inhibitor of TXNIP.
Because the expression of TXNIP is directly regulated by Ca2+ influx (Yamanaka et al., 2000), a recent study demonstrated that calcium channel blockers and verapamil in particular can effectively inhibit TXNIP expression in cardiomyocyte apoptosis in streptozotocin- and obesity-induced diabetic mice (Chen et al., 2009). Verapamil is an L-type calcium channel blocker from the phenylalkylamine class that has been used routinely in the treatment of hypertension, angina pectoris and cardiac arrhythmia (Yedinak, 1993). Here, we have assessed the neuroprotective effects of verapamil by reducing the expression of TXNIP. The study elucidates multiple pathways by which TXNIP can cause retinal inflammation and trigger RGC death by a mechanism involving induced release of the inflammatory mediators as TNF-α and IL-1β, altered antioxidant status and disrupted Trx-ASK-1 inhibitory complex leading to activation of p38 MAPK/JNK apoptotic pathway in a rat model, using intravitreal injection of NMDA.
Methods
Animals
All animal care and experimental procedures were in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the Charlie Norwood VA Medical Center Animal Care and Use Committee. Neurotoxicity was induced by intravitreal injection of NMDA, as previously described (El-Remessy et al., 2003; Al-Gayyar et al., 2010). Three sets of animals were prepared for a total of 36 male Sprague–Dawley rats (∼250 g body weight) from Harlan Laboratories (Indianapolis, IN, USA) that were divided into four groups: one group received a single intravitreal injection of NMDA, 40 nmol ≈ 5.9 µg dissolved in normal saline, Sigma-Aldrich, St. Louis, MO, USA); the second group received a single intravitreal injection of both NMDA (40 nmol) and an oral gavage of verapamil dissolved in distilled water (10 mg·kg−1, p.o.); the third group was intravitreally injected with N-methyl-L-aspartate (NMLA, 40 nmol ≈ 5.9 µg dissolved in normal saline, Sigma-Aldrich), which is the inactive isomer of NMDA and served as a control group; and the last group was intravitreally injected with NMLA (40 nmol) and received an oral gavage of verapamil (MP Biomedicals, Solon, OH, USA) (10 mg·kg−1, p.o.; ) to serve as treated control group. The animals were given a booster dose of verapamil 12 h after NMDA injection. The doses of NMDA and verapamil were selected based on previous studies (Bonhomme-Faivre et al., 2002; Al-Gayyar et al., 2010). Rats were killed 24 h after injection using a CO2 chamber and the eyes were enucleated and processed for further analyses.
Immunolocalization studies
Optimal cutting temperature (OCT)-frozen sections (Sakura Finetek USA Inc., Torrance, CA, USA) (15 µm) of eyes were fixed using 2% paraformaldehyde (PFA) (Electron Microscopy Sciences, Hatfield, PA, USA) in PBS and reacted with polyclonal anti-glial fibrillary acidic protein (GFAP) antibody (for glial activation), polyclonal anti-Iba1 is a marker of microglial cell activation (Santa Cruz Biotechnology, Santa Cruz, CA, USA), polyclonal IL-1β (R&D Systems, Minneapolis, MN, USA), monoclonal anti-NFκB p65 (BD Bioscience Pharmingen, San Diego, CA, USA) or monoclonal anti-TNF-α (Santa Cruz Biotechnology) overnight. All the primary antibodies were used in 1:200 dilutions in 1% BSA in 0.3% Triton X-100 in PBS. The sections were blocked using 5% goat serum dissolved in 1% BSA in 0.3% Triton X-100 in PBS. Then the sections were incubated with 1:500 dilutions of Texas-red or Oregon-green-conjugated goat anti-mouse or goat anti-rabbit antibodies (Invitrogen, Carlsbad, CA, USA) for 2 h. Images (n = 6 in each group) were collected using AxioObserver.Z1 Microscope (Carl Zeiss MicroImaging, Thornwood, NY, USA).
Quantitative real time PCR (qRT-PCR)
Retinal mRNA was prepared according to the manufacturer's instructions as described in our previous study (Ali et al., 2008). The One-Step qRT-PCR Invitrogen kit was used to amplify 10 ng retinal mRNA from each sample. PCR primers were designed to amplify Trx: 5'GCCAAAATGGTGAAGCTGAT3' and reverse primer 5'TGATCATTTTGCAAGGTCCA3'. TXNIP primers 5'AAGCTGTCCTCAGTCAGAGGCAAT3' and reverse primer 5'ATGACTTTCTTGGAGCCAGGGACA3'. Amplification of 18 S rRNA was used as an internal control. Quantitative PCR was performed using a Realplex Master cycler (Eppendorf North American Inc., Westbury, NY, USA). Trx and TXNIP expression was normalized to the 18S level in each sample and expressed as relative expression to control.
Western blot analysis and immuno-precipitation
Retinas were homogenized in radioimmunoprecipitation assay (RIPA) buffer (Millipore, Temecula, CA, USA) to examine the expression of various proteins as described previously (Ali et al., 2008; Al-Gayyar et al., 2010). The total amount of protein was determined by protein assay (Bio-Rad Laboratories Inc., Hercules, CA, USA). Samples (30 µg protein) were separated by SDS-PAGE and electroblotted to nitrocellulose membrane, and the membranes were blocked in PBS, 0.02% Tween 20 (PBST) containing 5% non-fat milk. Antibodies for Trx, TXNIP, ASK-1, TNF-α, JNK, phosphorylated-JNK (p-JNK; Santa Cruz Biotechnology), IL-1β (R&D Systems), p-p38, p38, cleaved caspase-3 (Cell Signaling, Danvers, MA, USA), cleaved poly-ADP-ribose polymerase (PARP) and NF-κB p65 (BD Bioscience Pharmingen) were used in 1:500 dilutions in PBST containing 5% non-fat milk and applied overnight at 4°C. Membranes were reprobed with 1:2000 β-actin (Sigma-Aldrich) in PBST containing 5% non-fat milk for 2 h to confirm equal loading. Primary antibody was detected by 1:5000 dilutions of horseradish peroxidase-conjugated sheep anti-mouse or anti-rabbit antibodies in PBST and enhanced-chemiluminescence (GE Healthcare, Piscataway, NJ, USA). For immuno-precipitation, 200 µg of retinal protein was diluted with RIPA buffer and incubated with 3 µg of anti-Trx antibody. Samples were rocked at 4°C overnight, followed by 40 µL agarose beads and rocked at 4°C for 4 h. The final extract was boiled, processed and analysed as described above. Membranes were probed with antibodies for ASK-1, TXNIP or Trx for equal loading. The band intensity was quantified using densitometry software (Alpha Innotech, San Leandro, CA, USA) and expressed as relative optical density (ROD).
Measurements of markers of oxidative and nitrosative stress
Slot-blot analysis was performed to quantify retinal levels of 4-hydroxy-nonenal (4-HNE) and nitrotyrosine as described previously (Ali et al., 2008; Abdelsaid et al., 2010). Samples of rat retinal homogenate (5 µg) were immobilized onto a nitrocellulose membrane. After blocking with PBST containing 5% non-fat milk, membranes were reacted with antibodies against nitrotyrosine (Calbiochem, San Diego, CA, USA) or 4-HNE (Alpha Diagnostics, San Antonio, TX, USA) using 1:500 dilutions in PBST containing 5% non-fat milk and applied overnight at 4°C. Primary antibody was detected by 1:5000 dilutions of horseradish peroxidase-conjugated sheep anti-rabbit antibodies in PBST and enhanced-chemiluminescence (GE Healthcare, Piscataway, NJ). The optical density of various samples was quantified using densitometry software (Alpha Innotech) and compared with that of controls and expressed as ROD.
Determination of levels of inflammatory mediators in rat vitreous
Rat vitreous concentration of TNF-α and IL-1β was determined using commercially available elisa kits (R&D Systems), following the manufacturer's procedure as described before (El-Remessy et al., 2008). Standards and samples were pipetted into the wells, and TNF-α or IL-1β present was bound by the immobilized antibody. After washing away any unbound substances, an enzyme-linked polyclonal antibody specific for TNF-α or IL-1β was added to the wells, followed by a substrate solution to develop colour in proportion to the amount of TNF-α and IL-1β bound in the initial step. The colour development was stopped and the intensity of the colour was measured at 450 nm. The concentrations of the samples were normalized to protein content and relative to the control.
Determination of Trx activity
Trx reductase activity was measured using a colorimetric assay kit (Sigma-Aldrich). Briefly, retinas were homogenized in a phosphate buffer (pH 7.4) and the lysate was used to test the Trx activity. Tissue Trx reductase was used to reduce 5,5'-dithiobis(2-nitrobenzoic) acid with NADPH to 5-thio-2-nitrobenzoic acid, which produces a strong yellow colour that can be measured at 412 nm. Because several enzymes present in biological samples can reduce the substrate, the enzyme activity was measured in the presence or absence of the Trx reductase inhibitor (provided by the manufacturer) and expressed as relative U/min/µg protein.
Evaluation of neuronal cell death in rat retina
Terminal dUTP nick-end labelling (TUNEL) was performed using the ApopTAG in situ apoptosis detection kit-fluorescein (Millipore, Billerica, MA, USA), following the manufacturer's directions. Briefly, OCT-frozen eye sections (15 µm) from each group were fixed using paraformaldehyde and ethanol:acetic acid (2:1). Then, the samples were incubated with terminal deoxynucleotidyl transferase enzyme followed by incubation with anti-digoxigenin conjugate. Propidium iodide (1 µg·mL−1) was added as a nuclear counter-stain, and cover slips were applied using Vectashield mounting medium for fluorescence (Vector Laboratories, Burlingame, CA, USA). Six rats from each group and six sections for each animal were used. Each section was systematically scanned and counted for positive green fluorescent cells in retinal layers indicating apoptosis. Images were obtained using an AxioObserver.Z1 Microscope (Zeiss) with 200 × magnification.
Counting number of neuronal cells in the ganglion cell layer (GCL)
OCT frozen retinal sections were stained with haematoxylin and eosin (H/E) for light microscopy. The nuclei in the GCL, not including nuclei in the vessels, were counted in four locations in the retina [both sides of the optic nerve (posterior) and mid-retina (central)] in a masked manner as described previously (Zheng et al., 2007). Four to six animals from each group and two fields for each location were used. Our previous study using the specific RGC marker, Brn-3a, showed that cells in the GCL include displaced amacrine cells (30–40%) in addition to RGC cells (Al-gayyar et al., 2010). Retinas were imaged by AxioObserver.Z1 Microscope (Zeiss).
Statistical analysis
The results were expressed as mean ± SEM. Differences among experimental groups were evaluated by anova followed by the Tukey–Kramer Multiple comparison test. Significance was defined as P < 0.05.
Results
Verapamil blocks TXNIP expression in NMDA-injected retinas
Because there is no direct pharmacological inhibitor of TXNIP but its expression is directly regulated by Ca2+ (Yamanaka et al., 2000), calcium channel blockers and verapamil in particular have been used effectively to inhibit TXNIP expression (Chen et al., 2009). In our experiments, real-time PCR analysis showed a 2.9-fold increase in the mRNA level of TXNIP in NMDA-injected rat retinas, compared with NMLA-controls (Figure 1A). In parallel, Western blot analysis of rat retinal lysates showed a 2.3-fold increase in TXNIP expression in NMDA-injected retinas compared with NMLA-controls (Figure 1B). Co-treatment with verapamil (10 mg·kg−1, p.o.) blocked TXNIP expression at protein and mRNA level in NMDA-treated rats, but not in those given NMLA.
Figure 1.
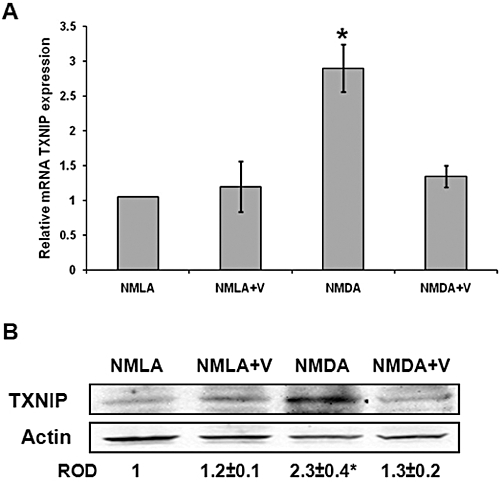
Verapamil blocks TXNIP expression in NMDA-injected retinas. (A) Real-time PCR analysis showed 2.9-fold increase in mRNA level of TXNIP in NMDA-injected rat retinas compared with NMLA-controls (n = 4–6). (B) Western blot analysis of rat retinal lysates showed 2.3-fold increase in TXNIP expression in NMDA-injected retinas, compared with NMLA-controls (n = 4–6). Treatment with verapamil (V; 10 mg·kg−1, p.o.) blocked TXNIP protein and mRNA level in NMDA injected retina but did not alter TXNIP in treated controls. Data expressed as fold value of control and represented as mean ± SEM. *P < 0.05, significantly different from the other groups. ROD, relative optical density.
Inhibiting TXNIP expression blocks activation of Muller and microglial cells and NF-κB expression
We examined the protective action of inhibiting TXNIP expression using verapamil on glial activation. As shown in Figure 2A, NMDA-injected rats showed a substantial increase in the intensity of GFAP immuno-reactivity in the filaments of Müller cells that extended from the nerve fibre layer and inner plexiform layer into the outer nuclear layer of retina, compared with NMLA-controls. In addition, intravitreal NMDA injection induced numerous Iba1-positive cells (activated microglial cells) that appeared hypertrophic or amoeboid, and were observed in the GCL, inner nuclear layer or frequently clustered around the perivascular region. Co-treatment of rats with verapamil (10 mg·kg−1, p.o.) blocked these effects in NMDA-injected rats but did not affect NMLA-controls. Next, we examined the effect of NMDA on the expression of NF-κB. Retinal lysate from NMDA-injected animals showed a 1.9-fold increase in NF-κB expression, compared with NMLA-controls (Figure 2B). The results of Western blot analysis was confirmed by immunohistochemistry. NMDA injection resulted in prominent immunolocalization of NF-κB in the GCL, inner nuclear layer and outer plexiform layer as compared with NMLA-controls (Figure 2B). Treatment of rats with verapamil (10 mg·kg−1, p.o.) blocked the increase in NF-κB in NMDA-injected rats, but did not affect NMLA-controls.
Figure 2.
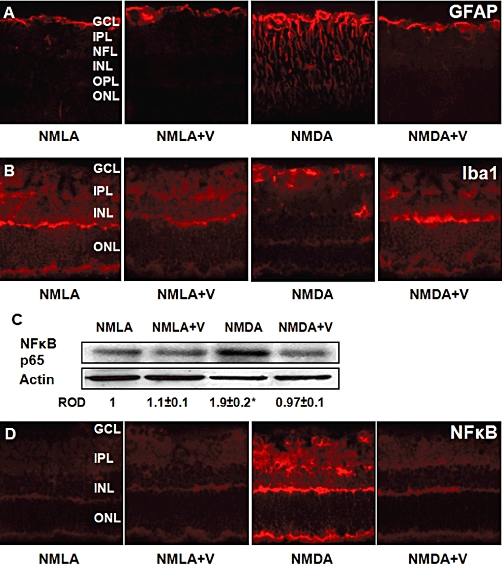
Inhibiting TXNIP expression blocks activation of Müller and microglial cells and NF-κB expression. (A) Representative images showing a substantial increase in the intensity of GFAP immuno-reactivity in the filaments of Müller cells that extended from the nerve fiber layer (NFL) and inner plexiform layer (IPL) into the outer nuclear layer (ONL) of retina, compared with NMLA-controls (400 × magnification). (B) Representative images showing microglial activation as indicated by numerous Iba1-positive cells that appeared hypertrophic or amoeboid in the ganglion cell layer (GCL) and inner nuclear layer (INL) in rats injected with NMDA, compared with NMLA-controls (400 × magnification). (C) Western blot analysis of rat retinal lysate showing a 1.9-fold increase in NF-κB p65 in NMDA-injected retinas, compared with NMLA-controls (n = 6). Data expressed as fold value of control and represented as mean ± SEM. (D) Representative images showing prominent immunolocalization of NF-κB in the GCL, INL and outer plexiform layer (OPL), compared with NMLA-controls (400 × magnification). Treatment of rats with verapamil (V; 10 mg·kg−1, p.o.) blocked these effects in NMDA-injected rats but did not affect NMLA-controls. *P < 0.05, significantly different from the other groups.. ROD, relative optical density.
Inhibiting TXNIP expression blocks release of inflammatory mediators
Western blot analysis showed 2.1- and 2.2-fold increases in TNF-α and IL-Iβ, respectively, in retinal lysate from NMDA-injected rats, compared with NMLA-controls (Figure 3A, B). In parallel, we measured the vitreous level of both TNF-α and IL-Iβ using elisa. NMDA-injected retinas showed 2.2- and 2.4-fold increases in TNF-α and IL-Iβ, respectively, as compared with NMLA-controls (Figure 3C). Treatment of rats with verapamil (10 mg·kg−1, p.o.) blocked the increase in inflammatory cytokines in NMDA-injected rats, but did not affect NMLA-controls.
Figure 3.
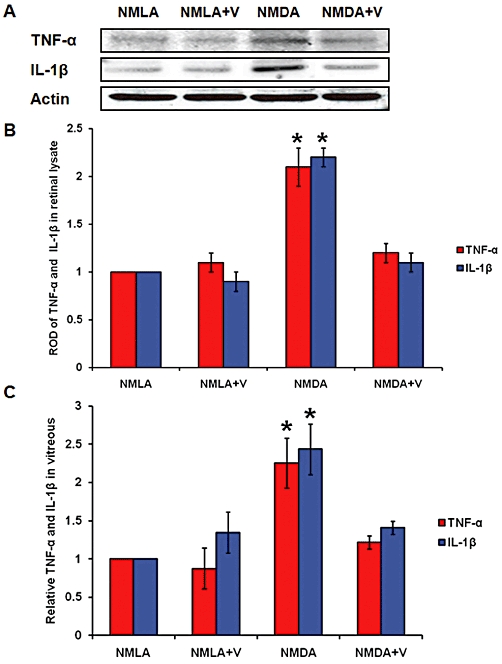
Inhibiting TXNIP expression blocks expression and release of inflammatory mediators. (A, B) Western blot analysis and statistical analysis of retinal lysate showed 2.1- and 2.2-fold increases in TNF-α and IL-Iβ, respectively, in NMDA-injected rats, compared with NMLA-controls (P < 0.05, n = 4–6). (C) Statistical analysis of the vitreous levels of TNF-α and IL-Iβ, using elisa. NMDA-injected retinas showed 2.2- and 2.4-fold increases in TNF-α and IL-Iβ, respectively, compared with NMLA-controls (n = 6–7). Treatment of rats with verapamil (V; 10 mg·kg−1, p.o.) blocked these effects in NMDA-injected rats but did not affect NMLA-controls. Data expressed as fold value of control and represented as mean ± SEM. *P < 0.05, significantly different from the other groups..; ROD, relative optical density.
Inhibiting TXNIP expression restors Trx activity and significantly reduces oxidative and nitrosative stress
In response to oxidative insult, cellular antioxidant defences can be up-regulated. Interestingly, NMDA injection caused about a two-fold increase in the Trx mRNA and a 2.6-fold increase in protein expression, compared with NMLA-control (Figure 4A, B). However, the Trx activity was reduced by 60% compared with NMLA-controls (Figure 4C). Treatment of the NMDA-injected rats with verapamil (10 mg·kg−1, p.o.) maintained high levels of Trx mRNA and protein expression, but restored the Trx activity of NMDA-injected rats to 80% of the NMLA-controls. Verapamil did not affect either Trx mRNA expression or Trx activity in NMLA-controls. In addition, slot-blot analysis of retinal lysate samples showed a 1.7-fold increase in 4-HNE adduct formation (a marker of oxidative stress) and a 1.6-fold increase in nitrotyrosine formation (a marker of peroxynitrite and nitrosative stress) in NMDA-injected retinas as compared with NMLA-controls (Figure 4D, E). Treatment of NMLA-injected rats with verapamil (10 mg·kg−1, p.o.) blocked these effects in NMDA-injected rats, but did not affect NMLA-controls.
Figure 4.
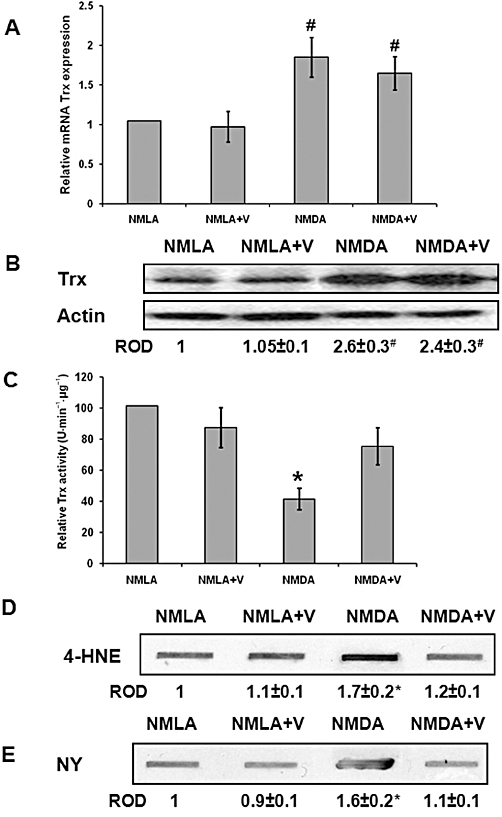
Inhibiting TXNIP expression restores Trx activity and significantly reduces oxidative and nitrosative stress. (A) Real-time PCR analysis showed about two-fold increase in mRNA level of Trx in NMDA-injected rat retinas, compared with NMLA-controls, that was not reversed by verapamil (V) treatment (n = 4–6, data expressed as percent value of control and represented as mean ± SEM). (B) Western blot analysis showed 2.6-fold increase in the expression of Trx protein in NMDA-injected retinas, compared with NMLA-controls that was not reversed by verapamil treatment (n = 4). (C) Statistical analysis of Trx activity, NMDA injection caused 60% reduction in Trx activity as compared with NMLA-controls (n = 4–6, Data expressed as percent value of control and represented as mean ± SEM). (D, E) Slot-blot analysis of retinal lysate samples showed a 1.7-fold increase in 4-HNE adduct formation (a marker of oxidative stress) and a 1.6-fold increase in nitrotyrosine formation (a marker of peroxynitrite) in NMDA-injected rats compared with NMLA-controls (n = 4–6, data expressed as fold value of control and represented as mean ± SEM). Treatment with verapamil (V; 10 mg·kg−1, p.o.) blocked these effects in NMDA-injected rats but did not affect NMLA-controls. *P < 0.05, significantly different from the other groups.. #P < 0.05, significantly different from the controls. ROD, relative optical density.
Inhibiting TXNIP expression preserves the TRX-ASK-1 ‘inhibitory complex’ and prevents release of ASK-1
Under resting conditions, Trx binds to and inhibits the apoptotic protein ASK-1 by forming the Trx-ASK-1 ‘inhibitory complex’. The interaction between Trx and ASK-1 was investigated using immunoprecipitation assays and the results showed that NMDA injection decreased the association between Trx and ASK-1 to only 40% of the basal level, compared with NMLA-controls (Figure 5A). Meanwhile, the association between Trx and TXNIP showed a 1.6-fold increase in NMDA-injected rats, compared with NMLA-controls (Figure 5B). In addition, NMDA injection caused a1.9-fold increase in ASK-1 expression in rat retina, compared with the NMLA-controls (Figure 5C). Co-treatment of animals with verapamil (10 mg·kg−1, p.o.) blocked these effects in NMDA-injected rats, but did not alter protein interactions in NMLA-controls.
Figure 5.
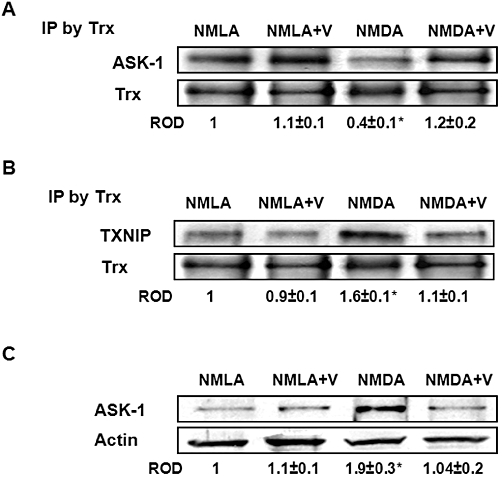
Inhibiting TXNIP expression preserves the TRX-ASK-1 ‘inhibitory complex’ and prevents release of ASK-1. (A) Immunoprecipitation (IP) of Trx and immunoblotting with ASK-1 showed 60% reduction in interaction between Trx and ASK-1 in NMDA-injected rats, compared with NMLA-controls (n = 5). (B) Immunoprecipitation of Trx and immunoblotting with TXNIP showed a 1.6-fold increase in interaction between Trx and TXNIP in NMDA-injected retinas as compared with NMLA-control (n = 5). (C) Western blot analysis showing NMDA injection caused a 1.9-fold increase in ASK-1 expression in rat retina as compared with the NMLA-controls (n = 4–5). Co-treatment of animals with verapamil (V; 10 mg·kg−1, p.o.) blocked these effects in NMDA-injected rats but did not alter protein interaction in NMLA-controls. Data expressed as fold value of control and represented as mean ± SEM. *P < 0.05, significantly different from the other groups.. ROD, relative optical density.
Inhibiting TXNIP expression blocks activation of p38 MAPK/JNK apoptotic pathway
Western blot analysis showed a 1.8-fold increase in p38 MAPK phosphorylation in NMDA-injected retinas, compared with NMLA-controls (Figure 6A). In addition, we found a 2.5-fold increase in phosphorylation of p-JNK-1 and p-JNK-2 as compared with NMLA-controls (Figure 6B). Apoptosis was further confirmed by the expression of cleaved caspase-3 and cleaved PARP, a marker of energy failure. NMDA-injected retinas showed 1.8- and 2.6-fold increases in cleaved PARP and cleaved caspase-3 expression, respectively, compared with NMLA-controls (Figure 6C, D). Co-treatment with verapamil (10 mg·kg−1, p.o.) significantly reduced phosphorylation of p38 MAPK and p-JNK, and blocked expression of cleaved PARP and cleaved caspase-3 in NMDA-injected animals but did not alter the basal levels in NMLA-controls (Figure 6).
Figure 6.
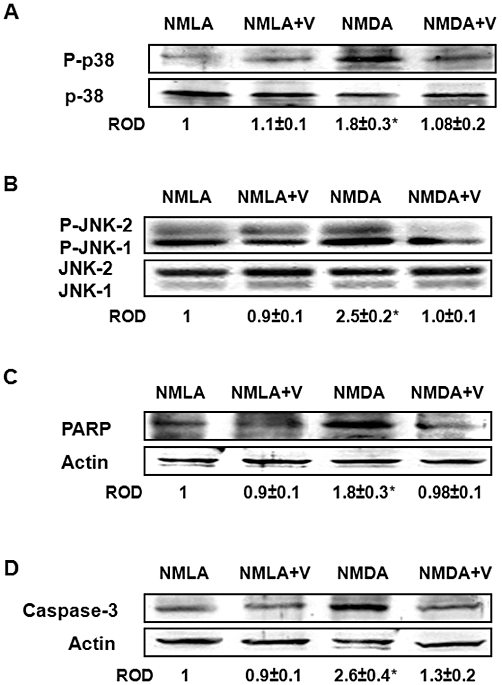
Inhibiting TXNIP expression blocks activation of p38 MAPK/JNK apoptotic pathway. (A) Western blot analysis showed a 1.8-fold increase in p38 MAPK phosphorylation in NMDA-injected retinas as compared with NMLA-controls (n = 4–5). (B) Western blot analysis showing a 2.5-fold increase in phosphorylation of p-JNK-1 and p-JNK-2 in NMDA-injected retinas, compared with NMLA-controls (n = 4–6). (C, D) Western blot analysis showing 1.8- and 2.6-fold increases in cleaved PARP and cleaved caspase-3 expression, respectively, in NMDA-injected retinas, compared with NMLA-controls (n = 4). Co-treatment with verapamil (V; 10 mg·kg−1, p.o.) significantly reduced phosphorylation of p38 MAPK and p-JNK and blocked expression of cleaved PARP and cleaved caspase-3 in NMDA-injected animals but did not alter the basal levels in NMLA-controls. Data expressed as fold value of control and represented as mean ± SEM. *P < 0.05, significantly different from the other groups. ROD, relative optical density.
Inhibiting TXNIP expression prevents death and loss of RGC
Intravitreal injection of NMDA induced severe RGC death as indicated by about 8-fold increase of TUNEL-labelled cells (arrows) mainly in RGC and the inner nuclear layer of the rat retina, compared with NMLA-controls. Co-treatment with verapamil (10 mg·kg−1, p.o.) blocked neuronal death in NMDA-injected animals (Figure 7A, B). In parallel, the protective effects of verapamil in NMDA-induced neuronal damage were assessed histologically by counting the number of neuronal cells in the GCL in retina sections stained with H/E (Figure 7C). Intravitreal NMDA injections resulted in the loss of 30% of cells in the GCL in the posterior and in the central regions of the retina. Co-treatment with verapamil (10 mg·kg−1, p.o.) prevented loss of neuronal cells and restored their number to the control levels. Treatment with verapamil did not affect NMLA-injected controls.
Figure 7.
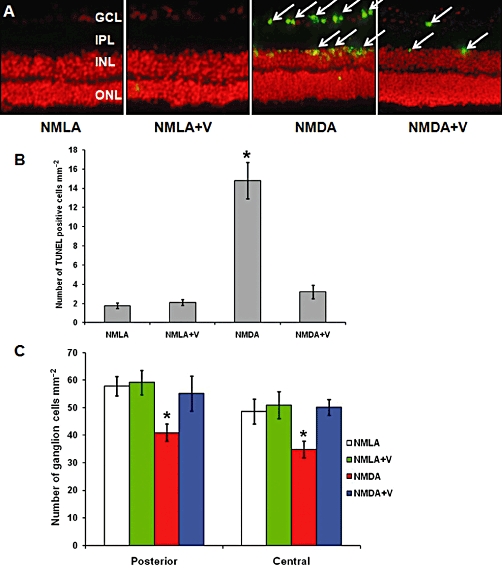
Inhibiting TXNIP expression prevents death and loss of RGC. (A, B) Representative images and statistical analysis showing that intravitreal injection of NMDA induced extensive RGC death as indicated by about 8-fold increase of TUNEL-labelled cells (arrows) mainly in RGC and inner nuclear layer (INL) of the rat retina compared with NMLA-controls (200 × magnification). (C) Statistical analysis of the number of neuronal cells in the ganglion cell layer (GCL) in retina sections stained with H/E. Intravitreal NMDA injections resulted in loss of 30% of cells in the GCL in the posterior and in the central regions of the retina. Co-treatment with verapamil (V) blocked these effects in NMDA-injected retinas but did not affect NMLA-injected controls. *P < 0.05, significantly different from the other groups..IPL, inner plexiform layer; ONL, outer nuclear layer; ROD, relative optical density.
Discussion
The main findings of the current study using the NMDA model were that elevated TXNIP expression contributed to retinal neurotoxicity by at least three pathways: (i) inducing retinal inflammation, indicated by enhanced expression of NF-κB and release of proinflammatory cytokines such as TNF-α and IL-1β; (ii) modulating antioxidant defence and increasing oxidative stress; and (iii) altering the inhibitory complex Trx/ASK-1 leading to release of ASK-1. Together, these pathways activated the apoptotic p38 MAPK/JNK pathway leading to RGC cell death. The proposed mechanism is outlined in Figure 8. The results of the current study indicated that TXNIP could be a therapeutic target for neuroprotection and revealed a novel neuroprotective role of verapamil. The clinical significance of these results is highlighted by the list of retinal neurodegenerative diseases, in which RGC death plays a critical role and which awaits development of effective neuroprotective agents. These diseases include glaucoma, diabetic retinopathy, traumatic optic neuropathy and retinal vein occlusion (see Osborne et al., 1999; Schmidt et al., 2008; Whitmire et al., 2011).
Figure 8.
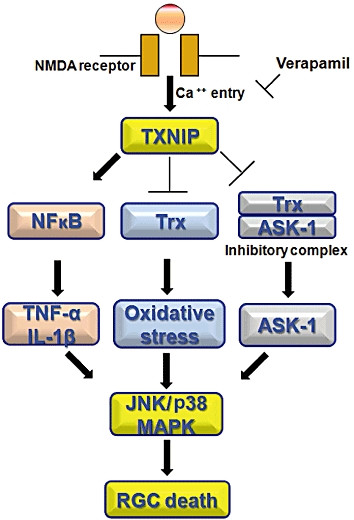
Schematic representation of the mechanism of action of verapamil in neuroprotection. Enhanced TXNIP expression contributes to retinal neurotoxicity by multiple pathways: (A) inducing retinal inflammation as indicated by enhanced expression of NF-κB and release of pro-inflammatory cytokines, such as TNF-α and IL-1β; (B) modulating antioxidant defence and increasing oxidative stress; and (C) altering the inhibitory complex Trx/ASK-1 leading to activation of ASK-1. Together, these pathways can activate the apoptotic p38 MAPK/JNK pathway leading to RGC cell death.
Retinal inflammation and glial activation have been demonstrated in human and experimental models of glaucoma, optic neuropathy and retinal ischaemia (Laabich et al., 2002; Wang et al., 2002; Nakazawa et al., 2007). NF-κB is one of the most ubiquitous transcription factors and functions as a central player in inflammatory disease. NMDA-induced retinal inflammation was evident by marked up-regulation of NF-κB p65 expression, which colocalized with the glio-filaments, GFAP, in retinal Müller cells. Our results were compatible with recent findings from other groups showing that over-expression of TXNIP stimulates NF-κB p65 expression and that it colocalizes with retinal Müller cells (Lebrun-Julien et al., 2009; Perrone et al., 2009). In parallel, microglial activation is a protective mechanism regulating tissue repair and recovery in the early phase of neurodegeneration (Streit, 2005). However, excessive or sustained activation of microglia often contributes to acute and chronic neuroinflammatory responses in the brain and the retina (Hanisch and Kettenmann, 2007). In agreement, NMDA-induced neurotoxicity was associated with the activation of microglial cells as indicated by numerous Iba1 positive cells (Figure 2B) that colocalized with NF-κB (data not shown). The increase in the activation of Müller and microglial cells were blocked by inhibiting TXNIP expression using verapamil, suggesting a potential role of TXNIP in mediating retinal inflammation. These results lend further support to earlier data from isolated retinal microglial cells showing that elevated hydrostatic pressure induced an influx of extracellular Ca2+ that preceded the activation of NF-κB (Sappington and Calkins, 2008). Several anti-inflammatory drugs have been shown to diminish neuroinflammation, but only few with direct functional effects on microglial activity have been elucidated (Tikka et al., 2002; Thomas and Kuhn, 2005). However, we believe that this is the first study that examined microglial activation in this NMDA model of neurotoxicity. Based on the evidence that NF-κB has a well-conserved cysteine residue, NF-κB activity is tightly linked with its redox regulation (Brodsky et al., 2010). Although, we did not assess the activity of NF-κB, our results showed that NMDA-enhanced p65 expression was associated with significant decreases in Trx activity supporting the notion that NF-κB is redox-regulated. NF-κB is activated by various stimuli including lipid peroxides (Arkan et al., 2005) and 4-HNE, a marker of oxidative stress (Malone and Hernandez, 2007; Shelton et al., 2007; 2009;). In support, our results showed a significant increase of the product of lipid peroxidation, 4-HNE, in NMDA-injected retinas, compared with controls.
Under basal conditions, NF-κB is present in the cytoplasm as an inactive heterotrimer consisting of the subunits p50, p65 and its inhibitor, Iκ−Bα, but upon activation and degradation of Ik-Bα, p50/p65 is allowed to enter the nucleus, where it induces transcription of pro-inflammatory genes such as plasminogen activator inhibitor-I, TNF-α, IL-6 and IL-Iβ (Rutledge and Adeli, 2007). In agreement, we found a greater than two-fold increase in expression and release of TNF-α and IL-Iβ in retina and vitreous samples from NMDA-injected rats. Similar upregulation of NF-κB p65 and IL-1β has been previously shown in a variety of retinal injuries, including ischaemia/reperfusion (Chen et al., 2003) and NMDA-neurotoxicity models (Kido et al., 2001; Kitaoka et al., 2007). Previous studies have shown also that increases in TNF-α can facilitate excitotoxic damage (Noh et al., 2005; Leonoudakis et al., 2008; Lebrun-Julien et al., 2009). The finding that inhibiting TXNIP expression using verapamil blocked the enhanced expression of NF-κB and release of TNF-α, IL-1β lend further support to previous studies showing that verapamil reduced the expression of vascular cell adhesion molecule-1 (Yamaguchi et al., 1997) and intercellular adhesion molecule-1 (Bukowska et al., 2008) along with production of TNF-α, in chronic lymphocytic leukaemia (Berrebi et al., 1994) and macrophages (Brown et al., 2004). However, to the best of our knowledge, this the first study that demonstrates a causal role of enhanced TXNIP expression and the anti-inflammatory effects of verapamil by modulating the expression of NF-κB, IL-1β and TNF-α in a neurotoxicity model.
Oxidative and nitrosative stress have been well documented in NMDA neurotoxicity models (El-Remessy et al., 2003; Nakajima et al., 2008; Inokuchi et al., 2009; Al-Gayyar et al., 2010). Although NMDA induced Trx expression (more than two-fold) possibly as a part of the host defence response, Trx activity was reduced to 40% of the basal level, suggesting a significant decrease in retinal antioxidant defence. Our results showed that verapamil restored Trx activity and reversed the increases in 4-HNE and nitrotyrosine back to normal levels. The neuroprotective effects of verapamil can be attributed to antioxidant effects either by restoring Trx activity and hence, restoring antioxidant defence, or by directly blocking oxidative stress. Previous reports have demonstrated that restoring Trx activity can prevent RGC death (Inomata et al., 2006; Munemasa et al., 2009) and that verapamil can block oxidative stress and ameliorate mitochondria dysfunction via blocking L-type calcium channels (Henrich et al., 2004; Schild et al., 2006).
A critical role of TXNIP in mediating stress-induced cellular apoptosis could be attributed to the fact that TXNIP binds reduced-Trx through its redox active site and inhibits its activity, thus releasing ASK-1 (Yamawaki et al., 2005; Chen et al., 2008; Al-Gayyar et al., 2010). Our analyses using immunoprecipitation assays showed a 1.6-fold increase of Trx association with TXNIP and ∼60% reduction with ASK-1, as well as increased ASK-1 expression. These effects were mitigated after inhibition of TXNIP expression with verapamil. We next evaluated the effects of enhanced TXNIP expression in triggering apoptosis in NMDA-injected rat retinas. Activation of the p38 MAPK/JNK pathway can be induced by several pathways including ASK-1, oxidative stress and proinflammatory cytokines (Lebrun-Julien et al., 2009; Munemasa et al., 2009). Indeed, our results showed significant increases in phosphorylation of JNK and p38 MAPK, as well as expression of cleaved PARP and caspase-3 that were associated with increased expression of TXNIP in NMDA-injected animals. In support, previous studies showed similar activation of the p38 MAPK/JNK apoptotic pathway in NMDA neurotoxicity model (Inomata et al., 2006; Inokuchi et al., 2009). Nevertheless, apoptosis is still a major cell death mechanism in this model based on the widespread TUNEL labelling observed by us and others (El-Remessy et al., 2003; Nakazawa et al., 2005; Barnett et al., 2009; Al-Gayyar et al., 2010; Ma et al., 2010). The neuroprotective effects of verapamil were evident by preventing activation of the apoptotic pathway, significant reduction of TUNEL-positive nuclei in GCL and restoration of neuronal cell count in GCL at both central and posterior retina back to the normal level.
In conclusion, the results of the current study support the potential role of TXNIP as a therapeutic target for neuroprotection in several ocular diseases. Increases in intraocular pressure (IOP) and subsequent loss of RGC have been documented in clinical and experimental models of glaucoma (Cone et al., 2010; Danesh-Meyer et al., 2010; Soto et al., 2011). Of note, this NMDA-model is a chemically induced RGC death model and does not induce increases in IOP. Interestingly, while chronic topical administration of verapamil can reduce IOP, systemic treatment with verapamil has little effect in lowering IOP (Liu et al., 1996; Mikheytseva et al., 2004; Shayegan et al., 2009). Here, we have demonstrated the systemic neuroprotective effects of verapamil via inhibiting TXNIP expression. We believe that our experimental results can be readily translated to clicncal use for several reasons. Verapamil has been used successfully for chronic cardiovascular diseases for many years. The drug was administered orally to rats at a dose that gives maximum plasma concentrations and total exposure (as area under the curve), comparable to those after oral administration of 80 mg in humans (Chung et al., 2009). The neuroprotective dose identified in the current study (80 mg·day−1) is significantly lower than the oral dose of verapamil commonly used in cardiovascular diseases (240–480 mg·day−1), and hence, alleviates the concern about the cardiovascular effects of verapamil use in patients without cardiovascular disease. However, the neuroprotective effects of verapamil in the current study were demonstrated using an acute model of neurotoxicity. The neuroprotective effects of verapamil in chronic optic neuropathy models warrant investigation.
Acknowledgments
This work was supported by Career Development Award 3–2008-149 from JDRF (ABE) and a Grant from Vision Discovery Institute (VDI), Georgia Health Science University (ABE).
Glossary
Abbreviations
- ASK-1
apoptosis signal-regulating kinase 1
- GCL
ganglion cell layer
- INL
inner nuclear layer
- IPL
inner plexiform layer
- NFL
nerve fiber layer
- NMLA
N-methyl-L-aspartate
- ONL
inner plexiform layer into the outer nuclear layer
- PARP
poly-ADP-ribose polymerase
- RGC
retinal ganglion cell
- ROD
relative optical density
- Trx
thioredoxin
- TUNEL
terminal dUTP nick-end labelling
- TXNIP
thioredoxin interacting protein
Conflicts of interest
None.
References
- Abdelsaid MA, Pillai BA, Matragoon S, Prakash R, Al-Shabrawey M, El-Remessy AB. Early intervention of tyrosine nitration prevents vaso-obliteration and neovascularization in ischemic retinopathy. J Pharmacol Exp Ther. 2010;332:125–134. doi: 10.1124/jpet.109.157941. [DOI] [PubMed] [Google Scholar]
- Al-Gayyar MMH, Abdelsaid MA, Matragoon S, Pillai BA, El-Remessy AB. Neuro and vascular protective effect of FeTPPs in NMDA-model: similarities to diabetes. Am J Pathol. 2010;177:1187–1197. doi: 10.2353/ajpath.2010.091289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali TK, Matragoon S, Pillai BA, Liou GI, El-Remessy AB. Peroxynitrite mediates retinal neurodegeneration by inhibiting NGF survival signal in experimental and human diabetes. J Diabetes. 2008;57:889–898. doi: 10.2337/db07-1669. [DOI] [PubMed] [Google Scholar]
- Ali TK, Al-Gayyar MMH, Matragoon S, Pillai BA, Abdelsaid MA, Nussbaum JJ, et al. Diabetes-induced peroxynitrite impairs the balance of ProNGF/NGF and causes neurovascular injury. Diabetologia. 2011;54:657–668. doi: 10.1007/s00125-010-1935-1. [DOI] [PubMed] [Google Scholar]
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Barnett EM, Zhang X, Maxwell D, Chang Q, Piwnica-Worms D. Single-cell imaging of retinal ganglion cell apoptosis with a cell-penetrating, activatable peptide probe in an in vivo glaucoma model. Proc Natl Acad Sci USA. 2009;106:9391–9396. doi: 10.1073/pnas.0812884106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrebi A, Shtalrid M, Klepfish A, Bassous L, Kushnir M, Shulman L, et al. Verapamil inhibits B-cell proliferation and tumor necrosis factor release and induces a clinical response in B-cell chronic lymphocytic leukemia. Leukemia. 1994;8:2214–2216. [PubMed] [Google Scholar]
- Bessero AC, Chiodini F, Rungger-Brandle E, Bonny C, Clarke PG. Role of the c-Jun N-terminal kinase pathway in retinal excitotoxicity, and neuroprotection by its inhibition. J Neurochem. 2010;113:1307–1318. doi: 10.1111/j.1471-4159.2010.06705.x. [DOI] [PubMed] [Google Scholar]
- Bonhomme-Faivre L, Forestier F, Auchere D, Soursac M, Orbach-Arbouys S, Farinotti R. Chronic administration of verapamil, ketoconazole and carbamazepine: impact on immunological parameters. Int J Pharm. 2002;238:133–137. doi: 10.1016/s0378-5173(02)00056-x. [DOI] [PubMed] [Google Scholar]
- Brodsky M, Halpert G, Albeck M, Sredni B. The anti-inflammatory effects of the tellurium redox modulating compound, AS101, are associated with regulation of NFkappaB signaling pathway and nitric oxide induction in macrophages. J Inflamm (Lond) 2010;7:3. doi: 10.1186/1476-9255-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DM, Donaldson K, Borm PJ, Schins RP, Dehnhardt M, Gilmour P, et al. Calcium and ROS-mediated activation of transcription factors and TNF-alpha cytokine gene expression in macrophages exposed to ultrafine particles. Am J Physiol Lung Cell Mol Physiol. 2004;286:L344–L353. doi: 10.1152/ajplung.00139.2003. [DOI] [PubMed] [Google Scholar]
- Bukowska A, Schild L, Keilhoff G, Hirte D, Neumann M, Gardemann A, et al. Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp Biol Med (Maywood) 2008;233:558–574. doi: 10.3181/0706-RM-155. [DOI] [PubMed] [Google Scholar]
- Chen CL, Lin CF, Chang WT, Huang WC, Teng CF, Lin YS. Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood. 2008;111:4365–4374. doi: 10.1182/blood-2007-08-106336. [DOI] [PubMed] [Google Scholar]
- Chen J, Cha-Molstad H, Szabo A, Shalev A. Diabetes induces and calcium channel blockers prevent cardiac expression of proapoptotic thioredoxin-interacting protein. Am J Physiol Endocrinol Metab. 2009;296:E1133–E1139. doi: 10.1152/ajpendo.90944.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Zhang C, Chiang SK, Wu T, Tso MO. Increased nuclear factor-kappa B p65 immunoreactivity following retinal ischemia and reperfusion injury in mice. J Neurosci Res. 2003;72:125–131. doi: 10.1002/jnr.10548. [DOI] [PubMed] [Google Scholar]
- Chung JH, Choi DH, Choi JS. Effects of oral epigallocatechin gallate on the oral pharmacokinetics of verapamil in rats. Biopharm Drug Dispos. 2009;30:90–93. doi: 10.1002/bdd.644. [DOI] [PubMed] [Google Scholar]
- Cone FE, Gelman SE, Son JL, Pease ME, Quigley HA. Differential susceptibility to experimental glaucoma among 3 mouse strains using bead and viscoelastic injection. Exp Eye Res. 2010;91:415–424. doi: 10.1016/j.exer.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danesh-Meyer HV, Boland MV, Savino PJ, Miller NR, Subramanian PS, Girkin CA, et al. Optic disc morphology in open-angle glaucoma compared with anterior ischemic optic neuropathies. Invest Ophthalmol Vis Sci. 2010;51:2003–2010. doi: 10.1167/iovs.09-3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Remessy AB, Khalil IE, Matragoon S, Abou-Mohamed G, Tsai NJ, Roon P, et al. Neuroprotective effect of (-)Delta9-tetrahydrocannabinol and cannabidiol in N-methyl-D-aspartate-induced retinal neurotoxicity: involvement of peroxynitrite. Am J Pathol. 2003;163:1997–2008. doi: 10.1016/s0002-9440(10)63558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Remessy AB, Tang Y, Zhu G, Matragoon S, Khalifa Y, Liu EK, et al. Neuroprotective effects of cannabidiol in endotoxin-induced uveitis: critical role of p38 MAPK activation. Mol Vis. 2008;14:2190–2203. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Goebel DJ, Winkler BS. Blockade of PARP activity attenuates poly(ADP-ribosyl)ation but offers only partial neuroprotection against NMDA-induced cell death in the rat retina. J Neurochem. 2006;98:1732–1745. doi: 10.1111/j.1471-4159.2006.04065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Henrich M, Paddenberg R, Haberberger RV, Scholz A, Gruss M, Hempelmann G, et al. Hypoxic increase in nitric oxide generation of rat sensory neurons requires activation of mitochondrial complex II and voltage-gated calcium channels. Neuroscience. 2004;128:337–345. doi: 10.1016/j.neuroscience.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Holmgren A. Thioredoxin structure and mechanism: conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure. 1995;3:239–243. doi: 10.1016/s0969-2126(01)00153-8. [DOI] [PubMed] [Google Scholar]
- Ichijo H, Nishida E, Irie K, Ten Dijke P, Saitoh M, Moriguchi T, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Inokuchi Y, Imai S, Nakajima Y, Shimazawa M, Aihara M, Araie M, et al. Edaravone, a free radical scavenger, protects against retinal damage in vitro and in vivo. J Pharmacol Exp Ther. 2009;329:687–698. doi: 10.1124/jpet.108.148676. [DOI] [PubMed] [Google Scholar]
- Inomata Y, Nakamura H, Tanito M, Teratani A, Kawaji T, Kondo N, et al. Thioredoxin inhibits NMDA-induced neurotoxicity in the rat retina. J Neurochem. 2006;98:372–385. doi: 10.1111/j.1471-4159.2006.03871.x. [DOI] [PubMed] [Google Scholar]
- Junn E, Han SH, Im JY, Yang Y, Cho EW, Um HD, et al. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol. 2000;164:6287–6295. doi: 10.4049/jimmunol.164.12.6287. [DOI] [PubMed] [Google Scholar]
- Kido N, Inatani M, Honjo M, Yoneda S, Hara H, Miyawaki N, et al. Dual effects of interleukin-1beta on N-methyl-D-aspartate-induced retinal neuronal death in rat eyes. Brain Res. 2001;910:153–162. doi: 10.1016/s0006-8993(01)02706-8. [DOI] [PubMed] [Google Scholar]
- Kitaoka Y, Munemasa Y, Nakazawa T, Ueno S. NMDA-induced interleukin-1beta expression is mediated by nuclear factor-kappa B p65 in the retina. Brain Res. 2007;1142:247–255. doi: 10.1016/j.brainres.2007.01.097. [DOI] [PubMed] [Google Scholar]
- Laabich A, Li G, Cooper NG. Enhanced expression of TNF-R1 protein in NMDA-mediated cell death in the retina. Brain Res Mol Brain Res. 2002;109:239–246. doi: 10.1016/s0169-328x(02)00553-3. [DOI] [PubMed] [Google Scholar]
- Lam TT, Abler AS, Kwong JMK, Tso MOM. N-Methyl-D-Aspartate (NMDA)-Induced Apoptosis in Rat Retina. Invest Ophthalmol Vis Sci. 1999;40:2391–2397. [PubMed] [Google Scholar]
- Lebrun-Julien F, Duplan L, Pernet V, Osswald I, Sapieha P, Bourgeois P, et al. Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J Neurosci. 2009;29:5536–5545. doi: 10.1523/JNEUROSCI.0831-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. 2008;28:2119–2130. doi: 10.1523/JNEUROSCI.5159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Araujo SV, Spaeth GL, Katz LJ, Smith M. Lack of effect of calcium channel blockers on open-angle glaucoma. J Glaucoma. 1996;5:187–190. [PubMed] [Google Scholar]
- Ma J, Yu W, Wang Y, Cao G, Cai S, Chen X, et al. Neuroprotective effects of C-type natriuretic peptide on rat retinal ganglion cells. Invest Ophthalmol Vis Sci. 2010;51:3544–3553. doi: 10.1167/iovs.09-5049. [DOI] [PubMed] [Google Scholar]
- Malone PE, Hernandez MR. 4-Hydroxynonenal, a product of oxidative stress, leads to an antioxidant response in optic nerve head astrocytes. Exp Eye Res. 2007;84:444–454. doi: 10.1016/j.exer.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheytseva IN, Kashintseva LT, Krizhanovsky GN, Kopp OP, Lipovetskaya EM. The influence of the calcium channel blocker verapamil on experimental glaucoma. Int Ophthalmol. 2004;25:75–79. doi: 10.1023/b:inte.0000031737.08988.b0. [DOI] [PubMed] [Google Scholar]
- Munemasa Y, Ahn JH, Kwong JM, Caprioli J, Piri N. Redox proteins thioredoxin 1 and thioredoxin 2 support retinal ganglion cell survival in experimental glaucoma. Gene Ther. 2009;16:17–25. doi: 10.1038/gt.2008.126. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Inokuchi Y, Nishi M, Shimazawa M, Otsubo K, Hara H. Coenzyme Q10 protects retinal cells against oxidative stress in vitro and in vivo. Brain Res. 2008;1226:226–233. doi: 10.1016/j.brainres.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Shimura M, Endo S, Takahashi H, Mori N, Tamai M. N-Methyl-D-Aspartic acid suppresses Akt activity through protein phosphatase in retinal ganglion cells. Mol Vis. 2005;11:1173–1182. [PubMed] [Google Scholar]
- Nakazawa T, Takahashi H, Nishijima K, Shimura M, Fuse N, Tamai M, et al. Pitavastatin prevents NMDA-induced retinal ganglion cell death by suppressing leukocyte recruitment. J Neurochem. 2007;100:1018–1031. doi: 10.1111/j.1471-4159.2006.04274.x. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999;274:21645–21650. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc Natl Acad Sci USA. 2005;102:12230–12235. doi: 10.1073/pnas.0505408102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne NN, Ugarte M, Chao M, Chidlow G, Bae JH, Wood JP, et al. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv Ophthalmol. 1999;43(Suppl. 1):S102–S128. doi: 10.1016/s0039-6257(99)00044-2. [DOI] [PubMed] [Google Scholar]
- Perrone L, Devi TS, Hosoya K, Terasaki T, Singh LP. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol. 2009;221:262–272. doi: 10.1002/jcp.21852. [DOI] [PubMed] [Google Scholar]
- Rutledge AC, Adeli K. Fructose and the metabolic syndrome: pathophysiology and molecular mechanisms. Nutr Rev. 2007;65:S13–S23. doi: 10.1111/j.1753-4887.2007.tb00322.x. [DOI] [PubMed] [Google Scholar]
- Sappington RM, Calkins DJ. Contribution of TRPV1 to microglia-derived IL-6 and NFkappaB translocation with elevated hydrostatic pressure. Invest Ophthalmol Vis Sci. 2008;49:3004–3017. doi: 10.1167/iovs.07-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild L, Bukowska A, Gardemann A, Polczyk P, Keilhoff G, Tager M, et al. Rapid pacing of embryoid bodies impairs mitochondrial ATP synthesis by a calcium-dependent mechanism–a model of in vitro differentiated cardiomyocytes to study molecular effects of tachycardia. Biochim Biophys Acta. 2006;1762:608–615. doi: 10.1016/j.bbadis.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt KG, Bergert H, Funk RH. Neurodegenerative diseases of the retina and potential for protection and recovery. Curr Neuropharmacol. 2008;6:164–178. doi: 10.2174/157015908784533851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayegan MR, Boloorian AA, Kianoush S. Comparative study of topical application of timolol and verapamil in patients with glaucoma within 6 months. J Ocul Pharmacol Ther. 2009;25:551–553. doi: 10.1089/jop.2009.0005. [DOI] [PubMed] [Google Scholar]
- Shelton MD, Kern TS, Mieyal JJ. Glutaredoxin regulates nuclear factor kappa-B and intercellular adhesion molecule in Muller cells: model of diabetic retinopathy. J Biol Chem. 2007;282:12467–12474. doi: 10.1074/jbc.M610863200. [DOI] [PubMed] [Google Scholar]
- Shelton MD, Distler AM, Kern TS, Mieyal JJ. Glutaredoxin regulates autocrine and paracrine proinflammatory responses in retinal glial (muller) cells. J Biol Chem. 2009;284:4760–4766. doi: 10.1074/jbc.M805464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto I, Pease ME, Son JL, Shi X, Quigley HA, Marsh-Armstrong N. Retinal ganglion cell loss in a rat ocular hypertension model is sectorial and involves early optic nerve axon loss. Invest Ophthalmol Vis Sci. 2011;52:434–441. doi: 10.1167/iovs.10-5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Brain Res Rev. 2005;48:234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Kuhn DM. MK-801 and dextromethorphan block microglial activation and protect against methamphetamine-induced neurotoxicity. Brain Res. 2005;1050:190–198. doi: 10.1016/j.brainres.2005.05.049. [DOI] [PubMed] [Google Scholar]
- Tikka TM, Vartiainen NE, Goldsteins G, Oja SS, Andersen PM, Marklund SL, et al. Minocycline prevents neurotoxicity induced by cerebrospinal fluid from patients with motor neurone disease. Brain. 2002;125:722–731. doi: 10.1093/brain/awf068. [DOI] [PubMed] [Google Scholar]
- Wang L, Cioffi GA, Cull G, Dong J, Fortune B. Immunohistologic evidence for retinal glial cell changes in human glaucoma. Invest Ophthalmol Vis Sci. 2002;43:1088–1094. [PubMed] [Google Scholar]
- Whitmire W, Al-Gayyar MMH, Abdelsaid MA, Yousufzai BK, El-Remessy AB. Alteration of growth factors and neuronal death in diabetic retinopathy: what we have learned so far. Mol Vis. 2011;17:300–308. [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Suwa H, Miyasaka M, Kumada K. Selective inhibition of vascular cell adhesion molecule-1 expression by verapamil in human vascular endothelial cells. Transplantation. 1997;63:759–764. doi: 10.1097/00007890-199703150-00024. [DOI] [PubMed] [Google Scholar]
- Yamanaka H, Maehira F, Oshiro M, Asato T, Yanagawa Y, Takei H, et al. A possible interaction of thioredoxin with VDUP1 in HeLa cells detected in a yeast two-hybrid system. Biochem Biophys Res Commun. 2000;271:796–800. doi: 10.1006/bbrc.2000.2699. [DOI] [PubMed] [Google Scholar]
- Yamawaki H, Pan S, Lee RT, Berk BC. Fluid shear stress inhibits vascular inflammation by decreasing thioredoxin-interacting protein in endothelial cells. J Clin Invest. 2005;115:733–738. doi: 10.1172/JCI200523001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedinak KC. Use of calcium channel antagonists for cardiovascular disease. Am Pharm. 1993;NS33:49–64. doi: 10.1016/s0160-3450(15)30720-0. quiz 64-6. [DOI] [PubMed] [Google Scholar]
- Zheng L, Gong B, Hatala DA, Kern TS. Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest Ophthalmol Vis Sci. 2007;48:361–367. doi: 10.1167/iovs.06-0510. [DOI] [PubMed] [Google Scholar]